

Abstract
Background
Immunodysregulation polyendocrinopathy enteropathy x-linked(IPEX) syndromeis a monogenic autoimmune disease caused by FOXP3 mutations. Because it is a rare disease, the natural history and response to treatments, including allogeneic hematopoietic stem cell transplantation (HSCT) and immunosuppression (IS), have not been thoroughly examined.
Objective
This analysis sought to evaluate disease onset, progression, and long-term outcome of the 2 main treatments in long-term IPEX survivors.
Methods
Clinical histories of 96 patients with a genetically proven IPEX syndrome were collected from 38 institutions worldwide and retrospectively analyzed. To investigate possible factors suitable to predict the outcome, an organ involvement (OI) scoring system was developed.
Results
We confirm neonatal onset with enteropathy, type 1 diabetes, and eczema. In addition, we found less common manifestations in delayed onset patients or during disease evolution. There is no correlation between the site of mutation and the disease course or outcome, and the same genotype can present with variable phenotypes. HSCT patients (n = 58) had a median follow-up of 2.7 years (range, 1 week-15 years). Patients receiving chronic IS (n = 34) had a median follow-up of 4 years (range, 2 months-25 years). The overall survival after HSCT was 73.2% (95% CI, 59.4-83.0) and after IS was 65.1% (95% CI, 62.8-95.8). The pretreatment OI score was the only significant predictor of overall survival after transplant (P = .035) but not under IS.
Conclusions
Patients receiving chronic IS were hampered by disease recurrence or complications, impacting long-term disease-free survival. When performed in patients with a low OI score, HSCT resulted in disease resolution with better quality of life, independent of age, donor source, or conditioning regimen.
Keywords: IPEX, primary immune deficiency, FOXP3, Treg cells, hematopoietic stem cell transplantation, immunosuppression, rapamycin, enteropathy, neonatal diabetes, genetic autoimmunity
GRAPHICAL ABSTRACT
The immunodysregulation-polyendocrinopathy-enteropathy x-linked (IPEX) syndrome is a primary immunodeficiency caused by hemizygous mutations in the gene FOXP3, which encodes an essential transcription factor required for maintenance of immunologic tolerance by thymus-derived regulatory T (Treg) cells. Since its first clinical description in 19821 and its genetic characterization in 2001,2,3 IPEX syndrome has gathered the attention of scientists and physicians as the prototype of a monogenic autoimmune disease and immune deficiency affecting the immune regulatory compartment.4,5 Significant advances have been made in elucidating the complex disease pathogenesis.6–9 The typical manifestations of severe enteropathy, type 1 diabetes (T1D) and eczema have been extensively reported, but IPEX still poses a significant therapeutic challenge. Single reports of young adults affected by “atypical” or “late onset” IPEX forms have suggested heterogeneity in both the clinical presentation and their response to therapy.10–12 Immunosuppressive therapy, the first-line treatment for IPEX patients, has changed considerably in recent years with the introduction of new drugs with an immune suppressive and modulatory action. Nevertheless, the use and efficacy of these therapies are largely undocumented in IPEX patients. Currently, the only potentially curative therapy for IPEX syndrome is allogeneic hematopoietic stem cell transplantation (HSCT) with the longest published follow-up of 8 years.13,14 Both HLA-identical and matched-unrelated HSCT can be successful. However, while there is a general understanding to treat early, a comprehensive study comparing different transplant protocols and long-term outcomes is not available. In addition, a significant number of patients cannot undergo HSCT, due to limited donor availability and high risk-benefit ratio. Finally, older patients with a mild disease phenotype, in whom the clinical manifestations are often not severe enough to justify HSCT, raise concerns regarding the appropriate treatment.15
This international multicenter retrospective analysis of IPEX patients aims to provide a comprehensive view of the disease, its evolution, and outcomes of different therapeutic strategies. We aim to unveil limitations of case reports or small case series by providing a more comprehensive analysis of a large patients’ cohort. Ultimately, our work aims to improve the diagnosis and the long-term treatment for IPEX syndrome, with the goal of achieving definitive cure, which could apply to other immunodeficiencies with autoimmunity.
METHODS
Patients’ cohort and definitions
A retrospective multicenter study was performed in which data were collected from 38 institutions worldwide. All patients included were diagnosed with IPEX syndrome based on the presence of a FOXP3 mutation (detected but not specified for 5 patients). Data were collected through a detailed survey. Each center notified to their institutional review board and, if required, obtained the approval for sharing the data included in the present study. Data were transferred in a completely deidentified form.
Clinical manifestations were defined as “autoimmune” based on exclusion of other causes, the presence of specific autoantibodies or presence of other pathological findings suggestive of autoimmune etiology, or ex juvantibus (positive response to immunosuppressive therapy). Based on the number of organs or systems impaired by the autoimmune damage or by secondary complications, before undergoing immunosuppression (IS) or HSCT, we established an organ involvement (OI) scoring system ranging between 0 and 5; 1 point was assigned for the presence of each of the following: intractable diarrhea, malnutrition, liver dysfunction, respiratory impairment, kidney dysfunction. The selection of these parameters was dictated by their relevance in affecting disease morbidity and survival. The presence of clinical manifestations was assessed by the caring physicians, according to standard of care definitions.
The effect of IS was considered “beneficial” when the physician observed a decrease (partial benefit) or complete disappearance (benefit) of signs and symptoms of the disease.
