

Abstract
Recent advances in molecular characterization of tumors have made possible the emergence of new types of cancer therapies where traditional cytotoxic drugs and nonspecific chemotherapy can be complemented with targeted molecular therapies. One of the main revolutionary treatments is the use of monoclonal antibodies (mAbs) that selectively target the disseminated tumor cells while sparing normal tissues. mAbs and related therapeutics can be efficiently radiolabeled with a wide range of radionuclides to facilitate preclinical and clinical studies. Non-invasive molecular imaging techniques, such as Positron Emission Tomography (PET), using radiolabeled mAbs provide useful information on the whole-body distribution of the biomolecules, which may enable patient stratification, diagnosis, selection of targeted therapies, evaluation of treatment response, and prediction of dose limiting tissue and adverse effects. In addition, when mAbs are labeled with therapeutic radionuclides, the combination of immunological and radiobiological cytotoxicity may result in enhanced treatment efficacy. The pharmacokinetic profile of antibodies demands the use of long half-life isotopes for longitudinal scrutiny of mAb biodistribution and precludes the use of well-stablished short half-life isotopes. Herein, we review the most promising PET radiometals with chemical and physical characteristics that make the appealing for mAb labeling, highlighting those with theranostic radioisotopes.
1 | INTRODUCTION
Monoclonal antibodies (mAbs) have become indispensable tools for the modern clinical management of cancer. Currently, approximately 76 mAbs or antibody-related therapeutics have been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of several primary and metastatic cancer types. Some of the advantages of mAbs as therapeutic agents include an exquisite affinity and specificity for their cognate antigen, relatively long circulation half-lives, and the ability to elicit mAb-mediated cell killing.1,2 Additionally, the process of generating cancer-specific mAbs is relatively straightforward compared with their small molecule counterparts. In contrast to conventional chemotherapy drugs, which are non-specific and incur serious toxicities, mAb-targeted antigens over-express in cancer cells compared with normal tissues.3 This broadens the therapeutic window of these agents while reducing the incidence of severe side effects. However, the effectiveness of mAb therapies depends on the careful selection of likely responders based on expression of the target of interest. Therefore, the parallel development of noninvasive, reliable methods to scrutinize the expression of a given molecular target is vital to the efficacious implementation of mAb regimes.
Positron emission tomography (PET) imaging is a versatile nuclear medicine technique to investigate the expression of molecular targets noninvasively. PET imaging tracks the spatial distribution of a positron-emitting radionuclide that is typically conjugated to a targeting molecule. Due to the high sensitivity of PET, concentrations of radiotracers as low as 10−12 M can be detected, facilitating noninvasive functional imaging with minimal pharmacological effects.4 A plethora of positron-emitting radionuclides with diverse chemistries and decay properties are available for conjugation to biologically active molecules ranging from simple molecules like glucose to more complex macromolecules such as proteins and polymers.
The radiolabeling of mAbs with positron emitters for PET imaging (immunoPET) may provide valuable information about the in vivo biodistribution of these molecules and their related therapeutics.5 ImmunoPET imaging can elucidate drug target expression via quantification of tracer uptake in the tumor, describe tumor saturation and heterogeneity, and provide data to support drug development, particularly regarding patient selection, stratification, and monitoring of treatment response.1 In fact, extensive preclinical and clinical studies highlight the increasing importance of immunoPET as a diagnostic tool in oncology.6,7 In addition, mAbs can also be labeled with therapeutic radionuclides (eg, 177Lu, 67Cu, and 90Y) to combine immunological and radiobiological cytotoxicity.5,8 Within this context, the use of diagnostic surrogate radioisotopes will facilitate quantification of the therapeutic agents’ biodistribution and dosimetry.
For each application, the selection of the optimal radioisotope is crucial. It starts by matching the half-life of the radionuclide with the pharmacokinetic profile of the mAb in vivo. This step is essential to radiotracers’ “smart” design and ensures that the time course of the radioactivity matches that of the mAb.7 Typically, due to prolonged circulation half-lives, antibodies’ accumulation in tumors tends to peak days after injection, which makes necessary the use of long half-life isotopes (eg, 89Zr, 64Cu, and 86Y) instead of more traditional choices such as 11C, 18F, or 68Ga. In instances where the conventional isotopes do not suit the desired application, other interesting radionuclides have been investigated which offer more appropriate chemical or decay properties. Notable examples of such attractive radionuclides include 52Mn, 55Co, 152Tb, 90Nb, 66Ga, 72As, and 69Ge. The utilization of these relatively long-lived PET isotopes often requires the leveraging of inorganic metal complexation chemistry with bifunctional chelators (BFCs) containing both a polydentate radiometal ligand and a bioconjugation functional group. The conjugation of many such BFC moieties to the free -COOH, -NH2, or -SH groups in mAb amino acid side chains allows efficient labeling of mAbs with a wide range of radionuclides. In this review, we present an overview of the most promising radiometals for immunoPET focusing on those that also possess an isotopic pair that could be used for theranostic applications.
1.1 | Zr-89
Currently, 89Zr (t1/2 = 78.4 hours, 22.7% β+, Eβ + ave = 396 keV; Figure 1A and Table 1) is utilized in clinical trials much more extensively than any other PET-radiometal.9 Due to its relatively long half-life, 89Zr is particularly well suited for centralized production and national and international transport. It decays via positron emission and electron capture to 89mY (t1/2 = 15.7 s), which decays via γ ray emission (909 keV, Iγ = 99.1%) to stable 89Y. The 3-day half-life of 89Zr matches the typical pharmacokinetic time scales of mAbs as mentioned previously. 89Zr’s relatively soft positron emissions are attractive in terms of achievable PET resolution. Additionally, 89Zr is a residualizing radionuclide: upon internalization and degradation, “free” 89Zr is trapped inside tumor cells, which results in improved tumor retention and enhanced tumor-to-normal tissue compared with non-residualizing radionuclides such as 124I.10–12
FIGURE 1.

A, Simplified decay scheme of 89Zr (data taken from the National Nuclear Data Center: www.nndc.bnl.gov), B, schematic overview of 89Zr-labeled antibody using DFO as chelator, and C, biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer (reprinted with permission from Dijkers EC, et al Clinical Pharmacology & Therapeutics 2010; 87: 586–592)
TABLE 1.
Decay properties, production methods, chelators, and theranostic pair for potential PET-radiometals. The gamma spectrum for each radiometal listed below is provided as Supporting Information (Figure S1)
| Radionuclide | t1/2 | Average β+Energy/ Iβ+ (%) | Principle γ Emissions / Iγ (%) | Optimal Production Method | Typical Chelators | Theranostic Partner Isotope |
|---|---|---|---|---|---|---|
| 89Zr | 78.4 h | 396 keV/ 23% | 909 keV/ 99% | Cyclotron | DFO HOPO DOTA |
- |
| 64Cu | 12.7 h | 278 keV/ 18% | - | Cyclotron | NOTA DOTA TETA SAR family | 67Cu |
| 86Y | 14.7 h | 660 keV/ 32% | 1076 keV/83% 628 keV/33% 1153 keV/31% 777 keV/22% 1921 keV/21% 1854 keV/17% |
Cyclotron | DOTA DTPA |
90Y |
| 52Mn | 5.6d | 242 keV/ 29% | 1434 keV/ 100% 935 keV/95% 744 keV/90% |
Cyclotron | DOTA | - |
| 55Co | 17.5 h | 570 keV/76% | 931 keV/75% 1409 keV/17% |
Cyclotron | DOTA HBED TETA NOTA |
58mCo |
| 152Tb | 17.5 h | 1140 keV/20% | - | Proton-induced spallation | DOTA | 161Tb |
| 90Nb | 14.6 h | 620 keV/51% | 1129 keV/93% 2319 keV/82% 141 keV/67% 2186 keV/18% |
High energy cyclotron | DFOA | - |
| 66Ga | 9.3 h | 1750 keV/57% | 1039 keV/37% 2752 keV/23% 4295 keV/4% |
Cyclotron | DOTA NOTA |
71Ga |
| 72As | 26.0 h | 1170 keV/88% | 834 keV/81% 630 keV/8% |
Cyclotron | Trithiol / lipoic acid | 77As |
| 69Ge | 39.1 h | 490 keV/24% | 1107 keV/36% 574 keV/13% 872 keV/12% |
Cyclotron | Metal oxide nanoparticles | - |
89Zr is mainly produced in small biomedical cyclotrons via the transmutation reaction natY(p,n)89Zr and proton irradiation of natural-abundance yttrium foils with yields of ~50 MBq/μAh.13 Following irradiation 89Zr is separated from the target material using a commercially available hydroxamic acid functionalized resin (hydroxamate resin), whose development built upon previously reported methodologies.14,15 89Zr is conveniently eluted in small volumes as 89Zr-oxalate; however, it can be reprocessed as 89ZrCl4 if desired.14,16 Our recent results have shown the possibility of obtaining 89ZrCl4 using a direct chloride-based 89Zr/natY separation strategy which might simplify radiochemical automation and improve the chemical purity and the 89Zr recovery.17 89Zr is typically obtained with high radiochemical yields (~90%) and effective molar (specific) activities of around 60 GBq/μmol, as measured by desferrioxamine (DFO) titration.14
Currently, DFO is the most commonly employed chelator for 89Zr-radiolabeling (Figure 1B).9,18 However, it is now generally accepted that DFO is not optimal for 89Zr coordination, as bone uptake-evidence of 89Zr transchelation-has been observed in preclinical in vivo experiments regardless of the DFO-based radio-tracer.9,19,20 Emerging reports have shown that extended DFO (DFO* or DFO-Sq) are superior because they allow for higher coordination numbers with the metal ion despite showing low radiolabeling efficiency and poor water solubility.9 Other promising alternatives are chelators featuring hydroxypyridinone moieties including HOPO and 2,3-HOPO which display higher in vitro and in vivo stabilities compared with DFO. Due to the well-known macrocyclic effect,21 macrocyclic chelators have been proposed as a viable alternative to less stable open chelators. However, the synthesis of chelators incorporating 4 hydroxamate or hydroxypyridinone moieties into a macrocyclic ring has proven challenging. Recent work has shown an excellent stability of 89Zr- DOTA complexes compared with linear chelators, but the synthesis of such Zr-chelates requires high temperatures and the use of 89Zr in the form of 89ZrCl4.18 The necessity for a heating step precludes the direct 89Zr labeling of the mAb using DOTA as chelator. Nonetheless, the long half-life of 89Zr is conducive to the implementation of a post-radiolabeling conjugation strategy, where 89Zr chelation is carried out prior to the BFC-mAb bioconjugation step, potentially overcoming the limitations imposed by the elevated temperatures needed for radiosynthesis.
