

Abstract
The synthesis of glycogen represents a key pathway for the disposal of excess glucose while its degradation is crucial for providing energy during exercise and times of need. The importance of glycogen metabolism is also highlighted by human genetic disorders that are caused by mutations in the enzymes involved. In this review, we provide a basic summary on glycogen metabolism and some of the clinical aspects of the classical Glycogen Storage Diseases. Disruptions in glycogen metabolism usually result in some level of dysfunction in the liver, muscle, heart, kidney and/or brain. Furthermore, the spectrum of symptoms observed is very broad, depending on the affected enzyme. Finally, we briefly discuss an aspect of glycogen metabolism related to the maintenance of its structure that seems to be gaining more recent attention. For example, in Lafora progressive myoclonus epilepsy, patients exhibit an accumulation of inclusion bodies in several tissues, containing glycogen with increased phosphorylation, longer chain lengths and irregular branch points. This abnormal structure is thought to make glycogen insoluble and resistant to degradation. Consequently, its accumulation becomes toxic to neurons, leading to cell death. Although the genes responsible have been identified, studies in the past two decades are only beginning to shed light into their molecular functions.
Glycogen is a branched polysaccharide consisting of glucose units found primarily in animals, fungi, and bacteria (Adeva-Andany, et al. 2016). Over a century of research on this macromolecule has led to many accomplishments. From the Glycogen Storage Diseases, congenital disorders arising from mutations in enzymes controlling glycogen metabolism, we have obtained a clear picture in the cellular pathways involved. Along the way, this led to three Nobel prizes in physiology. The first was in 1947, awarded to Carl Ferdinand Cori and Gerty Theresa Cori, for their work on the catalytic conversion of glycogen (Cori, et al. 1937; Cori and Cori 1929; Cori, et al. 1939). The second was awarded to Earl Wilbur Sutherland in 1971 for demonstrating how epinephrine initiates a signal cascade to trigger glycogen breakdown by the enzyme glycogen phosphorylase (Berthet, et al. 1957; Rall, et al. 1956; Sutherland and Wosilait 1955, 1956; Wosilait and Sutherland 1956). Finally, Edmond Henri Fischer and Edwin Gerhard Krebs were awarded the prize in 1992 for demonstrating how reversible protein phosphorylation could control the activity of glycogen phosphorylase (Fischer and Krebs 1955; Krebs, et al. 1958).
During the past 20 years, we have continued to broaden our understanding of the regulation of glycogen metabolism, as well as its importance to human disease. This review will attempt to touch upon both the clinical as well as the basic biochemical aspects of glycogen metabolism in normal and diseased states and provide a glimpse into what future studies may hold.
The biochemistry at a glance
In humans, glycogen is the main storage form of glucose and the primary means of non-oxidative glucose disposal into muscle and liver tissues (Shulman and Rothman 2001), although significant amounts are also found elsewhere, such as the brain and kidney (Adeva-Andany et al. 2016). During times of need, glycogen is rapidly broken down to produce glucose. In the muscle, this serves as an immediate and important source of energy for exercise during the first 30 minutes (Mul, et al. 2015). In the liver, the breakdown of glycogen during fasting conditions contributes to hepatic glucose production that is crucial for maintaining blood glucose levels, to support the needs of other tissues (Petersen, et al. 2017). Although glycogen is mobilized during fasting in both the liver and muscle, the latter tissue lacks key metabolic enzymes to allow it to transport glucose back into the bloodstream (briefly discussed below).
Glycogen synthesis and degradation are highly regulated multi-step processes involving distinct sets of enzymatic reactions (Adeva-Andany et al. 2016). It is generally believed that for glycogen synthesis to occur, an oligosaccharide primer is required that is first produced through the self-glucosylation of the protein glycogenin (Roach and Skurat 1997). This process requires the nucleotide sugar, uridine diphosphate glucose (UDP-glucose), as the donor substrate (Figure 1A). Up to seven glucose units can be added to glycogenin, and this is thought to be critical because the key enzyme that synthesizes glycogen, namely glycogen synthase (GS), can only extend an existing glucose chain.
Figure 1.
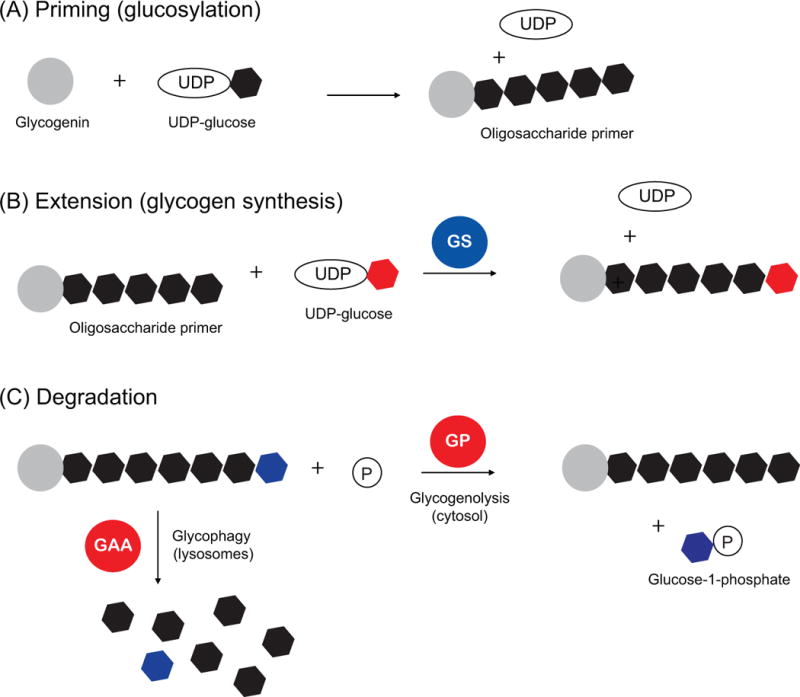
Schematic of the enzymes involved in the priming, synthesis and breakdown of glycogen. Hexagons denote glucose monomers, with various colors added for clarity. Glycogen degradation occurs via distinct mechanisms in the cytosol (glycogenolysis) and lysosomes (glycophagy). Abbreviations: glycogen synthase (GS), glycogen phosphorylase (GP), acid alpha glucosidase (GAA).
GS catalyzes the addition of glucose units (via UDP-glucose) to an existing glucose chain through α(1→4) glycosidic bonds (Figure 1B). The activity of GS is considered to be the rate-limiting step in glycogen synthesis, and GS is known to be tightly controlled in multiple ways, including phosphorylation, allosteric activation and subcellular localization (Greenberg, et al. 2006).
