

Abstract
Deciphering disulfide bond patterns in proteins remains a significant challenge. In the present study, inter-linked disulfide bonds connecting peptide chains are homolytically cleaved with 193 nm ultraviolet photodissociation (UVPD). Analysis of insulin showcased the ability of UVPD to cleave multiple disulfide bonds and provide sequence coverage of the peptide chains in the same MS/MS event. For proteins containing more complex disulfide bonding patterns, an approach combining partial reduction and alkylation mitigated disulfide scrambling and allowed assignment of the array of disulfide bonds. The four disulfide bonds of lysozyme and the 19 disulfide bonds of serotransferrin were characterized through LC-UVPD-MS analysis of non-reduced and partially reduced protein digests.
Keywords: Disulfide bonds, ultraviolet photodissociation, cysteine, protein
Graphical Abstract
Introduction
Disulfide bridges connecting cysteine residues define structure, provide stability, and regulate biological activity of proteins.1–3 Since improper disulfide bonding or disulfide scrambling can have detrimental effects on a protein’s function, determining cysteine disulfide bonding partners is a critical part of understanding protein folding as well as deciphering three dimensional and quaternary structures of proteins.4,5 Advances in biotherapeutics containing disulfide bonds have further driven the need for analyzing this post-translational modification to ensure correct disulfide bonding patterns.6,7 Conventional methods for analysis of disulfide bond connectivities include nuclear magnetic resonance (NMR) and X-ray crystallography.8–11 Neither of these methods directly probe disulfide bonds and instead assign disulfide bonding partners inferred from three dimensional structures using probabilistic models.12 In addition to requiring relatively large amounts of purified protein, the NMR and X-ray methods are not capable of analyzing proteins containing multiple disulfide linkage patterns or scrambled disulfide bonds. Mass spectrometry is an alternative analytical tool for disulfide linkage analysis that circumvent some of these shortcomings.13–15 Additionally, MS/MS methods are readily integrated with separation methods suitable for analyzing peptides possessing different disulfide connectivities.16,17
Conventional bottom-up proteomics workflows incorporate reduction and alkylation to cleave disulfide bonds and cap the resulting free cysteines; this process improves the subsequent proteolytic digestions of proteins at the expense of revealing information about the original disulfide bond locations and connectivities. Proteolysis without prior reduction or alkylation produces peptides bound by pseudo-intermolecular disulfide bonds if proteolytic cleavage sites are found between disulfide-bound cysteines.13 MS/MS is used to compare the peptides from the resulting reduced and non-reduced digests and identify disulfide-linked peptides. This method is successful only when at most one pseudo-intermolecular disulfide bond is present per peptide. A strategy utilizing partial reduction and alkylation has become commonplace for creating linear peptides linked by a single disulfide bond.5,18 Other strategies integrate partial reduction with sequential alkylation,19 electrochemical reduction of disulfide bonds,20 or in-source reduction for elucidation of disulfide connectivities.21 However, since the energy barrier for the cleavage of a disulfide bond is higher than the energy required to cleave an amide bond, activation of disulfide-bound proteolytic peptides by collision induced dissociation (CID) generally leads to product ions in which the disulfide bonds remain intact.22 As a result, collisional activation methods often fail to provide disulfide connectivity information and high sequence coverage. Activation methods that offer higher energy deposition or rely on different mechanisms, such as electron transfer dissociation (ETD),23,24 electron capture dissociation (ECD),25 have been shown to cleave disulfide bonds while providing backbone cleavages. 157 nm Ultraviolet photodissociation (UVPD) and 266 nm UVPD26–28 have been reported to selectively cleave disulfide bonds but require subsequent MS3 events to sequence the released peptides. Hybrid activation methods, such as pre-activation with 266 nm UV laser irradiation followed by ECD, have been reported to improve sequence coverage of disulfide-bound proteins.29 The hybrid method electron-transfer higher-energy collision dissociation (EThcD) provides both disulfide linkage information and diagnostic fragment ions of disulfide-bound peptide pairs.30 The activation period required for EThcD (50 ms) is similar to that required for ETD alone, and thus this hybrid method is gaining popularity for characterization of peptides with PTMs.31,32 To our knowledge disulfide-bound peptides have not been analyzed with 193 nm UVPD, which requires as little as one or two 5 ns laser pulses applied during a 2–4 ms activation period, making it an attractive alternative to electron based dissociation methods.
Assignment of fragment ions of disulfide-containing peptides is more difficult than assignment of fragment ions of peptides without disulfide bonds. The challenge arises from the production of fragment ions from peptide backbone cleavages as well as those resulting from the cleavage of a backbone bond in conjunction with either C-S or S-S bond cleavage. The resulting ions produced by the latter type of fragmentation contain mass shifts corresponding to cysteine aldehyde (−2 Da), cysteine persulfide (+32 Da), or dehydroalanine (−34 Da).33 Along the same lines, ETD of peptides containing intramolecular disulfide bonds has been reported to result in production of c/z ions shifted by +32 Da or −33 Da.23 Product ions containing more than one cysteine may have a combination of these variations, thus further complicating analysis.
