

Abstract
Glioblastoma (GBM) is an invasive brain cancer with tumor cells that disperse from the primary mass, escaping surgical resection and invariably giving rise to lethal recurrent lesions. Here we report that PTP-PEST, a cytoplasmic protein tyrosine phosphatase, controls GBM cell invasion by physically bridging the focal adhesion protein Crk-associated substrate (Cas) to valosin containing protein (Vcp), an ATP-dependent protein segregase that selectively extracts ubiquitinated proteins from multiprotein complexes and targets them for degradation via the ubiquitin proteasome system. Both Cas and Vcp are substrates for PTP-PEST, with the phosphorylation status of tyrosine 805 (Y805) in Vcp impacting affinity for Cas in focal adhesions and controlling ubiquitination levels and protein stability. Perturbing PTP-PEST-mediated phosphorylation of Cas and Vcp led to alterations in GBM cell invasive growth in vitro and in pre-clinical mouse models. Collectively, these data reveal a novel regulatory mechanism involving PTP-PEST, Vcp, and Cas that dynamically balances phosphorylation-dependent ubiquitination of key focal proteins involved in GBM cell invasion.
Keywords: extracellular matrix, vascular basement membrane, p130Cas, microenvironment, ubiquitin proteasome system, itgb8, glioma
Introduction
Patients diagnosed with the malignant cancer GBM have a median survival time of less than two years after diagnosis (1). This poor prognosis is largely due to invasive GBM cells that escape surgical resection and give rise to recurrent lesions that are resistant to chemotherapy such as temozolomide. Targeted therapies such as the anti-vascular endothelial growth factor (VEGF) blocking antibody bevacizumab have yielded disappointing results in GBM clinical trials, with no improvements in overall patient survival. Many patients treated with bevacizumab develop acquired resistance leading to lethal recurrent lesions associated with robust tumor cell invasion (2). While a great deal is known about genes and pathways that promote GBM growth and neovascularization, relatively little is understood about mechanisms that drive GBM cell invasion during progression who following anti-angiogenic therapy.
PTP-PEST is a 110 kilo-Dalton (kDa) cytosolic phosphatase that contains a 30 kDa N-terminal catalytic domain and a C-terminus with several proline, glutamate, serine and threonine-rich (PEST) sequences. PTP-PEST plays important roles in promoting tissue morphogenesis, with deletion of the murine PTP-PEST gene (Ptpn12) in all cells leading to embryonic lethality (3). Structural studies of the PTP-PEST catalytic domain reveal that it recognizes phosphotyrosine (pY) motifs in diverse substrates (4), including Rho GEFs, GAPs and focal adhesion proteins such as paxillin and focal adhesion kinase (FAK). Cultured PTP-PEST-/- cells show defective polarity and migration due, in part, to abnormal activation of Rho GTPase signaling and imbalances in cell-ECM adhesion (5,6).
Focal adhesions are multiprotein complexes that connect the cytoskeleton to the extracellular matrix (ECM) via integrins (7). Integrin-ECM adhesions continually develop and disassemble as a cell moves, with intermediate structures (nascent adhesions) forming and growing into larger focal adhesions at the leading edge, and subsequently disassembling under the cell body (8). A key regulatory event in the formation and disassembly of focal adhesions is post-translational tyrosine phosphorylation, related to activities of tyrosine kinases such as Src and FAK (9). Crk-associated substrate (Cas) is a 130 k-Da protein that was originally identified as a substrate of Src (10). There are five members of the Cas protein family: Cas, also known as breast cancer anti-estrogen resistance (Bcar1), Nedd9, Cass4, and embryonal Fyn substrate (Efs) (11). Cas is a core component of focal adhesions where it bridges multiple signaling proteins to modulate adhesion and motility (12). Cas-deficient cells show normal focal adhesion assembly, but dramatically impaired disassembly, leading to defective migration and invasion (13).
Phosphorylation and ubiquitination are tightly coupled processes, with ‘phosphodegron’ sequences in target proteins recruiting E3 ubiquitin ligases and other proteins involved in degradation by the ubiquitin proteasome system (14). Proteins are covalently tagged with ubiquitin via the activities of three enzymes, termed E1, E2 and E3 (15). Ubiquitinated proteins within multicellular complexes are selectively removed via chaperone activities associated with Vcp, a 97 kDa evolutionarily conserved protein (16). Vcp catalyzes the segregation of ubiquitinated proteins from organelles, chromatin, and multiprotein complexes, and promotes destruction by the proteasome (17). Vcp protein contains two AAA+ adenosine triphosphatase (ATPase) domains and an N-domain, which interacts with lipids in the plasma membrane and other proteins, including E2 and E3 enzymes (18). A Peptide:N-glycanase/UBA or UBX (PUB) domain-interaction sequence (PBS) in the Vcp C-terminus mediates associations with PUB domain-containing proteins and other factors (19). Src phosphorylation of Vcp tyrosine 805 (Y805) in the PBS blocks interactions with ubiquitinated PUB domain-containing proteins (20).
Here, we report that PTP-PEST dynamically regulates GBM cell invasive growth via the formation of a complex with Cas and Vcp. PTP-PEST enzymatic activities create phosphodegrons in Cas that target it for extraction by Vcp, thus stabilizing focal adhesions and balancing GBM cell invasive growth. Cells expressing PTP-PEST are less invasive due to lower levels of phosphorylated Cas and the presence of more stable focal adhesions. GBM cells that express low levels of PTP-PEST display enhanced invasion due to increased Cas phosphorylation and more dynamic focal adhesion disassembly. Collectively, these results not only elucidate novel signaling pathways that link the phosphorylation and ubiquitination pathways.
Materials and Methods
Ethics statement
Approval for the use of human specimens was obtained from the Institutional Review Board (IRB) at the University of Texas MD Anderson Cancer Center. The IRB waived the requirement for informed consent for previously collected residual tissues from surgical procedures stripped of unique patient identifiers according to the Declaration of Helsinki guidelines. All animal procedures and experiments conducted in this study were reviewed and approved by the University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee (IACUC).
GBM cell culturing and analysis
Primary human GBM cells from patient samples were cultured in the following growth media: DMEM-F12 (Mediatech), 20 ng/ml EGF and bFGF (Gibco), B27 supplement (Life Technologies) and one unit per ml penicillin-streptomycin (Gibco). After 7 to 10 days spheroids were passaged by accutase (Sigma #A 6964) treatment and mechanical disruption using a 1 ml syringe and a 23-gauge needle, and dissociated cells were re-plated in fresh growth media. LN229 GBM cells and HEK 293T cells were purchased from the American Type Culture Collection (Manassas, VA). GBM cell lines were cultured in DMEM (Mediatech; Manassas, VA) supplemented with 10% fetal bovine serum (Atlanta Biologics) and antibiotics. All cells were authenticated by DNA short tandem repeat profiling in an institutional Characterized Cell Line Core Facility. In addition, all cultured cells were routinely tested for mycoplasma using commercially available kits (Thermo Fisher), and only those cells deemed mycoplasma-free were used for experiments.
PTPN12-dependent cell viability was quantified using the Celltiter-Glo luminescent assay kit (Promega) according to the manufacture’s protocol. Briefly, control of PTPN12 KO spheres were dissociated as detailed above and 5 × 103 GSCs were added per well in a 96-well format. At 48, 72, and 96 hours after plating Celltiter-Glo reagent was added to each well. Plates were incubated at room temperature for 10 min and the luminescence intensity was measured with a microplate reader.
To analyze adherent cells by immunofluorescence, spheroids were dissociated as detailed above and GSCs were plated on glass coverslips coated with laminin for 24 hours (10 μg/ml) prior to fixation and permeabilization. For Matrigel invasion assays 5 × 104 GBM cells (control or PTPN12 KO) were added in serum-free media (GSCs) or 1% serum-containing media (LN229 GBM cells) in the upper chambers of Matrigel (BD Biosciences, 8 μm pore sizes) invasion transwell systems (n=3 wells per cell type). For RNAi-mediated silencing experiments, LN229 GBM cells were added to Matrigel transwell chambers 22-24 hours after transfection with siRNAs. Normal growth medium containing 10% FBS was added to the lower wells to stimulate cell motility. GBM cell invasion was quantified after 24 hours by staining the excised filters with Protocal HEMA3 stain followed by counting cells on the underside of the transwell filters.
For pharmacological treatment experiments, CB-5083 (Selleckchem) was dissolved in DMSO (10 mM stock solution) and diluted in growth media before addition to adherent LN229 GBM cells (5 × 103 cells per well) in a 96-well format. At 24, 48 or 72 hours after treatment, Celltiter Glo reagent was added to each well and viability was quantified based on luminescence readings. GraphPad Prism was used to analyze and viability and determine IC50 concentrations. For studies of focal adhesion proteins, LN229 cells were treated with DMSO or CB-5083 (0.4 μM) dissolved in DMSO for 24 hours followed by lysis with RIPA buffer and immunoblotting.