On the other side, the breakdown of glycogen occurs through two distinct pathways, one occurring in the cytosol (herein referred to as “glycogenolysis”), and one in the lysosomes (sometimes referred to as “glycogen autophagy” or “glycophagy” in the field). Glycogenolysis is carried out by the enzyme glycogen phosphorylase (GP), which catalyzes the release of glucose-1-phosphate (G1P) from the ends of glycogen branches (Figure 1C) (Johnson 1992). As with GS, the activity of GP is also regulated allosterically and by phosphorylation. The G1P produced can then be converted to glucose-6-phosphate (G6P) and funneled through glycolysis and other metabolic pathways (Agius, et al. 2002).
During glycolysis, G6P is ultimately converted to pyruvate which acts as a key metabolic intermediate for other pathways (Figure 2). For example, the conversion of pyruvate to acetyl-coA generates an important substrate for the tricarboxylic acid (TCA) cycle and fatty acid synthesis. Under certain conditions, the muscle can convert pyruvate to lactate (during anaerobic glycolysis) or alanine (during muscle breakdown) which are transported via the bloodstream to the liver, where they are then converted back to pyruvate (Cori cycle and Alanine cycle). In the liver, pyruvate can also be converted back to glucose via gluconeogenesis. This pathway is similar to the reverse direction of glycolysis but requires unique enzymes to bypass a few endergonic reactions. For example, in the liver and kidney, the unique presence of the enzyme glucose-6-phosphatase (G6Pase) converts G6P to glucose, allowing it to be released into the bloodstream to support other tissues (Petersen et al. 2017). This step (Step 3 of gluconeogenesis in Figure 2) is crucial as the conversion of glucose to G6P by the enzyme hexokinase is energetically unfavorable in the reverse direction.
Figure 2.
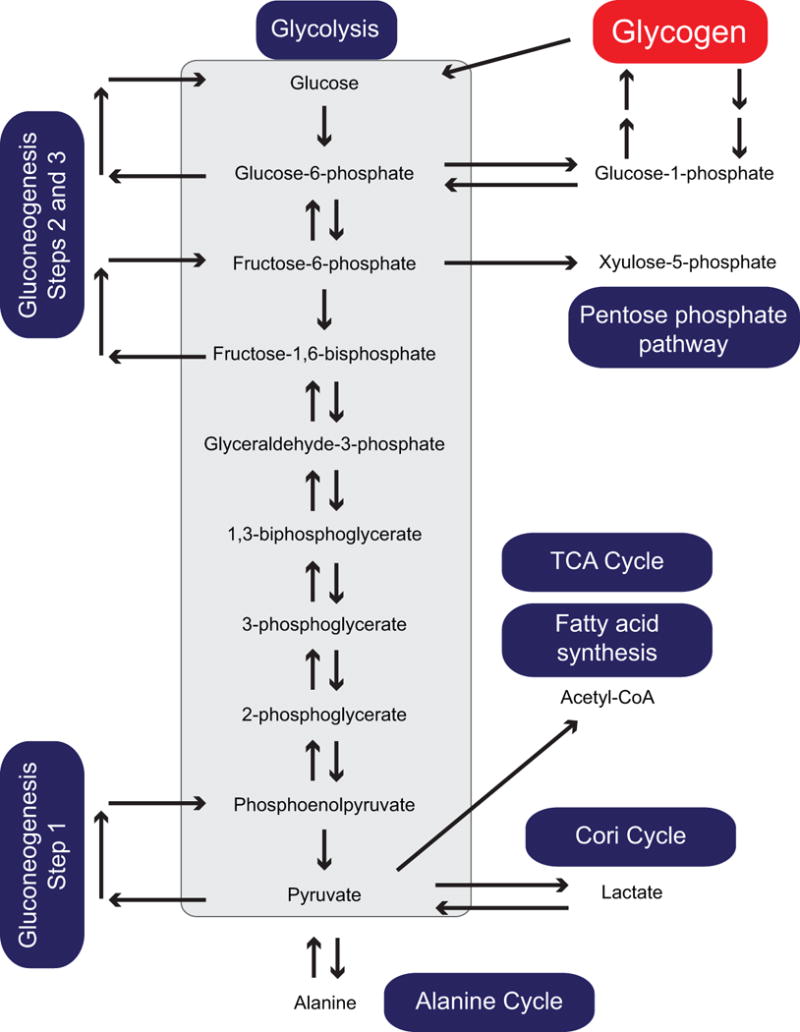
Schematic of the pathways linked to glycogen metabolism. Glycogen breakdown produces glucose-1-phosphate (via glycogenolysis) and glucose (via glycophagy and debranching enzyme activity). Both products enter into the glycolytic pathway giving rise to pyruvate which acts as a key precursor for the TCA cycle, fatty acid synthesis and gluconeogenesis. The interconversion of pyruvate to lactate and alanine further integrate the metabolism of the liver and muscle tissues. Additionally, fructose-6-phosphate generated in glycolysis can also shunt to the pentose phosphate pathway for nucleotide synthesis.
The second pathway for glycogen degradation requires the enzyme acid α-glucosidase (GAA) (Lim, et al. 2014). While initially assembled in the endoplasmic reticulum (ER), GAA is transported to lysosomes via the Golgi to degrade lysosomal glycogen via an autophagy dependent pathway, also known as glycophagy (Kaur and Debnath 2015). Although studies in Drosophila suggest glycophagy and glycogenolysis can compensate for each other, both pathways seem to be required for maximal glycogen degradation (Zirin, et al. 2013).
The significance of having two distinct pathways for the degradation of glycogen is not entirely clear. The majority of the studies have focused on the regulation of GP, especially in the context of hormonal regulation (e.g. insulin, glucagon, adrenaline) in response to feeding status and exercise. In contrast, much less is known about glycophagy, although one leading theory suggests that it plays a critical role for neonatal survival (Schiaffino, et al. 2008). At birth, animals have shown the presence of many glycogen containing autophagosomes, which are quickly mobilized within the first few hours. Thus, glycophagy may provide an immediate source of energy for the functioning of key tissues in the newborn, such as the heart and diaphragm muscles.
In addition to the above processes, glycogen is a branched polysaccharide, and requires the branching enzyme, GBE1, to generate regularly spaced branch points (every 10-20 residues) through α(1→6) glycosidic bonds (Figure 3A) (Adeva-Andany et al. 2016). The obvious advantage of glycogen branching is to provide multiple sites for glycogenolysis to occur in a more rapid fashion. However, glycogen branching is also important for increasing the solubility and decreasing the osmotic impact of the molecule.
Figure 3.