Disulfide-linked peptides can be assigned by evaluation of extracted ion chromatograms of cysteine-containing proteolytic peptides using an appropriate MS/MS filter.34 In essence, co-eluting peptides are assigned as disulfide-bound peptides, and their linkage is confirmed by the complementary tandem mass spectra. Several strategies have been pursued to develop algorithms capable of automating analysis of disulfide-bound peptides by mapping their connectivities.13,35 For example, DBond was developed to target fragment ions containing cysteine, cysteine persulfide (+32 Da) and cysteine dehydroalanine (−34 Da) residues that result from homolytic cleavage of a disulfide bond or cleavage of a C-S bond.36 Another program, SlinkS (now XlinkX), assigns ions resulting from S-S and C-S bond cleavage as well as fragment ions containing disulfide bonds and standard backbone fragment ions.30 MS2DB+ was developed to include a, b, c, x, y, and z fragment ions to facilitate the analysis of disulfide-linked peptides.37 Kojak-MS has been used for crosslinking analysis of protein complexes and allows the user to search for disulfide-containing peptides by considering each disulfide bond as a crosslink with a mass shift of −2.00965 Da per disulfide bond (−2H).38
In the present study, 193 nm UVPD is used to provide insight into the fragmentation patterns of disulfide-linked peptides. Partial reduction and alkylation are employed after proteolytic digestion to create peptides linked by at most one disulfide bond. The 193 nm UVPD strategy is assessed based on analysis of the benchmark proteins insulin and lysozyme and then human serotransferrin, a protein containing 19 disulfide bonds. 193 nm UVPD is shown to cleave pseudo-intermolecular disulfide bonds created by proteolytic digestion while simultaneously providing diagnostic fragment ions for both linked peptides in a high throughput mode well-suited for HPLC-MS/MS workflows.
Experimental
Sample preparation
Insulin was prepared in 50% methanol 0.1% formic acid at a concentration of 10 μM for static infusion electrospray ionization (nanoESI). Lysozyme and serotransferrin were digested and/or subjected to partial reduction and alkylation. Additional details are provided in the Supporting Information.
Mass spectrometry, liquid chromatography, and photodissociation
All experiments were conducted on a Thermo Scientific Instruments Orbitrap Fusion Lumos tribrid mass spectrometer (San Jose, CA) custom fitted with a 500 Hz Coherent ExciStar XS 193 nm ArF excimer laser (Santa Cruz, CA) to perform UVPD as previously described for a Fusion Orbitrap mass spectrometer.39 Insulin was introduced by nanoESI using a spray voltage of 1 kV and a capillary temperature of 275˚C. Protein digests were separated using an Ultimate 3000 UHPLC system (Sunnyvale, CA).
Data dependent acquisition was performed for all LC-MS/MS methods, in which each MS1 scan (resolution 30,000 at m/z 200) was followed by 10 consecutive ion activation events undertaken on the most abundant ions found in the MS1 spectrum. For UVPD experiments, ions were activated using two 5 ns laser pulses during an activation period of 4 ms at a laser energy of 3 mJ per pulse in the low pressure linear ion trap. For HCD in the ion routing multipole, a normalized collision energy (NCE) of 30% was applied.
Data processing
Insulin data were processed using UV-POSIT (Ultraviolet Photodissociation Online Structure Interrogation Tools), a suite of customized tools designed to facilitate interpretation of UVPD mass spectrometry experiments (available on-line at http://uv-posit.cm.utexas.edu).40 All LC-MS/UVPD data were processed with Kojak, an algorithm for crosslink assignment, to assign disulfide linkages and all spectra were confirmed manually.38
Results and Discussion
The general workflow is illustrated in Scheme S1. Linked peptides containing only one disulfide bond can be unambiguously assigned without prior reduction or alkylation. Linked peptides containing multiple disulfide bonds are first partially reduced and alkylated to achieve confident assignments of disulfide-bound cysteine partners.