The first 89Zr-labeled mAb (89Zr-labeled anti-EpCam antibody 323/A3) was successfully used to visualize human OVCAR-3 xenografts in immunodeficient mice in 1997.22 Less than a decade later, the first clinical 89Zr- labeled mAbs showed that primary head and neck squamous cell carcinomas might be detected by PET imaging using 89Zr-labeled chimeric anti-CD44v6 antibody U36.10 Since then, numerous 89Zr-labeled mAbs have been developed targeting several tumor-associated antigens, i.e., EGFR, HER2, PSMA, CD44v6, CD20, and VEGF-A.7,9,23 For example, 89Zr-trastuzumab has been used for the detection of lung, liver, bone, and brain metastatic lesions in patients with HER2-positive breast cancer (Figure 1C).11 Within this context, immunoPET was able to unequivocally diagnose patients with suspected liver and mediastinal metastases, which could not be properly confirmed by biopsies.24 Additionally, 89Zr-trastuzumab has also been employed for the prediction and monitoring of therapy response.25,26 Despite recent advancements in the field indicating a need for the optimization of the chelators and bioconjugation approaches used for clinical immunoPET with 89Zr immunoconjugates, the increasing number of successful preclinical studies using 89Zr- labeled mAbs for cancer imaging has established this radiometal as one of the most promising for antibody labeling.7,9
1.2 | Cu-64
64Cu (t1/2 = 12.7 hours, 17.4% β+, Eβ + ave = 278 keV; Figure 2A and Table 1) represents another convenient alternative for antibody and protein labeling as it has an intermediate half-life and negligible contaminating gamma emissions. Due to the favorable excitation function of the 64Ni (p,n)64Cu reaction at low energies (11 MeV), 64Cu can be economically produced by proton irradiation of isotopically enriched 64Ni targets with yields of 0.37 GBq/μAh.27 Our group and others have developed efficient methods for radiochemical isolation of produced 64Cu from Ni and Co targets and the subsequent recovery of the enriched 64Ni.27
FIGURE 2.

A, Simplified decay scheme of 64Cu (data taken from the National Nuclear Data Center: www.nndc.bnl.gov), B, schematic overview of 64Cu-labeled antibody using DOTA as chelator, and C, 64Cu-DOTA-trastuzumab PET images of HER2-positive metastatic brain lesions (arrows) (This research was originally published in JNM. Tamura K et al. 64Cu-DOTA-Trastuzumab PET Imaging in Patients with HER2-Positive Breast Cancer. J Nucl Med 2013; 54:1869–1875. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.)
The chemistry of the intermediate-hard metal ion Cu2+ is well understood, particularly its coordination with chelators with amino and carboxylic acid functional groups.28 Many macrocyclic chelators have been used for the coordination of Cu2+ isotopes with excellent kinetic and thermodynamic stabilities including DOTA (Figure 2B), DTPA, TETA, NOTA, and its derivatives.28,29 Another family of chelators, the hexaazamacrobicyclic cage-type ligands (namely SAR ligands) have shown excellent conjugation efficiencies at room temperature within minutes and much faster chelation kinetics compared with those with TETA derivatives (CB-TE1A1P and CB-TE2P).30 Despite the number of emerging Cu2+chelators, most conventional ligands have acceptable in vivo stabilities within a 48-hour time-frame post- injection (PI), and NOTA remains the gold standard for the radiolabeling of Cu isotopes.29,31
The intermediate half-life of 64Cu makes it an appropriate radioactive tag for a wide variety of molecules with a range of molecular weights and pharmacokinetic profiles.5 64Cu has been used for immunoPET imaging to scrutinize the expression of several cancer-specific receptors including the epidermal growth-factor receptor (EGFR; cetuximab),32 CD105 (TRC105),31 human αvβ3 integrin (etaracizumab),33 epithelial cell adhesion molecule (EpCAM; mAb7),34 CD20 (rituxi-mab),35 human epidermal growth factor receptor 2 (HER2; trastuzumab),5,36 and CD146 (YY146).37 An example of 64Cu-DOTA-Trastuzumab PET images of HER2-positive metastatic brain lesions is shown in Figure 2C. These studies have demonstrated the feasibility of immunoPET using 64Cu-labeled mAb for quantitative, noninvasive evaluation of the expression of cancer- specific molecules and pointed to the advantages of 64Cu over 89Zr in terms of procedural radiation exposure.38 Despite being appropriate for most preclinical immunoPET applications, the intermediate half-life of 64Cu limits the implementation of longitudinal or delayed imaging studies that require imaging time points beyond 48 hours PI.
The availability of theranostic isotope pairs is of great interest because image-guided, targeted radionuclide therapy (TRT) can be seamlessly implemented. 64Cu not only possesses intrinsic theranostic properties due to its mixed β+ and β− (38.5%, Eβ − ave = 191 keV) decay modes, but can also serve as surrogate for treatment planning and dose estimation of TRT using 67Cu (t1/2 = 2.58 days, 100% β− , Eβ − ave = 141 keV). Owing to a half-life similar to the biological half-life of many mAbs, a lower whole-body dose to the patient, and simple radiolabeling procedures, 67Cu is well-suited to radioimmunotherapy (RIT).36 Several mAbs have been labeled with 67Cu, including chCE7, an anti-L1-cell adhesion molecule mAb for the treatment of neuroblastoma, ovarian cancer, and renal carcinoma; Lym-1 for non- Hodgkin’s lymphoma; C595 an anti-MUC1 for bladder cancer treatment39; and trastuzumab for the treatment of HER2 positive tumors.40 Currently, the requirement for high energy charged particle bombardments, or high intensity neutral particle irradiations limits the availability of 67Cu. Notably, the development of the Facility for Rare Isotope Beams (FRIB) at the Michigan State University (MSU) will allow the harvesting of 67Cu in GBq-scale quantities from the aqueous beam dump produced at this projectile fragmentation facility.41 With further development of novel facilities and methods for the production of 67Cu, 64Cu will receive additional consideration for mAb-based theranostic applications.
1.3 | Y-86
Another isotope with promising characteristics for immunoPET is 86Y (t1/2 = 14.7 hours, 31.9% β+, Eβ + ave = 660 keV; Figure 3 and Table 1). This radionuclide can be produced in a small biomedical cyclotron by the proton-induced transmutation reaction 86Sr(p,n)86Y at less than 18 MeV.8 Solid targets of isotopically enriched 86SrCO3 are required to produce 86Y with a radionuclidic purity higher than 98%. This constitutes a challenge in the production of 86Y because the elevated market price of 86SrCO3 ($9/mg) mandates the recycling of the target material, which involves a series of precipitation/reconstitution steps.42 The target material is problematic due to its low thermal conductivity and difficulty coupling it to backing materials in geometries that can dissipate more than a few tens of watts of deposited beam power, limiting production yields to a few hundred MBq. Several methods have been reported for the separation of 86Y from the irradiated 86SrCO3 targets, including co-precipitation42,43 and ion exchange,44–46 electrolysis,47,48 single column chromatography,49 multiple column chromatography,50,51 and solvent extraction.45 Although each method has its own advantages and disadvantages, in general, all have achieved the efficient separation of the radionuclide. Unfortunately, 86Y emits a large number of gamma rays with nontrivial intensities and energies greater than a MeV, limiting injectable quantities to approximately 100 MBq.
FIGURE 3.

Simplified decay scheme of 86Y (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
The pseudo-lanthanide Y3+ has hard-acid character and tends to form complexes with ligands containing hard donor atoms, displaying high coordination numbers, usually 8 or 9. Consequently, the labeling of antibodies and proteins with 86Y has been performed using mainly polyaminocarboxylic ligands, for example DTPA, DOTA, or its derivatives. DOTA and DOTA-derivatives show particularly high thermodynamic stability and enhanced kinetic inertness because the cavity size of DOTA is well matched to the ionic radii of the yttrium trivalent metal ion.5,28
86Y has been used for longitudinal immunoPET recordings of the biodistribution of mAbs such as cetuximab.52 Compared with 64Cu and 89Zr, 86Y production rates are typically lower, and its decay properties, namely half-life and a cluttered gamma emission spectrum, have affected the quantity of literature studies using 86Y as a PET isotope.53 The main advantage of 86Y is the potential of its well-established therapeutic radioisotope: 90Y (t1/2 = 64.0 hours, 100% β− , Eβ − ave = 934 keV). In fact, the theranostic concept in nuclear medicine was first coined for the use of the radionuclide pair 86Y/90Y at the Research Center Jülich, Germany in 1992,8 which allowed a combination of PET and TRT.
90Y has been extensively used as a therapeutic radio-nuclide in the treatment of various malignancies, including lymphoma, ovarian, colorectal, leukemia, pancreatic, and bone cancers.8 In fact, one of the most efficacious TRT agents reported to date is 90Y-labeled mAb, 90Y- ibritumomab tiuxetan (Zevalin®, Spectrum Pharmaceuticals, Henderson, NV, USA), which was approved in 2002 by the US Food and Drug Administration for the targeting of CD20 in Non-Hodgkin’s lymphoma patients and remains a part of the standard of care today.5 Following the success of Zevalin, several proof-of-concept studies exploited the potential of the 86Y/90Y theranostic pair. For example, 90Y-labeled cetuximab combined with External Beam Radiotherapy (EBRT) was tracked by 86Y-cetuximab immunoPET.52 Due to a high positron branching ratio, convenient physical half-life, and the success of therapies employing 90Y, 86Y represents a valuable choice for PET imaging that is currently limited primarily by the scalability of its production. For additional discussion, the reader is referred to the recent review by the Jülich group.8
2 | EMERGING RADIOISOTOPES
2.1 | Mn-52
52Mn (t1/2 = 5.59 days, 29.4% β+, Eβ + ave = 242 keV; Figure 4 and Table 1) may offer advantages over conventional 89Zr or 64Cu in situations where treatment response monitoring at late time-points (2–3 weeks) is desired.54 In cases where RIT is initiated with long-lived nuclides, multi-week treatment time-courses can be monitored by 52Mn PET. Additionally, due to the abundance of coincident high energy gamma emission, 52Mn is one of the relatively few nuclides that may be used in third-gamma coincidence PET for either dual nuclide event tagging or combined Compton telescope PET tomography.55 Despite this, the clinical translation must be carried out with caution due to the preponderance of the coincident high energy gammas: 744 keV (90%), 935 keV (95%), and 1434 keV (100%). Together with the countless biological roles of manganese, which may lead to prolonged retention in critical organs, the biodistribution and dosimetry of 52Mn-labeled agents require evaluation prior to clinical translation.