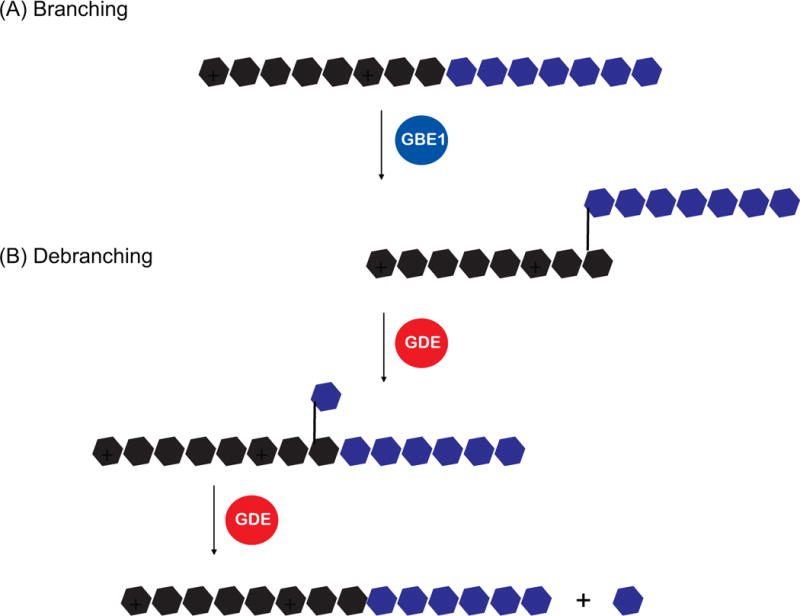
Schematic of the enzymes involved in the branching and debranching of glycogen. Hexagons denote glucose monomers, with various colors added for clarity. For simplicity, glycogenin has been omitted in this figure. Abbreviations: glycogen branching enzyme 1 (GBE1), glycogen debranching enzyme (GDE).
An additional enzyme that needs to be introduced is the glycogen debranching enzyme (GDE) (Adeva-Andany et al. 2016). Although GP is the key enzyme in glycogenolysis, it is sterically hindered when it reaches a branch point that is four glucose residues away. At this point, debranching occurs through the two catalytic activities of GDE (Figure 3B). First, one of the branches is transferred onto the other chain, but leaves a single glucose unit at the branch point. This is then removed to produce a single free glucose moiety. In contrast to the activities of GS and GP, little is known concerning the regulation of branching/debranching activities.
Classical Glycogen Storage Diseases (GSDs)
It is well established that glycogen metabolism plays an important role in exercise and blood glucose regulation. In the fed state, insulin stimulates glycogen storage in muscle and liver by simultaneously promoting glycogen synthesis and inhibiting glycogen breakdown. Conversely, in the fasted state, or during exercise, glucagon and catecholamines promote glycogen breakdown while inhibiting glycogen synthesis. The importance of glycogen is further highlighted by the fact that there are several congenital disorders caused by abnormalities in the function of the enzymes that control the synthesis, regulation, and degradation of glycogen. This section will cover some aspects of these disorders, collectively termed the Glycogen Storage Diseases (GSDs) (Hicks, et al. 2011). Cumulatively, the incidence of GSDs is rare (<1:20,000), and all are inherited in an autosomal recessive fashion, with the exception of GSD type IX and Danon disease (X-linked recessive). Although abnormal glycogen storage is a hallmark, there is a large spectrum of phenotypes associated with these disorders, with the age of onset ranging from in utero to adulthood. GSDs are primarily classified according to the affected enzyme, and some brief information is summarized in Table 1.
Table 1.
A list of the classical Glycogen Storage Diseases, Lafora disease and Danon disease depicting the enzyme/pathway involved and the Online Mendelian Inheritance in Man (OMIM) number.
| Type | Alternate names or subtype | Affected Enzyme/Pathway | Gene | OMIM* Phenotype no. |
|---|---|---|---|---|
| 0 | 0a | Liver glycogen synthase | GYS2 | 240600 |
| 0b | Muscle glycogen synthase | GYS1 | 611556 | |
| I | Ia; von Gierke | Glucose-6-phosphatase α | G6PC | 232200 |
| Ib; von Gierke | Glucose-6-phosphate transporter | SLC37A4 | 232220 | |
| II | Pompe | Acid α-glucosidase | GAA | 232300 |
| III | Cori/Forbes | Glycogen debranching enzyme | AGL | 232400 |
| IV | Andersen | Glycogen branching enzyme | GBE1 | 232500 |
| V | McArdle | Muscle glycogen phosphorylase | PYGM | 232600 |
| VI | Hers | Liver glycogen phosphorylase | PYGL | 232700 |
| VII | Tarui | Muscle phosphofructose kinase | PFKM | 232800 |
| IX | IXa | Phosphorylase kinase (α2 subunit) | PHKA2 | 306000 |
| IXb | Phosphorylase kinase (β subunit) | PHKB | 261750 | |
| IXc | Phosphorylase kinase (γ subunit) | PHKG2 | 613027 | |
| IXd | Phosphorylase kinase (α1 subunit) | PHKA1 | 300559 | |
| X | – | Muscle phosphoglycerate mutase | PGAM2 | 261670 |
| XI# | Fanconi-Bickel | Glucose transporter 2 | SLC2A2 | 227810 |
| XII | – | Aldolase A | ALDOA | 611881 |
| XIII | – | β-enolase | ENO3 | 612932 |
| XV | – | Glycogenin-1 | GYG1 | 603942 |
|
| ||||
| Danon disease | Lysosomal-associated membrane protein 2 | LAMP2 | 300257 | |
| Lafora disease | 2A | Laforin | EPM2A | 254780 |
| 2B | Malin | NHLRC1 | 254780 | |
OMIM (Online Mendelian Inheritance in Man)
In earlier sources, GSD type XI was associated with a deficiency in Lactate dehydrogenase A (OMIM 612933)
Type 0: Glycogen Synthase Deficiency
Traditionally, GSDs are characterized by an abnormal accumulation of glycogen storage. GSD type 0, due to a deficiency in glycogen synthase, is an exception, as glycogen storage is usually lacking (Weinstein, et al. 2006). The two genes that encode different isoforms of glycogen synthase (GYS1 and GYS2) are differentially expressed in tissues (Browner, et al. 1989; Nuttall, et al. 1994). While GYS1 is primarily expressed in cardiac and skeletal muscles, GYS2 expression is mainly restricted to the liver. Thus, symptoms will vary depending on which gene is affected. For GYS1, the defect in glycogen storage can lead to cardiomyopathy and exercise intolerance (Kollberg, et al. 2007). In the liver, a deficiency in GYS2 expression, prevents postprandial glycogen storage, and can cause hyperglycemia and hyperlipidemia (Weinstein et al. 2006). Postprandial hyperlactatemia and fasting ketotic hypoglycemia are typical features of GSD type 0. Other symptoms may include osteopenia, delayed development and growth.
