

Abstract
Cyclic peptide scaffolds are key components of signal transduction pathways in both prokaryotic and eukaryotic organisms since they act as chemical messengers that activate or inhibit specific cognate receptors. In prokaryotic organisms these peptides are utilized in non-essential pathways, such as quorum sensing, that are responsible for virulence and pathogenicity. In the more evolved eukaryotic systems, cyclic peptide hormones play a key role in the regulation of the overall function of multicellular organisms, mainly through the endocrine system. This review will highlight several prokaryote and eukaryote systems that use cyclic peptides as their primary signals and the potential associated with utilizing these scaffolds for the discovery of novel therapeutics for a wide range of diseases and illnesses.
Keywords: Cyclic Peptides, Signal Transduction, Quorum Sensing, Prokaryotes, Eukaryotes, Hormones, Autoinducers
TOC
INTRODUCTION
Peptides, biological molecules generally comprised of 2 to 50 amino acids, regulate a multitude of processes in simple prokaryote cells as well as complex multicellular organisms by acting as chemical messengers that initiate signal transduction pathways through the activation or inhibition of specific receptors.[1] Large peptide, 15 to 50 amino acids, and protein, greater than 50 amino acids, scaffolds possess optimal pharmacological properties required for dynamic signal transduction messengers since they form a high number of contacts with their target receptor, leading to high affinity and specificity, while at the same time they are efficiently inactivated and cleared from the environment to prevent over-stimulation of the pathway. Unfortunately, in many cases it is challenging to transport these scaffolds to the site of action due to their size. On the other hand, short peptide sequences are easier to transport, but they lack specificity and only exhibit moderate affinity to their target due to their ability to adopt a high number of conformations. Therefore, modifications that restrict their conformational space are required to convert these scaffolds into optimal signal transduction mediators.[2] Peptide cyclization is a widely used method of creating unique conformational constraints necessary for effective and specific receptor binding. Biologically active cyclic peptides are naturally formed through diverse ring-closure methods, including head-to-tail, side-chain-to-side-chain, head-to-side-chain, and side-chain-to-tail cyclization, and by utilizing various chemical bonds such as amide, lactone, thioester, or disulfide bonds.[3] This review will focus on cyclic peptides that regulate signal transduction pathways in both simple prokaryote cells and complex multicellular organisms to highlight the potential of such scaffolds as therapeutic agents in diverse clinical settings.
1. Cyclic Peptide Regulators of Non-Essential Bacterial Communication Pathways
Cyclic peptides are used as chemical signals, or pheromones, in prokaryotic signal transduction systems. In particular, many genera of Gram-positive bacteria utilize cyclic peptides to assess their population density, and at high cell number synchronize to initiate group behavior phenotypes.[4–6] These group behaviors regulate symbiotic relationships with a bacterial host, such as bioluminescence and root nodulation, or coordinate bacterial pathogenicity including swarming, motility, competence, biofilm formation, and virulence factor production.[7–9] The synchronization of bacteria to act as a group through the production, secretion, and detection of signaling molecules, such as cyclic peptides, is known as quorum sensing (QS).
QS was first identified by Nealson and Hastings in the 1970’s when researchers were attempting to understand how the symbiotic relationship between the Hawaiian squid (Euprymna scolopes) and the bioluminescent marine bacterium (Vibrio fischeri) resulted in the ability of the squid to use bioluminescence to evade its predators.[10, 11] The phenomenon of bioluminescence was observed only when the bacteria reached high cell density. This phenotype was suggested to be regulated by a signaling mechanism dependent on cell density that was mediated by small signaling molecules.[10, 12] The most basic of these signaling mechanisms consists of a synthase and a receptor. The synthase produces small signaling molecules that are secreted outside of the cell, and upon reaching a threshold concentration (corresponding to high bacterial density), the signaling molecules effectively bind and activate the cognate receptor to trigger the expression of genes involved in group-related phenotypes.[5, 13, 14] After observing this phenomenon of group behavior in other bacterial species, the term QS was used to define these cell-to-cell communication mechanisms that enable bacteria to alter their behavior at high cell densities using chemical signals.[15]
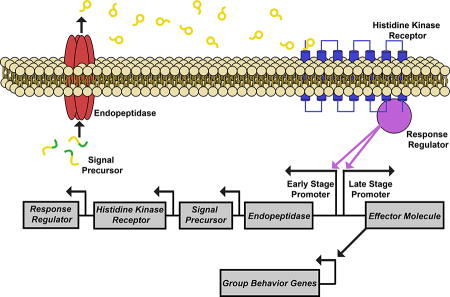
The chemical signals used for QS are usually referred to as autoinducers. These are defined as small molecules used by bacteria to measure the population density; the concentration of the signal increases outside of the bacterial cell in proportion to the increasing cell population.[5, 16] There are two main classes of autoinducers that regulate QS: small signal molecules, such as N-acyl-homoserine lactones (AHL), and oligopeptide autoinducers.[6, 17] In general, AHL autoinducers bearing different fatty acid tails are utilized for intraspecies communication by Gram-negative bacteria, while oligopeptide autoinducers of varying sequences are utilized for intraspecies communication by Gram-positive bacteria. Furthermore, autoinducer 2 (AI-2), a furanosyl borate diester, is used by both Gram-negative and Gram-positive bacteria for interspecies signaling.[6] The diversity of these autoinducers can thus allow specific bacterial species to assess their own cell density within a complex bacterial population and differentiate between other Gram-positive or Gram-negative species.[17] Generally, the Gram-positive QS circuits consist of four different components, the oligopeptide autoinducer, an endopeptidase, a transmembrane histidine kinase receptor, and a response regulator. The oligopeptide autoinducer is processed by the endopeptidase and exported to the extracellular environment. When the autoinducer reaches a minimum sensory concentration it binds to the histidine kinase receptor in a density dependent fashion. This binding triggers a signal cascade that involves the response regulator and results in the transcription of group behavior genes that are often key for the survival or pathogenicity of the Gram-positive bacteria (Figure 1).[18] Furthermore, Gram-positive oligopeptide autoinducers can be subdivided into linear and cyclic peptides. This review will focus on four Gram-positive genera that utilize cyclic peptides to communicate group behavior phenotypes: staphylococcus, enterococcus, clostridium, and lactobacillus, due to their wide therapeutic relevance.
Figure 1.

Generalized QS circuit utilized by Gram-positive species. A) The signal precursor is processed by the endopeptidase to afford the mature AIP; B) The endopeptidase then exports the AIP to the extracellular environment; C) Once the extracellular concentration of the AIP reaches a minimal sensory level, it is detected by the histidine kinase receptor; D) When the AIP binds to the histidine kinase receptor it autophosphorylates, causing a phosphate group to be transferred to the response regulator; E) The response regulator activates the transcription of early gene products; F) The response regulator triggers transcription of the effector molecule, which is responsible for the activation of a multitude of group behavior genes.
1.1. Staphylococcus
Staphylococci are the most studied Gram-positive genus that utilize QS to cause bacterial infections in their human hosts.[4, 19–25] Overall, staphylococci are commensal organisms; various species colonize in specific niches of the human body including the nasal passage, axillae, and skin.[26] However, many species of staphylococci are opportunistic pathogens that cause infections such as skin infections, nosocomial infections of surgical wounds, food poisoning, toxic shock syndrome (TSS), urinary tract infections, and other serious ailments that may result in death, if left untreated.[19, 26] Pathogenic staphylococci species cause disease due to their ability to colonize within host tissues and secrete toxins that not only create damage, but also impair the host immune system.[22, 26] The pathogenicity of staphylococci is regulated by the accessory gene regulator (agr) QS circuit that controls, among others, the expression of surface proteins, immune evasion factors and virulence factors.[4, 19, 23, 24, 27] The agr circuitry is conserved across staphylococci and is centered on a cyclic peptide signal (autoinducing peptide, AIP) that is secreted by the bacteria.[4, 21, 23, 24, 27]
The agr locus is composed of two divergent operons that are under the control of promoters, P2 and P3, that produce RNAII and RNAIII transcripts, respectively.[4, 25] The P2 operon controls the expression of four genes—AgrA, AgrB, AgrC, and AgrD—that constitute the four components of the agr QS circuit and regulate the expression of the effector molecule of the circuitry, RNAIII.[20] The premature AIP is encoded by AgrD, which is proteolytically cleaved and secreted out of the cell by AgrB.[4, 25, 28–31] AgrB is critical in the maturation of the chemical signal in staphylococci by catalyzing the cyclization of the AIP signal. The mature AIP is a short 7–12 amino acid peptide with a five-amino acid cyclic ring connected via a thiolactone bridge between the C-terminus of the peptide and a conserved cysteine (in the case of Staphylococcus intermedius the AIP was identified as a lactone-containing signal, Figure 2).[4, 21, 25, 31] When the AIP signal reaches a sufficiently high concentration it binds to the transmembrane receptor, AgrC, to trigger a signal transduction cascade. The macrocycle part of the AIP signals was found to be critical in triggering QS in staphylococci as the linear counterparts were found to be largely inactive in Staphylococcus aureus.[4, 21, 32] Furthermore, the synthesis of a cyclic peptide with a thioester bond is proposed to create more stability against peptide/protein degrading enzymes such as trypsin, chymotrypsin, proteinase K, and other proteases. Nonetheless, the bond is labile enough to allow for degradation so that the bacteria are able to accurately measure its fluctuating cell density through AIP concentration.[33] AgrC and AgrA work together to form a two-component signal transduction system (TCSTS) consisting of a transmembrane histidine kinase receptor and a response regulator, respectively. The activation of the TCSTS first upregulates RNAII transcription, resulting in autoinduction of the QS circuitry necessary for behavioral change synchronization. At a later stage, the TCSTS also activates the P3 promoter, leading to RNAIII transcription and the expression of group behavior genes including hemolysins and exoproteins.[4, 25] The agr system is activated during the post-exponential growth phase and is responsible for triggering staphylococcal pathogenesis.[4, 19] Thus, this QS circuitry has attracted significant attention as a potential anti-infective target for drug development.
Figure 2.

The AIP signals utilized by different staphylococcal species.
1.1.1. Staphylococcus aureus
S. aureus is a major human pathogen that causes a wide range of skin infections such as styes, boils, osteomyelitis, endocarditis, and furnunculosis in addition to other severe problems such as post-operative surgical infections, food poisoning, and TSS.[26] Due to the severity of diseases associated with S. aureus, coupled with the rise of multidrug resistant strains such as methicillin resistant S. aureus (MRSA), significant attention was given to study this pathogen and understand its ability to mitigate the effectiveness of antibiotics.[34] More specifically, early studies revealed that virulence was heavily controlled by the agr locus since agr-null S. aureus mutants had reduced ability to secrete toxins and other exoproteins.[35–38] Furthermore, activation of the agr circuitry by the AIP signal was found to trigger virulence factor production in S. aureus.[4]
Structural analysis of the chemical signal used by S. aureus was first reported by Novick and co-workers (1995). The chemical signal was identified as an octapeptide containing a thiolactone macrocyclic portion that is required for QS activation in S. aureus.[4, 24] Soon after, three additional AIP signals were identified in S. aureus, leading to the classification of four specificity groups within the species (termed agr-I through -IV).[21, 39, 40] These specific groups were found to possess unique AIP signals (Figure 2), AgrC sensory domains and AgrB endopeptidases.[30] Interestingly, Novick and co-workers have found that most cross-group interactions are inhibitory, suggestive of competition, mediated by agr interference, between the S. aureus specificity groups.[21, 41] Moreover, the four groups were found to strive in different niches and express different virulence factors, resulting in distinct disease profiles associated with each agr group. Groups -I and -II are predominately found to cause invasive infections, Group -III is predominantly responsible for TSS, and group -IV is considered rare and is mainly involved in skin infections.[42–46] The identification of agr interference has sparked numerous studies focused on the attenuation of QS in S. aureus as a potential therapeutic strategy by utilizing QS interference.[47] Structure-activity relationship (SAR) studies of the AIP-I, -II and –III signals conducted by Williams and co-workers,[40, 48] Muir and Novick and co-workers,[18, 23, 24, 49–52] and Blackwell and co-workers,[45, 53–56] respectively, identified similar activity trends and required structural features for AgrC activation and inhibition. These groups found that the macrocycle part of the AIP signal was involved in the initial receptor recognition, while the exocyclic tail region was largely involved in receptor activation.[50, 53, 55] Indeed, several truncated AIP analogs bearing only the macrocyclic structural feature were found to effectively inhibit the AgrC receptors and thus the agr QS circuitry in all S. aureus specificity groups.[24, 45, 56]
From a medicinal standpoint, over the years numerous AIP-based QS inhibitors of the S. aureus agr circuitry were identified and assessed for their therapeutic potential as anti-infective agents. The first pan-group AgrC inhibitor to be reported was an AIP-I analog bearing a single Asp to Ala mutation in the macrocyclic region, AIP-I D5A (Figure 3).[40] Using QS reporter strains of all four agr groups, this peptide was found to competitively inhibit all four AgrC receptors at low nM concentrations (IC50 values of 5, 8, 0.3, and 3 nM against AgrC-I, -II, -III, and -IV respectively).[24] Since the macrocycle portion was found to be critical for initial AgrC binding, the truncated version of AIP-I D5A, tAIP-I D2A (Figure 3), bearing the same Asp to Ala modification but lacking the exocyclic tail was introduced and exhibited similar inhibitory activity (IC50 values of 5, 5, 0.1, and 5 nM against AgrC-I, -II, -III, and -IV respectively).[24] In addition, the truncated version of AIP-II, tAIP-II (Figure 3), was also found to be a pan-group QS inhibitor, although with higher IC50 values (IC50 values of 260, 230, 4, and 150 nM against AgrC-I, -II, -III, and -IV respectively).[24] By far, the most progress in the development of an AIP-based therapeutic agent was conducted by Blackwell and co-workers: they found that an Asp to Ala mutation in AIP-III (AIP-III D4A, Figure 3), similar to the mutation in AIP-I, converts the peptide into a picomolar global QS inhibitor (IC50 values of 485, 429, 50.6, and 34.9 pM against AgrC-I, -II, -III, and -IV respectively). In addition, they found that removal of the exocyclic tail in that analog to form tAIP-III D2A (Figure 3) does not reduce the potency of the peptide (IC50 values of 257, 900, 329, and 95.7 pM against AgrC-I, -II, -III, and -IV respectively), making these two peptide analogs the most potent QS inhibitors known to date.[45] The researchers further assessed the ability of these peptides to attenuate virulence factor production and have shown that these peptides inhibit hemolysin and TSST-1 production at similar concentrations to the IC50 values obtained from the reporter strains.[45] To address the lability of the thioester linkage, which is prone to hydrolysis, in these AIP analogs, Blackwell and co-workers devised materials-based strategies to deliver and release these QS modulators in their active form to potential infection sites.[57, 58] Lastly, they have also replaced the thioester linkage with an amide to afford a lactam ring-containing analogs, AIP-III D4A Amide and tAIP-III D2A Amide (Figure 3), and have shown that these analogs maintain their high receptor affinities while enhancing their pharmacological properties, including metabolic and hydrolytic stability as well as water solubility.[56] Combined, these studies highlight the potential of targeting the agr QS circuit in S. aureus as a therapeutic strategy. Future studies in this species should include the administration of agr QS inhibitors in different animal infection models.