FIGURE 4.
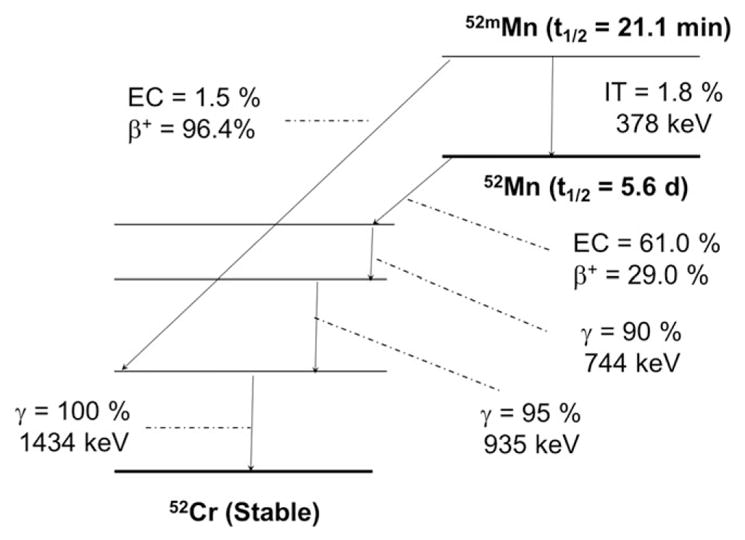
Simplified decay scheme of 52Mn (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
52Mn can be produced in small biomedical cyclotrons at low proton energies using pressed natural chromium targets.56 For incident proton energies of 16 MeV and “thick” targets, average production yields of 6.2 MBq/μAh are typical. Importantly, due to the high natural abundance of 52Cr (83.8%) and low coproduction yields of Mn radioisotopic impurities, natCr targets are a viable, inexpensive alternative to the enriched target material for preclinical studies.54 The main radionuclidic impurity from the irradiation of natCr is 54Mn (t1/2 = 312 days), representing 0.1% to 0.4% of the 52Mn activity at the end of a short bombardment at 16 MeV.54 Adjustment of incident energy affords some reduction in the relative contamination of 54Mn but cannot eliminate its contamination. The most recently reported separations of 52Mn from the target material use solid-phase anion exchange in ethanolic HCl and recover >90% of the 52Mn with 105–6 decontamination factors from chromium, copper, iron, cobalt, and zinc.56
Similar to other hard transition metals, Mn2+ forms highly stable complexes with polyaminocarboxylic acid chelators. From those, DOTA has been the most common choice of chelator for the manganese chelation.54,56 DOTA can be rapidly and quantitatively radiolabeled with 52Mn reaction times less than 1 minute, and the resulting complex shows excellent in vitro stability.56 These results have motivated the use of 52Mn in several preclinical investigations.57–59
Numerous applications of 52Mn have been reported, including myocardial perfusion tracer,60 neural tractography,61 stem cell tracking,62 and biological toxicity assays.54,63 However, the extension to immunoPET requires high-specific-activity 52Mn in a chemical state suitable for macromolecule labeling. Thanks to the contributions of our group and the group at the Hevesy Laboratory, 52Mn has been produced with sufficient specific activity for the labeling of a mAb for immunoPET.54,56 Using a DOTA-based BFC, TRC105, an anti-CD105 mAb was radiolabeled (52Mn-DOTA-TRC105) for the imaging of angiogenesis in a syngeneic 4T1 xenograft model of breast adenocarcinoma. The elevated stability of 52Mn- DOTA-TRC105 over the course of several days proves the usefulness of 52Mn as a radiotracer for immunoPET.54
2.2 | Co-55
55Co (t1/2 = 17.5 hours, 76% β+, Eβ + ave = 570 keV, 24% EC; Figure 5 and Table 1) offers a high positron yield and moderate abundance of co-emitted gamma-rays.64 55Co can be produced via either the 58Ni(p,α)55Co or the 54Fe(d,n)55Co nuclear reactions at low energy biomedical cyclotrons and possesses a half-life permissive of centralized production and distribution.64,65 After irradiation, the produced 55Co can be separated from the target material using an extraction resin functionalized with N,N,N′, N′-tetrakis-2-ethylhexyldiglycolamide, or branched DGA (Eichrom, Illinois).64 As with many of the radiometals discussed herein, radiocobalt can be efficiently coordinated using common hard donor chelators such as such as DOTA, HBED, TETA, and NOTA,66–68 but to date, strategies for the labeling of mAbs or proteins with 55Co have not been well established.
FIGURE 5.
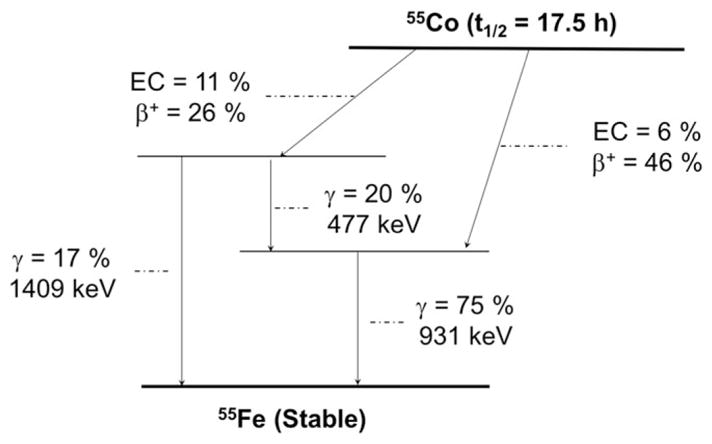
Simplified decay scheme of 55Co (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
Nevertheless, 55Co has been used as a PET imaging agent in diverse applications such as ischemic stroke,69 imaging of renal function,70 and multiple sclerosis.71 Recent reports show that high-resolution PET images can be obtained when labeling small peptides such as bombesin with radiocobalt.67,72 More recently, this radio-nuclide has shown promise for labeling affibodies. The DOTA-Z2395-C affibody was efficiently and stably labeled with 57Co.68 The intercellular retention of radiocobalt- labeled DOTA-Z2395-C was comparable to the retention of its 111In-labeled counterpart in vitro. In vivo, the radiocobalt label provided better tumor-to-organ ratios than the radionuclide 111In. We anticipate future studies with DOTA-coupled affibody molecules will show efficient 55Co radiolabeling and be used for in vivo PET imaging applications.
The recent realization of the therapeutic potential of the Auger electron emitter 58mCo (t1/2 = 9.10 hours, 100% IC) has revived interest in 55Co as an imaging surrogate of 58mCo for theranostic applications.64,73,74 Thisgaard et al were the first to demonstrate the possibility of producing therapeutic quantities of 58mCo using a small biomedical cyclotron.73 In a more recent study, Valdovinos and colleagues showed the viability of obtaining high specific activity 58mCo conducive to the quantitative radiolabeling of mAb.64 However, so far, the therapeutic potential of 58mCo has not been demonstrated in vivo. Nonetheless, the availability of this pair of radionuclides is compelling to the implementation of TRT with Auger electrons.
2.3 | Tb-152
Terbium offers 4 clinically interesting radioisotopes with complementary physical decay characteristics: 149Tb, 152Tb, 155Tb, and 161Tb.75 The identical chemical characteristics of these radioisotopes allow the preparation of radiopharmaceutical isosteres for PET (152Tb) and SPECT (155Tb) imaging, and for α (149Tb) and β− (161Tb) therapy. In addition, the half-life of 152Tb (t1/2 = 17.5 hours, 20.3% β+, Eβ + ave = 1140 keV; Figure 6 and Table 1) makes this radionuclide in conjunction with 161Tb (t1/2 = 6.7 days, Eβ − ave = 134 keV) a potential isotopic pair for mAb-based theranostic applications. 152Tb can be produced by proton-induced spallation of tantalum targets, and carrier-free terbium radioisotopes can be obtained after purification in mass separators, with activities around 600 MBq.75 Being a lanthanide, Tb3+ has chemical properties that resemble those of group III elements (Sc and Y), including the formation of stable coordination complexes with DOTA and DOTA-like chelators, which facilitates the radiolabeling of well-established targeting agents.75–78
FIGURE 6.
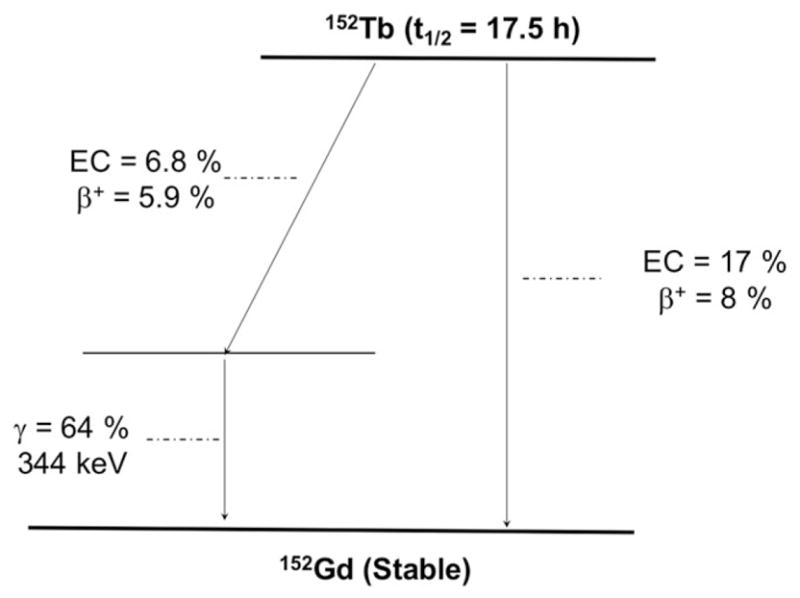
Simplified decay scheme of 152Tb (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
Because of its unique production facility requirements, only a limited number of studies using 152Tb have been reported. The first reported study using 152Tb as PET radionuclide radiolabeled a peptide (DOTANOC) for in vivo PET imaging of somatostatin receptor expression in AR42J tumor-bearing mice.77 The results of this study paved the way for a more recent first-in-human multi-disciplinary PET/CT study using this promising radionuclide.78 To this end, 152Tb-DOTATOC longitudinal PET scans were acquired over a period of 24 hours, allowing the visualization of even small metastases with excellent tumor-to-background contrast ratios. However, to the best of our knowledge, the use of this radionuclide for the labeling of mAbs has not yet been reported.