GSD type 0 is possibly under-diagnosed due to the wide spectrum of symptoms (Gitzelmann, et al. 1996). This is usually dependent on the time from their last meal. This disease should be considered in any child with asymptomatic hyperglycemia or glycosuria. Diagnosis can be made by biopsy which reveals reduced glycogen synthase activity but only slightly reduced glycogen stores. One possible explanation for this observation is that alternative pathways for glycogen synthesis may be present (Orho, et al. 1998). A less invasive diagnosis can also be performed through mutational analysis on leukocyte DNA (Bachrach, et al. 2002).
Patients are usually subject to protein-rich diets and are advised to avoid fasting. Administration of complex glucose-containing polysaccharides (e.g. cornstarch) may help in maintaining blood glucose levels during sleep, as this is slowly digested to provide glucose over a longer period (Chen, et al. 1984).
Type I: Glucose 6-Phosphatase Deficiency (von Gierke Disease)
GSD type I is the most common and one of the most severe GSDs, with over 80% of the cases attributed to a deficiency in the enzyme glucose-6-phosphatase α (G6Pase α; Type Ia) (Chou 2001). As mentioned above, G6Pase catalyzes the conversion of G6P to glucose and inorganic phosphate. G6Pase is anchored onto the ER and requires the G6P transporter (G6PT) to bring G6P into the ER lumen before it is hydrolyzed (Foster and Nordlie 2002). A defect in this translocase activity accounts for about 10% of the cases (Type Ib). Additional causes of GSD type I are suspected to exist, and candidates might include transcription factors that control the expression of G6Pase (Cheng and Saltiel 2009; Chopra, et al. 2008).
In GSD type I, the impaired ability to produce glucose from glycogenolysis (and gluconeogenesis) results in hypoglycemia, as well as elevated production of lactic acid and triglycerides. A rounded “doll-like” face is a typical feature due to the deposition of fat. In infants, GSD type I typically presents with hypoglycemia after nighttime sleeping or when a normal feeding schedule is disrupted. In rare cases, repeated hypoglycemic seizures can affect neurological development. Without treatment, GSD type I can lead to a failure to thrive, an enlarged liver, abdominal swelling, and delayed motor development. In addition to these features, GSD type Ib patients can also exhibit neutrophil dysfunction and an increased risk of developing inflammatory bowel diseases (Kishnani, et al. 2014).
GSD type I is usually suspected in patients with hypoglycemia, hypertriglyceridemia, hyperuricemia, and hepatomegaly. Diagnosis can be made on the basis of clinical symptoms as well as biochemical studies. However, current methods utilizing DNA mutational analysis eliminates the need to perform liver biopsies.
Before the 1970s, GSD type I was a fatal disease. However, dietary improvements have since changed this. Continuous dietary sources of glucose are employed to prevent hypoglycemia. Uncooked cornstarch is often used as it acts as an intestinal reservoir of glucose that is slowly absorbed into circulation. However, unpleasant effects of this treatment include diarrhea, increased flatulence, and excess weight gain. Moreover, long-term complications such as insulin resistance, liver tumors, kidney disease and osteopenia have resulted (Kishnani et al. 2014; Melis, et al. 2015).
Type II: Acid α-Glucosidase deficiency (Pompe Disease)
Although the breakdown of glycogen is primarily carried out by glycogen phosphorylase, a small amount occurs via lysosomal degradation by GAA, and is thought to involve autophagy (Nascimbeni, et al. 2012). A deficiency of GAA activity in GSD type II results in an intra-lysosomal accumulation of glycogen, resulting in lysosomal dysfunction and destruction (Lim et al. 2014). Rupture of the lysosomal membranes releases the hydrolytic material causing cellular damage.
GSD type II can be manifested either as a congenital subtype or later-onset subtype. Hypoglycemia is not a hallmark of this disease, as the contribution of glucose production from the lysosomal degradation of glycogen is insignificant. The severity of the disease correlates with the amount of residual GAA activity. GSD type II patients tend to develop progressive muscle weakness with or without cardiac involvement. The infantile form occurs when GAA levels are less than 1%, and the presentation of hypertrophic cardiomyopathy and liver enlargement are typical. Affected infants may also display feeding difficulties, delayed motor development, and a failure to thrive.
The later-onset forms of GSD type II occur when GAA activity lies between 1-30%. Cardiac involvement is rare, but patients may display difficulty with exercise, motor delay, clumsiness, myopathic gait, and obstructive sleep apnea. A more prolonged course in the disease occurs. Ultimately respiratory failure becomes a concern due to the involvement of the diaphragm muscles.
For the early onset GSD type II, a diagnosis is suspected in an infant with significant hypotonia and cardiac insufficiency. Routine elevations of creatine kinase, lactate dehydrogenase, suggesting muscle damage, are commonly observed. The delayed-onset form of GSD type II is usually suspected in individuals with respiratory deficiency and progressive proximal weakness in the limb-girdle area. An electromyogram is performed to find evidence of myopathic discharges. Patients may also exhibit reduced performance in certain pulmonary function tests.
Diagnostic tests involve the assessment of GAA activity in leukocytes or fibroblasts. A muscle biopsy, showing vacuolated myopathy with excessive lysosomal glycogen accumulation, can also be used to confirm the presence of GSD type II. If a family history with a mutation is known, prenatal genetic screening is possible. Otherwise GAA activity can be assessed from samples obtained from aminocytes.
In the absence of any treatment, death occurs in the infantile subtype, and becomes more likely in the adult-onset subtype. Intravenous enzyme replacement therapy is the primary means of treatment, with approximately a third of patients benefiting from the procedure (Broomfield, et al. 2016). The care team involves multiple aspects such as physical rehabilitation, occupational and speech therapy, as well as respiratory support. Although these treatments do not represent a cure, mortality and the quality of life are significantly improved. The potential of gene therapy holds promise but is still under investigation.
Type III: Glycogen debranching enzyme deficiency (Cori/Forbes Disease)
In the absence of the glycogen debranching enzyme (GDE), glycogenolysis is stalled when glycogen phosphorylase encounters a branch point around four glucose residues away. Consequently, an accumulation of abnormal glycogen with very short outer chains appears in patients with GSD type III (Shin 2006). This disorder is divided into four subtypes (IIIa-d) based on the tissue affected and the type of activity that is deficient. The majority of affected individuals lacking GDE in both liver and muscle are designated as GSD type IIIa, while a smaller proportion lacking hepatic GDE only are considered to be type IIIb. The more rare forms, types IIIc and IIId, are due to selective loss of glucosidase or transferase activity respectively.