Comparing activation methods for the analysis of insulin
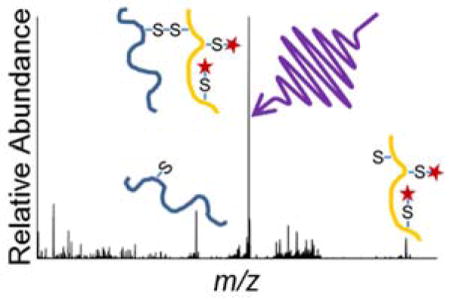
Insulin contains two intermolecular disulfide bonds connecting the A and B chains as well as one intramolecular disulfide bond on the A chain (Scheme S2). Owing to the number and nature of the disulfide bonds, insulin was chosen as a benchmark protein to evaluate the potential of 193 nm UVPD to characterize disulfide-linked peptides. For peptides linked with an intermolecular disulfide bond, diagnostic ions can be produced by a combination of backbone cleavage, disulfide bond cleavage, or a combination of the two (Scheme 1). Representative fragment ions arising from only a single backbone cleavage in a region that does not encompass an intermolecular disulfide bond are outlined in the red boxes. Ions resulting from a backbone cleavage in conjunction with a disulfide cleavage are outlined in green boxes. Diagnostic ions that result from backbone cleavage of one peptide chain while the other chain remains intact and bound by a disulfide bond to the first chain are also produced (orange and teal boxes). For example, a fragment ion from the A-chain that remains connected to the B-chain is outlined in an orange box, and a fragment ion from the B-chain bound to the A-chain is shown in a teal box. Finally, homolytic cleavage of the inter-linked disulfide bond without any concomitant backbone cleavage is possible and is shown in purple. This complex array of fragment ion types contributes to the challenges of deciphering the sequences and connectivities of disulfide-bonding proteins and at the same time offers the potential for comprehensive structure determination if all types of ions are successfully produced and assigned.
Scheme 1.

Generalized fragmentation pathways for inter-linked disulfide-bound peptides activated by 193 nm UVPD.
In the case of insulin, sequence ions can originate from cleavage of a backbone bond in conjunction with one or both intermolecular disulfide bonds, resulting in a number of combinatorial possibilities that must be considered for proper assignment of the ions and inclusion in searches. HCD, EThcD, and UVPD were used to characterize insulin (5+ charge state), and examples of the resulting deconvoluted mass spectra are shown in Figure 1. The need for alternative MS/MS methods for analysis of disulfide-bound peptides is evident when considering the HCD spectrum of insulin (Figure 1a). Higher energy collisional activation does not cause cleavage of the disulfide bonds and fails to produce many diagnostic sequence ions, and thus the resulting HCD mass spectrum exhibits some sequence ions but low coverage of each chain. EThcD of insulin liberates the A and B chains through homolytic cleavage of both intermolecular disulfide bonds while also providing sequence ions (Figure 1b). In contrast to HCD, two of the most abundant products observed upon UVPD correspond to the masses of the individual A and B chains, confirming that 193 nm UVPD preferentially and simultaneously cleaves the two intermolecular disulfide bonds of insulin (Figure 1c). The liberation of the A and B chains is due to homolytic cleavage of both intermolecular disulfide bonds, a phenomenon also observed for inter-linked disulfide bound peptides analyzed using 266 nm UVPD.28 The resulting liberated chains are anticipated to contain radical thiols on the cysteine side-chains. This feature is reflected in the resulting spectrum as the mass of the A-chain was found to be 2333.93 Da (compared to 2337.96 Da expected for cysteines containing free thiols) and the mass of the B-chain was found to be 3395.64 Da (compared to 3397.66 expected for cysteines containing free thiols). Additionally an expansive array of diagnostic sequence ions are produced, including ones from each individual chain as well as ones which retain the other chain as summarized in the cleavage map shown in Figure 1c. With respect to sequence coverages, both UVPD and EThcD gave excellent performance: UVPD yielded 70% coverage of the A chain and 93% coverage of the B chain, whereas EThcD provided 85% coverage of the A chain and 97% coverage of the B chain. HCD yielded lower coverage of both chains: 20% for A and 52% for B. The production of internal fragment ions has been reported for disulfide-bound peptides subjected to collisional activation,41 and this type of process may account for some of the unassigned peaks in the HCD mass spectrum. Similarly, it is likely that internal fragments are also generated in the EThcD and UVPD spectra. However, owing to the number of possible fragment ions generated by UVPD (any combination of a, b, c, x, y, or z ions), these ions were not considered in this study. The rather poor sequence coverage from HCD renders this an ineffective method for analyzing disulfide-bound peptides. EThcD provides excellent sequence coverage of both chains of insulin, similar to that returned by UVPD, at the cost of an activation period over an order of magnitude greater than HCD or UVPD.
Figure 1.

Deconvoluted (a) HCD, b) EThcD, and c) UVPD mass spectra of insulin (5+, m/z 1146.9) with sequence coverage maps. Sequence coverages for each chain are denoted in parentheses. Fragment ions denoted with a blue dot contain the intact mass of the B-chain. Fragment ions denoted with a red dot contain the intact mass of the A-chain. EThcD and UVPD spectra are magnified 5X.
For the EThcD and UVPD mass spectra, the liberated A and B peptide chains (originated from homolytic cleavage of both intermolecular disulfide bonds) comprised 34% and 37%, respectively, of the total product abundances. Identified sequence ions consisted of 66% and 63% of the total identified products for EThcD and UVPD, respectively. A-chain ions generated by UVPD are about twice as abundant as B-chain ions generated by UVPD (Figure S1a). This difference in abundances may be due to different propensities for secondary dissociation of the chain upon UVPD. The distributions of the fragment ion type abundances produced by EThcD (Figure S1b,c) and UVPD (Figure S1d,e) of insulin were evaluated. Owing to the single basic site of the A-chain localized at the N-terminus, A-chain sequence ions resulting from homolytic cleavage of one or both intermolecular disulfide bonds in conjunction with peptide backbone cleavage favor N-terminal fragment ions. When fragment ions containing the mass of the B-chain are considered, a significant portion of C-terminal fragments are also identified, primarily y-type. B-chain fragment ions resulting from homolytic cleavage of disulfide bonds are also well distributed among a/x, b/y, and c/z ions. The results confirming the diversity of diagnostic ions both with and without homolytic disulfide bond cleavage provided motivation to utilize the ability of UVPD to cleave inter-chain disulfide bonds for the purpose of mapping disulfide connectivities of larger proteins.