Experimental mice
All animal experiments were reviewed and approved prior to animal use under the guidance of the IACUC and the MD Anderson Subcommittee on Animal Studies, both AAALAC accredited institutions. NCR-nu/nu mice were purchased from Jackson Laboratories and used for all experiments involving intracranial injections of mouse astrocytoma cells and human GBM cells. Healthy male NCR-nu/nu mice between 6-12 weeks of age were purchased from Jackson Laboratories. NCR-nu/nu mice were injected intracranially with 1-2 × 105 GBM cells as previously described (21). Mice were anesthetized by intraperitoneal injection of a mixture of Ketamine/Xylazine/Acepromazine (100mg/kg + 2.5mg/kg + 2.5mg/kg body weight) using a 1 ml syringe and sterile 22 G needle. At these dosages, the mice remain deeply anesthetized for approximately 30 minutes, which allows sufficient time to perform the intracranial injections. At 5 minutes post-anesthesia, lower limb reflexivity was checked: deeply anesthetized animals were not responsive to a toe pinch. Corneal reflex was also used to assess depth of anesthesia during surgery. After surgery, mice were kept warm with gauze pads and a heat lamp and monitored until recovering fully from the procedure. No animals were exposed to unreasonable pain or distress. Analgesia in the form of subcutaneous injections of Buprenorphine SR was administered as necessary based on routine veterinary care guidelines. Mice were monitored daily for the development of brain tumor-related deficits, including lack of coordination, lethargy and cachexia.
Upon the development of tumor-related deficits in the first animal, all mice in the cohort were immediately euthanized by intraperitoneal injection of a lethal dose of Avertin (1 mg/10g body weight). The chest cavity was exposed by an incision above the diaphragm. A 22G needle was inserted into the left ventricle followed by continued perfusion with 4% PFA/PBS. Brains were post-fixed overnight followed by experimental analyses. To compare the PTPN12-dependent size of tumors, FFPE brain sections were stained with hematoxylin and eosin. The cross-sectional area of each tumor was measured at its largest point. Measurements from at least five tumors were averaged from either condition. The TCGA-based coincidental expression analysis for VCP and PTPN12 in GBM samples was performed using Project Betastasis (http://www.betastasis.com/). For quantitation of mitotic cancer cells in control and PTPN12 KO tumor sections (n=3 different mice per tumor type), randomly selected fields (n=3 per section) in fixed paraffin embedded brain sections were labeled with anti-pHH3 and anti-GFP. Percentages of pHH3+/GFP+ cells per field were quantified and plotted.
For subcutaneous tumor selection experiments, 5 × 105 U87 GBM cells were implanted into the flank of NCR-nu/nu athymic mice (n = 5) in a 100 μl suspension of growth factor-reduced matrigel (BD Biosciences) and serum free culture media as previously described (22). To develop U87-bevR and -bevS xenografts, five subcutaneous U87 tumors were treated with bevacizumab (10 mg/kg) or IgG from human serum (Sigma) for four weeks. The largest tumor was extracted, dissociated and then reinjected subcutaneously (n = 5 NCR-nu/nu mice), at which point the process was repeated two more times. Mice with tumors were monitored daily. Euthanasia occurred when mice reached institutional euthanasia criteria (2.1 cm maximal dimension or tumor symptoms). The final tumor lysates collected from bevacizumab-treated and IgG-treated mice represent the U87-bevR and U87-bevS xenografts, respectively.
Antibodies
Immunoblotting was performed on detergent-soluble lysates using standard protocols. The following primary antibodies were used: mouse anti-beta Actin antibody AC-15 (Abcam #ab6276, 1:5000), chicken anti-GFP (Aves Labs GFP-1020, 1:5000), anti-phosphotyrosine PY99 (Santa Cruz sc-7020, 1:1000), rabbit-anti-GST (Novus NB600326, 1:2000), rabbit anti-Vcp (GeneTex GTX101089, 1:2000), and mouse anti-ubiquitin (FK2 Millipore 04-263, 1:800). The following antibodies were purchased from Cell Signaling Technologies: anti-p130 Cas (#13846), anti-phospho-p130 Cas pY410 (#4011), anti-FAK (#3285), anti-phospho-FAK pY397 (#8556), anti-phospho-FAK pY925 (#3284), anti-phospho-FAK pY576/577 (#3281, 1:1000), FAK (BD810087), anti-paxillin pY118 (2541S, 1:1000), and rabbit anti-PTP-PEST (D4W7W, 1:1000), anti-EGFR (#4267, 1:1000), anti-EGFR pY1068 (#2234, 1:1000) and pY992 (#2235, 1:1000), anti-Her2 (#2242, 1:500), and anti-Her2 pY1196 (D66B7, 1:1000). Immunoblots were overlaid with donkey anti-rabbit 800, donkey anti-mouse 680 secondary antibodies purchased from Licor (1:10,000). Antibodies were added in blocking buffer comprising 3% bovine serum albumin (BSA) in Tris buffer saline containing 0.1% Tween-20 (TBST). Immunofluorescence and immunohistochemistry was performed on formalin-fixed paraffin-embedded (FFPE) tissue according to standard protocols using the following primary antibodies: rat anti-mouse CD31 (BD Biosciences, 1:200), chicken anti-Nestin (Neuromics, 1:50), rabbit anti-GFAP (DAKO, 1:500), and rabbit anti-laminin (Sigma, 1:500), anti-PTP-PEST (Abcam, Ab76942, 1:250), and rabbit anti-phospho-Histone H3 pSer10 (Cell Signaling Technologies, 1:100). Donkey anti-chicken 488, Alexa-conjugated goat anti-rabbit 488, goat anti-rabbit 594, and goat anti-mouse 594 secondary antibodies (1:500) were used. Antibodies were incubated in blocking buffer comprising 1% bovine serum albumin (BSA) in PBS containing 0.1% TritonX-100 (PBST).
Mass spectrometry experiments
Soluble GST-tagged recombinant PTP-PEST proteins from bacterial lysates were fractionated using glutathione-Agarose prior to mixing with detergent-soluble cell lysates as described previously (23). Glutathione-bound fractions were resolved on an SDS polyacrylamide gel under reducing conditions. Control and experimental bands were cut from the gel, and subjected to in-gel digestion with trypsin. Peptides were extracted and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Peptides were resuspended in 2.5% formic acid (FA), 2.5% acetonitrile (MeCN) and were loaded using a Micro AS autosampler (Thermo Electron) onto a microcapillary column of 100 μm inner diameter packed with 12 cm of reversed-phase Magic C18 packing material (5 μm, 200 Å; Michrom Bioresources, Inc., Auburn, CA, USA). SEQUEST matches were filtered by XCorr scores to a less than 1% false discovery rate when the control proteins were eliminated and protein matches were required to have three spectral matches, no identifiable false positive peptides remained.
CRISPR/Cas9 gene editing and siRNA transfections
To target PTPN12 using CRISPR/Cas9 methods three different synthetic guide DNAs (gDNAs) were designed using open source platforms (http://crispr.mit.edu). The PTP-PEST guide DNA sequences are as follows:4A2 (KO-1), 5′-GAGCGCTATTGGCCTTTGTA-3′, and 5B2 (KO-2), 5′-TCATCAAGAGGCAAGCGGTC-3′. 6C2 (KO-3), 5′ GTAAGATCGGAATGGAGTGA 3′. The vector LentiCRISPR (Addgene, #57818) were digested using BsmBI, and a pair of annealed oligos was cloned into the vector to make lentivirus using 293 (F)T and Infected target GSCs. Infected cells were selected by FACS based on GFP expression and individual clones were screened. PTPN12 editing was confirmed by immunoblotting and qRT-PCR. For quantitative RT-PCR experiments, total RNA was extracted using the RNAeasy Mini Kit (Qiagen, Hilden, Germany) and 500 ng of RNA were reversed-transcribed using SuperScript VILO Master Mix (ThermoFisher). Primer sequences used for human PTP-PEST 5′-AAATACTGCAGCCACCGGAAC-3′ and 5′-GCAACACTGGCTTTGGATGG -3′, for human VCP qRT-PCR reactions are 5′-AATTGCAGTTGTTCCGAGGT-3′ and 5′-TTGCCGTACTTCACATCAGG-3′.
To silence VCP gene expression, LN229 GBM cells were cultured in 6-well plates until 50%-60% confluent. Cells were transfected with targeting siRNAs or non-targeting controls at a final concentration 50nM using Lipofectamine RNAiMAX according to the manufacturer’s instructions. For transient silencing of VCP the following siRNA sequences (Sigma) were used: Duplex 1, 5′-GAAUAGAGUUGUUCGGAAU-3′ and 5′-AUUCCGAACAACUCUAUUC-3′; Duplex 2, 5′-GGAGGUAGAUAUUGGAAUU-3′ and 5′AAUUCCAAUAUCUACCUCC-3′. The non-targeting control duplex was 5′-UGGUUUACAUGUCGACUAA-3′ and 5′-UUAGUCGACAUGUAAACCA-3′. The effect of siRNA on cell viabilty was measured using the Celltiter-Glo luminescent cell viability assay kit according to the manufacture’s protocol. Briefly, at 24 hours after transfection, cells were collected and 5×103 cells were plated into 96-well plates. At 48, 72, and 96 hours post-transfection respectively, Celltiter-Glo reagent was added to each well that contained culture medium. Plates were incubated at room temperature for 10 min and the luminescence intensity was measured with a microplate reader.