GSD type III patients possess a spectrum of symptoms based on which tissues are involved. In children, the symptoms may be very similar to GSD I, but at a much milder level. Since gluconeogenesis is intact, there is no fasting hyperlactatemia. Renal disease is not observed, but osteopenia has been reported (Melis, et al. 2016). Unlike GSD type I, fasting ketosis is prominent and liver enzymes (i.e. ALT and AST) are much more elevated. Hepatomegaly, hypoglycemia, hyperlipidemia, and delayed growth may be observed. Although the hepatic symptoms typically improve with age, in rare cases progressive liver cirrhosis and failure can occur. In addition, end-stage liver cirrhosis can also lead to hepatocellular carcinoma (Demo, et al. 2007). In GSD type IIIa, creatine kinase levels are also significantly elevated.
Diagnostic DNA testing for mutations in GDE are available. Confirmation can also be carried out by assessing GDE activity in liver, muscle, or fibroblasts. Because the symptoms of GSD type III and GSD type I are similar in children, other distinguishing biochemical tests are often performed. These often focus on the potential for muscle and cardiac involvement.
Prevention of hypoglycemia by dietary management is often a key treatment. Normoglycemia can be achieved with the frequent consumption of carbohydrate-rich meals or by using nasogastric tube feeding. A high protein diet is also helpful, as the breakdown into amino acids can feed into the pathway for hepatic gluconeogenesis (Petersen et al. 2017). However, because there is no treatment for myopathy, many patients end up wheelchair bound. Finally, in the rare cases of end stage liver cirrhosis, liver transplantation will be required.
Type IV: Glycogen branching enzyme deficiency (Andersen’s Disease)
The deficiency of glycogen branching activity in GSD type IV causes an accumulation of abnormal glycogen with fewer branch points, often referred to as polyglucosans (Shin 2006). Buildup of this material within a variety of cells can lead to their dysfunction and even cellular death.
The ways in which GSD type IV may present are very numerous. Symptoms can begin as early as in utero (intrauterine hydrops) and perinatal death can occur due to hypotonia and severe cardiomyopathy. Beyond this point, patients may present with a failure to thrive, hepatosplenomegaly, and progressive liver cirrhosis, resulting in death by 5 years of age. Fasting hypoglycemia is not typical unless there is significant liver cirrhosis.
The adult form of GSD type IV is milder and can present as an isolated myopathy or as a multisystem disorder. These individuals exhibit signs of upper and lower motor neuron involvement. Typically, this can lead to bladder incontinence, followed by gait disturbance and lower limb paresthesias. A subset of individuals (usually beyond 30 years of age) with decreased GBE1 activity, may present with Adult Polygluosan Body Disease, affecting the central and peripheral nervous systems (Paradas, et al. 2014).
The diagnosis of GSD type IV requires a biopsy demonstrating abnormal glycogen by histological staining and by an analysis using electron microscopy. Confirmation can be achieved by the absence of the branching enzyme activity in liver, muscle, fibroblasts, leukocytes or erythrocytes. Mutational analysis can also be performed.
There is no cure for GSD type IV, and medical care focuses to maintain normoglycemia. Suitable nutrition may help improve liver and muscle function, as well as improve long-term outcomes for in these patients. For those with liver disease, a transplantation may be necessary.
Type V: Muscle glycogen phosphorylase deficiency (McArdle Disease)
There are two isoforms of glycogen phosphorylase encoded by two separate genes. Deficiency in the muscle isoform (encoded by PYGM) results in impaired glycogenolysis and leads to GSD type V (Nogales-Gadea, et al. 2015a), the most common GSD affecting the muscle.
Although symptoms may occur early in life, a diagnosis is usually made in adulthood. Individuals with GSD type V show exercise intolerance, muscle weakness, cramping, as well as pain. Patients possess the ability to resume exercise if they rest briefly, a phenomenon termed “second wind”. The majority of patients exhibit elevated baseline levels of creatine kinase (~5000 U/L), which can further reach to 1,000,000 U/L during rhabdomyolysis (0-400 U/L is considered normal). Additionally, about half of the patients experience myoglobinuria. In addition to the expected myopathy, a smaller percentage of patients may exhibit cardiovascular problems.
The preferred method of diagnosis of GSD type V is through genetic analysis (Nogales-Gadea, et al. 2015b). While there is no cure for this disease, a study has shown that ingestion of sucrose before exercise might be beneficial on work capacity (Andersen, et al. 2008). Additionally, moderate aerobic exercise may be beneficial for improving muscle function and outlook for patients.
Type VI: Liver glycogen phosphorylase deficiency (Hers’ Disease)
GSD type VI is caused by a deficiency in the activity of the liver isoform of glycogen phosphorylase (encoded by PYGL) (Burda and Hochuli 2015). Originally, this covered patients with mutations in the gene that encodes this isoform. However, GSD type VI now also includes individuals with mutations in other genes that affect the function of PYGL.
Noticeably, the symptoms of these patients differ dramatically when compared to GSD type V. Patients with GSD type VI exhibit normal creatine kinase and uric acid levels. The disease can start in early childhood but is significantly more benign. Individuals typically present with growth retardation and hepatomegaly. Because liver glycogenolysis is impaired, ketotic hypoglycemia is common, as well as hyperlipidemia. Often these conditions improve with age and treatment is often unnecessary. As a brief note, individuals with GSD type IX exhibit very similar phenotypes to those with GSD type VI. This is not surprising, as GSD type IX is caused by a deficiency in the hepatic isoform of glycogen phosphorylase kinase, the enzyme that activates PYGL.
Other GSDs and re-classification of previous GSDs
As the growing amount of information consolidated, several former GSDs (VIII, XI and XIV) were re-classified into other disorders. GSD type VIII is now grouped with GSD type VI or IX (Shin 2006), while GSD type XIV is re-classified as a congenital disorder of glycosylation (Peanne, et al. 2017). There is very little information for the remaining GSDs, and the genes involved are listed in Table I. For information not detailed in this review, readers can be directed elsewhere (Toscano and Musumeci 2007).
Due to the limited number of studies, there might be some disagreement concerning the nomenclature for GSD type XI. According to the Online Mendelian Inheritance in Man (OMIM) and Orphanet websites, GSD type XI is associated with either a deficiency in Lactate Dehydrogenase A (LDHA; OMIM #612933; ORPHA #284426), or the Glucose Transporter 2 (GLUT2; OMIM #227810; ORPHA #2088).