Disulfide mapping of lysozyme
Lysozyme (14.3 kDa, containing four disulfide bonds, Figure S2) was chosen to evaluate the ability to ascertain the disulfide connectivities of larger proteins by UVPD. The creation of peptides containing single intramolecular disulfide bonds or peptide pairs containing single intermolecular disulfide bonds is paramount to unambiguous assignment of disulfide bonds. This requirement is critical because UVPD has been shown to cleave more than one intermolecular disulfide bond, as exemplified by its simultaneous cleavage of both disulfide bonds of insulin. Thus, peptides containing multiple disulfide bonds may lead to equivocal assignments. One approach to enhance production of the disulfide-bound peptides is to digest the protein without reduction/alkylation. Trypsinolysis of lysozyme prior to reduction and alkylation cleaves lysozyme into three products containing the four disulfide bonds of lysozyme (see Table S1) as well as other conventional tryptic peptides that do not contain any disulfide bonds (not listed). Since each of the peptide pairs containing disulfide bonds 1 (Cys6-Cys127) and 2 (Cys30-Cys115) only contain cysteines involved in disulfide bonds (i.e. no other disulfide pairing possibilities), unequivocal assignment of those disulfide bonds can be facilitated by UVPD without further sample pre-treatment. In order to characterize the connectivities of disulfide bonds 3 (Cys64-Cys80) and 4 (Cys76-Cys94), a method was developed to partially reduce disulfide bonds of tryptic peptides using TCEP followed by alkylation of the reduced thiols with NEM (Scheme S3). The TCEP/NEM treatment converts the disulfide-containing tryptic peptides into ones that are non-reduced, partially reduced (for those that contain more than one disulfide bond), or fully reduced (no surviving disulfide bonds). In addition, the trackable mass shift associated with the NEM alkylation tag (+125 Da) allows differentiation of cysteines that are disulfide-bound from those that have been reduced.
The resulting mixture was analyzed by LC-MS/UVPD. Extracted ion chromatograms of m/z values corresponding to non-reduced and partially reduced samples were used to identify disulfide-bound peptide pairs (Figure 2a). The product containing disulfide bonds 3 (inter-chain) and 4 (intra-chain) was found to be orders of magnitude more abundant than the peptide pair containing disulfide bond 3 and the reduced/alkylated peptide 4 after a 30 minute partial reduction with TCEP. While longer reduction reaction times might yield a greater portion of the desired partially reduced peptide pair, longer reaction times also risk the depletion of the peptide pair containing disulfide bond 1 which was found to be the least abundant of the four disulfide-bound peptide pairs analyzed.
Figure 2.

(a) Extracted ion chromatograms of the four disulfide-bound peptide pairs of partially reduced lysozyme tryptic digest. Deconvoluted UVPD spectra of peptide pair containing (b) disulfide bond 1 (Cys6-Cys127) (Table S1) (3+) and (c) disulfide bond 2 (Cys30-Cys115) (Table S1) (3+). Fragment ions denoted with a blue dot contain the intact mass of the B-chain. Fragment ions denoted with a red dot contain the intact mass of the A-chain. Spectra magnified 5X.
Two pairs of peptides containing either disulfide bond 1 (Cys6-Cys127) or 2 (Cys30-Cys115) are readily characterized with UVPD (Figure 2b,c). Akin to the phenomenon seen with UVPD of insulin, homolytic cleavage of the inter-chain disulfide bond liberates the two peptide chains of each peptide pair (labelled as A and B chain in Figure 2b,c). Additionally, readily identified diagnostic sequence ions from each chain are produced. For example, the a5 ion of CELAAAMK is present both with and without its disulfide-bonded peptide partner GCR (Figure 2b). Similarly, the y8 ion of the peptide GYSLGNWVCAAK is identified both with and without its disulfide-bonded peptide partner CK (Figure 2c). EThcD and HCD were also evaluated for their performance in characterizing these disulfide-bound peptides. While it is evident that EThcD cleaved disulfide bond 1, no fragment ions were identified in conjunction with a disulfide bond cleavage (Figure S3a) and there were fewer than half the number of fragment ions identified by UVPD. Similarly, HCD provided very little diagnostic fragmentation for the peptide pair containing disulfide bond 1 (Figure S3b). Additionally, UVPD was the only activation method that generated a diagnostic ion from the B-chain of that particular peptide pair. EThcD of the peptide pair containing disulfide bond 2 provided very comparable sequence coverages of both chains as well as the liberated A and B-chains (Figure S4a). While HCD provided a number of sequence ions for the A-chain of this peptide pair, this activation method failed to generate any diagnostic fragmentation leading to the identification of the B-chain (Figure S4b).