Few studies have also reported the use of the γ-rays co- emission of 161Tb (48.9 keV-17%, 57.2 keV-1.8%, 74.6–10%) for monitoring the in-vivo behavior of radio-labeled bio-molecules and their effects on malignant tissue.75 It can be produced by neutron capture and subsequent decay via the reaction chain 160Gd(n, γ)161Gd→β-decay→161Tb in nuclear reactors with activities in the range of 15 GBq.75 Preliminary results using this radionuclide suggested that 161Tb-labeled compounds may elicit more robust therapeutic effects than other therapeutic radionuclides like 177Lu when applied at similar activity levels.75,79 Based on these preliminary results, 152Tb/ 161Tb seems to be a compelling pair for mAb-based theranostic applications. However, in our opinion, the complexity and high cost of the production of 152Tb constitute major limitations to the widespread implementation of this radioisotopes.
2.4 | Nb-90
Another interesting radionuclide with suitable decay properties as a radiometal for immunoPET is 90Nb (t1/2 = 14.6 hours, 51.2% β+, Eβ + ave = 620 keV, 24% EC; Figure 7 and Table 1), which may allow the noninvasive, high-resolution visualization and quantification of the whole-body distribution of macromolecules including mAb, mAb fragments, polymers, and nanoparticles.80 90Nb can be produced via 90Zr(p,n)90Nb nuclear reaction using low-energy proton (20 MeV) irradiation of natural zirconium targets.81 No-carrier-added 90Nb can be isolated by a multi-step separation procedure involving liquid extraction and ion-exchange chromatography.81 Recent advancements in optimization of the separation process of 90Nb from irradiated targets have allowed efficient (>90%) and fast (less than 1 hour) recovery of 90Nb with a radionuclidic purity higher than 97% (decay corrected to EoB) and molar (specific) activity suitable for the radiolabeling of mAbs at 4.5 TBq/mmol.81 However, the co-production of other long half-life Nb radioisotopes such as 92mNb (t1/2 = 10.2 days), 95mNb (t1/2 = 3.6 days), 95Nb (t1/2 = 35 days), or 96Nb (t1/2 = 23.4 hours) may make enriched 90Zr targets necessary for clinical applications.
FIGURE 7.
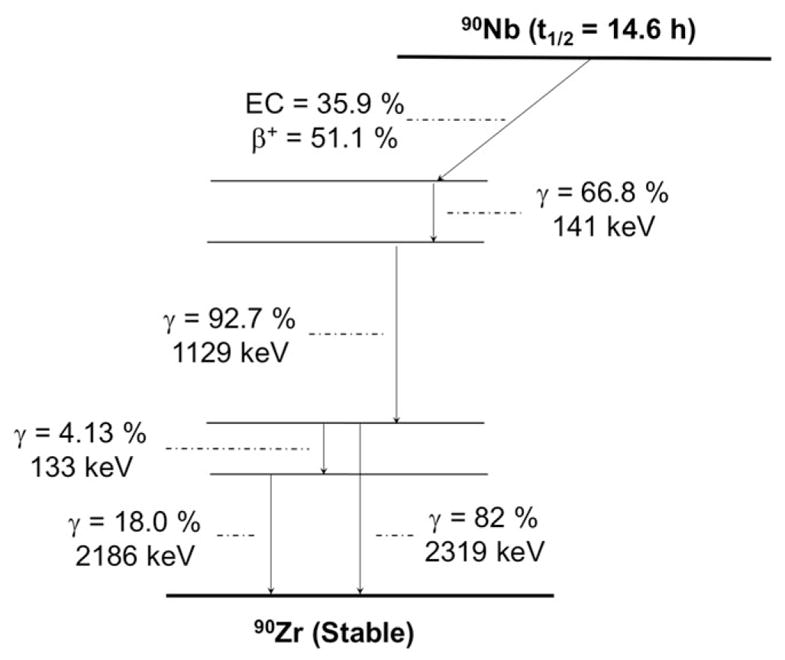
Simplified decay scheme of 90Nb (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
The coordination of 90Nb with different chelators has been evaluated in terms of radiolabeling efficiency and stability of the radiolabeled Nb5+ complex.82 Results indicate that DFO shows the best properties as it is able to form quantitative complexes at a wide range of pHs (4–7) at room temperature. In addition, it was verified that bi-functionalization does not affect the complex formation parameters of the DFO and that the complex remains stable in vivo.82 In follow-up studies, the mAb rituximab was radiolabeled with 90Nb, and stability measurements revealed that the complex was more than 99% stable over a prolonged period of 18 days.83 Recently, as a proof of concept, 90Nb was used to label the mAb bevacizumab (Avastin®), and in vitro and in vivo stability was evaluated in normal swiss mice and tumor-bearing SCID mice.81 In- vivo PET imaging in tumor-bearing SCID mice after the injection of 90Nb-bevacizumab showed avid localization of the radiotracer in the tumor, while uptake in the liver, spleen, kidneys, or bones remained low. Overall, these results indicate the feasibility of 90Nb-labeled antibodies for immunoPET. Nevertheless, the need for enriched targets for 90Nb production may be a significant drawback for future clinical implementation.
2.5 | Ga-66
The radionuclide 66Ga (t1/2 = 9.3 hours, 56.5% β+, Eβ + ave = 1750 keV, 43.5% EC; Figure 8 and Table 1) constitutes an intriguing alternative to 67Ga (t1/2 = 78.3 hours) and 68Ga (t1/2 = 68.3 minutes), which are used for SPECT and PET imaging, respectively.84 Particularly, the relatively long half-life of 66Ga makes it a more practical radiolabel for antibodies and proteins, whose slower in- vivo kinetics are poorly matched by the much shorter half-life of 68Ga.
FIGURE 8.
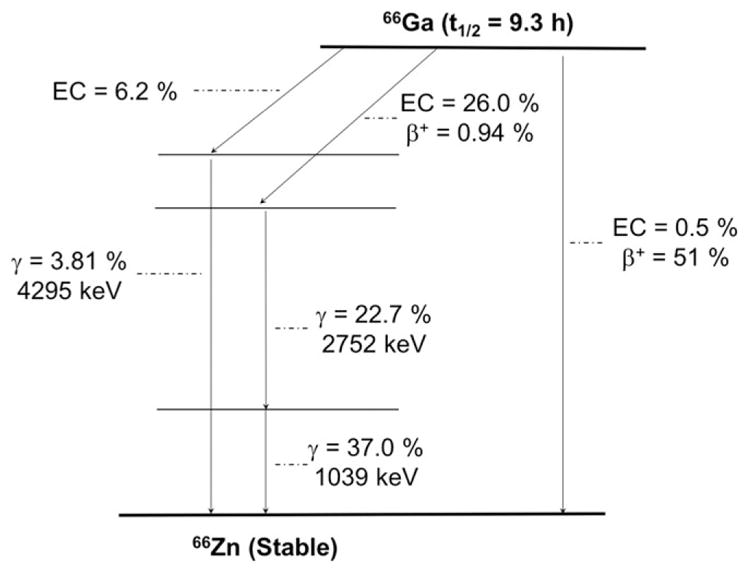
Simplified decay scheme of 66Ga (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
Lewis et al described the irradiation of natural Zn and enriched 66Zn targets to produce 66Ga, which was then purified by cation exchange chromatography and solvent extraction.85 This separation method has been used in most 66Ga-based studies to date; however, modest effective specific activities of 66Ga were obtained (4.6 GBq/ μmol). More recently, our own laboratory in collaboration with researchers at the Autonomous University of Mexico (UNAM) reported the production of high specific activity 66Ga (>70 GBq/μmol) from the natZn(p,n)66Ga and 66Zn(p,n)66Ga nuclear reactions, using a small biomedical cyclotron and energies of 16 → 7 MeV.86 The subsequent separation of 66Ga was accomplished using anion exchange chromatography.87 This result increases the potential of this non-conventional radionuclide for cancer imaging applications.
The coordination chemistry of radiogallium has been extensively studied due to the availability of 68Ge/68Ga generator-produced 68Ga. The choice of chelator for the radiolabeling of 66Ga has mirrored that of the 68Ga, with quantitative labeling being achieved with the macrocyclic chelators DOTA and NOTA.84,88 Although, limited studies have been reported on 66Ga-based immunoPET imaging including the work describing the radiolabeling of the mAb TRC105 with 66Ga for the in-vivo annotation of CD105 expression in a murine model of breast cancer.84 TRC105, a chimeric IgG1 mAb which binds to both human and murine CD105, was conjugated with NOTA and efficiently radiolabeled with 66Ga. PET imaging revealed fast, prominent, and CD105- specific tumor targeting in mice bearing 4T1 tumor xenografts. Such successful immunoPET imaging studies demonstrate the feasibility of using 66Ga to expand the spectrum of accessible radiogallium-based imaging agents to include macromolecules with slower pharmacokinetic profiles. Among the main disadvantages of 66Ga are the high energy of the positron (Eβ + ave = 1.75 MeV), which decreases the achievable quality of acquired PET images-especially in the preclinical setting-and the abundance of high-energy gamma rays: 1039 keV (37%), 2752 (23%), and 4295 keV (4%) that impose a challenge from the dosimetric point of view. These facts, together with the relatively low effective molar activities, achieved using the commonly reported methods are the main reasons for the limited number of studies using 66Ga. However, it remains the only option for long time-point PET studies of the accumulation of Ga-labeled vectors.