Figure 3.

Select synthetic S. aureus AIP-based antagonists synthesized by Williams and co-workers,[40] Novick, Muir and co-workers,[24] and Blackwell and co-workers.[45, 56] Shaded amino acids and groups indicate modification from the native peptide sequence. Ac, Acetyl; Dap, Diaminopropionic acid.
1.1.2. Staphylococcus epidermidis
S. epidermidis, while originally believed to be harmless, has been identified as one of the major causative agents of nosocomial infections.[59] Most commonly, these infections are caused by the insertion of implanted biomedical devices and indwelling catheters. S. epidermidis often attaches to and forms robust biofilms on these devices resulting in chronic infections that are very hard to eradicate due to impaired penetration of antibiotics and decreased effectiveness of the host immune response. Biofilm formation in S. epidermidis is regulated by the agr QS circuit, presenting an opportunity to significantly attenuate S. epidermidis virulence by intercepting this circuitry. Structural analysis of the AIP-I chemical signal used by the S. epidermidis QS circuit was first reported by Otto and co-workers (1998). The chemical signal was identified as an octapeptide containing a thioester linkage between the central cysteine and the C-terminal carboxyl group (Figure 2).[60] The thioester linkage and its three-amino acid tail were found to be required for the activation of the AgrC-I receptor.[32] After the discovery of this first S. epidermidis AIP, two additional agr specificity groups were identified in S. epidermidis, and their AIPs were characterized.[61, 62] It was determined that both AIP-II and AIP-III were 12-amino acid peptides, with a ring size equivalent to AIP-I (Figure 2). The characterization of these two new AIPs allowed the assessment of cross talk between the different S. epidermidis agr specificity groups. It was determined that agr interference occurred between groups –I and –II, and –I and –III, suggesting that these groups are in competition for the same niches and resources.[62]
Modification of the AIP-I pheromone was first performed by Otto and co-workers (Figure 4), where it was discovered that the thioester linkage was critical for agonistic activity (2001). In this study the S. epidermidis pheromone and its modified derivatives were tested as inhibitors against the S. aureus agr QS circuit. The native S. epidermidis peptide was found to be effective at inhibiting delta-toxin expression by S. aureus subgroups -I, -II, and -III, while it showed no effect on subgroup –IV.[63] In a more recent study to identify possible S. epidermidis QS modulators, the SAR of S. epidermidis AIP-I was assessed by Blackwell and co-workers.[64] Through alanine and D-amino acid substitution screening they determined that Tyr7, Phe8, and Ala5, in addition to the thioester linkage, were all critical for S. epidermidis AgrC-I recognition.[64] Furthermore, they found that Val3 was critical for activation of the S. epidermidis AgrC-I receptor, and that Asp1 and Ser6 could be modified to have increased agonistic activity compared to the native peptide.[64] By combining these observations, Blackwell and co-workers revealed several non-native QS modulators with one analog acting as a potent S. epidermidis agr-I biofilm formation inhibitor (IC50 2.08 nM) (Figure 4).[64] Overall, these studies emphasize the potential of targeting the agr QS circuit as a means to prevent the formation of S. epidermidis biofilms on surgical implants and highlight the importance of developing pan-group S. epidermidis QS modulators.
Figure 4.

Select synthetic S. epidermidis AIPs. The lactone and lactam derivatives were designed by Otto and co-workers, and displayed reduced agonistic activity compared to the native AIP-I peptide.[63] AIP-I D1AS6AV3A and AIP-1 D1AS6AV3AA5S were determined to be pan group AgrC inhibitors, while AIP-I D1AS6A was found to be a non-native agonist with improved potency over the native peptide.[64] Shaded amino acids indicate modification from the native peptide sequence. Dap, Diaminopropionic acid. * EC50 value not available.
1.2. Enterococcus
Enterococci are Gram-positive commensal bacteria, known to inhabit soil or the alimentary canals of humans and animals. Within humans, enterococci are predominantly found within the intestines and colon, however some species have been noted to colonize the female genital tract.[65] Many species of enterococci, mainly Enterococcus faecalis and Enterococcus faecium, are opportunistic pathogens and enterococci as a genus are the third most commonly isolated nosocomial pathogen.[66] In a clinical setting, enterococci are responsible for a wide range of infections including endocarditis, bacteremia, urinary tract infections, and pelvic and intra-abdominal infections.[67] These infections are caused by a wide range of enterococcal virulence factors, including adhesins, hemolysins, gelatinases, and biofilm formation.[68] One of the most notable traits of enterococci is their high level of intrinsic and acquired resistance to many antibiotics. Among these is an acquired resistance to the last resort antibiotic, vancomycin, which is generally used to treat multi-drug resistant pathogens like Clostridium difficile and MRSA.[69] The rising trend in vancomycin resistance among enterococci, coupled with the observed transfer of these antibiotic resistance genes to MRSA, makes enterococci a rapidly increasing threat in the clinical setting.[70, 71]
Among the various enterococcal species responsible for hospital infections, a QS circuit responsible for the regulation of virulence factors has only been characterized in E. faecalis. This QS circuit, dubbed the fsr circuit, possesses shared homology with the staphylococcal agr QS system.[72, 73] The fsr system possesses a regulatory locus comprised of four different genes, FsrA, FsrB, FsrC, and FsrD, all of which possess analogous roles to their agr counterparts. The centerpiece of this QS system is the gelatinase biosynthesis activating pheromone (GBAP), an 11-amino acid cyclic peptide possessing a lactone ring between the hydroxyl group of Ser3, and the α-carboxyl group found at the carboxy-terminus of the peptide (Figure 5).[74] FsrD was found to be the GBAP propeptide, as decreased FsrD translation resulted in decreased production of GBAP.[75] FsrD is processed and cyclized by FsrB before being exported from the cell as the mature GBAP cyclic peptide. Upon reaching an extracellular threshold concentration, the GBAP binds and activates FsrC, a transmembrane histidine kinase receptor that phosphorylates the response regulator, FsrA, to trigger the QS response.[74, 76, 77] This GBAP-mediated fsr QS system is responsible for the regulation of many enterococcal virulence factors including gelatinase, serine protease, biofilm formation, and other extracellular proteases, making this system a key target for the attenuation of E. faecalis virulence.[68]
Figure 5.

The gelatinase biosynthesis activating pheromone (GBAP) utilized by the fsr QS circuit in E. faecalis and several synthetic peptide analogs. Lactam-GBAP, thiolactone-GBAP, and CH2-lactone-GBAP all displayed agonistic activity, while disulfide-GBAP did not show any agonistic or antagonistic activity.[77] ZBzl-YAA5911 is the most potent fsr QS antagonist identified thus far.[78] Shaded amino acids and groups indicate modification from the native peptide sequence. Z, benzyloxycarbonyl; Bzl, benzyl; Dap, Diaminopropionic acid; Hse, homoserine. * EC50 value not available
The role of the fsr QS circuitry in E. faecalis pathogenicity has sparked several studies aimed at uncovering the SAR of the GBAP peptide with the hope of developing novel synthetic GBAP-based analogs that could be used to interrupt QS, and in turn virulence, in E. faecalis. Alanine scanning analysis of GBAP was utilized to identify key moieties involved in GBAP agonistic activity.[77] Through the alanine scanning, it was determined that GBAP’s entire ring region is involved in agonistic activity, while the tail region side chains are not strictly recognized. Specifically, Phe7 and Trp10 were identified as critical residues for receptor binding, as substitution of these residues almost completely abolished any activity against the FsrC receptor.[77] The importance of ring size and bridge chemistry to agonistic activity were also evaluated through the modification of the lactone linker (Figure 5). These experiments revealed that the ring size can be adjusted without a loss of agonistic activity, and that the carbonyl of the macrocycle linker is critical for agonistic activity. NMR analysis of the ring region showed that it is highly compact, taking on a hairpin-like fold. Combined, the biological assays and structural studies revealed that the Phe7 side chain was facing outside of the cyclic ring, and that this side chain is likely directly involved in binding to the FsrC receptor. In addition, the Trp10 side chain was found to contribute to both receptor binding and conformational stability of the cyclic peptide in its compact form. Lastly, it was found that the negatively charged π-electron cloud located on the aromatic rings of Trp10 is critical for agonistic activity of the peptide. Overall, the initial alanine scanning analysis of GBAP identified several peptides with lessened agonistic activity, however, none exhibited antagonistic activity against FsrC. In a later study by Nakayama and co-workers, a novel approach was utilized, termed reversed-alanine scanning, to identify possible FsrC antagonists.[78] In this approach, a receptor binding scaffold was created that possessed alanine substitutions along the entire GBAP region, with the exception of the Phe7 and Trp10 residues that were identified as crucial for receptor binding. Using an N-terminal benzyloxycarbonyl protected receptor binding scaffold, several novel peptides capable of FsrC inhibition were identified, with the most potent analog being [Ala5,9,11]Z-GBAP. Further development of [Ala5,9,11]Z-GBAP lead to the identification of [Tyr(Bzl)5,Ala9,11]Z-GBAP, later renamed ZBzl-YAA5911, which exhibited the highest inhibitory potency, with an IC50 value in the low nM range (IC50 26.2 nM) (Figure 5). ZBzl-YAA5911 was further tested in an aphakic rabbit endophthalmitis model and was found to significantly reduce retinal damage by enterococcal endophthalmitis through the inhibition of gelatinase expression. Further research would likely focus on the inhibition of the enterococcal fsr QS circuit, as this would permit the attenuation of enterococcal virulence without any resulting resistance and little to no side effects to the patients or their natural microflora, which should prove extremely useful in clinical settings.
1.3. Clostridium
Clostridium species are Gram-positive anaerobes that are commonly found in soil and sewage, but are also known to inhabit the human gastrointestinal tract.[79] These bacteria are known to form a durable endospore to survive in oxygen-rich and high temperature environments, and they are regarded among some of the most widely distributed pathogens in nature.[79, 80] In human infections, clostridium species are responsible for a wide range of diseases including gas gangrene, necrotizing enterocolitis, severe pseudomembranous colitis, and botulism.[79, 81–83] Recently, several different regulators of toxin production and endospore formation have been discovered in clostridium species.[84] Among these is an agr-like QS system that makes use of a cyclic peptide signaling molecule.[85, 86]
Several clostridium species including Clostridium perfringens, Clostridium botulinin, and Clostridium acetobutylicum contain agr QS-associated genes analogous to those seen within staphylococci.[84, 85, 87–89] All of these species encode unique analogs of AgrB and AgrD, which encode an integral membrane endopeptidase and a propeptide that will later be processed into the autoinducing cyclic peptide. Of these three species, only C. acetobutylicum possesses AgrC and AgrA, the transmembrane histidine kinase receptor and response regulator of the agr QS circuit, respectively.[88] It has been suggested that clostridium species like C. perfringens that lack the AgrC and AgrA components have the VirS/VirR two-component regulatory system, which has an analogous role to the AgrC/AgrA two-component system found within most agr-based QS circuits.[85, 90, 91] Several studies have highlighted the important role AgrB plays in toxin production and endospore formation of these clostridium species, revealing that a loss of AgrB significantly attenuates these aspects of clostridium pathogenesis.[87, 88, 92] As sporulation and toxin production are important not only for clostridium pathogenesis, but also for their survival, targeting the agr-based QS circuit, likely using cyclic peptide inhibitors derived from the native signaling peptide, could be utilized as a therapeutic pathway for the treatment and prevention of clostridium-based infections.
To date, no confirmed structures of the cyclic peptides utilized by the clostridium agr QS circuits have been published.[85, 88] However, sequence alignment of the AgrDs along with several synthetic peptides (Figure 6) that were able to activate the QS circuitry in C. perfringens and C. acetobutylicum provided generalized structural information about the native AIPs, including a proposed thiolactone-containing macrocyclic component. In both clostridium species, the agonistic synthetic peptides (Figure 6) were found to activate toxin production, sporulation, and granulose formation. More importantly, one of the synthetic peptides, peptide 6-R (Figure 6), was able to reduce or completely block toxin production in several different C. perfringens strains.[89] The ability to modulate many facets of clostridium pathogenesis using QS interference highlights the potential of cyclic peptide analogs that target non-essential pathways in clostridium species as therapeutic agents that are not likely to pressure the bacteria to develop resistance.
Figure 6.