The final pair of peptides is connected by one inter-chain disulfide bond (Cys64-Cys80), and the larger peptide in the pair (NLCNIPCSALLSSDITASVNCAK) also contains an intramolecular disulfide bond (Cys74-Cys96). UVPD of the non-reduced disulfide-bound tryptic peptide provides sequence coverage for both peptides but does not identify the disulfide connectivities (Figure S5). UVPD also cleaves the inter-chain disulfide bonds releasing the two bound peptides as product ions (see the A-chain and B-chain products in Figure S5). Partial reduction and NEM alkylation of lysozyme creates a new peptide pair bound by one inter-chain disulfide bond (Cys64-Cys80) in conjunction with reduction of the original intramolecular disulfide bond (Cys76-Cys94). UVPD of this partially reduced species allows unambiguous disulfide assignment (Figure 3). The mass shift associated with NEM alkylation (+125 Da) can be used to determine which cysteines are alkylated. For example, the b4 and b5 ions identified in Figure 3 (NLCNIPCSALLSSDITASVNCAK) display the characteristic +125 Da mass shift associated with NEM alkylation, confirming the alkylation of Cys76. Similarly y3 – y18 fragment ions from the larger chain also display a +125 Da mass shift associated with NEM alkylation localizing this alkylation to Cys94. Conversely, the a4 and a5 fragment ions identified in Figure S5 are created by the cleavage of the intramolecular disulfide bond (Cys76-Cys94) in conjunction with backbone cleavage. Because these ions do not display a mass shift, they cannot lead to the assignment of the cysteine bound to Cys76. Based on tracking the +125 Da mass shifts of the fragment ions from the peptide NLCNIPCSALLSSDITASVNCAK (such as mass shifts associated with y3 – y18 and b4 – b5 ions), the two cysteine residues found to be alkylated were Cys76 and Cys94. This result suggests that these two residues are naturally bound by a disulfide bond, an outcome consistent with that reported in Uniprot (#P00698). The other two cysteine residues (Cys64 and Cys80) contained in this peptide pair do not exhibit fragment ions containing the mass shift of an NEM alkylation (Figure 3), confirming that they are disulfide-bonding partners. EThcD and HCD were also evaluated and found to provide sequence information for both chains of this disulfide-bound peptide pair (Figure S6a,b). EThcD provided a greater number of diagnostic ions than HCD and generated the liberated A- and B-chains, thus providing comparable information to that of UVPD. While HCD generated many diagnostic fragment ions for the characterization of the A-chain, only two diagnostic ions were identified for the B-chain.
Figure 3.
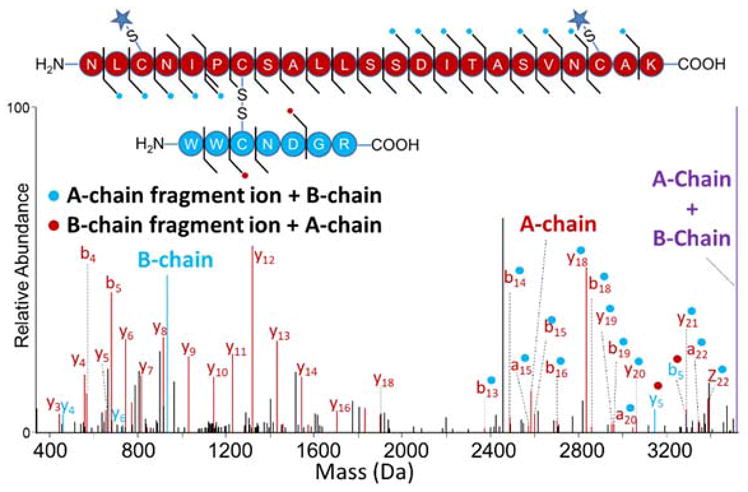
Deconvoluted UVPD spectrum of partially reduced peptide pair from lysozyme digest (4+). Disulfide bond 4 (Cys76-Cys94) was reduced and alkylated, while disulfide bond 3 (Cys64-Cys80) remained intact prior to UVPD (Table S1). Fragment ions denoted with a blue dot contain the intact mass of the B-chain. Fragment ions denoted with a red dot contain the intact mass of the A-chain. A star denotes an NEM alkylation. Spectrum magnified 5X.