2.6 | As-72
The radioisotopes of arsenic have played a role in nuclear medicine because the use of 74As (t1/2 = 17.77 days, 29% β+, Eβ + ave = 440 keV; Figure 9 and Table 1) in Sweet and Brownell’s pioneering work imaging intracranial lesions through positroencephalography.89 More recently, efforts have focused on utilizing the theranostic pair of 72As (t1/2 = 26.0 hours, 88% β+, Eβ + ave = 1170 keV) and 77As (t1/2 = 38.8 hours, 100% β− , Eβ − ave = 225 keV). Additionally, 71As (t1/2 = 65.3 hours, 28% β+, Eβ + ave = 350 keV) and 70As (t1/2 = 50.6 m, 91% β+, Eβ + ave = 980 keV) are alternative PET diagnostic isotopes for targeting vectors with longer or shorter biological half-lives and 73As (t1/2 = 80.3 days) has potential in Auger- emission TRT. This broad palette of radioisotopes, along with the metalloid element’s unique soft-Lewis-acid chemical properties, makes these radioisotopes compelling for use in theranostic applications with mAbs.
FIGURE 9.
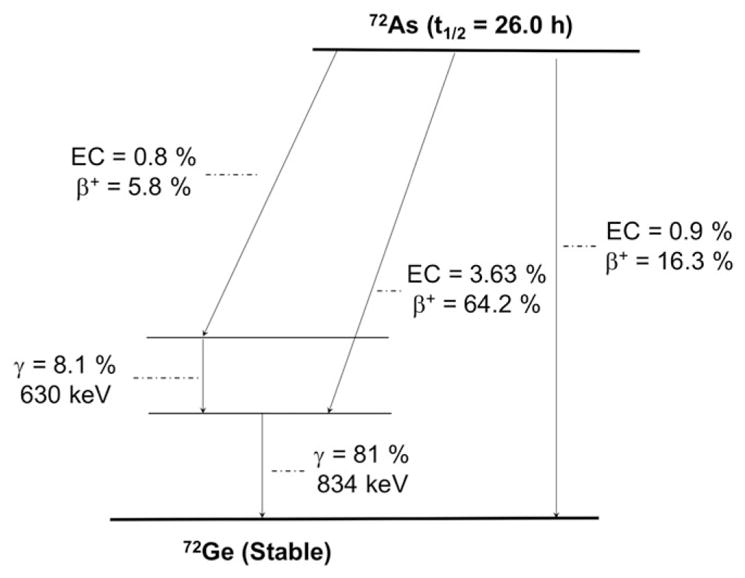
Simplified decay scheme of 72As (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
Radioarsenic can be produced through the irradiation of natGe(m) or natGeO2 using a small biomedical cyclotron with solid target capabilities.90,91 However, proton irradiation of natural enrichment germanium produces 72As with low radionuclidic purity, with significant end-of- bombardment contamination from 70As (600% of 72As), 76As (8% of 72As), and 74As (6% of 72As).91 More recently, isotopically enriched 72Ge(m) targets have been used to produce 72As with high yield (90 MBq/μAh) and radionuclidic purity (99.4%)92 with a biomedical cyclotron. The isolation of radioarsenic from germanium target material has been accomplished through a variety of methods, including precipitation,91,93,94 dry distillation,95 HCl distillation,90,92 solvent extraction,90,94 polystyrene- based solid phase extraction,92,96 and chromatography with anion exchange resins,90–92 silica,97,98 titania,99 and zirconia.100 Additionally, 72As can be produced as the radioactive decay product of 72Se (t1/2 = 8.5 days), allowing for the production of a 72Se/72As generator system, broadening the accessibility of 72As to sites without access to a solid-target-capable biomedical cyclotron.101,102
It is well known that arsenic is a very biologically reactive element with significant binding to proteins, as evidenced by its acute systemic toxicity estimated at approximately 0.6 mg/kg daily.103 This biological activity and protein binding capacity are largely due to the metalloid’s significant soft-Lewis-acid character that allows it to bind especially strongly to soft ligands such as the sulfhydryl groups on cysteine amino acid sidechains.104 Jennewein et al aimed to take advantage of this binding mechanism to label the anti-phosphatidylserine mAb, bavituximab, with 74As.105 Bavituximab was first modified using N- succinimidyl S-acetylthioacetate (SATA) to exhibit additional sulfhydryl functionality and then labeled using a trivalent 74AsI3 compound. The 74As-S-bavituximab radioimmunoconjugate appeared to exhibit good in-vitro stability and showed promising tumor-to-liver and tumor- to-muscle ratios in R3327-AT1 tumor-bearing rats.105 Despite this, it exhibited significant biological clearance with only 0.25% injected dose (ID)/g remaining in tumor and 0.125%ID/g in liver at 48 hours post injection. This clearance was dramatically faster than that of 64Cu-labeled bavituximab, which showed 3.2%ID/g in tumor and 20.6% ID/g in liver at 48 hours post injection in LNCaP-tumor bearing mice.106 This discrepancy is most likely explained by significant dearsenylation of the 74As-S-bavituximab. More recent work labeling the anti-CD105 mAb, TRC105, through similar direct sulfhydryl radioarsenic labeling methods also yielded rapid dearsenylation of the radioimmunoconjugate with the resulting radiotracer biodistribution indistinguishable from unlabeled radioarsenic.92 These studies demonstrate the need for the development of next generation methods for stable incorporation radioarsenic isotopes into radioimmunoconjugates. Several methods are currently under investigation for this, including the development of a trithiol containing chelators107 and the utilization of the dithiol containing mitochondrial enzyme cofactor, lipoic acid.92
2.7 | Ge-69
A much less common but potentially useful PET radioisotope is 69Ge (t1/2 = 39.05 hours, 21% β+, Eβ + ave = 490 keV;Figure 10 and Table 1). Only a handful of studies have been reported on the production, separation, and radiochemistry of 69Ge and its potential therapeutic counter-part, the Auger-emitter 71Ge (t1/2 = 11.4 days). However, they constitute an intriguing isotopic pair whose application niche is not yet established.108
FIGURE 10.
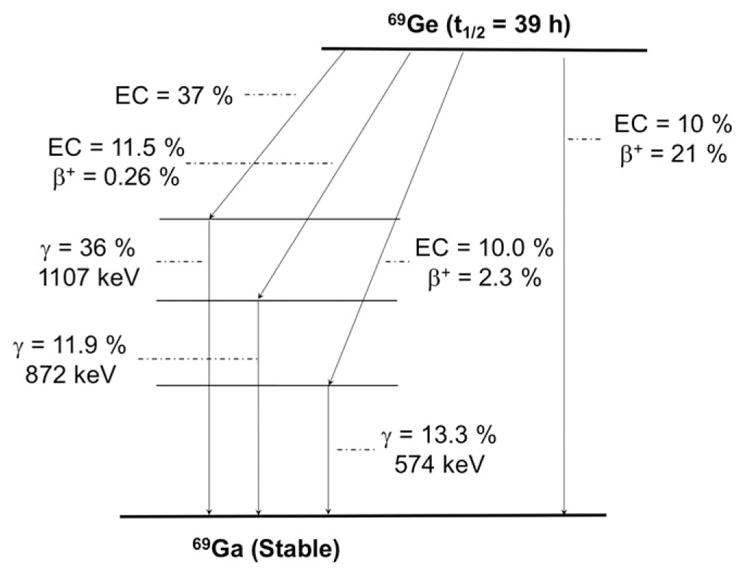
Simplified decay scheme of 69Ge (data taken from the National Nuclear Data Center: www.nndc.bnl.gov)
The production of this radionuclide can be accomplished in a simple and cost-effective manner via the bombardment of a Ga/Ni alloys, which are also proposed for the production of 68Ge with 11-MeV protons.108 Separation of 69Ge from target material is carried out by column chromatography using a DGA extraction resin in HNO3 media. One limitation is 69Ge’s complex chemical speciation in aqueous media, which has led to a lack of suitable radiolabeling techniques for the preparation of 69Ge- based agents. The distribution of germanium species depends on the total concentration and the pH of the medium with the 4 major hydrolysis products identified as Ge(OH)4, [GeO(OH)3]− , [GeO2(OH)2]2− , and[[Ge(OH)4]8(OH)3]3-,109 making it a major challenge for radiolabeling with 69Ge using traditional chelator-based methods.
In a recent effort to develop conjugation techniques for 69Ge, superparamagnetic iron oxide nanoparticles (SPION) were recently used as a matrix for chelator free radiolabeling of this nanoconstruct with 69Ge.108 The incorporation of Ge into metal oxide matrix (such as TiO2, ZrO2, CeO2, SnO2, Fe2O3, Fe3O4, Al2O3) has been extensively exploited for the preparation of the widely used 68Ge/68Ga generators.110,111 Therefore, SPION nanoparticles were employed as a platform to radiolabel 69Ge without the need for functionalized chelators. As an added benefit, given the magnetic properties of SPIONs simultaneous PET and magnetic resonance imaging (MRI) data were acquired using these constructs.112 Despite having nuclear properties favorable for mAb work, 69Ge remains underutilized as a PET radiolabel due to the lack of traditional BFC-based methods for incorporating it into targeting moieties of interest.
3 | CONCLUSIONS
The field of positron emission tomography imaging using labeled mAbs is rapidly progressing toward widespread clinical adoption. Radiolabeled immunoconjugates play an essential role in drug development and aid in patient stratification and monitoring of the treatment response. Moreover, the availability of theranostic isotopic pairs facilitates the implementation of TRT using therapeutic radionuclides that allow the combination of immunological and radiobiological cytotoxic effects for higher anti-tumor efficacy. Within this context, a parallel development of isotope production, separation, and radiochemistry methods is vital to nurture a steady development of both preclinical and clinical studies using different PET radiometals, which will ultimately result in the realization of radiolabeled mAbs as promising tools in the management of cancer. Nevertheless, especially for those emerging radiometals, the production, isolation, and labeling need to be optimized to efficiently fulfill the clinical requirements for human uses.
Supplementary Material
Biographies
 Eduardo Aluicio-Sarduy received his undergraduate education at the Higher Institute of Applied Sciences and Technology (InSTEC) in Havana, Cuba, where he completed with honors his BSc (2007) and MSc in Radiochemistry (2011). In 2016, he earned his PhD in Materials Engineering at Politecnico di Milano, and currently, he is a postdoctoral researcher at the University of Wisconsin-Madison working on the production and radiochemical isolation of long-lived radionuclides for theranostics applications.