Statistical analyses
All data represented herein were performed in replicates of three or more and are presented as the mean ± standard deviation, unless otherwise indicated. Differences among groups were analyzed using one-way analysis of variance. When overall analysis revealed significance among groups, means were compared and tested using Tukey’s post-hoc analysis. Statistical significance was set at P < 0.05. All statistical analyses were performed in SigmaPlot 12.0 software (Systat Software, Inc., San Jose, CA).
Results
Oncomine-based analysis for PTPN12 gene expression levels in open source datasets, including transcriptome sequences from GBM samples in The Cancer Genome Atlas (TCGA) and a large-scale cDNA microarray analysis of GBM samples, revealed approximately two-fold higher amounts of PTPN12 mRNA in GBM tissue versus normal human brain tissue (Figure 1A). In addition, querying the IVY GBM Atlas Project revealed that PTPN12 gene expression is spatially heterogeneous in different tumor samples. Most notably, invasive GBM regions expressed low levels of PTPN12 mRNA versus core regions of the tumor or the intratumoral vasculature, which expressed higher levels of PTPN12 mRNA (Figure 1B). Robust levels of PTP-PEST protein expression were detected in detergent-soluble lysates collected from 10 of 11 freshly resected GBM biopsies (Figure 1C). Next, we used an anti-PTP-PEST antibody to label fixed tissue sections from a normal human brain or from five different surgically resected GBM samples. All surgically resected specimens were from patients with primary GBM that had not received prior radiation or chemotherapy. As shown in Figure 1D-I, PTP-PEST protein was expressed at higher levels in GBM samples versus the non-cancerous brain. In all five GBM samples analyzed, PTP-PEST protein was detected in tumor cells as well as in vascular endothelial cells that comprise angiogenic GBM blood vessels. High levels of PTPN12 gene expression in patients with GBM or low-grade glioma, correlated with a reduced survival at three and five-year marks, based on analysis of Kaplan-Meier plots (Supplemental Figure S1A, B).
Figure 1. PTP-PEST expression is upregulated in human GBM.
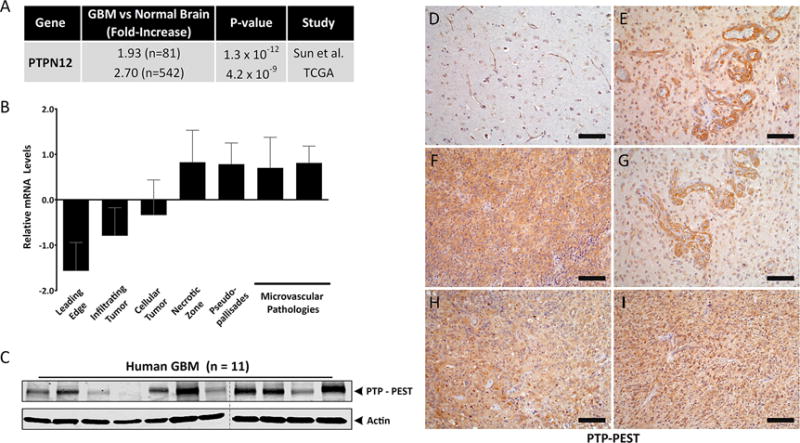
(A); Oncomine summary of two independent whole transcriptome data sets comparing PTPN12 mRNA levels in non-cancerous human brain samples versus primary GBM samples. (B); Differential expression of PTPN12 mRNA in various intratumoral regions based on analysis of the IVY GBM Atlas Project. Note that PTPN12 levels are lowest in invasive and infiltrating regions. (C); Comparative immunoblot analyses of PTP-PEST protein expression in 11 different detergent-soluble lysates from freshly resected human GBM samples. (D-I); Characterization of PTP-PEST protein expression patterns in the non-cancerous human brain (D) and in five different human GBM tissue sections (E-I) using an anti-PTP-PEST antibody. Scale bars, 50 μm.
Next, we analyzed levels of PTP-PEST protein in 9 different primary stem-like GBM cell (GSC) preparations that were cultured from freshly resected human tumor tissue (24). Human GSCs express the neural stem cell marker nestin and grow as neurosphere-like spheroids in serum-free media (Figure 2A). GSCs implanted into the brains of immunocompromised (NCR-nu/nu) mice generated malignant tumors that were well-vascularized and infiltrative (Figure 2B). The 9 GSCs analyzed expressed variable levels of PTP-PEST protein (Figure 2C). We selected GSC6-27 cells for more detailed functional analyses using Crisp/Cas9 gene editing, since these cells expressed robust levels of PTP-PEST protein and been reported previously to generate malignant brain tumors in mice (25). Select clinical features of patient tumors used for isolation of GSC6-27 cells and GSC7-2 cells are detailed in Supplemental Figure S1C. GSC6-27 spheroids were infected with recombinant lentiviruses expressing GFP and Cas9 (control lentivirus) or GFP and Cas9 in combination with guide RNAs (n=3) targeting different regions of PTPN12. Cas9-mediated targeting of PTPN12 in GSC6-27 cells resulted in a significant reduction in PTP-PEST protein expression, as determined by immunoblotting (Figure 2D) and immunofluorescence (Figure 2E, F). Analysis of viability and invasion revealed that GSC6-27 cells lacking PTP-PEST (KO) showed reduced viability (Figure 2G), but were significantly more invasive in comparison to ‘wild type’ (WT) PTPN12-expressing control cells (Figure 2H and Supplemental Figure S2A-F).
Figure 2. Genetically inhibiting PTP-PEST expression in human GSCs leads to enhanced invasion in vitro.
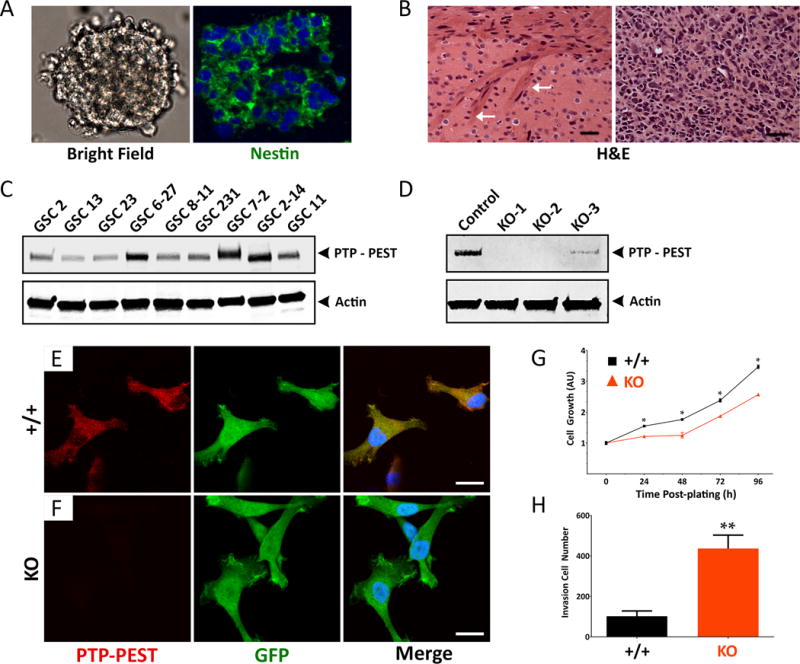
(A, B); Stem-like glioblastoma cells (GSCs) express the neural stem cell protein nestin and grow as free-floating spheroids in serum-free media (A). GSCs generate well-vascularized and invasive tumors after injection into the mouse brain (B). Arrows in the left panel (B) identify invasive GBM cells within white matter of the corpus callosum. Scale bars, 50 μm. (C); Comparative immunoblot showing PTP-PEST protein expression in detergent-soluble lysates from 9 different primary GSC cultures. (D); Anti-PTP-PEST immunoblot confirming CRISPR/Cas9 gene editing strategies to delete PTPN12 in GSC6-27 cells. Controls cell pools were generated with pLentiCRISPR lentivirus expressing GFP and Cas9 without targeting gDNAs, whereas KO cell pools were generated using lentivirus expressing GFP, Cas9 and gDNAs targeting three different regions of PTPN12. (E, F); Adherent control (E) and PTPN12 KO GSC6-27 cells (F) were labeled with anti-PTP-PEST and anti-GFP antibodies and visualized with fluorescent secondary antibodies. Scale bars, 200 μm. (G); Control and PTPN12 KO GSC6-27 spheroids were analyzed for viability over four days in vitro. Note that in comparison to control GSC spheroids, PTPN12 KO-1 spheroids show diminished viability, **p<0.001. Similar results were found with the PTPN12 KO-2 cells. (H); Analysis of invasive capacities of control and PTPN12 KO GSC6-27 cells. PTPN12 KO-2 spheroids were dissociated and three-dimensional cell invasion through Matrigel transwell assays, revealing increased invasion in cells with low levels of PTPN12 gene expression, **p<0.001.