Disulfide mapping of serotransferrin
Characterization of serotransferrin (75 kDa, sequence shown in Figure S7) is particularly challenging because it contains 38 cysteines with up to 19 disulfide bonds. Owing to the extreme complexity of the cysteine connectivities in serotransferrin, Lys-C was used in conjunction with trypsin to increase the efficiency of proteolysis without prior reduction of disulfide bonds. While disulfide-bound peptide pairs containing N-glycosylations could be identified based on matching the theoretical masses of glycopeptides to the experimental masses of ions in the MS1 spectra along with glycosidic cleavage fragments generated by UVPD, sequence coverage of glycosylated peptide pairs was poor (data not shown). Identification of N-glycosylation was not pursued further in this study, and rather the N-glycans were simply removed by addition of PNGase F prior to analysis. The complex array of disulfide-bound peptides produced in silico by exposure of serotransferrin to an enzyme cocktail of trypsin, Lys-C, and PNGase F is listed in Table S2. The 19 disulfide bonds of serotransferrin are contained in 29 peptides forming 13 disulfide-bound peptide products, reinforcing the need for partial reduction and alkylation to unambiguously identify the disulfide connectivities of all disulfide bonds.
Peptides from serotransferrin containing disulfide bonds can be categorized into four different groups based on whether the peptides contain one (simple) or more than one (complex) disulfide bond and whether the disulfide bond connects a contiguous stretch of residues (intra-linked) or two separate regions (inter-linked). The proteolytic serotransferrin products generated from the trypsin/Lys-C/PNGaseF cocktail were characterized using the LC-UVPD-MS strategy, and an example of a typical LC trace with assignment of peptides is shown in Figure S8. Examples of the resulting MS/MS spectra are shown in Figures 4 and S9–S24. Among the 17 disulfide-containing products identified by UVPD-MS, seven belong to the simple inter-linked category and connect seven different peptide pairs (Figure S9–S15). In addition, one simple intra-linked product (QQQHLFGSNVDCSGNFCLFR) was identified, with a disulfide between residues Cys615 and Cys620 (Figure S16). These eight peptide products, accounting for eight disulfide bonds, are the most straightforward because they require no reduction or alkylation and their disulfide connectivities can be determined unequivocally because they contain only two cysteine residues. Linear peptides containing an intramolecular disulfide bonds and peptide pairs bound by an intermolecular disulfide bond are readily identified with excellent sequence coverage without partial reduction and alkylation using this LC-UVPD-MS workflow.
Figure 4.

Deconvoluted UVPD spectra of (a) partially reduced peptide AVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFK (3+) (reduced/alkylated disulfide bonds 7 and 8, with intact disulfide C158-C174) and (b) extracted ion chromatograms of the precursor (purple, 5143.16 Da) and diagnostic y19 ion (red, 2248.96 Da) from serotransferrin. Deconvoluted UVPD spectra of (c) partially reduced peptide AVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFK (3+) (reduced/alkylated disulfide bonds 6 and 7, with intact disulfide C171-C179) and (d) extracted ion chromatograms of the precursor (purple, 5143.16 Da) and diagnostic b15 ion (red, 1690.59 Da) from serotransferrin from serotransferrin. The spectra in (a) and (c) were averaged over 26.15–26.80 minutes and 28.66–29.36 minutes respectively. Sequence coverage maps of each peptide are shown above their respective spectra/XIC. A star denotes an NEM alkylation site.
Of the remaining 11 disulfide bonds, eight occur in peptides classified as complex inter-linked. These disulfide bonds are found in four triplet-peptide products. For each of these sets of triplet-peptides, one peptide contains two cysteine residues that are each disulfide-bonded to cysteine residues on other peptides. To assign the disulfide bonds in these peptides, reduction of single (not multiple) disulfide bonds is required. Adapting the partial reduction and alkylation strategy described earlier, the key peptides were produced were characterized by UVPD-MS (Figure S17–S24). Localization of NEM alkylations to specific cysteine residues while providing sequence coverage of both chains leads to confident assignment of these disulfide bonds.
The remaining three disulfide bonds of serotransferrin were clustered in a single peptide containing residues 149–193 and is categorized as complex intra-linked. The expected disulfide connectivities of this peptide are Cys158-Cys174 (disulfide 6), Cys161-Cys177 (disulfide 7), and Cys171-Cys179 (disulfide 8). As a result of the proximity and linkage pattern of this series of three intra-chain disulfide bonds, assignment can be achieved by analyzing the fragmentation pattern of this peptide containing either one or two reduced and alkylated disulfide bonds. Since partial reduction cannot be tuned to reduce a specific disulfide bond, this particular peptide was targeted and analyzed in greater detail using a customized LC-MS/MS method defined in the experimental section. Partial chromatographic separation of isomeric products (3+) originating from reduction/alkylation of two disulfide bonds allowed unambiguous identification of the peptide containing disulfide bonds 6–8. Key features in the UVPD fragmentation patterns allowed confident localization of the sites of NEM alkylation (Figure 4). The y19 product ion in Figure 4a contains residues Cys177 and Cys179 along with two NEM alkylations (+250 Da), showing that neither cysteine residue is disulfide-bound. Sequence coverage between each cysteine residue resulted in confident assignment of disulfide bond 6 (Cys158-Cys174) and agrees with the connectivity reported in Uniprot (#P02787). Similarly, in Figure 4c product ion b15 contains two +125 Da mass shifts, localizing two NEM alkylations to Cys158 and Cys161. Another alkylation site is localized to Cys174 owing to fragment ions arising from backbone cleavages between Cys171 and Cys174. Despite determining three of the alkylated cysteines in Figure 4c confident assignment of the remaining disulfide bonds could not be unequivocally achieved. The peptides in Figure 4a,c are isomeric and do not exhibit baseline resolution upon chromatographic separation (Figure 4b,d). Extracted ion chromatograms (XIC) of each precursor peptide are shaded in purple, and key diagnostic fragment ions for each peptide are shaded in red for Figure 4b, 4d (y19 and b15 respectively). To optimize the UVPD analysis of only one specific isomer at a time, the spectra displaced in Figure 4a,c were obtained by averaging over a elution period when only one key diagnostic ion (y19 or b15) was detected. In Figure 4c NEM alkylations are localized to Cys158, Cys161, and Cys174, suggesting that Cys171 and Cys179 are disulfide-bound which is consistent with the protein in Uniprot (#P02787). However, for unequivocal assignment, sequence coverage between all cysteine residues is needed to localize all NEM alkylations.