Select synthetic Clostridium AIPs found to have either agonistic or antagonistic activity against the C. perfringens or C. acetobutylicum QS circuits. Peptides 8-R and 5-R were found to have agonistic activity on the C. perfringens QS circuit, while Peptide 6-R was found to be a C. perfringens QS antagonist.[89] Peptide R6T0 was determined to have high agonistic activity on the C. acetobutylicum QS circuit.[88]
1.4. Lactobacillus
Lactic acid bacteria are part of the genus Lactobacillus and are essential to the human gastrointestinal microflora, providing major health benefits by breaking down food and synthesizing essential vitamins.[93–95] Some species of lactobacillus use QS to enhance their ability to adapt to diverse environments and compete with other bacteria for various ecological niches.[96, 97] One such species is Lactobacillus plantarum, a bacterium that naturally colonizes the human gastrointestinal tract, which was found to possess multiple QS two-component regulatory systems (QS-TCSs) that include a transmembrane histidine kinase and response regulator.[96–99] One of these QS-TCSs is homologous to the staphylococcal agr and the enterococci fsr QS circuits and has been identified as the Lactobacillus agr-like module (lam) with four genes: LamA, LamB, LamC, and LamD that play analogous role to their agr and fsr counterparts.[99] The lam system of L. plantarum is proposed to begin with the synthesis of the AIP precursor peptide, LamD, which undergoes processing by the LamB protein. Maturation of the chemical signal occurs when the AIP is cleaved from LamD to form a cyclic thiolactone peptide (Figure 7). LamC is the histidine kinase receptor and LamA acts as the cognate response regulator of this QS circuit. Unlike the aforementioned staphylococci and enterococci, which use QS to trigger virulence, lactobacilli assess their cell density to regulate the production of adhesins used for biofilm formation as well as enzymes involved in the utilization of different sugars.[97, 99] Triggering the signaling cascade in L. plantarum by the AIP signal has been shown to affect the transcription of a set of late-phase genes that include adhesins, surface polysaccharide biosynthesis, cell envelope and sugar utilization enzymes. Moreover, de Vos and co-workers reported that LamA-null L. plantarum strains exhibited significantly reduced adherence ability, and down regulation of sugar metabolism.[99] Lastly, it was found that LamA mutant strains had up-regulated biosynthesis of surface polysaccharides. Although not much is known about the lam QS circuit in L. plantarum, the mutually symbiotic role of this bacterium in the human body provides an opportunity to improve human health by activating the lam QS circuitry and thus promoting productive processes that can benefit the human host.
Figure 7.

Structure of the AIP utilized by L. plantarum.
Thus far this review has demonstrated that in prokaryotic systems cyclic peptide scaffolds are used to modulate various signal transduction pathways that are responsible for a variety of group behaviors including biofilm formation, virulence and pathogenicity. Furthermore, it has been established that these peptides can be modified to afford scaffolds with enhanced agonistic or antagonistic properties. Despite their increased complexity, eukaryotic cyclic peptide scaffolds have also been extensively studied. In eukaryotic systems these cyclic peptide scaffolds have a more essential role, regulating the overall function of multicellular organisms. Within eukaryotic systems, modifications of these peptides have been explored in order to increase their stability and potential therapeutic utility. In the next section of this review, we will discuss several eukaryote cyclic peptide hormones and the main SAR analyses that were conducted to modulate their pharmacological properties.
2. Cyclic Peptide Modulators of Eukaryote Signal Transduction Pathways
The dynamic functions and processes occurring in multicellular organisms require constant activation and deactivation of many signal transduction pathways.[100] Hormones are the chemical messengers that mediate various signal transduction pathways and regulate the overall functions of multicellular organisms. The majority of hormones produced in multicellular organisms are released into the bloodstream and are part of the endocrine system, which consists of the following hormone-producing glands: liver, pineal, pituitary, hypothalamus, parathyroid, testes, ovaries, adrenal and thymus.[101] These glands secrete various hormones into the bloodstream so that the hormones can reach their target tissues or organs in a timely manner.
There are three structural groups of hormones: fat or phospholipid derived hormones (eicosanoids and steroids), amino acid derived hormones (monoamine derivatives), and polypeptide derived hormones (peptides and protein).[101] In general, all hormones have relatively short half-lives since they have a dynamic role of activating and deactivating various signal transduction pathways. Eicosanoids, polypeptide, and amino acid derived hormones have relatively short half-lives (seconds to minutes), while steroid hormones have moderately short half-lives (minutes to hours). Hormones are present in the blood at low concentrations, and the process of maintaining the proper concentration is referred to as the “feedback loop” where secretion of hormones can be either increased or decreased in response to changes in function. Most hormones are under a negative feedback system to maintain homeostasis where the secretion of hormones is decreased in response to activation of the hormone’s signal transduction pathway. On the other hand, some hormones, such as oxytocin, can operate under a positive feedback system where the secretion of the hormone increases in response to the hormone’s presence.[102] In some cases, certain hormones, such as estrogen, can induce a positive feedback system on other hormones.[103]
Polypeptide derived hormones are the most abundant class of hormones consisting of short to long polypeptide chains. Peptide hormones are translated as nascent polypeptides called preprohormones that contain a signal peptide sequence. According to the signal hypothesis proposed by Blobel and Sabatini, polypeptides contain a signal sequence at the N-terminus that aids in transporting the protein/peptide to the rough endoplasmic reticulum (ER) through the translocon.[104, 105] This signal peptide gets cleaved by proteases in a co-translational step to transport the resulting polypeptide (prohormone) to the ER, where it undergoes further processing, including proper folding and cyclization (Figure 8).[106, 107] Prohormones contain a sequence of amino acids that are required for proper folding but not function. Thus, upon folding, this region gets cleaved to afford the mature hormone.[108] The hormone will then translocate to the trans-Golgi network for packaging and secretion.
Figure 8.
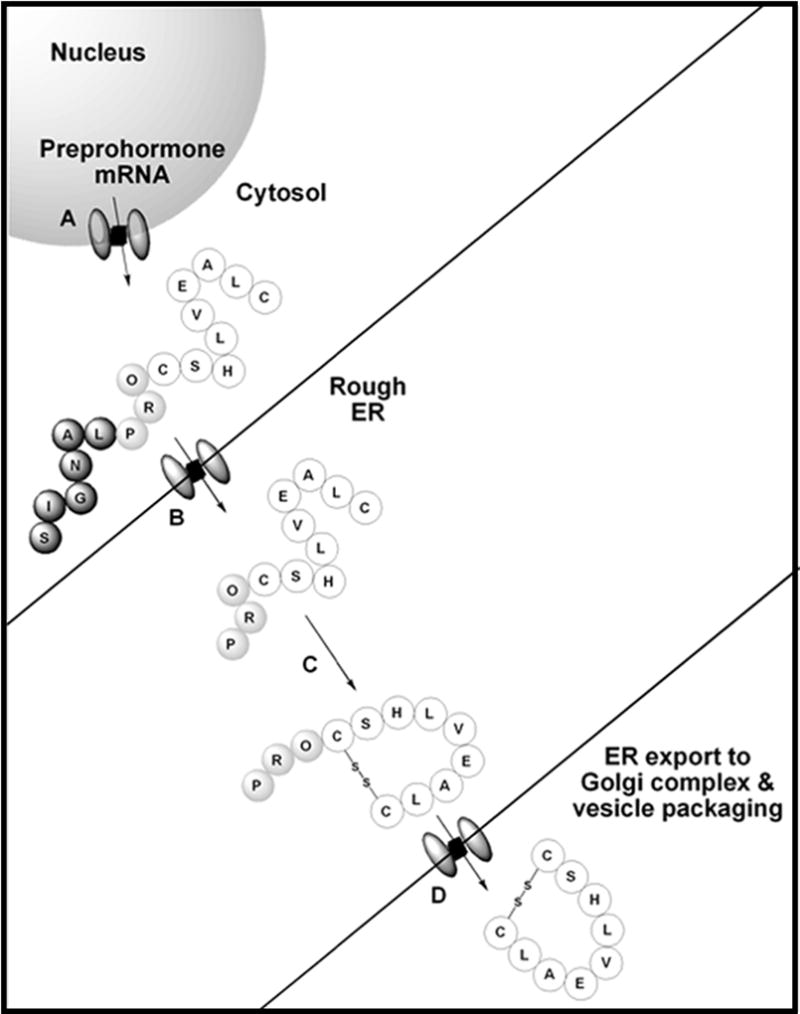
Diagram of hormone maturation process. A: Preprohormone mRNA is translated into preprohormone by the ribosome. B: Preprohormone has signal peptide cleaved as it passes through translocon into the rough ER. C: Prohormone folds into its correct structure in rough ER. D: Cleavage of prohormone into mature hormone prior to be transported to the Golgi complex for packaging and secretion.
Water soluble peptide hormones act as primary messengers because they cannot cross the cell membrane, thus they signal the target cells to initiate signal transduction cascades through the production or absorption of intracellular secondary messengers. Common secondary messengers include calcium, inositol trisphosphate, diacylglycerol, cyclic adenosine monophosphate, and cyclic guanosine monophosphate. The targets for the polypeptide hormones are thus transmembrane receptors bearing a sensory domain located on the extracellular domain of the plasma membrane.[109] Hormones must have high affinity and specificity to their particular receptors because they are secreted in low concentrations and should only activate their desired pathway. In the case of peptide hormones, this means the peptides should possess a relatively rigid structure that enables them to adopt only a small number of conformations. To achieve such structural rigidity, many peptide hormones are constrained by peptide cyclization. From a pharmacological standpoint, peptide cyclization generally results in increased half-life compared to the linear peptide counterpart.[32] This increased metabolic stability is attributed to the lower affinity of cyclic peptides to bind different peptidases.[110–114] That being said, the stability of different cyclic peptides can vary dramatically, for example oxytocin, vasopressin, and somatostatin have relatively short half-lives compared to insulin (Figure 9). This section will focus on a few cyclic peptide hormones, their biological roles, and potential therapeutic utility.
Figure 9.

Various cyclic peptide hormones associated with eukaryotes.
2.1. Somatostatin
Somatostatin was discovered in 1973 and has two biological active isoforms that exist either as a 14-amino acid (SST-14) or 28-amino acid (SST-28) cyclic peptide hormone (Figure 10). SST-28 differs from SST-14 by having an extended N-terminus. SST-28 makes up about 20–30% of somatostatin in the brain and is mainly found in the gastrointestinal tract.[115] SST-14 is the predominant isoform in the brain and is produced in the hypothalamus and pancreas. Somatostatin production starts as a 116-amino acid preprohormone, preprosomatostain, that has its signal peptide cleaved prior to entering the ER to form the 92-amino acid prohormone, prosomatostatin.[116, 117] The bioactive forms of somatostatin are located at the C-terminus of the prohormone and are formed by cleavage of the basic residues of the prohormone in the trans-golgi network.[106, 118] Cyclization of both bioactive forms is the result of an intramolecular disulfide bridge formation that occurs prior to the cleavage of the prohormone.[119] Somatostatin produced in the hypothalamus travels to the anterior pituitary gland where it inhibits the release of growth hormone, prolactin, and thyroid stimulating hormone, while somatostatin produced by the gastrointestinal tract and pancreas inhibits the secretion of various gastrointestinal hormones such as calcitonin, gastrin, insulin, glucagon and vasoactive intestinal peptide.[116, 117] Therefore, somatostatin regulates, directly or indirectly, many functions in the body including cell growth, cell regeneration, and stomach acid production by binding five different somatostatin receptor subtypes (SSTR1-5).[115, 120–122] Both bioactive isoforms of somatostatin can bind all five somatostatin receptor subtypes (SSTR1-5),[120] although with varying affinities.[123] Somatostatin is highly susceptible to endopeptidase degradation and thus has a short half-life of approximately 2–3 minutes.[118, 124] The broad spectrum of processes regulated by somatostatin has sparked numerous investigations aimed at utilizing its therapeutic potential by developing somatostatin-based drugs.
Figure 10.

Bioactive somatostatin isoforms along with a few of their synthetic analogs. Non-natural amino acids are shaded. Lower case letters indicate D-amino acids; D2* is 3-(2-Naphthyl)-D-alanine; Tol is L-Threoninol.
The use of somatostatin therapeutically is limited by its short half-life and has led to the development of analogs that are longer lasting. Structure-function relationship studies of somatostatin revealed that a beta turn formed by residues 7–10 in SST-14 (21–24 in SST-28) is required for receptor binding and that the disulfide bond creates a conformational constraint that stabilizes this conformation.[123] It was later determined that Lys6 and Trp7 in SST-14 (20 and 21 in SST-28) are essential for bioactivity and that the incorporation of D-amino acids in these positions further stabilizes the beta turn. These findings led to the development of the first FDA approved synthetic analog, Octreotide, which is used to treat acromegaly, various pituitary tumors, and pancreatic tumors by inhibiting the release of growth hormone, glucagon, and insulin.[125] Octreotide is a truncated eight-amino acid cyclic peptide containing D-Phe at position 1, D-Trp at position 4, and a L-Threoninol at position 8 (threonine containing an alcohol instead of a carboxylic acid terminus) along with a disulfide bridge between positions 2 and 7 (Figure 10). Octreotide has a longer half-life of 2 hours intravenously and also exists in a long-acting release form administered once every 28 days.[125, 126] A similar long-lasting FDA approved synthetic analog, Lanreotide, is administered once every 7–14 days and is used to treat acromegaly in addition to helping ease symptoms of neuroendocrine tumors. Lanreotide is also an eight-amino acid cyclic peptide with 3-(2-Naphthyl)-D-Ala at position 1, an amidated Thr at position 8, D-Trp at position 4, along with a disulfide bridge between positions 2 and 7 (Figure 10). Octreotide binds to SSTR2, SSTR5 and moderately to SSTR3 while Lanreotide binds to SSTR2 and SSTR5.[123, 125] Further studies revealed that somatostatin receptor agonists could be truncated further, forming a cyclohexapeptide core containing the disulfide bridge.[127] It was later determined that the conformational constraint induced by cyclization was necessary rather than the disulfide bridge itself, thus the potent analog, Seglitide, was identified. Seglitide contains a D-Trp at position 2 linked by an N-methyl Ala to the C-terminus of Phe at the end of the six amino acid sequence (Figure 10).[128] This synthetic analog was found to selectively bind to SSTR2 and led to the development of various cyclohexapeptide analogs.[129] The most successful cyclohexapeptide analog, Pasireotide, has a modified Pro containing an aminoethyl-carbamate connected to L-phenylglycyl, D-Trp, Lys, Tyr(Bzl) and Phe that is linked to the modified Pro (Figure 10). Pasireotide binds all SSTR subtypes except SSTR-4 and was FDA approved in 2012 to treat Cushing disease.[123] So far, long-acting somatostatin analogs have been used to treat various diseases associated with the dysregulation of growth hormone such as acromegaly, Cushing disease, and various neuroendocrine tumors.[122, 130] Current research efforts involving somatostatin analogs are aimed at taking advantage of the role somatostatin plays in inhibiting growth hormone, which is found to be upregulated in all cancer cells, for the development of novel strategies for anti-cancer therapy.