Intracranial injection of control or KO GSCs led to the development of tumor-related neurological deficits within 3 months, at which point all mice were sacrificed for comparative analyses. Control GSC6-27 cells that expressed PTP-PEST formed tumors in the injected brain hemisphere (Figure 3A-F) that displayed localized patterns of invasion (Figure 3C, F and Supplemental Figure S3A, B) as revealed by monitoring tumor cells for expression of GFP. In contrast, tumors derived from PTPN12 KO GSCs were significantly larger (Figure 3G-L) and displayed more invasive behaviors as evidenced by dispersive tumor cells in the contralateral hemisphere (Figure 3I, L and Supplemental Figure S3C, D). More detailed microscopic analyses of brains in xenograft mice by H&E staining, immunohistochemistry using a human-specific anti-vimentin antibody or double immunofluorescence staining with anti-GFP and anti-GFAP antibodies revealed significantly larger and more invasive tumors generated from PTPN12 KO GSCs versus control PTPN12-expressing GSCs, which displayed more localized invasion (Figure 3M-R). Immunohistochemical analysis of control and PTPN12 KO brain tumor sections (GSC6-27) using an antibody directed against PTP-PEST revealed significantly reduced levels of PTP-PEST protein expression in KO tumors versus control tumors (Supplemental Figure S4A, B). We also utilized CRISPR/Cas9 methods in a second primary GSC preparation (GSC7-2), which expresses PTP-PEST (Figure 2C) and generates malignant tumors in the mouse brain (25). Consistent with the GSC6-27 results, genetic deletion of PTPN12 in a second GSC cell culture (GSC7-2) led to larger, more invasive intracranial tumors (Supplemental Figure S5A-C).
Figure 3. PTP-PEST suppresses GBM cell invasive growth in vivo.
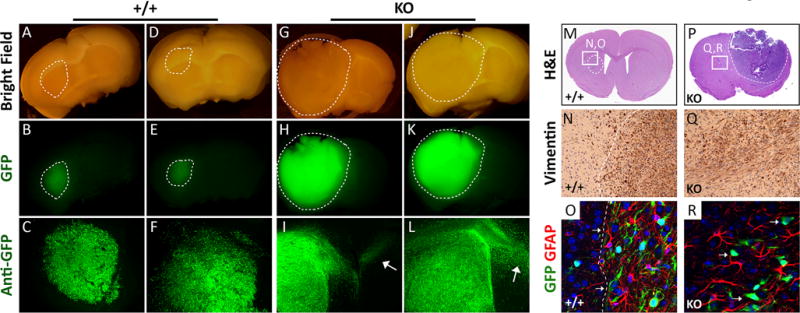
(A-L); Representative images of coronal sections from two different mouse brains harboring intracranial tumors derived from control (A-F) or PTPN12 KO-1 GSC6-27 cell pools (G-L). Note that control GSCs expressing PTP-PEST generate diffuse tumors that show localized patterns of invasion. In contrast, GSCs that lack PTP-PEST generate significantly larger tumors displaying robust patterns of invasion, with many GFP-expressing cells dispersing into the contralateral hemisphere. Images in upper and middle panels are of freshly cut brain sections. Images in lower panels are of anti-GFP immunofluorescence stains of vibratome-sectioned brains. (M-R); Coronal brain sections containing tumors generated from GSC6-27 control cells (M-O) or PTPN12 KO-1 cells (P-R) were stained with H&E (M, P), anti-vimentin antibody (N, Q), or anti-GFP and anti-GFAP antibodies in combination (O, P). Note that PTPN12 KO-1 cell pools generate significantly larger and more invasive intracranial brain tumors, as compared to control tumors that had more defined margins (dashed lines in N, O). PTPN12 KO tumors lack clearly defined margins due to their more dispersive behaviors.
Based on the in vitro viability and invasion data (Figure 2), we hypothesized that the larger tumors generated by PTPN12 KO GSCs in vivo were due to enhanced invasive growth behaviors. Therefore, we analyzed proliferative capacities of control and PTP12 KO tumors by immunofluorescence labeling with antibodies recognizing histone H3 phosphorylated on Ser10 (pHH3) and GFP to identify tumor cells in the mitotic stage of the cell cycle. PTPN12 KO GBM cells displayed similar rates of proliferation as control cells (Supplemental Figure S6A-C). Interestingly, comparison of tumor core regions by immunofluorescence staining for laminin, an abundant vascular basement membrane protein, revealed likely roles for tumor-expressed PTP-PEST in suppression of angiogenesis. While both control and PTPN12 KO tumors were well-vascularized, KO tumors contained abnormally large blood vessels with tortuous and telangiectasia-like morphologies (Supplemental Figure S7A, B).
Patients treated with ant-angiogenic agents such as the anti-VEGF neutralizing antibody bevacizumab, develop drug resistance that is frequently associated with robust GBM cell invasion (26,27). We analyzed roles for PTP-PEST in GBM cell invasion following treatment with bevacizumab using a xenograft mouse model in which human U87 cell-derived tumors are selected for acquired resistance to bevacizumab (Supplemental Figure S8A). Immunoblot analysis with a human-specific anti-PTP-PEST antibody, revealed robust levels of PTP-PEST protein in five different control (bevacizumab-sensitive) tumor samples. In contrast, three of five bevacizumab-resistant U87 tumor lysates showed reduced levels of PTP-PEST expression (Supplemental Figure S8B, C).
CRISPR/Cas9 gene editing strategies were incomplete in pools of GSCs, since low levels of PTP-PEST protein were detected (Figure 2D). Therefore, we performed selection experiments to identify GSC clones that displayed complete loss of PTP-PEST protein. Single GSCs were sorted based on GFP expression, deposited into individual wells of a 96-well plate, and allowed to grow as clonal spheroids in serum-free media. Protein lysates from expanded clones were then analyzed for PTPN12 expression by anti-PTP-PEST immunoblotting. In comparison to Cas9 controls, some clones expressing Cas9 and guide RNAs showed complete absence of PTP-PEST protein, whereas other clones expressed comparable levels of PTP-PEST (Supplemental Figure S9A-C). Quantitation of isolated clones did not reveal an essential role for PTPN12 in GSC self-renewal based on numbers of spheres formed (Supplemental Figure S9D). Lastly, in support of the tumor data for the GSC pools (Figures 2-3), PTPN12 KO cell clones (GSC6-27) generated larger brain tumors in vivo with prominent invasive growth features (Supplemental Figure S10A-F).
We next performed biochemical analyses of control and PTPN12 KO GSC6-27 cells. As shown in Figure 4A, immunoblots of detergent-soluble PTPN12 KO lysates revealed high levels of phosphotyrosine-containing proteins in comparison to control cell lysates. Immunofluorescence analysis of adherent PTPN12 KO cells also showed elevated levels of phosphotyrosine-containing proteins within cell-ECM attachment points (Figure 4B, C), suggesting hyperphosphorylation of PTP-PEST protein substrates. To identify and characterize PTP-PEST-regulated signaling pathways in GSCs, we performed a substrate-trapping screen. This strategy involves the use of a GST-tagged wild type PTP-PEST catalytic domain (GST-WT) or a catalytic domain containing a C231S point mutation (GST-C/S) that reduces enzymatic activities (23). Substrates that bind to wild type PTP-PEST protein are dephosphorylated, dissociate from the enzyme, and are not detected in the bound pull-down fraction, whereas substrates that interact with the mutant PTP-PEST protein are not rapidly dephosphorylated and remain in the bound fractions. In comparison to the GST control protein or the GST-WT recombinant protein (Figure 4D), the GST-C/S protein bound to multiple tyrosine-phosphorylated substrates from GSC lysates (Figure 4E). Next, we used GST-WT and GST-C/S recombinant proteins and fractionated interacting substrates from GSC6-27 lysates. Silver-stained SDS gels revealed several protein bands that interacted preferentially with GST-C/S PTP-PEST versus GST-WT PTP-PEST protein or the GST control protein (Figure 4F). Therefore, excised bands from silver-stained gels were enzymatically digested and analyzed by mass spectrometry, revealing human peptide sequences that bound preferentially to the GST-C/S PTP-PEST (Supplemental Table 1). The two major PTP-PEST substrates identified by mass spectrometry (based on total number of peptides with unique sequences) were human Vcp/p97 and Cas/Bcar1.
Figure 4. A substrate trapping screen to identify PTP-PEST protein substrates in GBM cells.