GSD type XI due to LDHA deficiency seems to have an earlier origin and is primarily associated with muscle dysfunction and exercise intolerance (Servidei and DiMauro 1989). In contrast, GSD type XI due to GLUT2 deficiency (also commonly referred to as Fanconi-Bickel Syndrome) was identified later, and presents mostly as a renal phenotype, which is not surprising given that GLUT2 plays a prominent role in renal glucose reabsorption (Santer, et al. 2002; Yan 2017).
For GSD type XV, the mutations in glycogenin-1 have only recently been described (Moslemi, et al. 2010). In this study, the patient exhibited a profound depletion of muscle glycogen stores, and the analysis supports the role of glycogenin-1 in the priming of glycogen synthesis. In contrast, other GSD type XV patients have demonstrated the accumulation of polyglucosans (Malfatti, et al. 2014). Interestingly, recent data demonstrate that the absence of glycogenin in mice, unexpectedly leads to increased glycogen storage in muscle tissues (Testoni, et al. 2017). One possibility is that other proteins may compensate for the absence of glycogenin. More studies will be needed to delineate the molecular causes of these findings and may suggest a more complicated role for glycogenin in glycogen metabolism.
Danon disease
Some disorders exhibiting abnormal glycogen accumulation can be classified as a GSD or otherwise, depending on the focus of the information. For example, the build-up of glycogen in the lysosomes of muscle in Danon disease originally led to its classification as a variant of GSD type II (Danon, et al. 1981). Danon disease is an X-linked disorder predominantly affecting the heart, with variable displays of skeletal muscle impairment and mental retardation (Endo, et al. 2015). Mutations in the Lysosomal Associated Membrane Protein 2 (LAMP2) were identified as the cause of this disorder (Nishino, et al. 2000), and subsequently it is now often referred to as a form of autophagic vacuolar myopathy (Sugie, et al. 2005). This seems appropriate given that LAMP2 serves a general lysosomal function (Alessandrini, et al. 2017), as opposed to a specific role in glycogen degradation.
Lafora disease
For the longest time, disruptions in glycogen metabolism were invariably associated with liver and/or muscle dysfunction. This is expected as the majority of glycogen stores in the human body is found in these tissues. However, there are appreciable amounts of glycogen in most cells, and some recent studies have focused on the importance of glycogen in the brain.
Lafora disease, an autosomal recessive form of epilepsy (Sullivan, et al. 2017), highlights this notion. A prominent clinical aspect of this disease is the presence of inclusion bodies (Lafora bodies) in most organs, including the brain. Lafora bodies contain high levels of polyglucosans that are thought to accumulate and cause cellular damage and death. The buildup of these bodies in the brain is believed to be responsible for the majority of the symptoms, such as seizures, ataxia, myoclonus, and the progressive development of severe dementia. Patients usually die by 30 years of age, and currently, there is no cure.
The two major genes implicated in Lafora disease encode Laforin, a dual specificity phosphatase (Gentry, et al. 2016); and Malin, an E3 ubiquitin ligase (Roma-Mateo, et al. 2012). How these two proteins regulate glycogen metabolism is only beginning to emerge. Early studies indicated that Lafora bodies contain about 10 times more phosphate than glycogen (Sakai, et al. 1970; Yokoi, et al. 1967), and evidence suggested that Laforin removes this modification. In the absence of Laforin, glycogen becomes hyperphosphorylated, which promotes the formation of Lafora bodies (Gentry et al. 2016; Tagliabracci, et al. 2008; Tagliabracci, et al. 2007; Worby, et al. 2006). More recently, data supports an alternative model with a role for Laforin in controlling glycogen chain length as opposed to glycogen phosphorylation (Nitschke, et al. 2017). Finally, the cellular function of Malin is unclear and thus the molecular role of these two proteins in glycogen metabolism remains to be established. Nevertheless, Lafora disease brings the structural regulation of glycogen to the forefront, and highlights the importance of glycogen in tissues other than the liver and muscle.
Conclusions
During the 20th century, the study of glycogen metabolism and their associated disorders have yielded a vast quantity of information on metabolic pathways. Here, we summarized the key pathways involved, and highlighted how perturbations in key enzymes can lead to metabolic disease. As the primary stores of glycogen are located in liver and muscle tissues, it is not surprising that the GSDs affect one or both of these sites. However, it is fascinating that the symptoms of different GSDs exist in a broad spectrum depending on the particular enzyme affected. While in some cases this may lead to lethality, in other cases the symptoms may be mild and improve with age.
Despite the enormous time and effort devoted to this subject, it seems that there are still many aspects of glycogen metabolism that have yet to be discovered. As we progress through the early part of the 21st century, studies from the past 20 years have revealed a novel molecular aspect of glycogen metabolism that has profound effects on the brain. Lafora disease has suggested to us that in addition to the synthesis and degradation of glycogen, the maintenance of its phosphorylation, its branched structure and chain length are also very important. Although the culprits have been identified (Laforin and Malin), we still do not have an established understanding of how they operate. Undoubtedly, further studies will reveal new insight into the regulation of glycogen metabolism.
Acknowledgments
This review summarizes the basic concepts of glycogen metabolism to a general audience, and we regret that some work from our colleagues could not be discussed.
Funding
This work was supported in part by the American Diabetes Association [#1-17-IBS-135 to AC]]. SSE was supported by the National Institutes of Health [T35 DK07293; PI: Klinge].