2.2. Insulin
Insulin is a 51-amino acid peptide hormone that is responsible for regulating blood glucose levels by activating the translocation of the glut4 symporter to the surface of the cell. This translocation allows cells to uptake glucose from the blood for energy, thus lowering blood glucose levels. Beta cells located in the islet of Langerhans of the pancreas produce a 110-amino acid pre-prohormone, preproinsulin, that is cleaved by signal peptidases into an 86-amino acid prohormone peptide, proinsulin.[131] Proinsulin folds into a cyclic prohormone containing three disulfide bridges and is then cleaved by prohormone convertases and a carboxypeptidase, forming a monomeric cyclic peptide hormone. The mature insulin hormone consists of two chains: the A chain (21 amino acids) and the B chain (30 amino acids), which are linked by two intermolecular disulfide bridges with an additional intramolecular disulfide bridge present on the A chain. As the concentration of insulin increases inside the beta cells, dimers are formed between the antiparallel beta-sheets located at the C-terminus of the B chain. Three dimers will combine to form a hexamer by coordinating two zinc atoms with the imidazole groups of three His residues located at position B10 that gets packaged with other hexamers into insulin granules.[131] The insulin granules containing the hexameric form are secreted into the blood and as the concentration of insulin granules decreases, the hexameric insulin dissociates to its dimer form, which eventually further dissociates into its active monomeric form. The active monomeric form binds to the insulin receptor located on the surface of cells and initiates a signal transduction cascade intracellularly that allows the entry of glucose into the cell. Hexameric insulin that is secreted into the bloodstream is cleared within an hour and the monomeric form has a half-life of 4–6 minutes.[132]
Therapeutic insulin and its analogs are mass produced using recombinant DNA technology for the treatment of diabetes mellitus. The development of insulin analogs has focused on creating analogs that dissociate either faster or slower than native insulin. Tightly controlled blood glucose levels are required for maintaining homeostasis, thus a combination of rapid-acting and long-lasting insulin analogs are needed to mimic the natural insulin secretion patterns in patients diagnosed with diabetes. The first FDA approved insulin analog, Lispro (Figure 11), is almost identical to native insulin with amino acids ProB28 and LysB29 reversed. This modification decreases the dimerization rate resulting in a rapid-acting insulin analog.[133] The development of Lispro has sparked substantial research efforts focused on shifting the oligomerization equilibrium of insulin to afford fast-acting analogs. For example, Aspart, another FDA approved insulin analog (Figure 11), is an insulin analog where ProB28 was changed to Asp creating a charge repulsion at the dimer interface, leading again to a rapid-acting insulin analog.[134, 135] Another example is Glulisine, a more recent FDA approved insulin analog (Figure 11), that bears two amino acid modifications: AsnB3 to Lys, and LysB29 to Glu. These two modifications alter the oligomerization equilibrium by decreasing the formation of the hexameric form, allowing it to exist as a dimer at pharmacological concentrations. Clinical studies have shown no significant differences between all three rapid-acting analogs with regards to the time of onset.[134, 136]
Figure 11.

Native insulin and various FDA approved analogs. Shaded amino acids and groups indicate modification from the native peptide sequence.
The problem with native insulin and rapid-acting analogs is their relative short half-lives, meaning patients with diabetes would have to receive multiple injections a day to regulate their blood glucose levels. This has led to the development of longer-lasting FDA approved analogs, such as Glargine and Detemir (Figure 11). Glargine is an insulin analog where the AsnA21 was replaced with Gly and two Arg residues were added to the C-terminus of the B chain. The addition of the two Arg residues shifted the pI of the molecule from 5.4 to 6.7 making it more soluble in acidic conditions and less soluble at physiological pH.[137] The substitution of AsnA21 with Gly prevents deamidation caused by the acidic conditions. Overall, these changes cause the molecule to microprecipitate in its hexameric form when injected, as it moves from acidic pH to the physiological neutral pH of the blood, helping maintain basal insulin levels for 24 hours. The acidic environment of Glargine preparations makes it difficult to combine with other insulin analogs, so the development of a long-acting analog capable of being mixed with fast-acting analogs was needed.[138] Detemir has the exact same peptide sequence as native insulin, with the exception of a fatty acid (myristic acid) acylated to the epsilon-amino group of LysB29, and a deleted ThrB30 residue.[139, 140] These modifications allows Detemir to bind to albumin in the blood, which causes it to dissociate slowly. The benefit of Detemir is that it can be combined with other, rapid-acting insulin analogs.
Current research regarding insulin analogs focuses on prolonging the bioactivity of the analogs, which would benefit diabetics by having them inject insulin less frequently.[135, 141, 142] Recently, a new FDA approved ultrastable analog was released to the market: Degludec (Figure 11), which is very similar to Determir, having the same ThrB30 deletion but possessing a different fatty acid, hexadecanedioic acid, attached to the epsilon-amino group of LysB29 by a γ-L-glutamic acid linker.[143] These modifications allows it to exist as dihexamers in pharmacological preparations that form a linear array of hexamers (multihexamer) when injected. These oligomers prolong the dissociation of the analogs to the monomeric form, giving it duration of action of more than 42 hours and allowing diabetic patients to inject it once every couple of days to help maintain basal insulin levels.[144] Researchers have also found an alternative method of delivering insulin in the form of inhalation, creating the first FDA approved ultra-rapid-acting insulin preparation, Afrezza.[145] Afrezza consists of powdered recombinant human insulin containing fumaryl diketopiperazine as an inert excipient. Another area of research that is currently being pursued involves creating thermostable analogs, which would make insulin more available to third-world countries that do not have access to electricity and refrigeration, a requirement for most insulin preparations currently used.[135] From its discovery in 1921, insulin and its derivatives were used to treat and save the lives of millions of diabetic patients worldwide. The projected increase in diabetics provides the driving force to the continuous efforts to develop novel insulin analogs that could be applied to better treat the growing diabetic population by closely mimicking the natural homeostasis.
2.3. Oxytocin
Neurohypophysial hormones refer to hormones secreted by the posterior pituitary (neurohypophysial) gland and are all nine-amino acid peptides containing a disulfide bridge between positons 1 and 6 along with a three-amino acid tail that is α-amidated at the C-terminus.[146, 147] The most studied neurohypophysial hormones are oxytocin and vasopressin. Oxytocin is produced mainly in the hypothalamus, but also produced in non-neuronal cells, such as the retina, Leydig cells, corpus lutea, adrenal medulla, pancreas, thymus and other peripheral tissues. Oxytocin starts as a 121-amino acid precursor polypeptide, where the signal peptide gets cleaved during protein synthesis to form a 102-amino acid prohormone, prooxytocin.[148] This prohormone then folds into its correct structure in the ER while forming multiple disulfide bridges for both oxytocin and its carrier protein neurophysin I.[148] Prooxytocin has a 93-amino acid carrier protein, neurophysin I, that gets cleaved from the prohormone and helps transport the mature oxytocin to the posterior pituitary gland where it gets secreted into the blood. Cleavage of neurophysin I is followed by an amidation step of the terminal glycine residue at the C-terminus creating the α-amidated glycine. Oxytocin has only one disulfide-bridge while neurophysin I has seven. Oxytocin regulates many social and nonsocial functions in the body by binding to the oxytocin receptor located in the uterus and central nervous system.[146, 149] The nonsocial effects of oxytocin include learning, memory, brain development, organ development, aging, appetite, metabolism, and childbirth and lactation in females. In addition to these functions, oxytocin plays a significant role in drug addiction and withdrawal since it regulates the mesolimbic dopamine pathway.[150] The social behaviors influenced by oxytocin include social bonding, depression, suppression of anxiety, and well-being, but it can also enhance fear and anxiety during negative or stressful experiences.[151, 152] In addition, due to the relative similarity to vasopressin, oxytocin can bind the subtype 2 vasopressin receptor (V2) and induce antidiuresis, but with relatively low affinity.[153, 154]
Similar to somatostatin, oxytocin has a short half-life of 3–6 minutes.[155, 156] The cyclic nature of oxytocin was determined to provide the conformational constraint necessary for receptor specificity.[146, 157] In addition, it was determined that the C-terminal region is involved in receptor specificity, thus synthetic analogs are usually designed without major truncations of the tail. NMR studies, along with analog assays, have provided a model for oxytocin:receptor binding where Ile3, Gln4, Pro7, and Leu8 are involved in the initial receptor recognition while Tyr2 and Asn5 are involved in receptor, and thus signaling pathway, activation.[158] The potential to use oxytocin as a therapeutic agent is limited due to its short half-life, yet it is currently being used intravenously or intranasally (longer half-life) to induce childbirth and lactation in women. A problem with using externally administered oxytocin during childbirth or lactation is that it can bind to the vasopressin subtype 2 (V2) receptor and cause antidiuresis, which can lead to hyponatremia.[158] To overcome the aforementioned problem, several synthetic analogs have been made that are longer lasting and more specific to the oxytocin receptor. Demoxytocin, a synthetic analog with similar potency and half-life as oxytocin, was created in the 1960’s to induce labor.[159–162] The benefit of Demoxytocin over oxytocin is that it can be delivered in a buccal tablet form instead of intravenously and is therefore commonly used in European countries. Demoxytocin has a similar structure to oxytocin except that it has a 3-mercaptopropionic acid at position 1 linked by a disulfide bridge to the Cys residue at position 6 (Figure 12). Another synthetic analog, Carbetocin, has a similar binding profile as oxytocin and longer half-life (85–100 minutes) than both oxytocin and Demoxytocin.[163–165] Unlike oxytocin and Demoxytocin, Carbetocin is used to control postpartum hemorrhaging for both humans and animals in certain countries, not including the U.S.[165, 166] This analog has 1-butanoic acid in position 1 that is linked to Cys at position 6 with a modified Tyr at position 2 containing a methoxy group on its side chain (Figure 12). The conversion of the liable disulfide bond to a more stable C-S bond through the incorporation of 1-butanoic acid at position 1, along with the addition of the methoxy group on Tyr at position 2, are responsible for the longer half-life of Carbetocin compared with oxytocin. This enhanced stability is attributed to reduced rate of macrocycle ring opening and prevention of chymotrypsin degradation, respectively.[167, 168] A recent analog that is currently in clinical trials, Merotocin, shows great potential for inducing lactation in mothers that fail to lactate after preterm birth.[158] The benefit of Merotocin over oxytocin is that it is highly selective to the oxytocin receptor and does not interact with the V2 receptor. Merotocin has 1-butanoic acid at position 1 that is linked to the Cys at position 6 with an N-4-fluorobenzyl glycine at position 7 (Figure 12). The improved selectivity and enhanced binding affinity of Merotocin (EC50 value of 0.08 nM compared with 2.3 nM for oxytocin) are attributed to the replacement of Pro with N-4-fluorobenzyl glycine at position 7.[158] Future studies are needed to shed light on how this modification increases receptor selectivity. Oxytocin and its analogs have many therapeutic uses related to childbirth and lactation, but it is also being investigated for its role in fighting drug addiction, social anxiety, social bonding, autism, and schizophrenia.[150, 151, 169, 170]
Figure 12.

Oxytocin and various synthetic analogs. Shaded amino acids and groups indicate modification from the native peptide sequence.
2.4. Vasopressin
Vasopressin, also known as antidiuretic hormone, is a neurohypophysial hormone that differs from oxytocin by two amino acids (Figure 9). Vasopressin is mainly known to regulate vasoconstriction, blood pressure, and water retention, but recently has been found to affect social behavior related to stress, memory, and anxiety.[171, 172] The hormone is produced as a 162-amino acid preprohormone that becomes the 143-amino acid prohormone when the signal sequence is cleaved. Provasopressin contains a 95-amino acid carrier protein (neurophysin II), vasopressin and a 39-amino acid glycosylated peptide (copeptin).[173] Just like prooxytocin, provasopressin folds into its correct structure, including the formation of eight disulfide bridges (seven in neurophysin II and one in vasopressin), in the ER. Cleavage of copeptin and neurophysin II is followed by an amidation step of the terminal glycine residue at the C-terminus creating the α-amidated glycine. Vasopressin binds to the vasopressin receptor, which has three subtypes (V1-3) that differ in their distribution within the body.[172] V1 receptors are found on vascular smooth muscles cells while V2 receptors are found in the kidneys, and V3 receptors are found in the pituitary gland.[174] Vasopressin can also bind to oxytocin receptors with an equal affinity as oxytocin and induce uterine contractions.[174, 175] Vasopressin differs from somatostatin and oxytocin by having a longer half-life of 10–35 minutes because it can only be cleared in the kidney and liver.[174]
Vasopressin’s role in water retention and blood pressure has made it a useful therapeutic for bleeding disorders, organ transplants, treating septic shock and cardiac arrest.[176] The relatively short half-life of vasopressin, like other peptide hormones, limits its therapeutic uses so the development of longer lasting analogs has been pursued.[174, 177, 178] Felypressin (Octapressin) is a synthetic vasopressin analog that was developed in 1960 and exhibited a localized vasoconstriction effect. Felypressin therapeutic use is limited, except as a vasoconstrictor adjunct used with local anesthetics typically associated with dentistry, because of its selectivity to V1 receptors.[179–181] Felypressin differs from vasopressin in positions 2 and 8 where it has Phe and Lys residues, respectively (Figure 13). Desmopressin, another synthetic analog designed to treat specific diseases, is selective to V2 receptors, which enables its use to retain water in patients without exerting a vasoconstriction effect.[182, 183] Desmopressin was developed in the mid 1960’s and was originally used to treat diabetes insipidus.[183–187] However, it was later found to be effective for the treatment of bleeding disorders like hemophilia A and von Willebrand disease due to the V2 receptors involvement in osmoregulation and the production of blood clotting factors.[183, 184] Desmopressin has a longer half-life of around 55 minutes and is very similar to vasopressin except that it has a deaminated N-terminus, along with a D-Arg at position 8 (Figure 13).[188] The deamination of Cys at position 1 was found to increase its solubility in lipids, which increased its antidiuretic effect and duration of action while the substitution with D-Arg diminished its affinity towards the V1 receptor reducing its vasoconstriction abilities.[189] Terlipressin is commonly used to treat septic shock, hepatorenal syndrome (kidney failure from cirrhosis), and esophageal varices.[190–192] This analog differs from vasopressin by the addition of three Gly residues at the N-terminus prior to the conserved Cys that occupies the first position in vasopressin, making this an 11-amino acid analog. In addition, Terlipressin contains an Arg to Lys substitution in the eighth position of vasopressin (11th position in Terlipressin; Figure 13). Terlipressin is not approved for use in the U.S, but is used in other countries.[193] Selepressin, a recent analog currently in phase 2 clinical trials, shows great potential as a therapeutic for treating septic shock.[194, 195] Selepressin differs from vasopressin by possessing Phe at position 2, Ile at position 3, homoglutamine at positon 4, and an ornithine at position 8 with an isopropyl group attached to the side chain amine (Figure 13). Synthetic analogs of vasopressin are being used therapeutically to treat septic shock, bedwetting, cardiac arrest, diabetes insipidus, von Willebrand disease and mild hemophilia A.[174, 196–198] Many analogs have been synthesized but only a limited number of analogs are used therapeutically, demonstrating the need to develop more therapeutically-relevant analogs given that there are three vasopressin subtype receptors that have different functions and distribution in the body. More research in exploiting these receptors through the development of selective analogs could possibly lead to better treatment options for diseases associated with water retention, bleeding disorders, in addition to social illnesses related to stress, memory, and anxiety.