Conditioning regimens were defined as fully myeloablative (full) or as reduced intensity transplant (RIT). Full conditioning regimens included busulfan plus cyclophosphamide, busulfan of > 14 mg/kg or cumulative area under the curve of 80 to 90 mg × h/L (when available) plus fludarabine, or treosulfan. RIT encompassed both reduced intensity conditioning (eg, fludarabine plus nonmyeloablative doses of busulfan, treosulfan, or melphalan) and minimal intensity conditioning (eg, fludarabine plus low dose radiation or cyclophosphamide).16
Neutrophil engraftment was defined as the first of 3 consecutive days with neutrophils >1000 per mm3 unsupported; platelet engraftment was defined as the first of 3 days with platelets >50,000 per mm3 unsupported. A “boost” was an additional infusion of HSC without conditioning, while “second or third” transplants were additional infusions of HSC from a different donor with conditioning. T-cell reconstitution (analyzed at 3, 6, and 12 months and at the last available follow-up) was defined as >1000 CD3+ T cells per mm3, >500 CD4+ T cells per mm3 and presence of proliferative response to mitogens (considered as positive when within the normal range of the laboratory). B-cell reconstitution was defined as independence from immunoglobulin replacement therapy. Full conditioning regimen donor chimerism was defined as >95% donor cells in peripheral blood (evaluated at 3, 6, and 12 months and at last follow-up). Primary graft failure was defined as absence of donor cells in peripheral blood posttransplant. Secondary graft failure was defined as reduction of neutrophil counts, occurring after engraftment, and absence of donor cells (below 5%), despite normal blood counts. The diagnosis and grading of acute or chronic graft-versus-host disease (GvHD) were based on previously published criteria and scoring system.17
Evaluation of quality of life was reported by the caring physicians without a standardized scoring system (yes/no questions).
Statistical analysis
Demographic, disease-related, or transplant-related characteristics were reported as frequencies for categorical variables, and median and range for quantitative variables. Associations between variables and disease outcome or type of treatment were compared with the use of the chi-square test (categorical variables) or unpaired t-test (continuous variables).
Probabilities of survival after treatment and disease-free survival (DFS) were computed with the use of the Kaplan-Meier estimator and compared with the log-rank test (Mantel-Cox). We then used a multivariable analysis to examine risk factors for transplantation outcomes and adjust for confounding factors using the Cox (proportional hazard) regression models with Breslow estimator; a P value of .05 or less was considered statistically significant. The variables considered in the Cox regression to model transplantation outcome were age at transplant, pre-HSCT OI score, donor type, degree of HLA-matching, intensity of conditioning regimen, graft source, and number of CD34 cells infused. For a small cohort of patients transplanted at <1 year, the variables used in the Cox regression were age at HSCT, score, and weight. We also examined DFS with a Cox regression model, where death is treated as a censoring event, using as predictor variables the score pre-HSCT, graft failure, and conditioning.
The patients whose mutations were known (n = 91) were grouped in 4 categories depending on type of mutation: missense, splice site, frameshift or in-frame deletions or insertions; and “others” including mutations in the untranslated region, promoter, or start codon. We furthermore explored each mutation effect on protein function through available algorithms such as PolyPhen-2 (providing predictions for amino acid substitutions) and PROVEAN (providing predictions for amino acid substitutions, insertions, and deletions), obtaining a numeric score and a qualitative prediction outcome such as “benign” or “neutral,” and “deleterious” or “probably damaging.” We examined associations among the predicted protein scores and each patient score before HSCT or IS and their survival outcome, by evaluating Spearman correlation coefficient and contingency tables.
RESULTS
Initial clinical manifestations, disease course, and genetics
Ninety-six patients with FOXP3 mutations were included in our study. The median age at disease onset was 2 months (range, birth to 11.3 years): 41% had onset within the first month of life, 46% between 1 month and 1 year, 10% after 1 year of age, and for 3% of patients, the time of onset was unavailable. The median time to diagnosis (elapsed time between onset of symptoms and genetic diagnosis) was 14 months (up to a maximum of 23.9 years) (Fig 1, A); however, the number of IPEX patients diagnosed per year shows an increased trend (Fig 1, B).
FIG 1.

Patients’ demographics and disease-related characteristics. A, Diagnostic delay scatter plot displaying correlation between age at diagnosis and age at onset (n = 96). Spearman’s rank correlation ρ = 0.456, P < .001. B, Histogram distribution of number of patients diagnosed between 2001 and 2015. Overlaid density distribution showed in blue. C, Heat map of number of symptoms at onset grouped by age at onset. Symptoms present in each age group (indicated by rows) were scaled (z-score or standardized score) and then converted to colors from yellow (low = less frequent) to red (high = highly frequent). Data were not available (NA) for 3 patients; 1 patient has the mutation but has not yet experienced the onset; and for 2 patients, the first symptom is unknown. D, Bar graph comparing frequency of symptoms at onset and later during disease evolution. Every bar indicates number of patients presenting each symptom. However, each patient can exhibit >1 symptom at once. E, Uncommon manifestations. Every bar indicates number of patients presenting each symptom. F, Scatter plot of FOXP3 gene mutations grouped by domain, indicating age at onset (circles) and number of symptoms at onset (asterisks), with median (n = 87; for 10 patients, cDNA FOXP3 mutations were not specified). Gene structure: N-terminal proline-rich (PRR) domain (orange), zinc-finger (ZF) domain (green), leucine-zipper (LZ) domain (blue), LZ-FKH loop (yellow), and FKH domain (red). Mutations were grouped as follows: <c.1 to c.570> 5 E1-5 N-terminal domain, <c.591 to c.666> = E5-6 ZF domain, <c.717 to c.780> = E6-7 LZ domain, <c.781 to c.1010> = E7-9 LZ-FKH loop domain, <c.1011 to c.1251> = E9-11 FKH domain. AIHA, Autoimmune hemolytic anemia; AIN, autoimmune neutropenia; AT, autoimmune thyroiditis; FTT, failure to thrive; ITP, idiopathic thrombocytopenic purpura; Neph, nephropathy; LN, lymphadenopathy; PRR, proline-rich region; UTR, untranslated region.