Eduardo Aluicio-Sarduy received his undergraduate education at the Higher Institute of Applied Sciences and Technology (InSTEC) in Havana, Cuba, where he completed with honors his BSc (2007) and MSc in Radiochemistry (2011). In 2016, he earned his PhD in Materials Engineering at Politecnico di Milano, and currently, he is a postdoctoral researcher at the University of Wisconsin-Madison working on the production and radiochemical isolation of long-lived radionuclides for theranostics applications.
 Jonathan Ward Engle is an assistant professor and cyclotron jockey in the departments of Medical Physics and Radiology at the University of Wisconsin. He has experience in radionuclide production, accelerator targetry, analytical and preparative radiochemistry for clinical and pre-clinical positron emission tomography (PET) scanning, and automation. He has degrees in Religion, Education, and Physics.
Jonathan Ward Engle is an assistant professor and cyclotron jockey in the departments of Medical Physics and Radiology at the University of Wisconsin. He has experience in radionuclide production, accelerator targetry, analytical and preparative radiochemistry for clinical and pre-clinical positron emission tomography (PET) scanning, and automation. He has degrees in Religion, Education, and Physics.
Footnotes
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- 1.Bailly C, Cléry PF, Faivre-Chauvet A, et al. Immuno-PET for clinical theranostic approaches. Int J Mol Sci. 2016;18(1):1–12. doi: 10.3390/ijms18010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fauvel B, Yasri A. Antibodies directed against receptor tyrosine kinases. MAbs. 2014 Jul;6(4):838–851. doi: 10.4161/mabs.29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright BD, Lapi SE. Designing the magic bullet? The advancement of immuno-PET into clinical use. J Nucl Med. 2013 Aug;54(8):1171–1174. doi: 10.2967/jnumed.113.126086. [DOI] [PubMed] [Google Scholar]
- 4.James ML, Gambhir SS. A molecular imaging primer: modalities, imaging agents, and applications. Physiol Rev. 2012;92(2):897–965. doi: 10.1152/physrev.00049.2010. [DOI] [PubMed] [Google Scholar]
- 5.Kraeber-Bodéré F, Rousseau C, Bodet-Milin C, et al. Tumor immunotargeting using innovative radionuclides. Int J Mol Sci. 2015;16(2):3932–3954. doi: 10.3390/ijms16023932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams S-P. Tissue distribution studies of protein therapeutics using molecular probes: molecular imaging. AAPS J. 2012 Sep;14(3):389–399. doi: 10.1208/s12248-012-9348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moek KL, Giesen D, Kok IC, et al. Theranostics using antibodies and antibody-related therapeutics. J Nucl Med. 2017;58(Supplement 2):83S–90S. doi: 10.2967/jnumed.116.186940. [DOI] [PubMed] [Google Scholar]
- 8.Rösch F, Herzog H, Qaim SM. The beginning and development of the theranostic approach in nuclear medicine, as exemplified by the radionuclide pair 86Y and 90Y. Pharmaceuticals. 2017;10(2):1–28. doi: 10.3390/ph10020056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heskamp S, Raavé R, Boerman OC, Rijpkema M, Goncalves V, Denat F. 89Zr-immunoPET in oncology: state of the art 89Zr-radio-chemistry. Bioconjug Chem. 2017 doi: 10.1021/acs.bioconjchem.7b00325. p. acs.bioconjchem.7b00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Börjesson PKE, Jauw YWS, Boellaard R, et al. Performance of immuno-positron emission tomography with zirconium-89- labeled chimeric monoclonal antibody U36 in the detection of lymph node metastases in head and neck cancer patients. Clin Cancer Res. 2006 Apr;12(7):2133. doi: 10.1158/1078-0432.CCR-05-2137. LP-2132140. [DOI] [PubMed] [Google Scholar]
- 11.Dijkers EC, Oude Munnink TH, Kosterink JG, et al. Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin Pharmacol Ther. 2010 May;87(5):586–592. doi: 10.1038/clpt.2010.12. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Baidoo KE, Brechbiel MW. Mapping biological behaviors by application of longer-lived positron emitting radionuclides. Adv Drug Deliv Rev. 2013;65(8):1098–1111. doi: 10.1016/j.addr.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellison PA, Valdovinos HF, Graves SA, Barnhart TE, Nickles RJ. Spot-welding solid targets for high current cyclotron irradiation. Appl Radiat Isot. 2016 Dec;118:350–353. doi: 10.1016/j.apradiso.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nucl Med Biol. 2009;36(7):729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meijs WE, Herscheid JDM, Haisma HJ, et al. Production of highly pure no-carrier added 89Zr for the labelling of antibodies with a positron emitter. Appl Radiat Isot. 1994 Dec;45(12):1143–1147. [Google Scholar]
- 16.Sato N, Wu H, Asiedu KO, Szajek LP, Griffiths GL, Choyke PL. 89Zr-Oxine complex PET cell imaging in monitoring cell-based therapies. Radiology. 2015;275(2):490–500. doi: 10.1148/radiol.15142849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graves SA, Kutyreff CJ, Hernandez R, Barnhart TE, Ellison PA, Nickles RJ, Engle JW. Evaluation of a chloride- based Zr isolation strategy. Manuscript in preparation. personal communication. 2017.
- 18.Pandya DN, Bhatt N, Yuan H, et al. Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiophar-maceutical development. Chem Sci. 2017 Mar;8(3):2309–2314. doi: 10.1039/c6sc04128k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dijkers ECF, Kosterink JGW, Rademaker AP, et al. Development and characterization of clinical-grade 89Zr-Trastuzumab for HER2/neu ImmunoPET imaging. J Nucl Med. 2009 Jun;50(6):974–981. doi: 10.2967/jnumed.108.060392. [DOI] [PubMed] [Google Scholar]
- 20.Laverman P, van der Geest T, Terry SYA, et al. Immuno-PET and immuno-SPECT of rheumatoid arthritis with radiolabeled anti-fibroblast activation protein antibody correlates with severity of arthritis. J Nucl Med. 2015 May;56(5):778–783. doi: 10.2967/jnumed.114.152959. [DOI] [PubMed] [Google Scholar]
- 21.Constable EC. Metals and Ligand Reactivity. KGaA: Wiley-VCH Verlag GmbH & Co; 1995. Cyclic ligands and the template effect; pp. 135–182. [Google Scholar]
- 22.Meijs WE, Haisma HJ, Klok RP, et al. Zirconium-labeled mono-clonal antibodies and their distribution in tumor-bearing nude mice. J Nucl Med. 1997 Jan;38(1):112–118. [PubMed] [Google Scholar]
- 23.Jauw YWS, Menke-van der Houven van Oordt CW, Hoekstra OS, et al. Immuno-positron emission tomography with zirconium-89-labeled monoclonal antibodies in oncology: what can we learn from initial clinical trials? Front Pharmacol. 2016 May;7:1–15. doi: 10.3389/fphar.2016.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaykema SBM, Brouwers AH, Hovenga S, Lub-de Hooge MN, de Vries EGE, Schröder CP. Zirconium-89-trastuzumab positron emission tomography as a tool to solve a clinical dilemma in a patient with breast cancer. J Clin Oncol. 2012 Feb;30(6):e74–e75. doi: 10.1200/JCO.2011.38.0204. [DOI] [PubMed] [Google Scholar]
- 25.Gebhart G, Lamberts LE, Wimana Z, et al. Molecular imaging as a tool to investigate heterogeneity of advanced HER2-posi-tive breast cancer and to predict patient outcome under trastuzumab emtansine (T-DM1): the ZEPHIR trial. Ann Oncol. 2016 Apr;27(4):619–624. doi: 10.1093/annonc/mdv577. [DOI] [PubMed] [Google Scholar]
- 26.Gaykema SBM, Schröder CP, Vitfell-Rasmussen J, et al. 89Zr- trastuzumab and 89Zr-bevacizumab PET to evaluate the effect of the HSP90 inhibitor NVP-AUY922 in metastatic breast cancer patients. Clin Cancer Res. 2014 Jul;20(15):3945. doi: 10.1158/1078-0432.CCR-14-0491. LP-3943954. [DOI] [PubMed] [Google Scholar]
- 27.Avila-Rodriguez MA, Nye JA, Nickles RJ. Simultaneous production of high specific activity 64Cu and 61Co with 11.4 MeV protons on enriched 64Ni nuclei. Appl Radiat Isot. 2007 Oct;65(10):1115–1120. doi: 10.1016/j.apradiso.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110(5):2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price EW, Orvig C. Matching chelators to radiometals for radio-pharmaceuticals. Chem Soc Rev. 2014;43(1):260–290. doi: 10.1039/c3cs60304k. [DOI] [PubMed] [Google Scholar]
- 30.Alt K, Paterson BM, Ardipradja K, et al. Single-chain antibody conjugated to a cage amine chelator and labeled with positron-emitting Copper-64 for diagnostic imaging of activated platelets. Mol Pharm. 2014 Aug;11(8):2855–2863. doi: 10.1021/mp500209a. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Hong H, Engle JW, et al. Positron emission tomography imaging of CD105 expression with a 64Cu-labeled monoclonal antibody: NOTA is superior to DOTA. PLoS One. 2011 Dec;6(12):e28005. doi: 10.1371/journal.pone.0028005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Dijk LK, Yim C-B, Franssen GM, et al. PET of EGFR with 64Cu-cetuximab-F(ab’) 2 in mice with head and neck squamous cell carcinoma xenografts. Contrast Media Mol Imaging. 2016 Jan;11(1):65–70. doi: 10.1002/cmmi.1659. [DOI] [PubMed] [Google Scholar]