Figure 13.

Vasopressin and various synthetic analogs. Shaded amino acids and groups indicate modification from the native peptide sequence. Lower case letters indicate D-amino acids. Hgn, homoglutamine.
CONCLUSION
Cyclic peptide scaffolds are widely used as key components in essential and non-essential signal transduction pathways found in both prokaryotic and eukaryotic organisms. The widespread utilization of cyclic peptides by nature emphasizes the pharmacological advantages of these privileged scaffolds and highlights their therapeutic potential. In the case of bacterial pathogens, such as staphylococcus, enterococcus, and clostridium species, these cyclic peptides have the potential to be applied as novel therapeutics aimed to inhibit non-essential pathways responsible for virulence and pathogenicity. In symbiotic species, such as lactobacillus, these peptides can be used to enhance the beneficial activity of commensal, QS utilizing, species. The modification of cyclic peptides in eukaryotic systems has been shown to result in increased half-life of important hormones and enhanced receptor binding affinities, creating drugs that are longer lasting and faster acting than their parent signals. Overall, we believe that the enormous potential of cyclic peptides in various practical applications will lead to continuously growing research efforts aimed at delineating the SARs of these scaffolds and the development of novel classes of therapeutics.
Table 1.
Various organisms and the processes governed by their cognate cyclic signaling peptides, as discussed in this review.
| Species | Cyclic Peptide |
Process governed by cyclic peptides |
|---|---|---|
|
| ||
| Prokaryotes | ||
| Staphylococcus aureus | AIP | Toxin and exoprotein secretion, virulence factor production |
| Staphylococcus epidermidis | AIP | Biofilm formation, virulence factor production |
| Enterococcus faecalis | GBAP | Gelatinase biosynthesis |
| Clostridium perfringens | AIP | Toxin production, sporulation, granulose formation |
| Clostridium acetobutylicum | AIP | Toxin production, sporulation, granulose formation |
| Lactobacillus plantarum | AIP | Adhesin and enzyme production |
| Eukaryotes | ||
| Mammalian | Somatostatin | Inhibition of hormone secretion in the pituitary gland, gastrointestinal tract, and pancreas |
| Mammalian | Insulin | Regulation of blood glucose levels |
| Mammalian | Oxytocin | Regulation of some social / nonsocial behaviors and regulation of the mesolimbic dopamine pathway |
| Mammalian | Vasopressin | Regulation of vasoconstriction, blood pressure, water retention, and some social behaviors |
Acknowledgments
The Nevada INBRE (NIH GM103440) is acknowledged for the generous support of research in our laboratory.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interest.
References
- 1.Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14(9):1027–1047. [PubMed] [Google Scholar]
- 2.Roxin A, Zheng G. Flexible or fixed: a comparative review of linear and cyclic cancer-targeting peptides. Future Med Chem. 2012;4(12):1601–1618. doi: 10.4155/fmc.12.75. [DOI] [PubMed] [Google Scholar]
- 3.Joo SH. Cyclic peptides as therapeutic agents and biochemical tools. Biomol Ther. 2012;20(1):19–26. doi: 10.4062/biomolther.2012.20.1.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji G, Beavis RC, Novick RP. Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc Natl Acad Sci U S A. 1995;92(26):12055–12059. doi: 10.1073/pnas.92.26.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 6.Ng W-L, Bassler BL. Bacterial quorum-sensing network architectures. Annu Rev Genet. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y-H, Tian X. Quorum sensing and bacterial social interactions in biofilms. Sensors. 2012;12(3):2519–2538. doi: 10.3390/s120302519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55(1):165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 9.Parsek MR, Greenberg E. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol. 2005;13(1):27–33. doi: 10.1016/j.tim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Nealson K, Hastings JW. Bacterial bioluminescence: its control and ecological significance. Microbiol Rev. 1979;43(4):496–518. doi: 10.1128/mr.43.4.496-518.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Visick KL, Foster J, Doino J, McFall-Ngai M, Ruby EG. Vibrio fischeri lux genes play an important role in colonization and development of the host light organ. J Bacteriol. 2000;182(16):4578–4586. doi: 10.1128/jb.182.16.4578-4586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi S, Greenberg E. The C-terminal region of the Vibrio fischeri LuxR protein contains an inducer-independent lux gene activating domain. Proc Natl Acad Sci U S A. 1991;88(24):11115–11119. doi: 10.1073/pnas.88.24.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerdt JP, Blackwell HE. Competition studies confirm two major barriers that can preclude the spread of resistance to quorum-sensing inhibitors in bacteria. ACS Chem Biol. 2014;9(10):2291–2299. doi: 10.1021/cb5004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bassler BL. How bacteria talk to each other: regulation of gene expression by quorum sensing. Curr Opin Microbiol. 1999;2(6):582–587. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 15.Carrolo M, Pinto FR, Melo-Cristino J, Ramirez M. Pherotypes are driving genetic differentiation within Streptococcus pneumoniae. BMC Microbiol. 2009;9(1):191–200. doi: 10.1186/1471-2180-9-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberhard A, Burlingame A, Eberhard C, Kenyon G, Nealson K, Oppenheimer N. Structural identification of autoinducer of Photobacterium fischeri luciferase. Biochemistry. 1981;20(9):2444–2449. doi: 10.1021/bi00512a013. [DOI] [PubMed] [Google Scholar]
- 17.Fuqua C, Parsek MR, Greenberg EP. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu Rev Genet. 2001;35(1):439–468. doi: 10.1146/annurev.genet.35.102401.090913. [DOI] [PubMed] [Google Scholar]
- 18.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW. Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc Natl Acad Sci U S A. 1999;96(4):1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng H, Novick R, Kreiswirth B, Kornblum J, Schlievert P. Cloning, characterization, and sequencing of an accessory gene regulator (agr) in Staphylococcus aureus. J Bacteriol. 1988;170(9):4365–4372. doi: 10.1128/jb.170.9.4365-4372.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novick R, Projan S, Kornblum J, Ross H, Ji G, Kreiswirth B, Vandenesch F, Moghazeh S. The agr P2 operon: An autocatalytic sensory transduction system in Staphylococcus aureus. Mol Gen Genet. 1995;248(4):446–458. doi: 10.1007/BF02191645. [DOI] [PubMed] [Google Scholar]
- 21.Ji G, Beavis R, Novick RP. Bacterial interference caused by autoinducing peptide variants. Science. 1997;276(5321):2027–2030. doi: 10.1126/science.276.5321.2027. [DOI] [PubMed] [Google Scholar]
- 22.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 23.Lyon GJ, Wright JS, Christopoulos A, Novick RP, Muir TW. Reversible and specific extracellular antagonism of receptor-histidine kinase signaling. J Biol Chem. 2002;277(8):6247–6253. doi: 10.1074/jbc.M109989200. [DOI] [PubMed] [Google Scholar]
- 24.Lyon GJ, Wright JS, Muir TW, Novick RP. Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry. 2002;41(31):10095–10104. doi: 10.1021/bi026049u. [DOI] [PubMed] [Google Scholar]
- 25.Ji G, Pei W, Zhang L, Qiu R, Lin J, Benito Y, Lina G, Novick RP. Staphylococcus intermedius produces a functional agr autoinducing peptide containing a cyclic lactone. J Bacteriol. 2005;187(9):3139–3150. doi: 10.1128/JB.187.9.3139-3150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster T. In: Medical Microbiology. 4. Baron S, editor. Galveston (TX): 1996. [Google Scholar]
- 27.Geisinger E, George EA, Muir TW, Novick RP. Identification of ligand specificity determinants in AgrC, the Staphylococcus aureus quorum-sensing receptor. J Biol Chem. 2008;283(14):8930–8938. doi: 10.1074/jbc.M710227200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saenz HL, Augsburger V, Vuong C, Jack RW, Götz F, Otto M. Inducible expression and cellular location of AgrB, a protein involved in the maturation of the staphylococcal quorum-sensing pheromone. Arch Microbiol. 2000;174(6):452–455. doi: 10.1007/s002030000223. [DOI] [PubMed] [Google Scholar]
- 29.Thoendel M, Horswill AR. Identification of Staphylococcus aureus AgrD residues required for autoinducing peptide biosynthesis. J Biol Chem. 2009;284(33):21828–21838. doi: 10.1074/jbc.M109.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Peptide signaling in the staphylococci. Chem Rev. 2011;111(1):117–151. doi: 10.1021/cr100370n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang B, Zhao A, Novick RP, Muir TW. Key driving forces in the biosynthesis of autoinducing peptides required for staphylococcal virulence. Proc Natl Acad Sci U S A. 2015;112(34):10679–10684. doi: 10.1073/pnas.1506030112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otto M, Süßmuth R, Vuong C, Jung G, Götz F. Inhibition of virulence factor expression in Staphylococcus aureus by the Staphylococcus epidermidis agr pheromone and derivatives. FEBS Lett. 1999;450(3):257–262. doi: 10.1016/s0014-5793(99)00514-1. [DOI] [PubMed] [Google Scholar]
- 33.Chan WC, Coyle BJ, Williams P. Virulence Regulation and Quorum Sensing in Staphylococcal Infections: Competitive AgrC Antagonists as Quorum Sensing Inhibitors. J Med Chem. 2004;47(19):4633–4641. doi: 10.1021/jm0400754. [DOI] [PubMed] [Google Scholar]
- 34.Chambers HF, DeLeo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7(9):629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Björklind A, Arvidson S. Mutants of Staphylococcus aureus affected in the regulation of exoprotein synthesis. FEMS Microbiol Lett. 1980;7(3):203–206. [Google Scholar]
- 36.Janzon L, Löfdahl S, Arvidson S. Evidence for a coordinate transcriptional control of alpha-toxin and protein A synthesis in Staphylococcus aureus. FEMS Microbiol Lett. 1986;33(2–3):193–198. [Google Scholar]
- 37.Janzon L, Löfdahl S, Arvidson S. Identification and nucleotide sequence of the delta-lysin gene, hld, adjacent to the accessory gene regulator (agr) of Staphylococcus aureus. Mol Gen Genet. 1989;219(3):480–485. doi: 10.1007/BF00259623. [DOI] [PubMed] [Google Scholar]
- 38.Recsei P, Kreiswirth B, O'reilly M, Schlievert P, Gruss A, Novick R. Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol Gen Genet. 1986;202(1):58–61. doi: 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- 39.Jarraud S, Lyon G, Figueiredo A, Gérard L, Vandenesch F, Etienne J, Muir T, Novick R. Exfoliatin-Producing Strains Define a Fourthagr Specificity Group in Staphylococcus aureus. J Bacteriol. 2000;182(22):6517–6522. doi: 10.1128/jb.182.22.6517-6522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDowell P, Affas Z, Reynolds C, Holden MT, Wood SJ, Saint S, Cockayne A, Hill PJ, Dodd CE, Bycroft BW, Chan WC, Williams P. Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus. Mol Microbiol. 2001;41(2):503–512. doi: 10.1046/j.1365-2958.2001.02539.x. [DOI] [PubMed] [Google Scholar]
- 41.Geisinger E, Muir TW, Novick RP. Agr receptor mutants reveal distinct modes of inhibition by staphylococcal autoinducing peptides. Proc Natl Acad Sci U S A. 2009;106(4):1216–1221. doi: 10.1073/pnas.0807760106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarraud S, Mougel C, Thioulouse J, Lina G, Meugnier H, Forey F, Nesme X, Etienne J, Vandenesch F. Relationships between Staphylococcus aureus genetic background, virulence factors, agr groups (Alleles), and human disease. Infect Immun. 2002;70(2):631–641. doi: 10.1128/IAI.70.2.631-641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holtfreter S, Grumann D, Schmudde M, Nguyen HTT, Eichler P, Strommenger B, Kopron K, Kolata J, Giedrys-Kalemba S, Steinmetz I, Witte W, Broker BM. Clonal distribution of superantigen genes in clinical Staphylococcus aureus isolates. J Clin Microbiol. 2007;45(8):2669–2680. doi: 10.1128/JCM.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Limbago B, Fosheim GE, Schoonover V, Crane CE, Nadle J, Petit S, Heltzel D, Ray SM, Harrison LH, Lynfield R, Dumyati G, Townes JM, Schaffner W, Mu Y, Fridkin SK. Characterization of Methicillin-Resistant Staphylococcus aureus Isolates Collected in 2005 and 2006 from Patients with Invasive Disease: a Population-Based Analysis. J Clin Microbiol. 2009;47(5):1344–1351. doi: 10.1128/JCM.02264-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J Am Chem Soc. 2013;135(21):7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]