The most common presentation suggestive of the diagnosis of IPEX syndrome remained the classical triad of enteropathy, T1D, and eczema for the majority of the 39 of 96 patients who had neonatal onset. Gastrointestinal involvement and eczema, but not T1D, were the dominant features of disease presentation after 1 month of age. However, failure to thrive was a hallmark of the disease and it was sometimes the sole initial manifestation after 1 month of age. After 1 year of age, the disease could also present with nephropathy or hepatitis, otherwise considered atypical at onset (Fig 1, C and D).
The symptoms observed over the course of the disease and their prevalence are listed (Fig 1, D): with the exception of diarrhea, eczema, and failure to thrive, nephropathy (autoimmune or secondary to malnutrition and medication) was the most common (33 of 96 patients), varying from lithiasis, nephrocalcinosis, or isolated proteinuria to more severe manifestations such as nephrotic syndrome (in some cases with a definite diagnosis of glomerulonephritis) or tubulopathy and interstitial nephritis. Hematological manifestations included autoimmune hemolytic anemia (25 of 96 patients), thrombocytopenia (13 of 96 patients), and neutropenia (6 of 96 patients). Other conditions included autoimmune thyroiditis (15 of 96 patients) and hepatitis (19 of 96 patients), while food allergies (13 of 96 patients), arthritis (8 of 96 patients), alopecia (8 of 96 patients), and lymphadenopathy (9 of 96 patients) were occasionally reported during disease progression. Interestingly, among other uncommon manifestations (Fig 1, E), neurological findings, of uncertain relation with FOXP3 mutations, were reported in 16 of 96 patients, including peripheral neuropathy, myopathies/ hypotonia, hemidiaphragmatic paralysis, eosinophilic meningitis, neurodevelopmental delay, seizures, and benign intracranial hypertension.
The 96 patients included in the study displayed 33 published mutations4,5 and 21 novel mutations. The forkhead (FKH) domain emerges as a mutational hotspot of the FOXP3 gene (Fig 1, F), although mutations were scattered along the gene. Mutations in the N-terminal and FKH domains correlated with high variability of age at onset. The number of symptoms at onset was independent from the site of the mutations.
Laboratory findings
Table I summarizes the biological findings characterizing this IPEX patients’ cohort before therapy. The distribution of lymphocyte subsets before IS or in patients treated with long-lasting IS or before HSCT was normal or seldom characterized by increased counts. The proportion of Treg cells, evaluated by flow cytometry, was available only in few patients and FOXP3 showed a wide range of expression. Gut biopsies frequently showed simultaneous involvement of several sites: the most common target was the small bowel, followed by large bowel, stomach, and esophagus. The inflammatory infiltrates were usually polymorphic with predominance of lymphocytes and eosinophils. Villous atrophy remained the hallmark of IPEX enteropathy, although other nonspecific lesions associated with inflammatory damage (ulcers, crypt hyperplasia, and abscesses) were present. Of note, 4 patients showed metaplastic lesions, either in the bowel or in the stomach, and 1 developed dysplastic lesions of the gastric mucosa. Antienterocyte, antiharmonin, or antivillin autoantibodies were positive in 33 of 42 patients with enteropathy tested (Table I). In contrast, in 9 of 42 cases these autoantibodies could not be detected despite the presence of enteropathy, a finding possibly dependent on the choice of antibodies that were tested. Remarkably, in 1 case, antiharmonin autoantibody positivity prompted the diagnosis in a patient with isolated IPEX-related gastritis. Antineuron antibodies were detected in 1 of the patients with neurological involvement. Finally, 12 patients screened positive for T1D-associated autoantibodies (2 of them had 2 positive auto-antibodies), without having T1D (Table I). Thus, the histological and serological markers were indicative of the diagnosis only when considered together with the clinical manifestations.
TABLE I.