- 33.White JB, Hu LY, Boucher DL, Sutcliffe JL. ImmunoPET imaging of αvβ6 expression using an engineered anti-αvβ6 Cys-diabody site-specifically radiolabeled with cu-64: considerations for optimal imaging with antibody fragments. Mol Imaging Biol. 2017 doi: 10.1007/s11307-017-1097-3. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh SC, Pinkston KL, Robinson H, et al. Comparison of DOTA and NODAGA as chelators for 64Cu-labeled immunoconjugates. Nucl Med Biol. 2015;42(2):177–183. doi: 10.1016/j.nucmedbio.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Xie Q, Zhu H, Wang F, et al. Establishing reliable cu-64 production process: from target plating to molecular specific tumor micro-PET imaging. Molecules. 2017;22(4) doi: 10.3390/molecules22040641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asabella AN, Cascini GL, Altini C, Paparella D, Notaristefano A, Rubini G. The copper radioisotopes: a systematic review with special interest to 64Cu. Biomed Res Int. 2014;2014:1–9. doi: 10.1155/2014/786463. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Kraeber-Bodere F, Bailly C, Chérel M, Chatal JF. ImmunoPET to help stratify patients for targeted therapies and to improve drug development. Eur J Nucl Med Mol Imaging. 2016;43(12):2166–2168. doi: 10.1007/s00259-016-3458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Kurihara H, Yonemori K, et al. 64Cu-DOTA- Trastuzumab PET imaging in patients with HER2-positive breast cancer. J Nucl Med. 2013 Nov;54(11):1869–1875. doi: 10.2967/jnumed.112.118612. [DOI] [PubMed] [Google Scholar]
- 39.Szymański P, Frączek T, Markowicz M, Mikiciuk-Olasik E. Development of copper based drugs, radiopharmaceuticals and medical materials. Biometals. 2012;25(6):1089–1112. doi: 10.1007/s10534-012-9578-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferreira CL, Yapp DTT, Crisp S, et al. Comparison of bifunctional chelates for 64Cu antibody imaging. Eur J Nucl Med Mol Imaging. 2010;37(11):2117–2126. doi: 10.1007/s00259-010-1506-1. [DOI] [PubMed] [Google Scholar]
- 41.Mastren T, Pen A, Peaslee GF, et al. Feasibility of isotope harvesting at a projectile fragmentation facility: 67Cu. Sci Rep. 2015;4(1):6706. doi: 10.1038/srep06706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avila-Rodriguez MA, Nye JA, Nickles RJ. Production and separation of non-carrier-added 86Y from enriched 86Sr targets. Appl Radiat Isot. 2008;66(1):9–13. doi: 10.1016/j.apradiso.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 43.Sadeghi M, Zali A, Avila M. A novel method for radiochemical separation of radioyttrium from Sr targets using precipitation technique. Radiochim Acta. 2010;98(7):437–439. [Google Scholar]
- 44.Rösch F, Qaim SM, Stöcklin G. Production of the positron emitting radioisotope 86Y for nuclear medical application. Appl Radiat Isot. 1993;44(4):677–681. [Google Scholar]
- 45.Kandil SA, Scholten B, Hassan KF, Hanafi HA, Qaim SM. A comparative study on the separation of radioyttrium from Sr- and Rb-targets via ion-exchange and solvent extraction techniques, with special reference to the production of no-carrier- added 86Y, 87Y and 88Y using a cyclotron. J Radioanal Nucl Chem. 2009;279(3):823–832. [Google Scholar]
- 46.Kettern K, Linse KH, Spellerberg S, Coenen HH, Qaim SM. Radiochemical studies relevant to the production of 86Y and 88Y at a small-sized cyclotron. Radiochim Acta. 2002;90(37591):845–849. [Google Scholar]
- 47.Reischl G, Rösch F, Machulla HJ. Electrochemical separation and purification of yttrium-86. Radiochim Acta. 2002;90(4):225–228. [Google Scholar]
- 48.Lukić D, Tamburella C, Buchegger F, Beyer GJ, Ćomor JJ, Seimbille Y. High efficiency production and purification of 86Y based on electrochemical separation. Appl Radiat Isot. 2009;67(4):523–529. doi: 10.1016/j.apradiso.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 49.Medvedev DG, Mausner LF, Srivastava SC. Irradiation of strontium chloride targets at proton energies above 35 MeV to produce PET radioisotope Y-86. Radiochim Acta. 2011;99(12):755–761. [Google Scholar]
- 50.Park LS, Szajek LP, Wong KJ, et al. Semi-automated 86Y purification using a three-column system. Nucl Med Biol. 2004;31(2):297–301. doi: 10.1016/j.nucmedbio.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Sadeghi M, Aboudzadeh M, Zali A, Mirzaii M, Bolourinovin F. Radiochemical studies relevant to 86Y production via 86Sr(p, n)86Y for PET imaging. Appl Radiat Isot. 2009;67(1):7–10. doi: 10.1016/j.apradiso.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 52.Koi L, Bergmann R, Brüchner K, et al. Radiolabeled anti- EGFR-antibody improves local tumor control after external beam radiotherapy and offers theragnostic potential. Radiother Oncol. 2014;110(2):362–369. doi: 10.1016/j.radonc.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 53.Lubberink M, Herzog H. Quantitative imaging of 124I and 86Y with PET. Eur J Nucl Med Mol Imaging. 2011;38(1):10–18. doi: 10.1007/s00259-011-1768-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graves SA, Hernandez R, Fonslet J, et al. Novel preparation methods of 52Mn for ImmunoPET imaging. Bioconjug Chem. 2015;26(10):2118–2124. doi: 10.1021/acs.bioconjchem.5b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lang C, Habs D, Parodi K, Thirolf PG. Sub-millimeter nuclear medical imaging with high sensitivity in positron emission tomography using β + γ coincidences. J Instrum. 2014;9:P01008–P01008. [Google Scholar]
- 56.Fonslet J, Tietze S, Jensen AI, Graves SA, Severin GW. Optimized procedures for manganese-52: production, separation and radiolabeling. Appl Radiat Isot. 2017 Dec;121:38–43. doi: 10.1016/j.apradiso.2016.11.021. 2016. [DOI] [PubMed] [Google Scholar]
- 57.Wooten AL, Aweda TA, Lewis BC, Gross RB, Lapi SE. Biodistribution and PET imaging of pharmacokinetics of manganese in mice using Manganese-52. PLoS One. 2017 Mar;12(3):e0174351. doi: 10.1371/journal.pone.0174351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brunnquell CL, Hernandez R, Graves SA, et al. Uptake and retention of manganese contrast agents for PET and MRI in the rodent brain. Contrast Media Mol Imaging. 2016 Sep;11(5):371–380. doi: 10.1002/cmmi.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edmonds S, Volpe A, Shmeeda H, et al. Exploiting the metal-chelating properties of the drug cargo for in vivo positron emission tomography imaging of liposomal nanomedicines. ACS Nano. 2016 Nov;10(11):10294–10307. doi: 10.1021/acsnano.6b05935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daube ME, Nickles RJ. Development of myocardial perfusion tracers for positron emission tomography. Int J Nucl Med Biol. 1985;12(4):303–314. doi: 10.1016/0047-0740(85)90185-8. [DOI] [PubMed] [Google Scholar]
- 61.Inoue T, Majid T, Pautler RG. Manganese enhanced MRI (MEMRI): neurophysiological applications. Rev Neurosci. 2011;22(6):675–694. doi: 10.1515/RNS.2011.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim T, Momin E, Choi J, et al. Mesoporous silica-coated hollow manganese oxide nanoparticles as positive T 1 contrast agents for labeling and MRI tracking of adipose-derived mesenchymal stem cells. J Am Chem Soc. 2011;133(9):2955–2961. doi: 10.1021/ja1084095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crossgrove J, Zheng W. Manganese toxicity upon overexposure. NMR Biomed. 2004;17(8):544–553. doi: 10.1002/nbm.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valdovinos HF, Hernandez R, Graves S, et al. Cyclotron production and radiochemical separation of 55Co and 58mCo from 54Fe, 58Ni and 57Fe targets. Appl Radiat Isot. 2017 Jul;130:90–101. doi: 10.1016/j.apradiso.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garousi J, Anderson K, Dam JH, et al. The use of radiocobalt as a label improves PET imaging of EGFR using DOTA-conjugated affibody molecules. Eur J Nucl Med Mol Imaging. 2015;42(1):S244. doi: 10.1038/s41598-017-05700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mastren T, Marquez BV, Sultan DE, et al. Cyclotron production of high specific activity 55Co and in vivo evaluation of the stability of 55Co metal-chelate-peptide complexes. Mol Imaging. 2015;14(10):526–533. doi: 10.2310/7290.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dam JH, Olsen BB, Baun C, Høilund-Carlsen PF, Thisgaard H. In vivo evaluation of a bombesin analogue labeled with Ga-68 and co-55/57. Mol Imaging Biol. 2016;18(3):368–376. doi: 10.1007/s11307-015-0911-z. [DOI] [PubMed] [Google Scholar]
- 68.Garousi J, Andersson KG, Dam JH, et al. The use of radiocobalt as a label improves imaging of EGFR using DOTA-conjugated Affibody molecule. Sci Rep. 2017 Jul;7(1):5961. doi: 10.1038/s41598-017-05700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jansen HM, Pruim J, vd Vlieta M, et al. Visualization of damaged brain tissue after ischemic stroke with cobalt-55 positron emission tomography. J Nucl Med. 1994;35(3):456–460. [PubMed] [Google Scholar]
- 70.Goethals P, Volkaert A, Vandewielle C, Dierckx R, Lameire N. 55Co-EDTA for renal imaging using positron emission tomography (PET): a feasibility study. Nucl Med Biol. 2000;27(1):77–81. doi: 10.1016/s0969-8051(99)00077-3. [DOI] [PubMed] [Google Scholar]
- 71.Jansen HML, Willemsen ATM, Sinnige LGF, et al. Cobalt-55 positron emission tomography in relapsing-progressive multiple sclerosis. J Neurol Sci. 1995;132(2):139–145. doi: 10.1016/0022-510x(95)00139-s. [DOI] [PubMed] [Google Scholar]
- 72.Mitran B, Thisgaard H, Rosenström U, et al. High contrast PET imaging of GRPR expression in prostate cancer using cobalt-labeled bombesin antagonist RM26. 2017;2017 doi: 10.1155/2017/6873684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thisgaard H, Elema DR, Jensen M. Production and dosimetric aspects of the potent Auger emitter 58mCo for targeted radionuclide therapy of small tumors. Med Phys. 2011 Jul;38(8):4535–4541. doi: 10.1118/1.3608905. [DOI] [PubMed] [Google Scholar]