- 46.Goerke C, Kümmel M, Dietz K, Wolz C. Evaluation of intraspecies interference due to agr polymorphism in Staphylococcus aureus during infection and colonization. J Infect Dis. 2003;188(2):250–256. doi: 10.1086/376450. [DOI] [PubMed] [Google Scholar]
- 47.Gordon CP, Williams P, Chan WC. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J Med Chem. 2013;56(4):1389–1404. doi: 10.1021/jm3014635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott RJ, Lian LY, Muharram SH, Cockayne A, Wood SJ, Bycroft BW, Williams P, Chan WC. Side-chain-to-tail thiolactone peptide inhibitors of the staphylococcal quorum-sensing system. Bioorg Med Chem Lett. 2003;13(15):2449–2453. doi: 10.1016/s0960-894x(03)00497-9. [DOI] [PubMed] [Google Scholar]
- 49.Lyon GJ, Mayville P, Muir TW, Novick RP. Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase, AgrC. Proc Natl Acad Sci U S A. 2000;97(24):13330–13335. doi: 10.1073/pnas.97.24.13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright JS, Lyon GJ, George EA, Muir TW, Novick RP. Hydrophobic interactions drive ligand-receptor recognition for activation and inhibition of staphylococcal quorum sensing. Proc Natl Acad Sci U S A. 2004;101(46):16168–16173. doi: 10.1073/pnas.0404039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.George EA, Novick RP, Muir TW. Cyclic peptide inhibitors of staphylococcal virulence prepared by Fmoc-based thiolactone peptide synthesis. J Am Chem Soc. 2008;130(14):4914–4924. doi: 10.1021/ja711126e. [DOI] [PubMed] [Google Scholar]
- 52.Johnson JG, Wang B, Debelouchina GT, Novick RP, Muir TW. Increasing AIP Macrocycle Size Reveals Key Features of agr Activation in Staphylococcus aureus. ChemBioChem. 2015;16(7):1093–1100. doi: 10.1002/cbic.201500006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tal-Gan Y, Ivancic M, Cornilescu G, Cornilescu CC, Blackwell HE. Structural characterization of native autoinducing peptides and abiotic analogues reveals key features essential for activation and inhibition of an AgrC quorum sensing receptor in Staphylococcus aureus. J Am Chem Soc. 2013;135(49):18436–18444. doi: 10.1021/ja407533e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tal-Gan Y, Stacy DM, Blackwell HE. N-Methyl and peptoid scans of an autoinducing peptide reveal new structural features required for inhibition and activation of AgrC quorum sensing receptors in Staphylococcus aureus. Chem Commun. 2014;50(23):3000–3003. doi: 10.1039/c4cc00117f. [DOI] [PubMed] [Google Scholar]
- 55.Tal-Gan Y, Ivancic M, Cornilescu G, Blackwell HE. Characterization of structural elements in native autoinducing peptides and non-native analogues that permit the differential modulation of AgrC-type quorum sensing receptors in Staphylococcus aureus. Org Biomol Chem. 2016;14(1):113–121. doi: 10.1039/c5ob01735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tal-Gan Y, Ivancic M, Cornilescu G, Yang T, Blackwell HE. Highly Stable, Amide-Bridged Autoinducing Peptide Analogues that Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus. Angew Chem Int Ed Engl. 2016;55(31):8913–8917. doi: 10.1002/anie.201602974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Broderick AH, Stacy DM, Tal-Gan Y, Kratochvil MJ, Blackwell HE, Lynn DM. Surface Coatings that Promote Rapid Release of Peptide-Based AgrC Inhibitors for Attenuation of Quorum Sensing in Staphylococcus aureus. Adv Healthcare Mater. 2014;3(1):97–105. doi: 10.1002/adhm.201300119. [DOI] [PubMed] [Google Scholar]
- 58.Kratochvil MJ, Tal-Gan Y, Yang T, Blackwell HE, Lynn DM. Nanoporous superhydrophobic coatings that promote the extended release of water-labile quorum sensing inhibitors and enable long-term modulation of quorum sensing in Staphylococcus aureus. ACS Biomater Sci Eng. 2015;1(10):1039–1049. doi: 10.1021/acsbiomaterials.5b00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vuong C, Otto M. Staphylococcus epidermidis infections. Microbes Infect. 2002;4(4):481–489. doi: 10.1016/s1286-4579(02)01563-0. [DOI] [PubMed] [Google Scholar]
- 60.Otto M, Süßmuth R, Jung G, Götz F. Structure of the pheromone peptide of the Staphylococcus epidermidis agr system. FEBS Lett. 1998;424(1–2):89–94. doi: 10.1016/s0014-5793(98)00145-8. [DOI] [PubMed] [Google Scholar]
- 61.Dufour P, Jarraud S, Vandenesch F, Greenland T, Novick RP, Bes M, Etienne J, Lina G. High genetic variability of the agr locus in Staphylococcus species. J Bacteriol. 2002;184(4):1180–1186. doi: 10.1128/jb.184.4.1180-1186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, Dunman PM, Rohde H, Cech NB, Fey PD, Horswill AR. Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J Bacteriol. 2014;196(19):3482–3493. doi: 10.1128/JB.01882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Otto M, Echner H, Voelter W, Götz F. Pheromone Cross-Inhibition between Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun. 2001;69(3):1957–1960. doi: 10.1128/IAI.69.3.1957-1960.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang T, Tal-Gan Y, Paharik AE, Horswill AR, Blackwell HE. Structure-Function Analyses of a Staphylococcus epidermidis Autoinducing Peptide Reveals Motifs Critical for AgrC-type Receptor Modulation. ACS Chem Biol. 2016;11(7):1982–1991. doi: 10.1021/acschembio.6b00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10(4):266–278. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollenbeck BL, Rice LB. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence. 2012;3(5):421–569. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sood S, Malhotra M, Das BK, Kapil A. Enterococcal infections & antimicrobial resistance. Indian J Med Res. 2008;128(2):111–121. [PubMed] [Google Scholar]
- 68.Coque TM, Patterson JE, Steckelberg JM, Murray BE. Incidence of hemolysin, gelatinase, and aggregation substance among enterococci isolated from patients with endocarditis and other infections and from feces of hospitalized and community-based persons. J Infect Dis. 1995;171(5):1223–1229. doi: 10.1093/infdis/171.5.1223. [DOI] [PubMed] [Google Scholar]
- 69.Eliopoulos GM, Gold H. Vancomycin-resistant enterococci: mechanisms and clinical observations. Clin Infect Dis. 2001;33(2):210–219. doi: 10.1086/321815. [DOI] [PubMed] [Google Scholar]
- 70.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science. 2003;302(5650):1569–1571. doi: 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- 71.Cetinkaya Y, Falk P, Mayhall CG. Vancomycin-resistant enterococci. Clin Microbiol Rev. 2000;13(4):686–707. doi: 10.1128/cmr.13.4.686-707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sifri CD, Mylonakis E, Singh KV, Qin X, Garsin DA, Murray BE, Ausubel FM, Calderwood SB. Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect Immun. 2002;70(10):5647–5650. doi: 10.1128/IAI.70.10.5647-5650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hancock LE, Perego M. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J Bacteriol. 2004;186(17):5629–5639. doi: 10.1128/JB.186.17.5629-5639.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakayama J, Cao Y, Horii T, Sakuda S, Akkermans AD, De Vos WM, Nagasawa H. Gelatinase biosynthesis-activating pheromone: a peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol. 2001;41(1):145–154. doi: 10.1046/j.1365-2958.2001.02486.x. [DOI] [PubMed] [Google Scholar]
- 75.Nakayama J, Chen S, Oyama N, Nishiguchi K, Azab EA, Tanaka E, Kariyama R, Sonomoto K. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheromone propeptide corresponding to staphylococcal AgrD. J Bacteriol. 2006;188(23):8321–8326. doi: 10.1128/JB.00865-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakayama J, Cao Y, Horii T, Sakuda S, Nagasawa H. Chemical synthesis and biological activity of the gelatinase biosynthesis-activating pheromone of Enterococcus faecalis and its analogs. Biosci Biotechnol Biochem. 2001;65(10):2322–2325. doi: 10.1271/bbb.65.2322. [DOI] [PubMed] [Google Scholar]
- 77.Nishiguchi K, Nagata K, Tanokura M, Sonomoto K, Nakayama J. Structure-activity relationship of gelatinase biosynthesis-activating pheromone of Enterococcus faecalis. J Bacteriol. 2009;191(2):641–650. doi: 10.1128/JB.01029-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakayama J, Yokohata R, Sato M, Suzuki T, Matsufuji T, Nishiguchi K, Kawai T, Yamanaka Y, Nagata K, Tanokura M, Sonomoto K. Development of a peptide antagonist against fsr quorum sensing of Enterococcus faecalis. ACS Chem Biol. 2013;8(4):804–811. doi: 10.1021/cb300717f. [DOI] [PubMed] [Google Scholar]
- 79.Rood JI, Cole ST. Molecular genetics and pathogenesis of Clostridium perfringens. Microbiol Rev. 1991;55(4):621–648. doi: 10.1128/mr.55.4.621-648.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, Ogasawara N, Hattori M, Kuhara S, Hayashi H. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci U S A. 2002;99(2):996–1001. doi: 10.1073/pnas.022493799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hambleton P. Clostridium botulinum toxins: a general review of involvement in disease, structure, mode of action and preparation for clinical use. J Neurol. 1992;239(1):16–20. doi: 10.1007/BF00839205. [DOI] [PubMed] [Google Scholar]
- 82.Kelly CP, LaMont JT. Clostridium difficile infection. Annu Rev Med. 1998;49:375–390. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- 83.Leffler DA, Lamont JT. Clostridium difficile infection. N Engl J Med. 2015;372(16):1539–1548. doi: 10.1056/NEJMra1403772. [DOI] [PubMed] [Google Scholar]
- 84.Chen J, McClane BA. Role of the Agr-like quorum-sensing system in regulating toxin production by Clostridium perfringens type B strains CN1793 and CN1795. Infect Immun. 2012;80(9):3008–3017. doi: 10.1128/IAI.00438-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vidal JE, Ma M, Saputo J, Garcia J, Uzal FA, McClane BA. Evidence that the Agr-like quorum sensing system regulates the toxin production, cytotoxicity and pathogenicity of Clostridium perfringens type C isolate CN3685. Mol Microbiol. 2012;83(1):179–194. doi: 10.1111/j.1365-2958.2011.07925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee AS, Song KP. LuxS/autoinducer-2 quorum sensing molecule regulates transcriptional virulence gene expression in Clostridium difficile. Biochem Biophys Res Commun. 2005;335(3):659–666. doi: 10.1016/j.bbrc.2005.07.131. [DOI] [PubMed] [Google Scholar]
- 87.Cooksley CM, Davis IJ, Winzer K, Chan WC, Peck MW, Minton NP. Regulation of neurotoxin production and sporulation by a putative agrBD signaling system in proteolytic Clostridium botulinum. Appl Environ Microbiol. 2010;76(13):4448–4460. doi: 10.1128/AEM.03038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steiner E, Scott J, Minton NP, Winzer K. An agr quorum sensing system that regulates granulose formation and sporulation in Clostridium acetobutylicum. Appl Environ Microbiol. 2012;78(4):1113–1122. doi: 10.1128/AEM.06376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma M, Li J, McClane BA. Structure-function analysis of peptide signaling in the Clostridium perfringens Agr-like quorum sensing system. J Bacteriol. 2015;197(10):1807–1818. doi: 10.1128/JB.02614-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vidal JE, Chen J, Li J, McClane BA. Use of an EZ-Tn 5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS One. 2009;4(7):e6232. doi: 10.1371/journal.pone.0006232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohtani K, Hirakawa H, Tashiro K, Yoshizawa S, Kuhara S, Shimizu T. Identification of a two-component VirR/VirS regulon in Clostridium perfringens. Anaerobe. 2010;16(3):258–264. doi: 10.1016/j.anaerobe.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Chen J, Vidal JE, McClane BA. The Agr-like quorum-sensing system regulates sporulation and production of enterotoxin and beta2 toxin by Clostridium perfringens type A non-food-borne human gastrointestinal disease strain F5603. Infect Immun. 2011;79(6):2451–2459. doi: 10.1128/IAI.00169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaughan EE, de Vries MC, Zoetendal EG, Ben-Amor K, Akkermans AD, de Vos WM. Lactic Acid Bacteria: Genetics, Metabolism and Applications. Springer; 2002. In; pp. 341–352. [Google Scholar]