Biological findings characterizing the IPEX patients’ cohort before therapy
| Immunophenotype pre-IS | Median; range (no. Of patients tested) |
|---|---|
| Lymph tot/mmc | 2925; 900-7480 (22) |
| CD3+/mmc | 2047; 600-4838 (23) |
| CD3+CD4+/mmc | 1276; 370-3658 (24) |
| CD3+CD8+/mmc | 586; 210-2020 (23) |
| Ratio CD4:CD8 | 2; 0.5-3.9 (23) |
| CD19+/mmc | 276; 0-1715 (22) |
| CD16+CD56+/mmc | 170; 20-525 (21) |
| CD4+CD25+, % | 9; 0-35 (10) |
| CD4+CD25+CD127low, % | 3; 1-6.6 (5) |
| FOXP3+, % in CD4+CD25+CD127low | 36; 2.2-58 (5) |
| FOXP3+, % in CD4+ | 5; 0.8-7 (4) |
| Immunophenotype pre-HSCT | |
|---|---|
| Lymph tot/mmc | 3350; 440-8189 (39) |
| CD3+/mmc | 2185; 350-9388 (52) |
| CD3+CD4+/mmc | 1311; 150-7824 (52) |
| CD3+CD8+/mmc | 781; 106-2612 (51) |
| Ratio CD4:CD8 | 1.9; 0.3-10.1 (51) |
| CD19+/mmc | 572; 0-6245 (51) |
| CD16+CD56+/mmc | 200; 0-1112 (45) |
| CD4+CD25+, % | 13; 0-49 (18) |
| CD4+CD25+CD127low, % | 7.8; 3.3-20.2 (4) |
| FOXP3+, % in CD4+CD25+CD127low | 50; 1.3-98 (9) |
| FOXP3+, % in CD4+ | 0; 0-5 (3) |
| Gut biopsies before therapy (tot n = 61) | No. of cases |
|---|---|
| Number of sites involved | |
| Single | 26 |
| Multiple | 34 |
| Affected sites | |
| Esophagus | 7 |
| Stomach | 16 |
| Small bowel | 45 |
| Large bowel | 16 |
| All sites | 7 |
| Combinations of affected sites | |
| Small and large bowel | 11 |
| Stomach + small bowel | 9 |
| Stomach + large bowel | 1 |
| Esophagus + stomach | 1 |
| Esophagus + small bowel | 2 |
| Stomach + small and large bowel | 2 |
| Esophagus + stomach + small bowel | 1 |
| Esophagus + small and large bowel | 1 |
| Infiltrating cell type | |
| Eosinophils | 19 |
| Lymphocytes | 32 |
| Plasma cells | 8 |
| Polymorphonucleated cells | 6 |
| Polymorphic infiltrate including all cells | 8 |
| Unspecified inflammatory cell type | 4 |
| Histological lesions | |
| Villous atrophy | 42 |
| Loss of goblet cells | 3 |
| Crypt hyperplasia | 9 |
| Crypt abscesses | 8 |
| Ulcers | 9 |
| Gut biopsies before therapy (tot n = 61) | No. of cases |
|---|---|
| Loss of parietal cells | 2 |
| Metaplasia | 4 |
| Enteropathy-related autoantibodies | No. positive/No. of cases tested |
|---|---|
| Antienterocyte Abs | 25/34 |
| HAA | 13/24 |
| Antienterocyte and HAA | 4/5 |
| VAA | 1/12 |
| Other autoantibodies before therapy | |
|---|---|
| Tot no of patients tested for GAD | 41 |
| GAD among pts with T1D | 15/18 |
| GAD among pts without T1D | 9/23 |
| Tot no. of patients tested for IA | 27 |
| IA among pts with T1D | 9/15 |
| IA among pts without T1D | 0/12 |
| Tot no. of patients tested for IAA | 33 |
| IAA among pts with T1D | 10/13 |
| IAA among pts without T1D | 3/20 |
| Tot no. of patients tested for ZNT8 | 14 |
| ZNT8 among pts with T1D | 1/8 |
| ZNT8 among pts without T1D | 0/6 |
| Tot no. of patients tested for ICA | 6 |
| ICA among pts with T1D | 1/3 |
| ICA among pts without T1D | 3/3 |
GAD, Glutamic acid decarboxylase autoantibodies; HAA, antiharmonin autoantibodies; IA, islet antigen; IAA, anti-insulin autoantibodies; ICA, anti-islet cell antibodies; mmc, mm3; pts, patients; tot, total; VAA, antivillin autoantibodies; ZNT8, zinc transporter 8 autoantibodies.
Treatments
Of the 96 patients included in the study, 34 received IS and 58 underwent HSCT. Four patients with FOXP3 mutations improved spontaneously (1), did not require IS (2), or were still asymptomatic (1) and were thus excluded from this analysis. One patient had several manifestations of the disease (enteropathy, growth retardation, eczema, autoimmune cytopenia) that improved spontaneously, with supportive therapy and without any immunosuppressive drugs. Two other patients had persistent signs of the disease that, however, did not require IS (eczema and growth retardation in 1 case and T1D in the other). The last patient is still asymptomatic at 6 years of age, and he was diagnosed only due to familial history. Fig 2 provides a schematic view of the disease evolution of each patient and his mutation and reveals the variability of disease progression in our cohort.
FIG 2.