Conclusions
193 nm UVPD results in homolytic cleavage of the intermolecular disulfide bonds connecting the A and B-chains of insulin while simultaneously providing high sequence coverage of both chains. For proteins containing multiple disulfide bonds, a strategy combining partial reduction and alkylation with TCEP and NEM of enzymatically digested proteins in acidic conditions prevents disulfide scrambling and allows assignment of more complex disulfide patterns. This method proved successful for characterization of the four disulfide bonds of lysozyme and the 19 disulfide bonds of serotransferrin. By mitigating disulfide scrambling, this method has the potential for elucidating disulfide linkage patterns for proteins with no known disulfide connectivities.
Supplementary Material
Acknowledgments
We acknowledge the following funding sources: NSF (Grant CHE1402753) and the Welch Foundation (Grant F-1155). MMQ acknowledges support from the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM121714. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
Footnotes
Supporting Information: The following items are included: a more detailed experimental section, tables summarizing the theoretical disulfide-containing tryptic peptides from lysozyme and serotransferrin, ion distributions observed upon MS/MS of insulin, the sequences of lysozyme and serotransferrin, a chromatogram of serotransferrin tryptic digest, a workflow scheme, a reaction pathway for TCEP reduction and NEM alkylation, and numerous EThcD, HCD, and UVPD mass spectra for tryptic peptides from lysozyme and serotransferrin.
References
- 1.Hogg PJ. Trends Biochem Sci. 2003;28(4):210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, Pan H, Chen X. Mass Spectrom Rev. 2009;28(1):147–176. doi: 10.1002/mas.20190. [DOI] [PubMed] [Google Scholar]
- 3.Trivedi MV, Laurence JS, Siahaan TJ. Curr Protein Pept Sci. 2009;10(6):614–625. doi: 10.2174/138920309789630534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Biochemistry (Mosc) 2000;39(15):4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 5.Glocker MO, Arbogast B, Deinzer ML. J Am Soc Mass Spectrom. 1995;6(8):638–643. doi: 10.1016/1044-0305(95)00250-H. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Lu Q, Wu SL, Karger BL, Hancock WS. Anal Chem. 2011;83(8):3133–3140. doi: 10.1021/ac200128d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, et al. J Biol Chem. 2008;283(23):16206–16215. doi: 10.1074/jbc.M709988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A, et al. Nature. 2016;538(7625):329–335. doi: 10.1038/nature19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardi A, Galdes A, Florance J, Maniconte D. Biochemistry (Mosc) 1989;28(13):5494–5501. doi: 10.1021/bi00439a026. [DOI] [PubMed] [Google Scholar]
- 10.Walewska A, Skalicky JJ, Davis DR, Zhang MM, Lopez-Vera E, Watkins M, Han TS, Yoshikami D, Olivera BM, Bulaj G. J Am Chem Soc. 2008;130(43):14280–14286. doi: 10.1021/ja804303p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutertre S, Ulens C, Büttner R, Fish A, Elk R, van Kendel Y, Hopping G, Alewood PF, Schroeder C, Nicke A, et al. EMBO J. 2007;26(16):3858–3867. doi: 10.1038/sj.emboj.7601785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poppe L, Hui JO, Ligutti J, Murray JK, Schnier PD. Anal Chem. 2012;84(1):262–266. doi: 10.1021/ac203078x. [DOI] [PubMed] [Google Scholar]
- 13.Tsai PL, Chen SF, Huang SY. Rev Anal Chem. 2013;32(4):257–268. [Google Scholar]
- 14.Gorman JJ, Wallis TP, Pitt JJ. Mass Spectrom Rev. 2002;21(3):183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 15.Wiesner J, Resemann A, Evans C, Suckau D, Jabs W. Expert Rev Proteomics. 2015;12(2):115–123. doi: 10.1586/14789450.2015.1018896. [DOI] [PubMed] [Google Scholar]
- 16.Durand KL, Ma X, Plummer CE, Xia Y. Int J Mass Spectrom. 2013;343–344:50–57. [Google Scholar]