Footnotes
Declaration of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
References
- Adeva-Andany MM, Gonzalez-Lucan M, Donapetry-Garcia C, Fernandez-Fernandez C, Ameneiros-Rodriguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. doi: 10.1016/j.bbacli.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agius L, Centelles J, Cascante M. Multiple glucose 6-phosphate pools or channelling of flux in diverse pathways? Biochem Soc Trans. 2002;30:38–43. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Alessandrini F, Pezze L, Ciribilli Y. LAMPs: Shedding light on cancer biology. Semin Oncol. 2017;44:239–253. doi: 10.1053/j.seminoncol.2017.10.013. [DOI] [PubMed] [Google Scholar]
- Andersen ST, Haller RG, Vissing J. Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol. 2008;65:786–789. doi: 10.1001/archneur.65.6.786. [DOI] [PubMed] [Google Scholar]
- Bachrach BE, Weinstein DA, Orho-Melander M, Burgess A, Wolfsdorf JI. Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. J Pediatr. 2002;140:781–783. doi: 10.1067/mpd.2002.124317. [DOI] [PubMed] [Google Scholar]
- Berthet J, Rall TW, Sutherland EW. The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J Biol Chem. 1957;224:463–475. [PubMed] [Google Scholar]
- Broomfield A, Fletcher J, Davison J, Finnegan N, Fenton M, Chikermane A, Beesley C, Harvey K, Cullen E, Stewart C, et al. Response of 33 UK patients with infantile-onset Pompe disease to enzyme replacement therapy. J Inherit Metab Dis. 2016;39:261–271. doi: 10.1007/s10545-015-9898-5. [DOI] [PubMed] [Google Scholar]
- Browner MF, Nakano K, Bang AG, Fletterick RJ. Human muscle glycogen synthase cDNA sequence: a negatively charged protein with an asymmetric charge distribution. Proc Natl Acad Sci U S A. 1989;86:1443–1447. doi: 10.1073/pnas.86.5.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda P, Hochuli M. Hepatic glycogen storage disorders: what have we learned in recent years? Curr Opin Clin Nutr Metab Care. 2015;18:415–421. doi: 10.1097/MCO.0000000000000181. [DOI] [PubMed] [Google Scholar]
- Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen-storage disease. N Engl J Med. 1984;310:171–175. doi: 10.1056/NEJM198401193100306. [DOI] [PubMed] [Google Scholar]
- Cheng A, Saltiel AR. Von Gierke’s disease adopts an orphan (and its partner) Sci Signal. 2009;2:e8. doi: 10.1126/scisignal.258pe8. [DOI] [PubMed] [Google Scholar]
- Chopra AR, Louet JF, Saha P, An J, Demayo F, Xu J, York B, Karpen S, Finegold M, Moore D, et al. Absence of the SRC-2 coactivator results in a glycogenopathy resembling Von Gierke’s disease. Science. 2008;322:1395–1399. doi: 10.1126/science.1164847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY. The molecular basis of type 1 glycogen storage diseases. Curr Mol Med. 2001;1:25–44. doi: 10.2174/1566524013364112. [DOI] [PubMed] [Google Scholar]
- Cori CF, Colowick SP, Cori GT. The isolation and synthesis of glucose-1-phosphoric acid. J Biol Chem. 1937;121:465–477. [Google Scholar]
- Cori CF, Cori GT. Glycogen formation in the liver from d- and l-lactic acid. J Biol Chem. 1929;81:389–403. [Google Scholar]
- Cori CF, Schmidt G, Cori GT. The Synthesis of a Polysaccharide from Glucose-1-Phosphate in Muscle Extract. Science. 1939;89:464–465. doi: 10.1126/science.89.2316.464. [DOI] [PubMed] [Google Scholar]
- Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31:51–57. doi: 10.1212/wnl.31.1.51. [DOI] [PubMed] [Google Scholar]
- Demo E, Frush D, Gottfried M, Koepke J, Boney A, Bali D, Chen YT, Kishnani PS. Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J Hepatol. 2007;46:492–498. doi: 10.1016/j.jhep.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y, Furuta A, Nishino I. Danon disease: a phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 2015;129:391–398. doi: 10.1007/s00401-015-1385-4. [DOI] [PubMed] [Google Scholar]
- Fischer EH, Krebs EG. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J Biol Chem. 1955;216:121–132. [PubMed] [Google Scholar]
- Foster JD, Nordlie RC. The biochemistry and molecular biology of the glucose-6-phosphatase system. Exp Biol Med (Maywood) 2002;227:601–608. doi: 10.1177/153537020222700807. [DOI] [PubMed] [Google Scholar]
- Gentry MS, Brewer MK, Vander Kooi CW. Structural biology of glucan phosphatases from humans to plants. Curr Opin Struct Biol. 2016;40:62–69. doi: 10.1016/j.sbi.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitzelmann R, Spycher MA, Feil G, Muller J, Seilnacht B, Stahl M, Bosshard NU. Liver glycogen synthase deficiency: a rarely diagnosed entity. Eur J Pediatr. 1996;155:561–567. doi: 10.1007/BF01957905. [DOI] [PubMed] [Google Scholar]
- Greenberg CC, Jurczak MJ, Danos AM, Brady MJ. Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab. 2006;291:E1–8. doi: 10.1152/ajpendo.00652.2005. [DOI] [PubMed] [Google Scholar]
- Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011;35:183–196. doi: 10.3109/01913123.2011.601404. [DOI] [PubMed] [Google Scholar]
- Johnson LN. Glycogen phosphorylase: control by phosphorylation and allosteric effectors. FASEB J. 1992;6:2274–2282. doi: 10.1096/fasebj.6.6.1544539. [DOI] [PubMed] [Google Scholar]
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, Chung WK, Dagli AI, Dale D, Koeberl D, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014;16:e1. doi: 10.1038/gim.2014.128. [DOI] [PubMed] [Google Scholar]
- Kollberg G, Tulinius M, Gilljam T, Ostman-Smith I, Forsander G, Jotorp P, Oldfors A, Holme E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med. 2007;357:1507–1514. doi: 10.1056/NEJMoa066691. [DOI] [PubMed] [Google Scholar]
- Krebs EG, Kent AB, Fischer EH. The muscle phosphorylase b kinase reaction. J Biol Chem. 1958;231:73–83. [PubMed] [Google Scholar]
- Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci. 2014;6:177. doi: 10.3389/fnagi.2014.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfatti E, Nilsson J, Hedberg-Oldfors C, Hernandez-Lain A, Michel F, Dominguez-Gonzalez C, Viennet G, Akman HO, Kornblum C, Van den Bergh P, et al. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann Neurol. 2014;76:891–898. doi: 10.1002/ana.24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis D, Rossi A, Pivonello R, Del Puente A, Pivonello C, Cangemi G, Negri M, Colao A, Andria G, Parenti G. Reduced bone mineral density in glycogen storage disease type III: evidence for a possible connection between metabolic imbalance and bone homeostasis. Bone. 2016;86:79–85. doi: 10.1016/j.bone.2016.02.012. [DOI] [PubMed] [Google Scholar]
- Melis D, Rossi A, Pivonello R, Salerno M, Balivo F, Spadarella S, Muscogiuri G, Casa RD, Formisano P, Andria G, et al. Glycogen storage disease type Ia (GSDIa) but not Glycogen storage disease type Ib (GSDIb) is associated to an increased risk of metabolic syndrome: possible role of microsomal glucose 6-phosphate accumulation. Orphanet J Rare Dis. 2015;10:91. doi: 10.1186/s13023-015-0301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moslemi AR, Lindberg C, Nilsson J, Tajsharghi H, Andersson B, Oldfors A. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med. 2010;362:1203–1210. doi: 10.1056/NEJMoa0900661. [DOI] [PubMed] [Google Scholar]