- 94.Ouwehand AC, Salminen S, Isolauri E. Probiotics: an overview of beneficial effects. Antonie Van Leeuwenhoek. 2002;82(1–4):279–289. [PubMed] [Google Scholar]
- 95.Ahrné S, Nobaek S, Jeppsson B, Adlerberth I, Wold AE, Molin G. The normal Lactobacillus flora of healthy human rectal and oral mucosa. J Appl Microbiol. 1998;85(1):88–94. doi: 10.1046/j.1365-2672.1998.00480.x. [DOI] [PubMed] [Google Scholar]
- 96.Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrink HM, Fiers MW, Stiekema W, Lankhorst RM, Bron PA, Hoffer SM, Groot MN, Kerkhoven R, de Vries M, Ursing B, de Vos WM, Siezen RJ. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci U S A. 2003;100(4):1990–1995. doi: 10.1073/pnas.0337704100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sturme MH, Francke C, Siezen RJ, de Vos WM, Kleerebezem M. Making sense of quorum sensing in lactobacilli: a special focus on Lactobacillus plantarum WCFS1. Microbiology. 2007;153(12):3939–3947. doi: 10.1099/mic.0.2007/012831-0. [DOI] [PubMed] [Google Scholar]
- 98.Kleerebezem M, Quadri LE, Kuipers OP, De Vos WM. Quorum sensing by peptide pheromones and two-component signal-transduction systems in Gram-positive bacteria. Mol Microbiol. 1997;24(5):895–904. doi: 10.1046/j.1365-2958.1997.4251782.x. [DOI] [PubMed] [Google Scholar]
- 99.Sturme MH, Nakayama J, Molenaar D, Murakami Y, Kunugi R, Fujii T, Vaughan EE, Kleerebezem M, de Vos WM. An agr-like two-component regulatory system in Lactobacillus plantarum is involved in production of a novel cyclic peptide and regulation of adherence. J Bacteriol. 2005;187(15):5224–5235. doi: 10.1128/JB.187.15.5224-5235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berg JM, Tymoczko JL, Stryer L. Biochemistry. W.H. Freeman; New York: 2012. [Google Scholar]
- 101.Nussey SS, Whitehead SA. Endocrinology: an integrated approach. CRC Press; 2013. [PubMed] [Google Scholar]
- 102.Russell JA, Leng G, Douglas AJ. The magnocellular oxytocin system, the fount of maternity: adaptations in pregnancy. Front Neuroendocrinol. 2003;24(1):27–61. doi: 10.1016/s0091-3022(02)00104-8. [DOI] [PubMed] [Google Scholar]
- 103.Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne H-J, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE. Definition of Estrogen Receptor Pathway Critical for Estrogen Positive Feedback to Gonadotropin-Releasing Hormone Neurons and Fertility. Neuron. 2006;52(2):271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johnson AE, van Waes MA. The translocon: a dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol. 1999;15(1):799–842. doi: 10.1146/annurev.cellbio.15.1.799. [DOI] [PubMed] [Google Scholar]
- 105.Blobel G, Sabatini DD. Biomembranes. Springer; 1971. pp. 193–195. [Google Scholar]
- 106.Xu H, Shields D. Prohormone processing in the trans-Golgi network: endoproteolytic cleavage of prosomatostatin and formation of nascent secretory vesicles in permeabilized cells. J Cell Biol. 1993;122(6):1169–1184. doi: 10.1083/jcb.122.6.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perone M, Castro M. Prohormone and proneuropeptide synthesis and secretion. Histol Histopathol. 1997;12(4):1179–1188. [PubMed] [Google Scholar]
- 108.Cooper GM. The Cell: A Molecular Approach. 2. ASM Press; 2000. [Google Scholar]
- 109.Catt K, Dufau M. Peptide hormone receptors. Annu Rev Physiol. 1977;39(1):529–557. doi: 10.1146/annurev.ph.39.030177.002525. [DOI] [PubMed] [Google Scholar]
- 110.Hess S, Linde Y, Ovadia O, Safrai E, Shalev DE, Swed A, Halbfinger E, Lapidot T, Winkler I, Gabinet Y, Faier A, Yarden D, Xiang Z, Portillo FP, Haskell-Luevano C, Gilon C, Hoffman A. Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. J Med Chem. 2008;51(4):1026–1034. doi: 10.1021/jm701093y. [DOI] [PubMed] [Google Scholar]
- 111.Hess S, Ovadia O, Shalev DE, Senderovich H, Qadri B, Yehezkel T, Salitra Y, Sheynis T, Jelinek R, Gilon C, Hoffman A. Effect of structural and conformation modifications, including backbone cyclization, of hydrophilic hexapeptides on their intestinal permeability and enzymatic stability. J Med Chem. 2007;50(24):6201–6211. doi: 10.1021/jm070836d. [DOI] [PubMed] [Google Scholar]
- 112.Naveh S, Tal-Gan Y, Ling S, Hoffman A, Holoshitz J, Gilon C. Developing potent backbone cyclic peptides bearing the shared epitope sequence as rheumatoid arthritis drug-leads. Bioorg Med Chem Lett. 2012;22(1):493–496. doi: 10.1016/j.bmcl.2011.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Young TS, Young DD, Ahmad I, Louis JM, Benkovic SJ, Schultz PG. Evolution of cyclic peptide protease inhibitors. Proc Natl Acad Sci U S A. 2011;108(27):11052–11056. doi: 10.1073/pnas.1108045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lawson KV, Rose TE, Harran PG. Template-constrained macrocyclic peptides prepared from native, unprotected precursors. Proc Natl Acad Sci U S A. 2013;110(40):E3753–E3760. doi: 10.1073/pnas.1311706110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Patel YC. Somatostatin and Its Receptor Family. Front Neuroendocrinol. 1999;20(3):157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 116.Kumar U, Grant M. In: Cellular Peptide Hormone Synthesis and Secretory Pathways. Rehfeld FJ, Bundgaard RJ, editors. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. pp. 97–120. [Google Scholar]
- 117.Ten Bokum A, Hofland L, van Hagen PM. Somatostatin and somatostatin receptors in the immune system: a review. Eur Cytokine Netw. 2000;11(2):161–176. [PubMed] [Google Scholar]
- 118.Patel YC, S Galanopoulou A, Rabbani SN, Liu J-L, Ravazzola M, Amherdt M. Somatostatin-14, somatostatin-28, and prosomatostatin[1–10] are independently and efficiently processed from prosomatostatin in the constitutive secretory pathway in islet somatostatin tumor cells (1027B2)1. Mol Cell Endocrinol. 1997;131(2):183–194. doi: 10.1016/s0303-7207(97)00107-x. [DOI] [PubMed] [Google Scholar]
- 119.Patel YC, O'Neil W. Peptides derived from cleavage of prosomatostatin at carboxyl- and amino-terminal segments. Characterization of tissue and secreted forms in the rat. J Biol Chem. 1988;263(2):745–751. [PubMed] [Google Scholar]
- 120.Harris AG. Somatostatin and somatostatin analogues: pharmacokinetics and pharmacodynamic effects. Gut. 1994;35(3 Suppl):S1–S4. doi: 10.1136/gut.35.3_suppl.s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Larsson L-I, Goltermann N, de Magistris L, Rehfeld JF, Schwartz TW. Somatostatin Cell Processes as Pathways for Paracrine Secretion. Science. 1979;205(4413):1393–1395. doi: 10.1126/science.382360. [DOI] [PubMed] [Google Scholar]
- 122.Rai U, Thrimawithana TR, Valery C, Young SA. Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacol Therapeut. 2015;152:98–110. doi: 10.1016/j.pharmthera.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 123.Sun L, Coy DH. Somatostatin and its Analogs. Curr Drug Targets. 2016;17(5):529–537. doi: 10.2174/1389450116666141205163548. [DOI] [PubMed] [Google Scholar]
- 124.Mentlein R, Dahms P. Endopeptidases 24.16 and 24.15 are responsible for the degradation of somatostatin, neurotensin, and other neuropeptides by cultivated rat cortical astrocytes. J Neurochem. 1994;62(1):27–36. doi: 10.1046/j.1471-4159.1994.62010027.x. [DOI] [PubMed] [Google Scholar]
- 125.Lamberts SWJ, Van der Lely A-J, de Herder WW, Hofland LJ. Octreotide. N Engl J Med. 1996;334(4):246–254. doi: 10.1056/NEJM199601253340408. [DOI] [PubMed] [Google Scholar]
- 126.McKeage K, Cheer S, Wagstaff AJ. Octreotide Long-Acting Release (LAR) Drugs. 2003;63(22):2473–2499. doi: 10.2165/00003495-200363220-00014. [DOI] [PubMed] [Google Scholar]
- 127.Veber DF, Freidinger RM, Perlow DS, Paleveda WJ, Holly FW, Strachan RG, Nutt RF, Arison BH, Homnick C, Randall WC, Glitzer MS, Saperstein R, Hirschmann R. A potent cyclic hexapeptide analogue of somatostatin. Nature. 1981;292(5818):55–58. doi: 10.1038/292055a0. [DOI] [PubMed] [Google Scholar]
- 128.Veber DF, Saperstein R, Nutt RF, Freidinger RM, Brady SF, Curley P, Perlow DS, Paleveda WJ, Colton CD, Zacchei AG, Tocco DJ, Hoff DR, Vandlen RL, Gerich JE, Hall L, Mandarino L, Cordes EH, Anderson PS, Hirschmann R. A super active cyclic hexapeptide analog of somatostatin. Life Sciences. 1984;34(14):1371–1378. doi: 10.1016/0024-3205(84)90009-2. [DOI] [PubMed] [Google Scholar]
- 129.Robertson CR, Flynn SP, White HS, Bulaj G. Anticonvulsant neuropeptides as drug leads for neurological diseases. Nat Prod Rep. 2011;28(4):741–762. doi: 10.1039/c0np00048e. [DOI] [PubMed] [Google Scholar]
- 130.Shah P, Rahman SA, McElroy S, Gilbert C, Morgan K, Hinchey L, Senniappan S, Levy H, Amin R, Hussain K. Use of Long-Acting Somatostatin Analogue (Lanreotide) in an Adolescent with Diazoxide-Responsive Congenital Hyperinsulinism and Its Psychological Impact. Horm Res Paediatr. 2015;84(5):355–360. doi: 10.1159/000439131. [DOI] [PubMed] [Google Scholar]
- 131.Fu Z, Gilbert ER, Liu D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr Diabetes Rev. 2013;9(1):25–53. [PMC free article] [PubMed] [Google Scholar]
- 132.Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential 1. Endocr Rev. 1998;19(5):608–624. doi: 10.1210/edrv.19.5.0349. [DOI] [PubMed] [Google Scholar]
- 133.Holleman F, Hoekstra JBL. Insulin Lispro. N Engl J Med. 1997;337(3):176–183. doi: 10.1056/NEJM199707173370307. [DOI] [PubMed] [Google Scholar]
- 134.Homko C, Deluzio A, Jimenez C, Kolaczynski JW, Boden G. Comparison of Insulin Aspart and Lispro. Diabetes Care. 2003;26(7):2027–2031. doi: 10.2337/diacare.26.7.2027. [DOI] [PubMed] [Google Scholar]
- 135.Weiss M. Design of ultra-stable insulin analogues for the developing world. J Health Spec. 2013;1(2):59–70. [Google Scholar]
- 136.Garnock-Jones KP, Plosker GL. Insulin Glulisine. Drugs. 2009;69(8):1035–1057. doi: 10.2165/00003495-200969080-00006. [DOI] [PubMed] [Google Scholar]
- 137.Bolli BG, Di Marchi DR, Park DG, Pramming S, Koivisto AV. Insulin analogues and their potential in the management of diabetes mellitus. Diabetologia. 1999;42(10):1151–1167. doi: 10.1007/s001250051286. [DOI] [PubMed] [Google Scholar]
- 138.Vague P, Selam J-L, Skeie S, De Leeuw I, Elte JWF, Haahr H, Kristensen A, Draeger E. Insulin Detemir Is Associated With More Predictable Glycemic Control and Reduced Risk of Hypoglycemia Than NPH Insulin in Patients With Type 1 Diabetes on a Basal-Bolus Regimen With Premeal Insulin Aspart. Diabetes Care. 2003;26(3):590–596. doi: 10.2337/diacare.26.3.590. [DOI] [PubMed] [Google Scholar]
- 139.Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Larsen UD, Ribel U, Markussen J. Albumin binding of insulins acylated with fatty acids: characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem J. 1995;312(Pt 3):725–731. doi: 10.1042/bj3120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kurtzhals P. Engineering predictability and protraction in a basal insulin analogue: the pharmacology of insulin detemir. Int J Obes Relat Metab Disord. 2004;28(Suppl 2):S23–28. doi: 10.1038/sj.ijo.0802746. [DOI] [PubMed] [Google Scholar]
- 141.Atkin S, Javed Z, Fulcher G. Insulin degludec and insulin aspart: novel insulins for the management of diabetes mellitus. Ther Adv Chronic Dis. 2015;6(6):375–388. doi: 10.1177/2040622315608646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zinman B. Newer insulin analogs: advances in basal insulin replacement. Diabetes Obes Metab. 2013;15(s1):6–10. doi: 10.1111/dom.12068. [DOI] [PubMed] [Google Scholar]
- 143.Jonassen I, Havelund S, Hoeg-Jensen T, Steensgaard DB, Wahlund P-O, Ribel U. Design of the Novel Protraction Mechanism of Insulin Degludec, an Ultra-long-Acting Basal Insulin. Pharmaceut Res. 2012;29(8):2104–2114. doi: 10.1007/s11095-012-0739-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haahr H, Heise T. A Review of the Pharmacological Properties of Insulin Degludec and Their Clinical Relevance. Clin Pharmacokinet. 2014;53(9):787–800. doi: 10.1007/s40262-014-0165-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Klonoff DC. Afrezza Inhaled Insulin: The Fastest-Acting FDA-Approved Insulin on the Market Has Favorable Properties. J Diabetes Sci Technol. 2014;8(6):1071–1073. doi: 10.1177/1932296814555820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gimpl G, Fahrenholz F. The Oxytocin Receptor System: Structure, Function, and Regulation. Physiol Rev. 2001;81(2):629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- 147.Burbach JPH, Luckman SM, Murphy D, Gainer H. Gene Regulation in the Magnocellular Hypothalamo-Neurohypophysial System. Physiol Rev. 2001;81(3):1197–1267. doi: 10.1152/physrev.2001.81.3.1197. [DOI] [PubMed] [Google Scholar]