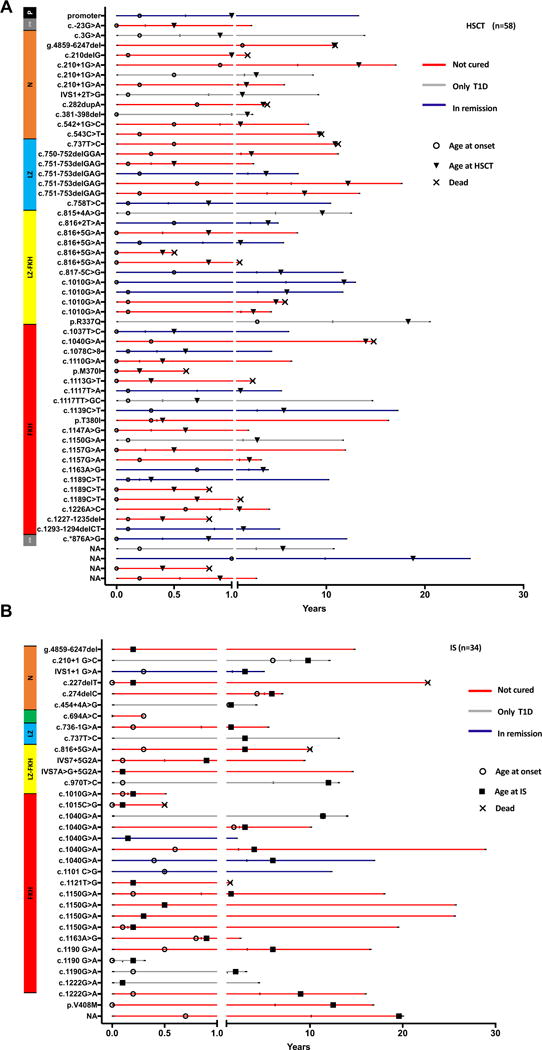
Timeline of natural history and disease evolution. Patients undergoing HSCT (A) and IS (B). Each line represents a patient identified by his FOXP3 mutation in order of localization on the gene. The end on the line represents the last day of follow-up, and different symbols represent age at onset (circles) and age at HSCT (triangles) or the beginning of IS. An X at the end of the line indicates the age of death. The color of the line indicates the disease status after treatment, whether the patient went into remission (blue), was still diabetic (gray) or not cured (red). NA, Not available.
Immunosuppressive therapy
The 34 patients who received IS and were not transplanted started treatment at a median age of 1.5 years (range, 1 month-19.6 years) with a median follow-up of 4 years (range, 1 month-25 years). Only 3 patients had a follow-up shorter than 7 months.
Fig 3 shows the response to each drug, to drug combinations, and the final outcome after IS. Twenty-five patients received systemic steroids (range, 2 months-22 years; median, 2 years). While steroid administration benefited 56% of the treated patients, it was often administered concomitantly with other immunosuppressive drugs (Fig 3, B), thus direct effects cannot be confirmed.
FIG 3.
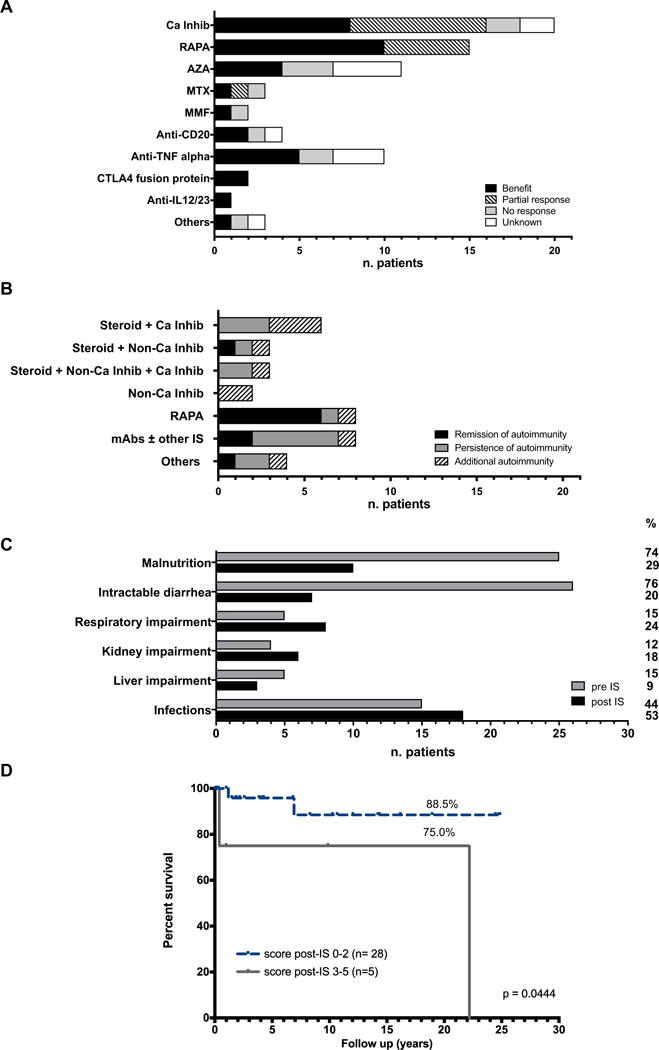
Immunosuppressive therapy. A, Bar graph indicating numbers of patients exhibiting response, partial benefit, or no response according to a specific immunosuppressive drug administered (n = 34; however, each patient received >1 drug). B, Bar graph indicating each patient’s outcome after treatment with a combination of drugs, as last treatment. Each bar represents the number of patients receiving the treatment, distinguishing those in remission from those with additional or persistent autoimmunity (n = 34). C, Pre- and post-IS patients’ conditions, each bar represents the number of patients presenting each condition. On the right side, the relative percentage is reported (n = 34; however, each patient could present with >1 condition at once). D, Percentage of survival for patients undergoing IS (n = 34) according to score post-IS (P = .0444). AZA, Azathioprine; Ca Inhib, calcineurin inhibitors; CTLA, cytotoxic T lymphocyte–associated antigen; MTX, methotrexate; MMF, mycophenolate mofetil; “others”, any different IS (eg, 6-mercaptopurina, mesalazine); RAPA, rapamycin.