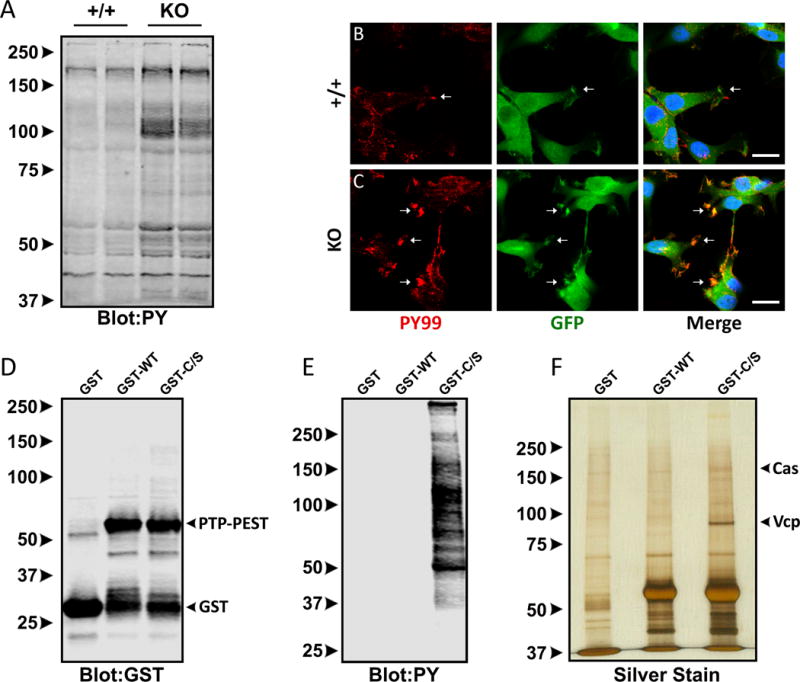
(A); Detergent-soluble lysates from control and PTPN12 KO GSC6-27 cells were immunoblotted with an anti-phosphotyrosine antibody, revealing increased levels of tyrosine phosphorylated proteins in KO-1 cells. (B, C); Control cells (B) and PTPN12 KO GSC6-27 cells (C) were immunofluorescently labeled with anti-phosphotyrosine (red) and anti-GFP (green) antibodies. Scale bars, 200 μm. (D); Anti-GST immunoblot showing expression of GST-tagged wild type and mutant (C231S) PTP-PEST recombinant proteins purified from bacteria. (E); Detergent-soluble lysates from control and PTPN12 KO-1 cells (GSC6-27) were incubated with recombinant GST-tagged wild type and mutant (C231S) PTP-PEST protein constructs. Interacting proteins were identified by anti-phosphotyrosine immunoblotting. (F); Silver-stained gel showing protein bands that differentially interact with GST-tagged wild type PTP-PEST versus catalytically inactive (C231S) PTP-PEST protein. Detergent-soluble lysates from GSC6-27 cells were used for the substrate-trapping screen.
PTP-PEST binding to Cas and Vcp was next confirmed by pull-down experiments with recombinant GST-C/S protein (Figure 5A). We also confirmed multiprotein complex formation between PTP-PEST, Cas and Vcp by co-immunoprecipitating these proteins from detergent-soluble WT and KO GSC6-27 lysates using different antibody combinations (Figure 5B-E). Notably, interactions between Cas and Vcp were not detected in KO lysates, revealing that PTP-PEST is necessary to effectively bridge Vcp and Cas in GSCs. Use of PTPN12 KO cells also supports the specificity of the anti-PTP-PEST antibody. Analysis of Vcp binding to phosphorylated Cas revealed reduced interactions in PTPN12 KO cells (Figure 5F). Furthermore, we detected tyrosine hyperphosphorylation of Vcp in PTPN12 KO cells (Figure 5F). In comparison to control GSC6-27 cells, PTPN12 KO cell lysates contained significantly higher levels of Cas phosphorylated on tyrosine 410 (Figure 5G). Increased levels of other tyrosine phosphorylated focal adhesion components, including FAK and paxillin, were also detected in PTPN12 KO cells, as revealed by immunoblotting with phospho-specific antibodies (Figure 5G). Immunofluorescence analyses revealed elevated levels of total paxillin protein with enrichment at the leading edge of PTPN12 KO GSCs (Supplemental Figure S11A-F), supporting possible roles for PTP-PEST in regulating focal adhesion substrate phosphorylation and/or focal adhesion protein stability. Prior reports have shown that PTP-PEST mediates the dephosphorylation of various receptor tyrosine kinases to suppress their signaling capacities in breast carcinoma cells (28). We analyzed the phosphorylation status of EGFR and Her2/ErbB2, two receptor tyrosine kinases with established roles in GBM initiation and progression. As shown in Figure 5H, increased tyrosine phosphorylation of these proteins was detected in PTPN12 KO cells versus control cells.
Figure 5. PTP-PEST forms a multi-protein complex with Cas and Vcp in GBM cells.
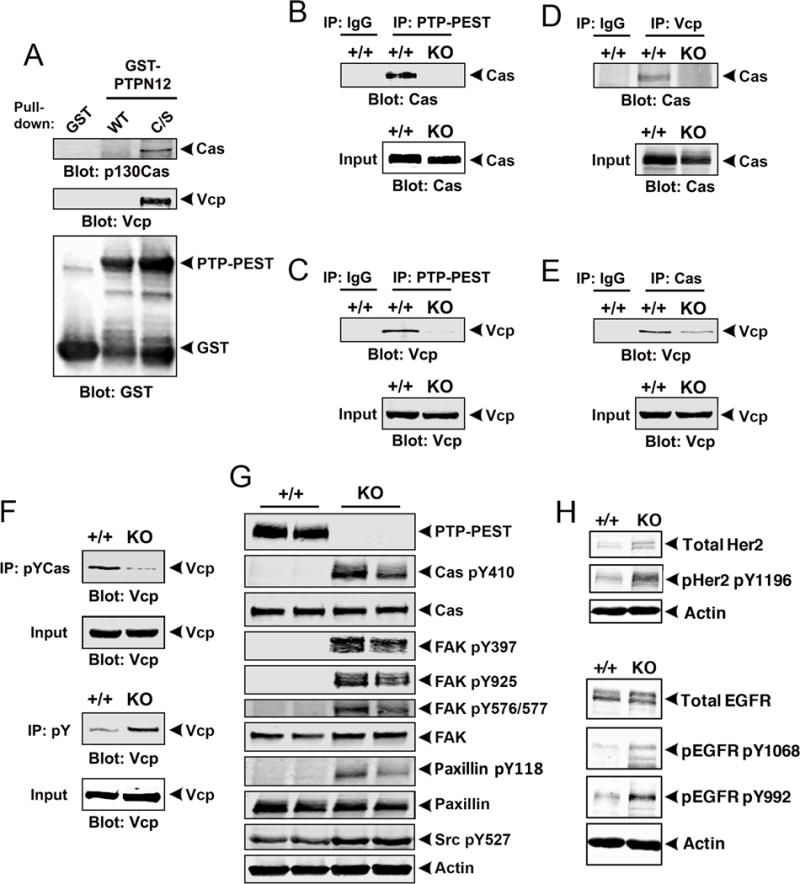
(A); Pull-down experiments with GST-tagged PTP-PEST proteins (WT and C/S) reveal interactions with Vcp and Cas in detergent-soluble GSC lysates. (B, C); Lysates from GSC6-27 control or PTPN12 KO-1 cell pools were immunoprecipitated with anti-PTP-PEST antibodies and resolved lysates were immunoblotted to analyze protein-protein interactions. Note that Cas (B) and Vcp (C) proteins interact in control lysates but not in PTPN12 KO lysates as revealed by co-immunoprecipitation. (D, E); Detergent-soluble lysates from GSC6-27 control or PTPN12 KO-1 cells were immunoprecipitated with anti-Vcp (D) or anti-Cas (E) antibodies and resolved lysates were immunoblotted to analyze protein-protein interactions. (F); Tyrosine phosphorylated Cas protein or total cellular phosphotyrosine-containing proteins were immunoprecipitated from control or PTPN12 KO GSC6-27 cells and Vcp interactions were analyzed by immunoblotting with anti-Vcp antibodies. Note that in comparison to control cells, Vcp is hyperphosphorylated in PTPN12 KO-1 cells, but also shows reduced interactions with phosphorylated Cas. (G); Detergent-soluble lysates from control (n=2) and PTPN12 KO GSC6-27 cells (n=2) were immunoblotted for phosphorylated Cas and other phospho-specific antibodies recognizing common focal adhesion proteins such as FAK and paxillin. (H); Control and PTPN12 KO GSC6-27 cell lysates were immunoblotted for total EGFR, total Her2/ErbB2, or specific cytoplasmic domain phosphorylated sites on each protein. Note the increased levels of EGFR and Her2/ErbB2 tyrosine phosphorylation in PTPN12 KO GSCs.
Src phosphorylates Vcp at Y805 and this impacts interactions with other proteins (20). Therefore, we generated point mutations at Vcp Y805 that would block tyrosine phosphorylation (Y805F) or partially mimic phosphorylation (Y805E). Co-immunoprecipitation experiments confirmed that full-length PTP-PEST interacts with Vcp, with the Y805E phosphomimetic diminishing interactions between PTP-PEST and Cas (Figure 6A). There were also differences in the subcellular distribution of EGFP-tagged Vcp proteins, with the Y805E mutant protein showing enriched localization at cell-ECM contacts, likely representing focal adhesions (Figure 6B). Analysis of subcellular localization of endogenous Vcp protein by immunofluorescence also revealed expression throughout control and PTPN12 KO cells. Interestingly, in PTPN12 KO cells we detected possible enrichment of Vcp at cell-ECM attachment points (Supplemental Figure S12A, B).
Figure 6. PTP-PEST-dependent phosphorylation of Vcp at Y805 regulates interactions with Cas and impacts protein ubiquitination in GSCs.