- 17.Durand KL, Tan L, Stinson CA, Love-Nkansah CB, Ma X, Xia Y. J Am Soc Mass Spectrom. 2017;28(6):1099–1108. doi: 10.1007/s13361-017-1595-1. [DOI] [PubMed] [Google Scholar]
- 18.Foley SF, Sun Y, Zheng TS, Wen D. Anal Biochem. 2008;377(1):95–104. doi: 10.1016/j.ab.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 19.Albert A, Eksteen JJ, Isaksson J, Sengee M, Hansen T, Vasskog T. Anal Chem. 2016;88(19):9539–9546. doi: 10.1021/acs.analchem.6b02115. [DOI] [PubMed] [Google Scholar]
- 20.Cramer CN, Haselmann KF, Olsen JV, Nielsen PK. Anal Chem. 2016;88(3):1585–1592. doi: 10.1021/acs.analchem.5b03148. [DOI] [PubMed] [Google Scholar]
- 21.Cramer CN, Kelstrup CD, Olsen JV, Haselmann KF, Nielsen PK. Anal Chem. 2017;89(11):5949–5957. doi: 10.1021/acs.analchem.7b00424. [DOI] [PubMed] [Google Scholar]
- 22.Lioe H, O’Hair RAJ. J Am Soc Mass Spectrom. 2007;18(6):1109–1123. doi: 10.1016/j.jasms.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Cole SR, Ma X, Zhang X, Xia Y. J Am Soc Mass Spectrom. 2012;23(2):310–320. doi: 10.1007/s13361-011-0300-z. [DOI] [PubMed] [Google Scholar]
- 24.Lakbub JC, Clark DF, Shah IS, Zhu Z, Su X, Go EP, Tolbert TJ, Desaire H. Anal Methods. 2016;8(31):6046–6055. doi: 10.1039/C6AY01248E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan X, Engler F, Qu J. J Mass Spectrom JMS. 2010;45(12):1477–1482. doi: 10.1002/jms.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu SL, Jiang H, Hancock WS, Karger BL. Anal Chem. 2010;82(12):5296–5303. doi: 10.1021/ac100766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fung YME, Kjeldsen F, Silivra OA, Chan TWD, Zubarev RA. Angew Chem. 2005;117(39):6557–6561. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 28.Agarwal A, Diedrich JK, Julian RR. Anal Chem. 2011;83(17):6455–6458. doi: 10.1021/ac201650v. [DOI] [PubMed] [Google Scholar]
- 29.Wongkongkathep P, Li H, Zhang X, Ogorzalek Loo RR, Julian RR, Loo JA. Int J Mass Spectrom. 2015;390:137–145. doi: 10.1016/j.ijms.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, van Breukelen B, Heck AJR. Mol Cell Proteomics MCP. 2014;13(10):2776–2786. doi: 10.1074/mcp.O114.039057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frese CK, Altelaar AFM, van den Toorn H, Nolting D, Griep-Raming J, Heck AJR, Mohammed S. Anal Chem. 2012;84(22):9668–9673. doi: 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- 32.Frese CK, Zhou H, Taus T, Altelaar AFM, Mechtler K, Heck AJR, Mohammed S. J Proteome Res. 2013;12(3):1520–1525. doi: 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho Sohn C, Gao J, Thomas AD, Kim T-Y, WAG, Beauchamp LJ. Chem Sci. 2015;6(8):4550–4560. doi: 10.1039/c5sc01305d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark DF, Go EP, Desaire H. Anal Chem. 2013;85(2):1192–1199. doi: 10.1021/ac303124w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu H, Zhang L, Freitas MA. J Proteome Res. 2008;7(1):138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Na S, Paek E, Choi J-S, Kim D, Jae Lee S, Kwon J. Mol Biosyst. 2015;11(4):1156–1164. doi: 10.1039/c4mb00688g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murad W, Singh R. IEEE Trans NanoBioscience. 2013;12(2):69–71. doi: 10.1109/TNB.2012.2212029. [DOI] [PubMed] [Google Scholar]
- 38.Hoopmann MR, Zelter A, Johnson RS, Riffle M, MacCoss MJ, Davis TN, Moritz RL. J Proteome Res. 2015;14(5):2190–2198. doi: 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein DR, Holden DD, Brodbelt JS. Anal Chem. 2016;88(1):1044–1051. doi: 10.1021/acs.analchem.5b04218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg J, Parker WR, Cammarata MB, Brodbelt JS. J Am Soc Mass Spectrom. doi: 10.1007/s13361-018-1918-x. Just Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark DF, Go EP, Toumi ML, Desaire H. J Am Soc Mass Spectrom. 2011;22(3):492–498. doi: 10.1007/s13361-010-0064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.