Twenty patients received calcineurin inhibitors, either cyclosporine-A or tacrolimus, with benefit in 40% (Fig 3, A). Six of them also received concomitant steroids and 3 both steroids and noncalcineurin inhibitors. Among patients treated with noncalcineurin inhibitors, 15 patients received rapamycin and 19 received other immunosuppressive agents comprising azathioprine, methotrexate, mycophenolate-mofetil, mesalazine, sulfasalazine, and 6-mercaptopurine. Combination of steroids with calcineurin inhibitor, noncalcineurin inhibitors, or both (12 of 21 patients) infrequently (1 of 12 patients) led to remission of autoimmunity (Fig 3, B). Rapamycin improved autoimmune manifestations in 67% of patients (Fig 3, A) and in 8 of 10 patients when it was used as monotherapy, with 6 of 8 patients achieving remission (Fig 3, B). The use of azathioprine was beneficial in 36% of cases (Fig 3, A). The use of mAbs, including anti-TNF-α, anti-CD20, and cytotoxic T lymphocyte–associated antigen 4 fusion protein (abatacept), was usually in addition to other IS; therefore, their efficacy as single drugs is not fully evaluable. Although treatment with a mAb was beneficial in the 52% of cases (Fig 3, A), mAb-based regimens rarely resulted in sustained remission of autoimmunity (2 of 8 patients) (Fig 3, B).
Prior to initiation of IS, 25 of 34 patients were malnourished (74%) and 26 of 34 had intractable diarrhea (76%). The prevalence of these clinical manifestations significantly diminished following IS, since malnutrition persisted in 10 cases (29%; P < .001) while diarrhea persisted in 7 cases (20%; P < .001). Similarly, 3 of 5 patients with autoimmune hepatitis improved after IS. Nevertheless, prevalence of respiratory impairment, kidney dysfunction and frequency of infections increased under IS although not significantly (P = .538, P = .511, and P = .627) (Fig 3, C).
Overall, 10 of 34 patients receiving IS completely controlled autoimmunity, while 24 patients still had autoimmune manifestations, mainly enteropathy (11 of 24). T1D remained the only autoimmune manifestation in 2 patients. In 11 patients new autoimmune manifestations arose while receiving IS, including T1D (4), thyroiditis (3), autoimmune cytopenia (2), autoimmune hepatitis (2), enteropathy (1), adrenal insufficiency (1), and arthritis (1). Overall, the OI score before starting IS mostly corresponded to the OI score at disease onset and did not significantly influence the outcome. Indeed, most of the patients improved in the short term; whereas in the long term, the OI score could worsen due to disease recurrence or progression and side effects of treatment, thus impacting survival (Fig 3, D), as discussed below.
Nutritional support (enteral, parenteral, or both) was often necessary for patients under IS (19 of 34), lasting for months or years. Recurrent hospitalization for disease complications and infections occurred a median of 4 times per year (ranging from 0 to 7 times per year). Thirty of 34 patients are alive at the last follow-up. Four patients died (at 5 months, and 1.5, 7, and 22.5 years, respectively). Causes of death were acute respiratory distress syndrome with multiple organ failure, pneumonia, idiopathic cardiac arrest, and sepsis. The estimated overall survival of patients under IS was 86.8% (95% CI, 62.8-95.8) at 15 years and 65.1% (95% CI, 18.3-89.7) at 24 years.
Thus, the merely symptomatic approach of long-term IS can be beneficial in the short term, but it did not prevent disease progression and development of complications in the majority of the patients and it could have an impact on patients’ survival.
Hematopoietic stem cell transplantation
Fifty-eight patients underwent HSCT. Seven patients required a second transplant and 1 patient received 3 HSCTs. In these patients, follow-up records refer to the last procedure. The median follow-up was 2.7 years (range, 2 weeks-15 years). The median age at onset for patients in this cohort was 1 month (range, birth-1 year) and transplant was performed at a median age of 1.4 years (range, 1 month-18.8 years), indicating that the majority of the transplants were performed at early age and close to disease onset (see Fig E1, A in this article’s Online Repository at www.jacionline.org). Despite the short time frame between onset and transplantation, 54 patients received IS prior to transplantation. Disease manifestations improved before HSCT in 27 of 54 patients, while 23 of 54 patients had partial or no control of the autoimmune manifestations and 4 of 54 patients could not be assessed for response. Application of the OI score at the time of transplantation demonstrated a strong association with outcome. The estimated overall survival after HSCT was 73.2% at 15 years, with significant differences for patients with either low or high OI score, as described below.