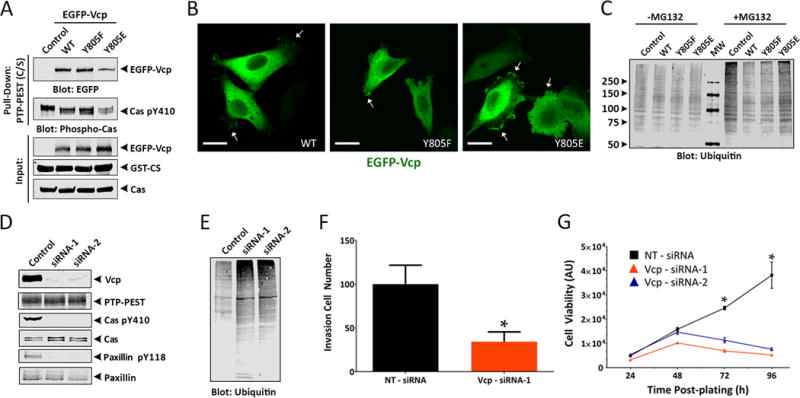
(A); GST pull-down experiments reveal that PTP-PEST C/S protein interacts with Vcp independently of Y805 phosphorylation status. HEK-293T cells were transiently transfected with plasmids expressing Flag-tagged PTP-PEST in combination EGFP-tagged wild type Vcp or Vcp mutant constructs containing Y805F or Y805E point mutations. Note that expression of the VcpY805E phosphomimetic point mutation leads to reduced interactions between Vcp and phosphorylated Cas. (B); HEK-293T cells, which do not express endogenous PTP-PEST protein, were transfected with plasmids expressing EGFP-tagged wild type Vcp or EGFP-Vcp constructs containing Y805F or Y805E point mutations. Anti-GFP immunofluorescence reveals that wild type and Y805F proteins display diffuse intracellular expression, whereas the Y805E protein shows enriched localization in focal adhesions. Scale bars, 20 μm. (C); Detergent-soluble lysates from cells transiently expressing either EGFP-tagged wild type Vcp, Vcp Y805F, or Vcp Y805E proteins were analyzed for total ubiquitination levels by immunoblotting with anti-ubiquitin antibodies after pre-treatment with the proteasome inhibitor MG132. Note that overexpression of the Vcp Y805E phosphomimetic mutant protein leads to higher levels of ubiquitinated cellular proteins, similar to the non-transfected control cells. (D, E); Transient RNAi-mediated targeting of VCP gene expression in LN229 GBM cells using two different siRNAs. (F, G); Transiently silencing VCP expression using two different transfected siRNAs leads to diminished LN229 GBM cell invasion (F) and growth (G) in vitro.
The C-terminal domain of Vcp interacts with various proteins involved in segregating ubiquitinated substrates for degradation by the proteasome (18), and these interactions can be modulated via phosphorylation of Y805 (20). Therefore, we analyzed levels of ubiquitinated proteins in cells expressing EGFP-tagged wild type Vcp, Y805F or Y805E mutant proteins. Expression of wild type Vcp or Y805F resulted in similar levels of ubiquitin-containing proteins, whereas cells expressing Vcp Y805E showed a noticeable increase in ubiquitinated proteins (Figure 6C). Next, we used siRNAs to transiently silence VCP gene expression in LN229 human GBM cells, which form invasive brain tumors in vivo (21). A significant reduction in Vcp gene expression was confirmed by quantitative RT-PCR with primers specific for VCP, as well as by immunoblotting detergent-soluble lysates with anti-Vcp antibodies (Figure 6D). Interestingly, silencing Vcp expression correlated with reduced levels of tyrosine phosphorylated Cas and paxillin, although the total levels of these proteins were not impacted (Figure 6D). Vcp has been shown to bind to multiple ubiquitinated targets and direct them for degradation via the proteasome. Along these lines, we detected increased levels of ubiquitinated proteins in siRNA-transfected GBM cells lacking VCP expression (Figure 6E), similar to levels detected after expressing the Vcp Y805E phosphomimetic protein (Figure 6C). Consistent with the reduced levels of phosphorylated focal adhesion proteins and increased ubiquitination, RNAi-mediated silencing of VCP resulted in impaired invasion as quantified in Matrigel transwell assays (Figure 6F) and diminished LN229 GBM cell viability (Figure 6G).
Analysis of fixed human tissue sections by immunohistochemical labeling with anti-Vcp antibodies revealed elevated levels of Vcp protein in human GBM cells in fixed tissue sections versus non-cancerous human brain tissue (Figure 7A-D). Interestingly, comparisons of VCP versus PTPN12 mRNA expression levels in microdissected tumor regions (IVY GBM Atlas Project) revealed an inverse relationship between the two genes in GBM cells, but not in intratumoral blood vessels (Figure 7E). GBM cells in proliferative zones expressed higher levels of PTPN12 and low levels of VCP, whereas GBM cells at the invasive margins expressed low PTPN12 and high VCP. These results are consistent with our analyses of GSCs and tumor invasiveness in pre-clinical animal models. To further establish links between Vcp and GBM cell focal adhesion dynamics, we used CB-5083, a pharmacologic inhibitor of Vcp ATPase activities (29). Treatment of LN229 GBM cells with of CB-5083 resulted in a significant increase in ubiquitinated proteins and a reduction in total cellular tyrosine phosphorylated proteins (Figure 7F, G). Analysis of focal adhesion proteins revealed that Vcp inhibition with CB-5083 resulted in a reduction in the phosphorylation of Cas and Paxillin and this correlated with reduced expression of PTP-PEST (Figure 7H). A major reduction in expression of total FAK and phosphorylation of FAK at Y935 was detected following CB-5083 treatment. Interestingly, expression of PTP-PEST was also reduced in CB-5083 treated cells. In contrast, Vcp protein levels were not impacted in cells treated with CB-5083. Lastly, in cells we detected PTPN12-dependent sensitivity to CB-5083. As shown in Figure 7I, high concentrations of CB-5083 impacted the viability of control LN229 cells (IC50 = 5 μM), whereas PTPN12 KO cells showed significantly reduced viability at much lower concentrations of CB-5083 (IC50 = 0.937 μM). Collectively, these genetic and pharmacologic data reveal that PTP-PEST in GBM cells precisely balances focal adhesion assembly and disassembly via regulation of the phosphorylation status of Vcp and key focal adhesion and cytoskeletal proteins such as Cas. These regulatory mechanisms control the stability of focal adhesion proteins such as Cas via phosphorylation-mediated ubiquitination, which balances GBM cell viability and growth versus invasion (Figure 7J, K).
Figure 7. PTPN12 and VCP gene expression are inversely correlated with GBM cell invasive behaviors.
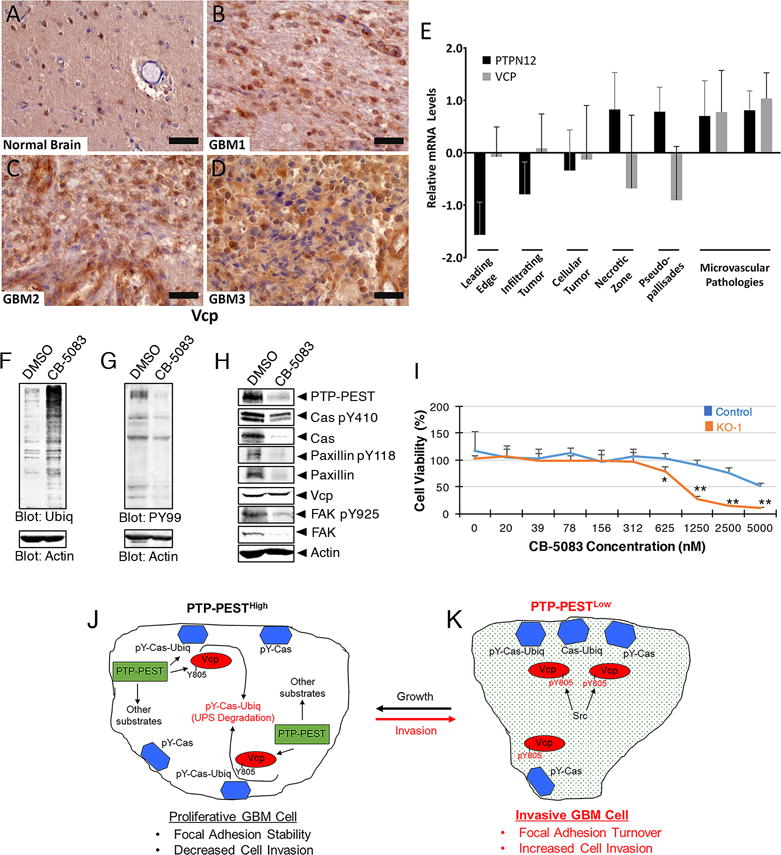
(A-D); Fixed normal human brain (A) or human GBM sections (B-D) were immunolabeled with anti-Vcp antibodies. Note the increased expression of Vcp protein in GBM cells of the tumor core in comparison to the non-cancerous brain region. Scale bars, 20 μm. (E); Querying the IVY GBM Atlas Project revealed inverse expression of PTPN12 and VCP mRNA levels in various GBM regions, including the invasive front. Notably, in comparison to intratumoral blood vessels, invasive GBM cells express significantly lower levels of PTPN12 mRNA and higher levels of VCP. (F, G); Detergent-soluble lysates from LN229 GBM cells treated with DMSO or 0.4 μM CB-5083 were immunoblotted with anti-ubiquitin (F) or anti-phosphotyrosine antibodies (G). Note that treatment with CB-5083 leads to higher levels of ubiquitinated proteins, but lower levels of tyrosine phosphorylated proteins. (H); LN229 GBM cell lysates were immunoblotted with antibodies recognizing specific focal adhesion proteins. Note the decreased levels of Cas, Paxillin and FAK tyrosine phosphorylation in cells treated with CB-5083. In addition, CB-5083 treatment leads to decreased levels of total PTP-PEST expression, but Vcp protein expression levels are not impacted. (I); Analysis of survival in control and PTPN12 KO LN229 GBM cells treated with varying concentrations of CB-5083. In contrast to control cells (IC50 = 5 μM), note that PTPN12 KO cells are more sensitive to CB-5083 (IC50 = 0.937 μM), *p=0.03 and **p<0.001. (J, K); Illustrative model showing that GBM cells with high levels of PTP-PEST are more proliferative (J). PTP-PEST interacts with Vcp (shown as a monomer) and ubiquitinated and phosphorylated Cas (pY-Cas-Ubiq) in focal adhesions (blue hexagons), leading to Vcp-mediated Cas protein segregation and degradation via the UPS. As a result, there is balanced focal adhesion assembly and disassembly, leading to more viable and less invasive cells. In GBM cells that express low levels of PTP-PEST (K), interactions between Vcp and pY-Cas-Ubiq are impaired, due in part to hyperphosphorylation at Vcp Y805 and altered phosphodegrons in Cas and probably other proteins. This leads to increased FA disassembly and robust tumor cell invasion in vivo.