- Mul JD, Stanford KI, Hirshman MF, Goodyear LJ. Exercise and Regulation of Carbohydrate Metabolism. Prog Mol Biol Transl Sci. 2015;135:17–37. doi: 10.1016/bs.pmbts.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimbeni AC, Fanin M, Masiero E, Angelini C, Sandri M. Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy. 2012;8:1697–1700. doi: 10.4161/auto.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- Nitschke F, Sullivan MA, Wang P, Zhao X, Chown EE, Perri AM, Israelian L, Juana-Lopez L, Bovolenta P, Rodriguez de Cordoba S, et al. Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. EMBO Mol Med. 2017;9:906–917. doi: 10.15252/emmm.201707608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales-Gadea G, Brull A, Santalla A, Andreu AL, Arenas J, Martin MA, Lucia A, de Luna N, Pinos T. McArdle Disease: Update of Reported Mutations and Polymorphisms in the PYGM Gene. Hum Mutat. 2015a;36:669–678. doi: 10.1002/humu.22806. [DOI] [PubMed] [Google Scholar]
- Nogales-Gadea G, Santalla A, Brull A, de Luna N, Lucia A, Pinos T. The pathogenomics of McArdle disease–genes, enzymes, models, and therapeutic implications. J Inherit Metab Dis. 2015b;38:221–230. doi: 10.1007/s10545-014-9743-2. [DOI] [PubMed] [Google Scholar]
- Nuttall FQ, Gannon MC, Bai G, Lee EY. Primary structure of human liver glycogen synthase deduced by cDNA cloning. Arch Biochem Biophys. 1994;311:443–449. doi: 10.1006/abbi.1994.1260. [DOI] [PubMed] [Google Scholar]
- Orho M, Bosshard NU, Buist NR, Gitzelmann R, Aynsley-Green A, Blumel P, Gannon MC, Nuttall FQ, Groop LC. Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0. J Clin Invest. 1998;102:507–515. doi: 10.1172/JCI2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradas C, Akman HO, Ionete C, Lau H, Riskind PN, Jones DE, Smith TW, Hirano M, Dimauro S. Branching enzyme deficiency: expanding the clinical spectrum. JAMA Neurol. 2014;71:41–47. doi: 10.1001/jamaneurol.2013.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, Perez B, Seta N, Thiel C, Van Schaftingen E, et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 2017 doi: 10.1016/j.ejmg.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13:572–587. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall TW, Sutherland EW, Wosilait WD. The relationship of epinephrine and glucagon to liver phosphorylase. III. Reactivation of liver phosphorylase in slices and in extracts. J Biol Chem. 1956;218:483–495. [PubMed] [Google Scholar]
- Roach PJ, Skurat AV. Self-glucosylating initiator proteins and their role in glycogen biosynthesis. Prog Nucleic Acid Res Mol Biol. 1997;57:289–316. doi: 10.1016/s0079-6603(08)60284-6. [DOI] [PubMed] [Google Scholar]
- Roma-Mateo C, Sanz P, Gentry MS. Deciphering the role of malin in the lafora progressive myoclonus epilepsy. IUBMB Life. 2012;64:801–808. doi: 10.1002/iub.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M, Austin J, Witmer F, Trueb L. Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology. 1970;20:160–176. doi: 10.1212/wnl.20.2.160. [DOI] [PubMed] [Google Scholar]
- Santer R, Steinmann B, Schaub J. Fanconi-Bickel syndrome–a congenital defect of facilitative glucose transport. Curr Mol Med. 2002;2:213–227. doi: 10.2174/1566524024605743. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Mammucari C, Sandri M. The role of autophagy in neonatal tissues: just a response to amino acid starvation?0. Autophagy. 2008;4:727–730. doi: 10.4161/auto.6143. [DOI] [PubMed] [Google Scholar]
- Servidei S, DiMauro S. Disorders of glycogen metabolism of muscle. Neurol Clin. 1989;7:159–178. [PubMed] [Google Scholar]
- Shin YS. Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol. 2006;13:115–120. doi: 10.1016/j.spen.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Shulman RG, Rothman DL. 13C NMR of intermediary metabolism: implications for systemic physiology. Annu Rev Physiol. 2001;63:15–48. doi: 10.1146/annurev.physiol.63.1.15. [DOI] [PubMed] [Google Scholar]
- Sugie K, Noguchi S, Kozuka Y, Arikawa-Hirasawa E, Tanaka M, Yan C, Saftig P, von Figura K, Hirano M, Ueno S, et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J Neuropathol Exp Neurol. 2005;64:513–522. doi: 10.1093/jnen/64.6.513. [DOI] [PubMed] [Google Scholar]
- Sullivan MA, Nitschke S, Steup M, Minassian BA, Nitschke F. Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18081743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland EW, Jr, Wosilait WD. Inactivation and activation of liver phosphorylase. Nature. 1955;175:169–170. doi: 10.1038/175169a0. [DOI] [PubMed] [Google Scholar]
- Sutherland EW, Wosilait WD. The relationship of epinephrine and glucagon to liver phosphorylase. I. Liver phosphorylase; preparation and properties. J Biol Chem. 1956;218:459–468. [PubMed] [Google Scholar]
- Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, Roach PJ. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283:33816–33825. doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, Depaoli-Roach AA, Roach PJ. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104:19262–19266. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testoni G, Duran J, Garcia-Rocha M, Vilaplana F, Serrano AL, Sebastian D, Lopez-Soldado I, Sullivan MA, Slebe F, Vilaseca M, et al. Lack of Glycogenin Causes Glycogen Accumulation and Muscle Function Impairment. Cell Metab. 2017;26:256–266 e254. doi: 10.1016/j.cmet.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Toscano A, Musumeci O. Tarui disease and distal glycogenoses: clinical and genetic update. Acta Myol. 2007;26:105–107. [PMC free article] [PubMed] [Google Scholar]
- Weinstein DA, Correia CE, Saunders AC, Wolfsdorf JI. Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol Genet Metab. 2006;87:284–288. doi: 10.1016/j.ymgme.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, Dixon JE. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281:30412–30418. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosilait WD, Sutherland EW. The relationship of epinephrine and glucagon to liver phosphorylase. II. Enzymatic inactivation of liver phosphorylase. J Biol Chem. 1956;218:469–481. [PubMed] [Google Scholar]
- Yan N. A Glimpse of Membrane Transport through Structures-Advances in the Structural Biology of the GLUT Glucose Transporters. J Mol Biol. 2017;429:2710–2725. doi: 10.1016/j.jmb.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Yokoi S, Austin J, Witmer F. Isolation and characterization of Lafora bodies in two cases of myoclonus epilepsy. J Neuropathol Exp Neurol. 1967;26:125–127. [PubMed] [Google Scholar]
- Zirin J, Nieuwenhuis J, Perrimon N. Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy. PLoS Biol. 2013;11:e1001708. doi: 10.1371/journal.pbio.1001708. [DOI] [PMC free article] [PubMed] [Google Scholar]