HSCT characteristics and complications
The majority of patients (33 of 58) received RIT (Table II; see also Table E1 in this article’s Online Repository at www.jacionline.org for further details about conditioning). Donor type included matched related (31 of 58 patients), matched unrelated (21 of 58 patients), haploidentical (5 of 58 patients, 3 of which had α/βT-cell depletion), and other (1 mismatched related of 58 patients). HSC sources included bone marrow (35 of 58 patients), peripheral blood stem cells (12 of 58 patients), and cord blood (13 of 58 patients). Antithymocyte globulin or alemtuzumab were frequently used (49 of 58 patients). GvHD prophylaxis mainly consisted of cyclosporine-A associated with mycophenolate-mofetil or steroids. Hematological recovery was obtained at a median of 16 days for neutrophils and 20 days for platelets. At 1 year after HSCT, 22 of 33 patients had achieved T-cell reconstitution. At a median of 14.5 months, 20 of 29 patients had positive proliferative response to mitogens. Independence from IVIG was reached at a median of 7 months in 30 of 45 patients. Transplant-related toxicity was observed in 11 cases (19%). Multiple infections were reported in 46 patients (85%).
TABLE II.
Characteristics, outcomes, and complications of HSCT
| No. patients | Percent | |
|---|---|---|
| Characteristics | ||
| Tot no. of patients | 58 | |
| Age (y) at transplant, median (range) | 1.4 (0.2-18.8) | |
| Patients who received multiple transplants | ||
| 2 HSCT | 7 | |
| 3 HSCT | 1 | |
| Conditioning | ||
| Full | 27 | 47 |
| RIT | 31 | 53 |
| Donor-related; unrelated | ||
| Matched | 10; 21 | 17; 36 |
| 1 MM | 1; 13 | 2; 22 |
| 2 MM | 0; 6 | 0; 10 |
| 3 MM | 1; 1 | 2; 2 |
| Haplo | 5; 0 | 9; 0 |
| HSC source | ||
| BM | 35 | 60 |
| PB | 11 | 19 |
| CB | 12 | 21 |
| Cell doses | ||
| BM (TNC × 108/kg), median (range) | 7.1 (0.01-91.3) | |
| PB (CD34 × 106/kg), median (range) | 11.4 (4.3-40) | |
| CB (TNC × 107/kg), median (range) | 9.8 (0.6-42) | |
| Serotherapy | ||
| ATG | 22 | 38 |
| Alm | 27 | 46 |
| None | 9 | 16 |
| GvHD prophylaxis | ||
| CSA + MMF | 18 | 31 |
| CSA + steroids | 10 | 17 |
| CSA | 6 | 10 |
| MTX + CSA (with or without short course of steroids) | 8 | 14 |
| MTX + FK506 1 steroids | 7 | 12 |
| Others | 9 | 16 |
| Bone marrow recovery | ||
| Neutrophils (days after HSCT), median (range) | 16 (3-33) | |
| Platelets (days after HSCT), median (range) | 20 (5-114) | |
| Immunoreconstitution | ||
| Patients with T cells > 1000/mmc at 1 y | 22 of 33 | |
| Positive PHA response (months after HSCT), median (range) | 14.5 (3-60) | |
| Independence from IVIg substitution (months after HSCT), median (range) | 7 (1-48) | |
| Use of donor stem cell boost | 3 | |
| Use of donor lymphocytes infusion | 3 | |
| Complications | ||
| Transplant-related toxicity* | 11 | 20 |
| Infections | 46 | 79 |
| Tot GvHD, | 21 | 36 |
| aGvHD (grade I-IV) | 19 | 33 |
| aGvHD (grade III-IV) | 9 | 16 |
| cGvHD | 6 | 10 |
| No GvHD | 37 | 64 |
| Deaths | 15 | 26 |
Alm, Alemtuzumab; aGvHD, acute graft-versus-host disease; ATG, antithymocyte globulin; BM, bone marrow; CB, cord blood; cGvHD, chronic graft-versus-host disease; CSA, cyclosporine; FK506, tacrolimus; Full, full conditioning regimen; IVIg, intravenous immunoglobulin; MM, mismatch; MMF, mycophenolate mofetil; MTX, methotrexate; PB, peripheral blood; PHA, phytohemagglutinine; TNC, total nucleated cells.
Toxicity after HSCT consisted of mucositis, pneumonitis, posterior reversible encephalopathy, undefined hypertrophic cardiomyopathy, nephropathy, and hepatic sinusoidal obstruction syndrome.
GvHD
Nineteen of the transplanted patients (33%) experienced acute GvHD, which was of grade III to IV in 9 patients. Among patients surviving >100 days, 6 of 52 developed chronic GvHD (10% of the transplanted patients). While not statistically significant, the incidence of acute GvHD was higher in patients who did not receive serotherapy (5 of 9; 55%) compared with those receiving antithymocyte globulin or alemtuzumab (16 of 47; 34%), for 2 patients GvHD could not be ascertained. Occurrence of acute GvHD was comparable (P = .2362) in related (5 of 17; 29%) and unrelated (15 of 31; 48%) donor transplants. Overall, the incidence and severity of GvHD for IPEX syndrome were similar to that described for other primary immune deficiencies.18
Chimerism
Full donor peripheral blood chimerism was detected in 31 of 53 patients evaluated for chimerism; 17 of them were alive and in remission (or with T1D only). Among the patients with full donor chimerism, 3 died and 11 had autoimmune manifestations (or GvHD). Mixed chimerism was detected in 18 of 53 patients and associated with disease remission in 9 of 18 (Fig E1, B). Importantly, the Treg cells were 100% of donor origin in 3 of 9 patients carrying mixed chimerism in remission. Moreover, 4 of 18 patients with mixed chimerism are alive with autoimmune manifestations (Fig E1, C) and 5 of 18 patients have died—all but 1 of infections, at different times post-HSCT (Fig E1, D). The occurrence of mixed chimerism was not related to the use of RIT and was also observed following fully myeloablative conditioning. Overall, the data show similar proportion of patients in remission among those with full (17 of 31, 54%) or mixed (9 of 18, 50%) chimerism. In addition, graft failure was observed in 4 of 53 patients (Fig E1, E). Two patients experienced secondary graft failure between 3 and 6 months posttransplantation, they are still alive but with disease relapse. The other 2 patients are dead, 1 with primary graft failure and the second with acute graft loss 40 days after transplant.