Discussion
There is precedent for the ubiquitin proteasome system in the regulation of cell-ECM adhesion and signaling dynamics. For example, SOCS7, an SH2 domain-containing component of Cullin-5 E3 ubiquitin ligase complex (CRL5), promotes degradation of FAK (30). In addition, the E3 ligase Trim15 binds to paxillin and promotes focal adhesion disassembly (31). Degradation of Cas in focal adhesions via SOCS6 and CRL5 is involved in Src-dependent cell transformation (32) and migration (33). Our data showing that PTP-PEST couples protein phosphorylation and ubiquitination are consistent with these prior reports, and suggest that PTP-PEST control of Vcp segregase activities is an additional regulatory step to control focal adhesion dynamics by selectively targeting Cas for degradation by the proteasome. Cas contains a substrate binding domain comprised of amino acids ~100-450, which contains 15 YxxP motifs that are phosphorylated by Src (34) that is involved in the recruitment of various SH2 domain-containing proteins. Cells expressing mutant Cas that lacks all 15 tyrosine phosphorylation sites (Y-F mutations) display defects in migration, but not proliferation (35). In epithelial cells mutant Cas protein that lack these Src phosphorylation motifs is resistant to degradation by the Cul5SOCS6 E3 ligase complex (33). We propose that PTP-PEST-mediated dephosphorylation of Cas creates phosphodegrons that lead to additional post-translational modifications via recruited ubiquitin ligase complexes. Vcp subsequently segregates Cas from focal adhesions and targets it to the proteasome for destruction.
The substrate trapping screen was performed with the isolated catalytic domain of PTP-PEST, revealing that this region alone can interact with Cas. However, it is likely that the SH3 domain of Cas is also important for interactions with proline-rich region of PTP-PEST based on a previous substrate trapping screen (36) as well as a more recent structural study showing that the isolated SH3 domain of Cas binds to PTP-PEST (37). Interactions between Vcp and PTP-PEST may also impact PTP-PEST functions; indeed, we detect reduced phosphorylated levels of the PTP-PEST substrates Cas and Paxillin in GBM cells that lack Vcp expression, suggesting that Vcp may normally suppress PTP-PEST enzymatic activities. However, silencing VCP in GBM cells does not impact total PTP-PEST protein levels. We have discovered an inverse relationship between VCP and PTP-PEST mRNA levels in different GBM regions, suggesting that PTPN12 and VCP expression in GBM regions may be linked to gene regulatory events. While we have characterized these signaling links in GBM cells, it is likely that tyrosine phosphorylation-mediated regulation of Vcp occurs in other cell types, and may actually be a general regulatory mechanism during cell migration. For example, a substrate-trapping screen using Ewing sarcoma cells identified Vcp as a substrate of PTPN13/PTPL1, with Y805 as the major site of dephosphorylation (38). However, the functional significance of the PTPL1-Vcp interaction was not elucidated.
While we have focused on GBM cell-intrinsic regulatory mechanisms underlying PTP-PEST control of invasion, there are extracellular cues that activate and inhibit PTP-PEST functions. For example, we have reported that the adhesion receptor β8 integrin is upregulated in invasive GBM cells and drives tumor cell invasion along blood vessels. In cultured neuroblasts we have found that β8 integrin is necessary for localization of a fraction of PTP-PEST protein to the leading edge (39). However, when we use RNAi strategies to inhibit β8 integrin gene expression in GBM cells or fractionate cancer cells that lack endogenous β8 integrin from resected GBM samples (40,41), tumor invasion in the mouse brain is diminished. This is in contrast to inhibition of PTP-PEST, which leads to enhanced GBM cell invasion. β8 integrin interacts with cell surface receptors such as Nrp1 to regulate ECM adhesion (22,42) and intracellular proteins such as RhoGDI1 and Spinophilin to control Rac1/Cdc42 signaling and actin dynamics to promote migration and invasion (21,43). Hence, it is likely that integrin signaling via PTP-PEST suppresses GBM cell invasion, whereas interactions with RhoGDI1 and Spinophilin promote cell invasion. PTP-PEST is also reported to have tumor suppressor-like functions in breast cancer via negative regulation of HER2/ErbB2 signaling (28), with loss of PTPN12 expression leading to enhanced tumor cell growth, migration and survival (44). In hepatocellular carcinoma, down-regulation of PTPN12 expression also enhances tumor growth via increased RTK signaling and increased ubiquitin proteasome-related activities (45). In contrast to these studies of epithelial cell-derived carcinomas, our data for PTPN12 in gliomas, which are mainly derived from neural stem cells, reveals that it suppresses migration/invasion but promotes growth and viability, with PTPN12 KO cells showing increased motility but diminished growth and survival, even though EGFR and Her2 are hyperphosphorylated. Hence, we propose that PTPN12 has cell type-specific roles in differentially regulating growth, survival and migration. Alternatively, in PTPN12 KO GBM cells cross-talk between receptor tyrosine kinases and β8 integrin or other receptors may be altered, leading to reduced growth/viability, but enhanced invasion.
The ubiquitin proteasome system is deregulated in GBM, with elevated expression of key proteasome components and increased enzymatic activities, which likely provide growth and survival advantages to tumor cells (46). Treatment with bortezomib/velcade (47) or the related compound carfilzomib (48), which both bind and inhibit the core catalytic site of the 26S proteasome, leads to diminished GBM cell proliferation and invasion. Unfortunately, bortezomib in combination with temozolomide has shown limited clinical efficacy in patients with recurrent GBM (49). Additional components of the ubiquitin proteasome system, such as Vcp, are also attractive targets for therapy. For example, CB-5083 induces the accumulation of ubiquitinated peptides in tumor cells resulting in proteotoxic crisis and apoptosis. Given our discovery of important functions for Vcp in phosphorylation-dependent ubiquitination of key focal adhesion proteins, targeting Vcp or its regulatory effectors with specific small molecule inhibitors may prove beneficial for blocking tumor cell invasive growth in patients with GBM.
Supplementary Material
Statement of significance.
PTP-PEST balances GBM cell growth and invasion by interacting with the ATP-dependent ubiquitin segregase Vcp/p97 and regulating phosphorylation and stability of the focal adhesion protein p130Cas.
Acknowledgments
We thank Dr. David Hawk for technical assistance with mass spectrometry analyses, Dr. Jordan Toutounchian for assistance with figure compilation, and Dr. Manish Aghi for providing the U87 xenograft lysates.
Financial Support
J.H. McCarty received grant support from NIH/NINDS (R01NS07635, R01NS078402 and R21NS085688), Cancer Prevention and Research Institute of Texas (RP140411), and NIH/NCI (P50CA127001).