- 148.Richter D. Molecular events in expression of vasopressin and oxytocin and their cognate receptors. Am J Physiol. 1988;255(2 Pt 2):F207–219. doi: 10.1152/ajprenal.1988.255.2.F207. [DOI] [PubMed] [Google Scholar]
- 149.Yang H-P, Wang L, Han L, Wang SC. Nonsocial functions of hypothalamic oxytocin. ISRN Neurosci. 2013;2013 doi: 10.1155/2013/179272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McGregor IS, Bowen MT. Breaking the loop: oxytocin as a potential treatment for drug addiction. Horm Behav. 2012;61(3):331–339. doi: 10.1016/j.yhbeh.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 151.Churchland PS, Winkielman P. Modulating social behavior with oxytocin: How does it work? What does it mean? Horm Behav. 2012;61(3):392–399. doi: 10.1016/j.yhbeh.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Guzman YF, Tronson NC, Jovasevic V, Sato K, Guedea AL, Mizukami H, Nishimori K, Radulovic J. Fear-enhancing effects of septal oxytocin receptors. Nat Neurosci. 2013;16(9):1185–1187. doi: 10.1038/nn.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Terashima Y, Kondo K, Oiso Y. Administration of oxytocin affects vasopressin V2 receptor and aquaporin-2 gene expression in the rat. Life Sci. 1999;64(16):1447–1453. doi: 10.1016/s0024-3205(99)00078-8. [DOI] [PubMed] [Google Scholar]
- 154.Joo KW, Jeon US, Kim G-H, Park J, Oh YK, Kim YS, Ahn C, Kim S, Kim SY, Lee JS, Han JS. Antidiuretic action of oxytocin is associated with increased urinary excretion of aquaporin-2. Nephrol Dial Transpl. 2004;19(10):2480–2486. doi: 10.1093/ndt/gfh413. [DOI] [PubMed] [Google Scholar]
- 155.Fabian M, Forsling ML, Jones J, Pryor J. The clearance and antidiuretic potency of neurohypophysial hormones in man, their plasma binding and stability. J Physiol. 1969;204(3):653. doi: 10.1113/jphysiol.1969.sp008937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Welches WR, Bridget Brosnihan K, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: Angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life Sci. 1993;52(18):1461–1480. doi: 10.1016/0024-3205(93)90108-f. [DOI] [PubMed] [Google Scholar]
- 157.Gimpl G, Reitz J, Brauer S, Trossen C. Oxytocin receptors: ligand binding, signalling and cholesterol dependence. Prog Brain Res. 2008;170:193–204. doi: 10.1016/S0079-6123(08)00417-2. [DOI] [PubMed] [Google Scholar]
- 158.Wisniewski K, Alagarsamy S, Galyean R, Tariga H, Thompson D, Ly B, Wisniewska H, Qi S, Croston G, Laporte R, Riviere PJ, Schteingart CD. New, potent, and selective peptidic oxytocin receptor agonists. J Med Chem. 2014;57(12):5306–5317. doi: 10.1021/jm500365s. [DOI] [PubMed] [Google Scholar]
- 159.Gazis D, Roy J, Schwartz IL. Prolonged Responses by the Rat Uterus in Vivo to Deamino-Oxytocin Disproportionate to Its Estimated Plasma Half-Life*. Endocrinology. 1980;106(3):805–810. doi: 10.1210/endo-106-3-805. [DOI] [PubMed] [Google Scholar]
- 160.du Vigneaud V, Winestock G, Murti VVS, Hope DB, Kimbrough RD. Synthesis of 1-β-mercaptopropionic acid oxytocin (desamino-oxytocin), a highly potent analogue of oxytocin. J Biol Chem. 1960;235(12):PC64–PC66. [PubMed] [Google Scholar]
- 161.Tyagi MG, Murugan MS. Influence of Demoxytocin on the Behavioural Actions of Melatonin. J Exp Sci. 2011;2(8):07–09. [Google Scholar]
- 162.Ulstein M, Sagen N, Eikhom SN. A comparative study of labor induced by oral prostaglandin E2 and buccal tablets of demoxytocin. Int J Gynaecol Obstet. 1978;17(3):243–245. doi: 10.1002/j.1879-3479.1979.tb00158.x. [DOI] [PubMed] [Google Scholar]
- 163.Gimpl G, Postina R, Fahrenholz F, Reinheimer T. Binding domains of the oxytocin receptor for the selective oxytocin receptor antagonist barusiban in comparison to the agonists oxytocin and carbetocin. Eur J Pharmacol. 2005;510(1):9–16. doi: 10.1016/j.ejphar.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 164.Engstrøm T, Barth T, Melin P, Vilhardt H. Oxytocin receptor binding and uterotonic activity of carbetocin and its metabolites following enzymatic degradation. Eur J Pharmacol. 1998;355(2):203–210. doi: 10.1016/s0014-2999(98)00513-5. [DOI] [PubMed] [Google Scholar]
- 165.Schramme AR, Pinto CRF, Davis J, Whisnant CS, Whitacre MD. Pharmacokinetics of carbetocin, a long-acting oxytocin analogue, following intravenous administration in horses. Equine Vet J. 2008;40(7):658–661. doi: 10.2746/042516408x334343. [DOI] [PubMed] [Google Scholar]
- 166.Borruto F, Treisser A, Comparetto C. Utilization of carbetocin for prevention of postpartum hemorrhage after cesarean section: a randomized clinical trial. Arch Gynecol Obstet. 2009;280(5):707–712. doi: 10.1007/s00404-009-0973-8. [DOI] [PubMed] [Google Scholar]
- 167.Morioka T, Loik ND, Hipolito CJ, Goto Y, Suga H. Selection-based discovery of macrocyclic peptides for the next generation therapeutics. Curr Opin Chem Biol. 2015;26:34–41. doi: 10.1016/j.cbpa.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 168.Vilhardt H, Atke A, Barthova J, Ubik K, Barth T. Interaction of chymotrypsin with carbetocin ([1-deamino-1-monocarba-2-O-methyltyrosine]-oxytocin) Pharmacol Toxicol. 1997;81(3):147–150. doi: 10.1111/j.1600-0773.1997.tb00045.x. [DOI] [PubMed] [Google Scholar]
- 169.Anagnostou E, Soorya L, Brian J, Dupuis A, Mankad D, Smile S, Jacob S. Intranasal oxytocin in the treatment of autism spectrum disorders: a review of literature and early safety and efficacy data in youth. Brain Res. 2014;1580:188–198. doi: 10.1016/j.brainres.2014.01.049. [DOI] [PubMed] [Google Scholar]
- 170.Heringa SM, Begemann MJ, Goverde AJ, Sommer IE. Sex hormones and oxytocin augmentation strategies in schizophrenia: a quantitative review. Schizophr Res. 2015;168(3):603–613. doi: 10.1016/j.schres.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 171.Rotondo F, Butz H, Syro LV, Yousef GM, Di Ieva A, Restrepo LM, Quintanar-Stephano A, Berczi I, Kovacs K. Arginine vasopressin (AVP): a review of its historical perspectives, current research and multifunctional role in the hypothalamo-hypophysial system. Pituitary. 2016;19(4):345–355. doi: 10.1007/s11102-015-0703-0. [DOI] [PubMed] [Google Scholar]
- 172.Holmes CL, Landry DW, Granton JT. Science Review: Vasopressin and the cardiovascular system part 1 – receptor physiology. Crit Care. 2003;7(6):427–434. doi: 10.1186/cc2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Acher R, Chauvet J, Rouille Y. Dynamic processing of neuropeptides. J Mol Neurosci. 2002;18(3):223–228. doi: 10.1385/JMN:18:3:223. [DOI] [PubMed] [Google Scholar]
- 174.Sharman A, Low J. Vasopressin and its role in critical care. Contin Educ Anaesth Crit Care Pain. 2008;8(4):134–137. [Google Scholar]
- 175.Chan WY, Wo NC, Manning M. The role of oxytocin receptors and vasopressin V 1a receptors in uterine contractions in rats: Implications for tocolytic therapy with oxytocin antagonists. Am J Obstet Gynecol. 1996;175(5):1331–1335. doi: 10.1016/s0002-9378(96)70050-9. [DOI] [PubMed] [Google Scholar]
- 176.Mitchell SLM, Hunter JM. Vasopressin and its antagonists: what are their roles in acute medical care? Br J Anaesth. 2007;99(2):154–158. doi: 10.1093/bja/aem204. [DOI] [PubMed] [Google Scholar]
- 177.Sawyer W, Pang P, Seto J, McEnroe M, Lammek B, Manning M. Vasopressin analogs that antagonize antidiuretic responses by rats to the antidiuretic hormone. Science. 1981;212(4490):49–51. doi: 10.1126/science.7209515. [DOI] [PubMed] [Google Scholar]
- 178.Manning M, Misicka A, Olma A, Bankowski K, Stoev S, Chini B, Durroux T, Mouillac B, Corbani M, Guillon G. Oxytocin and Vasopressin Agonists and Antagonists as Research Tools and Potential Therapeutics. J Neuroendocrinol. 2012;24(4):609–628. doi: 10.1111/j.1365-2826.2012.02303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Åkerman B. On Felypressin (Octapressin®) as an Adjunct to Lidocaine and Prilocaine – an Experimental Study in Animals. Acta Pharmacol Tox. 1966;24(4):377–388. [PubMed] [Google Scholar]
- 180.Inagawa M, Ichinohe T, Kaneko Y. Felypressin, but not epinephrine, reduces myocardial oxygen tension after an injection of dental local anesthetic solution at routine doses. J Oral Maxillofac Surg. 2010;68(5):1013–1017. doi: 10.1016/j.joms.2009.07.080. [DOI] [PubMed] [Google Scholar]
- 181.Cecanho R, De Luca LA, Jr, Ranali J. Anesth Prog. Anesthesia progress. 2006;53(4):119–125. doi: 10.2344/0003-3006(2006)53[119:CEOF]2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kaufmann JE, Iezzi M, Vischer UM. Desmopressin (DDAVP) induces NO production in human endothelial cells via V2 receptor-and cAMP-mediated signaling. J Thromb Haemost. 2003;1(4):821–828. doi: 10.1046/j.1538-7836.2003.00197.x. [DOI] [PubMed] [Google Scholar]
- 183.Ślusarz MJ, Ślusarz R, Ciarkowski J. Investigation of mechanism of desmopressin binding in vasopressin V2 receptor versus vasopressin V1a and oxytocin receptors: Molecular dynamics simulation of the agonist-bound state in the membrane–aqueous system. Biopolymers. 2006;81(5):321–338. doi: 10.1002/bip.20420. [DOI] [PubMed] [Google Scholar]
- 184.Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years. Blood. 1997;90(7):2515–2521. [PubMed] [Google Scholar]
- 185.Becker DJ, Foley TP. 1-Deamino-8-d-arginine vasopressin in the treatment of central diabetes insipidus in childhood. J Pediatr. 1978;92(6):1011–1015. doi: 10.1016/s0022-3476(78)80389-8. [DOI] [PubMed] [Google Scholar]
- 186.Seif SM, Zenser TV, Ciarochi FF, Davis BB, Robinson AG. DDAVP (l-Desamino-8-D-arginine-vasopressin) Treatment of Central Diabetes Insipidus—Mechanism of Prolonged Antidiuresis. J Clin Endocrinol Metab. 1978;46(3):381–388. doi: 10.1210/jcem-46-3-381. [DOI] [PubMed] [Google Scholar]
- 187.Delin K, Aurell M, Ewald J. Urinary concentration test with desmopressin. Br Med J. 1978;1(6115):757–758. doi: 10.1136/bmj.1.6115.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vilhardt H. Basic pharmacology of desmopressin. Drug Invest. 1990;2(5):2–8. [Google Scholar]
- 189.Cvetković RS, Plosker GL. Desmopressin. Drugs. 2005;65(1):99–107. doi: 10.2165/00003495-200565010-00008. [DOI] [PubMed] [Google Scholar]
- 190.Solanki P, Chawla A, Garg R, Gupta R, Jain M, Sarin SK. Beneficial effects of terlipressin in hepatorenal syndrome: A prospective, randomized placebo-controlled clinical trial. J Gastroenterol Hepatol. 2003;18(2):152–156. doi: 10.1046/j.1440-1746.2003.02934.x. [DOI] [PubMed] [Google Scholar]
- 191.Albanèse J, Leone M, Delmas A, Martin C. Terlipressin or norepinephrine in hyperdynamic septic shock: a prospective, randomized study. Crit Care Med. 2005;33(9):1897–1902. doi: 10.1097/01.ccm.0000178182.37639.d6. [DOI] [PubMed] [Google Scholar]
- 192.Ioannou GN, Doust J, Rockey DC. Terlipressin in acute oesophageal variceal haemorrhage. Aliment Pharm Therap. 2003;17(1):53–64. doi: 10.1046/j.1365-2036.2003.01356.x. [DOI] [PubMed] [Google Scholar]
- 193.Sharma P, Kumar A, Shrama BC, Sarin SK. An Open Label, Pilot, Randomized Controlled Trial of Noradrenaline Versus Terlipressin in the Treatment of Type 1 Hepatorenal Syndrome and Predictors of Response. Am J Gastroenterol. 2008;103(7):1689–1697. doi: 10.1111/j.1572-0241.2008.01828.x. [DOI] [PubMed] [Google Scholar]
- 194.He X, Su F, Taccone FS, Laporte R, Kjølbye AL, Zhang J, Xie K, Moussa MD, Reinheimer TM, Vincent J-L. A selective v1a receptor agonist, selepressin, is superior to arginine vasopressin and to norepinephrine in ovine septic shock. Crit Care Med. 2016;44(1):23. doi: 10.1097/CCM.0000000000001380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Asfar P, Russell JA, Tuckermann J, Radermacher P. Selepressin in Septic Shock: A Step Toward Decatecholaminization? Crit Care Med. 2016;44(1):234–236. doi: 10.1097/CCM.0000000000001441. [DOI] [PubMed] [Google Scholar]
- 196.Evans JH, Meadow SR. Desmopressin for bed wetting: length of treatment, vasopressin secretion, and response. Arch Dis Child. 1992;67(2):184–188. doi: 10.1136/adc.67.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hodge EK, Hughes DW, Attridge RL. Effect of Body Weight on Hemodynamic Response in Patients Receiving Fixed-Dose Vasopressin for Septic Shock. Ann Pharmacother. 2016 doi: 10.1177/1060028016656384. (In press) [DOI] [PubMed] [Google Scholar]
- 198.Miano TA, Crouch MA. Evolving Role of Vasopressin in the Treatment of Cardiac Arrest. Pharmacotherapy. 2006;26(6):828–839. doi: 10.1592/phco.26.6.828. [DOI] [PubMed] [Google Scholar]