- 74.Valdovinos HF, Hernandez R, Goel S, et al. Auger electron-based targeted radioimmunotherapy with 58mCo, a feasibility study. AIP Conf Proc. 2016:1747. [Google Scholar]
- 75.Muller C, Zhernosekov K, Koster U, et al. A unique matched quadruplet of terbium radioisotopes for PET and SPECT and for radionuclide therapy: an in vivo proof-of-concept study with a new receptor-targeted folate derivative. J Nucl Med. 2012;53(12):1951–1959. doi: 10.2967/jnumed.112.107540. [DOI] [PubMed] [Google Scholar]
- 76.Müller C, Reber J, Haller S, et al. Folate receptor targeted alpha-therapy using terbium-149. Pharmaceuticals. 2014;7(3):353–365. doi: 10.3390/ph7030353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Müller C, Vermeulen C, Johnston K, et al. Preclinical in vivo application of 152Tb-DOTANOC: a radiolanthanide for PET imaging. EJNMMI Res. 2016;6(1):35. doi: 10.1186/s13550-016-0189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baum RP, Singh A, Benečová M, et al. Clinical evaluation of the radiolanthanide terbium-152: first-in-human PET/CT with 152 Tb-DOTATOC. Dalt Trans. 2017;54:2121. doi: 10.1039/c7dt01936j. [DOI] [PubMed] [Google Scholar]
- 79.Haller S, Pellegrini G, Vermeulen C, et al. Contribution of Auger/conversion electrons to renal side effects after radionuclide therapy: preclinical comparison of 161Tb-folate and 177Lu-folate. EJNMMI Res. 2016:1–11. doi: 10.1186/s13550-016-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Radchenko V, Hauser H, Eisenhut M, Vugts DJ, van Dongen GA, Roesch F. 90Nb-a potential PET nuclide: production and labeling of monoclonal antibodies. Radiochimica Acta Int j chem asp nucl sci technol. 2012;100:857. [Google Scholar]
- 81.Radchenko V, Bouziotis P, Tsotakos T, et al. Labeling and preliminary in vivo assessment of niobium-labeled radioactive species: a proof-of-concept study. Nucl Med Biol. 2016;43(5):280–287. doi: 10.1016/j.nucmedbio.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 82.Radchenko V, Busse S, Roesch F. Desferrioxamine as an appropriate chelator for 90Nb: comparison of its complexation properties for M-Df-octreotide (M=Nb, Fe, Ga, Zr) Nucl Med Biol. 2014;41(9):721–727. doi: 10.1016/j.nucmedbio.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 83.Radchenko V, Filosofov DV, Bochko OK, et al. Separation of 90Nb from zirconium target for application in immunopet. Radiochim Acta. 2014;102(5):433–442. [Google Scholar]
- 84.Engle JW, Hong H, Zhang Y, et al. Positron emission tomography imaging of tumor angiogenesis with a (66)Ga-labeled monoclonal antibody. Mol Pharm. 2012;9(5):1441–1448. doi: 10.1021/mp300019c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewis MR, Reichert DE, Laforest R, et al. Production and purification of gallium-66 for preparation of tumor-targeting radiopharmaceuticals. Nucl Med Biol. 2002;29(6):701–706. doi: 10.1016/s0969-8051(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 86.Engle JW, Lopez-Rodriguez V, Gaspar-Carcamo RE, et al. Very high specific activity 66/68Ga from zinc targets for PET. Appl Radiat Isot. 2012;70(8):1792–1796. doi: 10.1016/j.apradiso.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.El-Azony KM, Ferieg K, Saleh ZA. Direct separation of 67Ga citrate from zinc and copper target materials by anion exchange. Appl Radiat Isot. 2003;59(5–6):329–331. doi: 10.1016/s0969-8043(03)00203-3. [DOI] [PubMed] [Google Scholar]
- 88.Lopez-Rodriguez V, Gaspar-Carcamo RE, Pedraza-Lopez M, et al. Preparation and preclinical evaluation of 66Ga-DOTA- E(c(RGDfK))2 as a potential theranostic radiopharmaceutical. Nucl Med Biol. 2015;42(2):109–114. doi: 10.1016/j.nucmedbio.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 89.Sweet WH, Brownell GL. Localization of intracranial lesions by scanning with positron-emitting arsenic. JAMA. 1955;157(14):1183–1188. doi: 10.1001/jama.1955.02950310009002. [DOI] [PubMed] [Google Scholar]
- 90.Jahn M, Radchenko V, Filosofov D, et al. Separation and purification of no-carrier-added arsenic from bulk amounts of germanium for use in radiopharmaceutical labelling. Radiochim Acta. 2010;98(12):807–812. [Google Scholar]
- 91.Ellison PA, Barnhart TE, Engle JW, Nickles RJ, DeJesus OT. Production and chemical isolation procedure of positron-emit-ting isotopes of arsenic for environmental and medical applications. AIP Conf Proc. 2012;1509:135–140. [Google Scholar]
- 92.Ellison PA, Barnhart TE, Chen F, et al. High yield production and radiochemical isolation of isotopically pure arsenic-72 and novel radioarsenic labeling strategies for the development of theranostic radiopharmaceuticals. Bioconjug Chem. 2016;27(1):179–188. doi: 10.1021/acs.bioconjchem.5b00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chattopadhyay S, Pal S, Vimalnath KV, Das MK. A versatile technique for radiochemical separation of medically useful no-carrier-added (nca) radioarsenic from irradiated germanium oxide targets. Appl Radiat Isot. 2007;65(11):1202–1207. doi: 10.1016/j.apradiso.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 94.Shehata MM, Scholten B, Spahn I, Coenen HH, Qaim SM. Separation of radioarsenic from irradiated germanium oxide targets for the production of 71As and 72As. J Radioanal Nucl Chem. 2011;287(2):435–442. [Google Scholar]
- 95.Tolmachev V, Lundqvist H. Separation of arsenic from germanium oxide targets by dry distillation. J Radioanal Nucl Chem. 2001;247(1):61–66. [Google Scholar]
- 96.Jennewein M, Qaim SM, Hermanne A, et al. A new method for radiochemical separation of arsenic from irradiated germanium oxide. Appl Radiat Isot. 2005;63(3):343–351. doi: 10.1016/j.apradiso.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 97.Caletka R, Kotas P. Separation of germanium from some elements by adsorption on silica gel. J Radioanal Chem. 1974;21(2):349–353. [Google Scholar]
- 98.Gott MD, DeGraffenreid AJ, Feng Y, et al. Chromatographic separation of germanium and arsenic for the production of high purity 77As. J Chromatogr A. 2016;1441:68–74. doi: 10.1016/j.chroma.2016.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chakravarty R, Ram R, Jagdeesan KC, Venkatesh M, Dash A. Polymer embedded nanocrystalline titania: a new generation sorbent for the separation of 77As from Ge for biomedical applications. Chromatographia. 2011;74(7–8):531–540. [Google Scholar]
- 100.Hussain Bokhari T, Ahmad M, Ullah Khan I. Separation of no-carrier-added arsenic-77 from neutron irradiated germanium. 2009;97 [Google Scholar]
- 101.Ballard B, Wycoff D, Birnbaum ER, et al. Selenium-72 formation via natBr(p,x) induced by 100MeV protons: steps towards a novel 72Se/ 72As generator system. Appl Radiat Isot. 2012;70(4):595–601. doi: 10.1016/j.apradiso.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 102.Chajduk E, Doner K, Polkowska-Motrenko H, Bilewicz A. Novel radiochemical separation of arsenic from selenium for 72Se/72As generator. Appl Radiat Isot. 2012;70(5):819–822. doi: 10.1016/j.apradiso.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 103.ATSDR (Agency for Toxic Substances and Disease Registry) Toxicological profile for arsenic. Agency Toxic Subst. Dis. Regist. U.S. Public Heal. Serv; Atlanta, GA: 1989. no. ATSDR/TP-88/02. [Google Scholar]
- 104.Shen S, Li XF, Cullen WR, Weinfeld M, Le XC. Arsenic binding to proteins. Chem Rev. 2013;113(10):7769–7792. doi: 10.1021/cr300015c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jennewein M, Lewis MA, Zhao D, et al. Vascular imaging of solid tumors in rats with a radioactive arsenic-labeled antibody that binds exposed phosphatidylserine. Clin Cancer Res. 2008;14(5):1377–1385. doi: 10.1158/1078-0432.CCR-07-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kumar A, Hao G, Liu L, et al. Click-chemistry strategy for labeling antibodies with copper-64 via a cross-bridged tetraazamacrocyclic chelator scaffold. Bioconjug Chem. 2015;26(4):782–789. doi: 10.1021/acs.bioconjchem.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.DeGraffenreid AJ, Feng Y, Barnes CL, Ketring AR, Cutler CS, Jurisson SS. Trithiols and their arsenic compounds for potential use in diagnostic and therapeutic radiopharmaceuticals. Nucl Med Biol. 2016;43(5):288–295. doi: 10.1016/j.nucmedbio.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chakravarty R, Valdovinos HF, Chen F, et al. Intrinsically germanium-69-labeled iron oxide nanoparticles: synthesis and in-vivo dual-modality PET/MR zimaging. Adv Mater. 2014;26(30):5119–5123. doi: 10.1002/adma.201401372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park H-J, Tavlarides LL. Germanium(IV) adsorption from aqueous solution using a Kelex-100 functional adsorbent. Ind Eng Chem Res. 2009 Apr;48(8):4014–4021. [Google Scholar]
- 110.Roesch F, Riss PJ. The renaissance of the 68Ge/68Ga radionuclide generator initiates new developments in 68Ga radiopharmaceutical chemistry. Curr Top Med Chem. 2010;10(16):1633–1668. doi: 10.2174/156802610793176738. [DOI] [PubMed] [Google Scholar]
- 111.Chakravarty R, Chakraborty S, Ram R, Dash A, Pillai MRA. Long-term evaluation of ‘BARC 68Ge/68Ga generator’ based on the nanoceria-polyacrylonitrile composite sorbent. Cancer Biother Radiopharm. 2013 Jun;28(8):631–637. doi: 10.1089/cbr.2012.1470. [DOI] [PubMed] [Google Scholar]
- 112.Guo J, Yang W, Wang C. Magnetic colloidal supraparticles: design, fabrication and biomedical applications. Adv Mater. 2013 Oct;25(37):5196–5214. doi: 10.1002/adma.201301896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.