Footnotes
COI Statement: The authors declare no potential conflicts of interest
References
- 1.Louis DN. Molecular pathology of malignant gliomas. Annual review of pathology. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 2.Batchelor TT, Reardon DA, de Groot JF, Wick W, Weller M. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:5612–9. doi: 10.1158/1078-0432.CCR-14-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sirois J, Cote JF, Charest A, Uetani N, Bourdeau A, Duncan SA, et al. Essential function of PTP-PEST during mouse embryonic vascularization, mesenchyme formation, neurogenesis and early liver development. Mech Dev. 2006;123:869–80. doi: 10.1016/j.mod.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, Yang F, Liu C, Xiao P, Xu Y, Liang Z, et al. Crystal Structure and Substrate Specificity of PTPN12. Cell reports. 2016;15:1345–58. doi: 10.1016/j.celrep.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Jamieson JS, Tumbarello DA, Halle M, Brown MC, Tremblay ML, Turner CE. Paxillin is essential for PTP-PEST-dependent regulation of cell spreading and motility: a role for paxillin kinase linker. J Cell Sci. 2005;118:5835–47. doi: 10.1242/jcs.02693. [DOI] [PubMed] [Google Scholar]
- 6.Sastry SK, Lyons PD, Schaller MD, Burridge K. PTP-PEST controls motility through regulation of Rac1. J Cell Sci. 2002;115:4305–16. doi: 10.1242/jcs.00105. [DOI] [PubMed] [Google Scholar]
- 7.Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. The Journal of cell biology. 2016;215:445–56. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devreotes P, Horwitz AR. Signaling networks that regulate cell migration. Cold Spring Harbor perspectives in biology. 2015;7:a005959. doi: 10.1101/cshperspect.a005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinschmidt EG, Schlaepfer DD. Focal adhesion kinase signaling in unexpected places. Current opinion in cell biology. 2017;45:24–30. doi: 10.1016/j.ceb.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tikhmyanova N, Little JL, Golemis EA. CAS proteins in normal and pathological cell growth control. Cellular and molecular life sciences: CMLS. 2010;67:1025–48. doi: 10.1007/s00018-009-0213-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deneka A, Korobeynikov V, Golemis EA. Embryonal Fyn-associated substrate (EFS) and CASS4: The lesser-known CAS protein family members. Gene. 2015;570:25–35. doi: 10.1016/j.gene.2015.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, et al. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015–26. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nature cell biology. 2004;6:154–61. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 14.Cooper JA, Kaneko T, Li SS. Cell regulation by phosphotyrosine-targeted ubiquitin ligases. Molecular and cellular biology. 2015;35:1886–97. doi: 10.1128/MCB.00098-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komander D, Rape M. The ubiquitin code. Annual review of biochemistry. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 16.Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochimica et biophysica acta. 2012;1823:130–7. doi: 10.1016/j.bbamcr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, et al. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016;351:871–5. doi: 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Madsen L, Seeger M, Semple CA, Hartmann-Petersen R. New ATPase regulators–p97 goes to the PUB. The international journal of biochemistry & cell biology. 2009;41:2380–8. doi: 10.1016/j.biocel.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Li G, Zhao G, Schindelin H, Lennarz WJ. Tyrosine phosphorylation of ATPase p97 regulates its activity during ERAD. Biochem Biophys Res Commun. 2008;375:247–51. doi: 10.1016/j.bbrc.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Reyes SB, Narayanan AS, Lee HS, Tchaicha JH, Aldape KD, Lang FF, et al. alphavbeta8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Molecular biology of the cell. 2013;24:474–82. doi: 10.1091/mbc.E12-07-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwiatkowski SC, Guerrero PA, Hirota S, Chen Z, Morales JE, Aghi M, et al. Neuropilin-1 modulates TGFbeta signaling to drive glioblastoma growth and recurrence after anti-angiogenic therapy. PloS one. 2017;12:e0185065. doi: 10.1371/journal.pone.0185065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Substrate-trapping techniques in the identification of cellular PTP targets. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res. 2010;16:461–73. doi: 10.1158/1078-0432.CCR-09-1983. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–46. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19:1773–83. doi: 10.1158/1078-0432.CCR-12-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22:21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144:703–18. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blythe EE, Olson KC, Chau V, Deshaies RJ. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP*NPLOC4*UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E4380–E8. doi: 10.1073/pnas.1706205114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu E, Cote JF, Vuori K. Negative regulation of FAK signaling by SOCS proteins. The EMBO journal. 2003;22:5036–46. doi: 10.1093/emboj/cdg503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uchil PD, Pawliczek T, Reynolds TD, Ding S, Hinz A, Munro JB, et al. TRIM15 is a focal adhesion protein that regulates focal adhesion disassembly. Journal of cell science. 2014;127:3928–42. doi: 10.1242/jcs.143537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teckchandani A, Laszlo GS, Simo S, Shah K, Pilling C, Strait AA, et al. Cullin 5 destabilizes Cas to inhibit Src-dependent cell transformation. J Cell Sci. 2014;127:509–20. doi: 10.1242/jcs.127829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teckchandani A, Cooper JA. The ubiquitin-proteasome system regulates focal adhesions at the leading edge of migrating cells. eLife. 2016:5. doi: 10.7554/eLife.17440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynolds AB, Kanner SB, Bouton AH, Schaller MD, Weed SA, Flynn DC, et al. SRChing for the substrates of Src. Oncogene. 2014;33:4537–47. doi: 10.1038/onc.2013.416. [DOI] [PubMed] [Google Scholar]
- 35.Patwardhan P, Shen Y, Goldberg GS, Miller WT. Individual Cas phosphorylation sites are dispensable for processive phosphorylation by Src and anchorage-independent cell growth. The Journal of biological chemistry. 2006;281:20689–97. doi: 10.1074/jbc.M602311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cote JF, Charest A, Wagner J, Tremblay ML. Combination of gene targeting and substrate trapping to identify substrates of protein tyrosine phosphatases using PTP-PEST as a model. Biochemistry. 1998;37:13128–37. doi: 10.1021/bi981259l. [DOI] [PubMed] [Google Scholar]
- 37.Gemperle J, Hexnerova R, Lepsik M, Tesina P, Dibus M, Novotny M, et al. Structural characterization of CAS SH3 domain selectivity and regulation reveals new CAS interaction partners. Scientific reports. 2017;7:8057. doi: 10.1038/s41598-017-08303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abaan OD, Hendriks W, Uren A, Toretsky JA, Erkizan HV. Valosin containing protein (VCP/p97) is a novel substrate for the protein tyrosine phosphatase PTPL1. Experimental cell research. 2013;319:1–11. doi: 10.1016/j.yexcr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HS, Cheerathodi M, Chaki SP, Reyes SB, Zheng Y, Lu Z, et al. Protein tyrosine phosphatase-PEST and beta8 integrin regulate spatiotemporal patterns of RhoGDI1 activation in migrating cells. Mol Cell Biol. 2015;35:1401–13. doi: 10.1128/MCB.00112-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerrero PA, Tchaicha JH, Chen Z, Morales JE, McCarty N, Wang Q, et al. Glioblastoma stem cells exploit the alphavbeta8 integrin-TGFbeta1 signaling axis to drive tumor initiation and progression. Oncogene. 2017 doi: 10.1038/onc.2017.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH. Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res. 2011;71:6371–81. doi: 10.1158/0008-5472.CAN-11-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirota S, Clements TP, Tang LK, Morales JE, Lee HS, Oh SP, et al. Neuropilin 1 balances beta8 integrin-activated TGFbeta signaling to control sprouting angiogenesis in the brain. Development. 2015;142:4363–73. doi: 10.1242/dev.113746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheerathodi M, Avci NG, Guerrero PA, Tang LK, Popp J, Morales JE, et al. The Cytoskeletal Adapter Protein Spinophilin Regulates Invadopodia Dynamics and Tumor Cell Invasion in Glioblastoma. Mol Cancer Res. 2016;14:1277–87. doi: 10.1158/1541-7786.MCR-16-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Davidson D, Martins Souza C, Zhong MC, Wu N, Park M, et al. Loss of PTPN12 Stimulates Progression of ErbB2-Dependent Breast Cancer by Enhancing Cell Survival, Migration, and Epithelial-to-Mesenchymal Transition. Mol Cell Biol. 2015;35:4069–82. doi: 10.1128/MCB.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kodama T, Newberg JY, Kodama M, Rangel R, Yoshihara K, Tien JC, et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2016;113:E3384–93. doi: 10.1073/pnas.1606876113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hede SM, Savov V, Weishaupt H, Sangfelt O, Swartling FJ. Oncoprotein stabilization in brain tumors. Oncogene. 2014;33:4709–21. doi: 10.1038/onc.2013.445. [DOI] [PubMed] [Google Scholar]
- 47.Styczynski J, Olszewska-Slonina D, Kolodziej B, Napieraj M, Wysocki M. Activity of bortezomib in glioblastoma. Anticancer research. 2006;26:4499–503. [PubMed] [Google Scholar]
- 48.Areeb Z, Stylli SS, Ware TM, Harris NC, Shukla L, Shayan R, et al. Inhibition of glioblastoma cell proliferation, migration and invasion by the proteasome antagonist carfilzomib. Medical oncology. 2016;33:53. doi: 10.1007/s12032-016-0767-3. [DOI] [PubMed] [Google Scholar]
- 49.Odia Y, Kreisl TN, Aregawi D, Innis EK, Fine HA. A phase II trial of tamoxifen and bortezomib in patients with recurrent malignant gliomas. J Neurooncol. 2015;125:191–5. doi: 10.1007/s11060-015-1894-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.