Survival after transplant
The estimated overall survival rate for transplanted patients at 15 years was 73.2%. The majority of deaths occurred in the first months after HSCT due to infections. Multivariable analysis showed that the type of conditioning, type of donor, and age at transplantation did not significantly influence survival (Fig 4, A-C). In fact, the pre-HSCT OI score was the only variable significantly influencing survival after HSCT. Indeed, the probability of survival was significantly lower in patients with a score between 3 and 5 as compared to patients with an initial score between 0 and 2 (P = .002) (Fig 4, D). In addition, the combined analysis of the score and of the conditioning regimen, showed that patients with scores 0 to 2 had better survival independently of the administration of full conditioning or RIT. Within the same analysis, patients with scores 3 to 5 who received full conditioning had better survival than the ones who received RIT (Fig 4, E). Similarly, the OI score affected the survival outcome among patients below or above 1 year of age, with patients <1 year and high score performing significantly worse (Fig 4, F). The majority of patients who were <1 year of age at the time of HSCTwere also at or below the third centile for weight (16 of 21 patients whose weight at transplantation was reported). Among these patients, we further observed that the patients who did not survive had lower weight and younger age, although the number of events was not sufficient to perform a multivariable analysis (see Fig E2, A and B in this article’s Online Repository at www.jacionline.org). DFS analysis showed that the probability of recurrent or new onset autoimmunity after transplantation was not dependent on the conditioning administered (Fig E2, C) or chimerism obtained after transplantation (Fig E2, D). Multivariable analysis did not identify a variable significantly affecting DFS after HSCT.
FIG 4.

Survival analysis of patients undergoing HSCT. Percentage of survival of patients undergoing HSCT (n = 58) according to conditioning (log-rank test, P = .234) (A), donor type (P = .886) (B), age at HSCT (P = .359) (C), score pre-HSCT (P = .003) (D), score and conditioning (P = .010) (E), and score and age at HSCT (P = .019) (F). A survival probability table accompanies those plots that show significant differences (time points: 6 months, and 1, 3, 5, and 10 years). Full C, Full conditioning regimen; MMRD, mismatched related donor; MMUCB, mismatched unrelated cord blood; MMUD, mismatched unrelated donor; MSD, matched sibling donor; MUD, matched unrelated donor.
Comparison of outcomes between transplanted and not transplanted patients
Survival rates at 15 years among children undergoing HSCTwere lower, although not significantly, as compared to those receiving chronic IS (73.2% vs 86.8%; P = .055) (Fig 5, A). This difference is largely due to the high mortality rate within 2.5 years following transplantation, with up 15% of patients dying by the first 100 days and 25% by the first 2.5 years (Fig 5, A). Patients who survive over 2.5 years after transplant do not show additional mortality up to 15 years later, and their probability of survival remains constant, whereas that of IS patients, who have a longer follow-up, drops as the disease progresses and treatment complications increase with time (survival rate at 24 years is 65.1%). Indeed, the survival of patients receiving IS does not depend on the OI score pre-IS but, in the long-term, a worsening of the OI score can negatively influence the survival in these patients. Thus, the OI score post-IS is significantly correlated with survival (P = .0444) (Fig 3, D).
FIG 5.
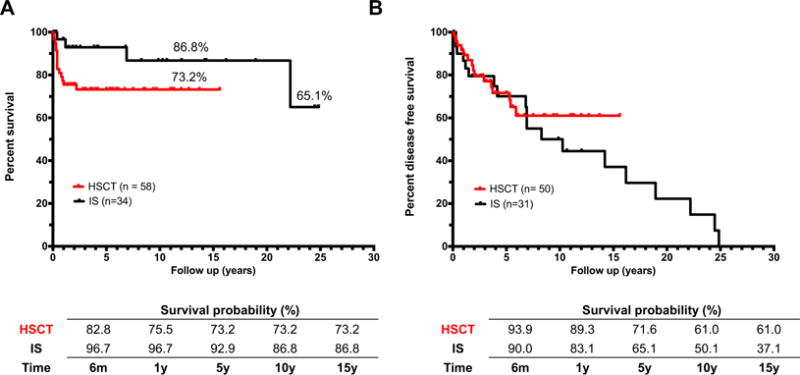
Probability of survival and disease status after treatment. A, Survival analysis of IPEX patients undergoing HSCT or IS (n = 92, P = .055). B, Disease-free survival analysis of IPEX patients undergoing IS or HSCT censored for deaths (n = 81, P = .419).
The percentage of patients who completely resolved autoimmunity (or had only T1D) was higher in alive transplanted patients than in those receiving IS, although not significantly (56% vs 36%, P =.083). Similarly, the persistence or the onset of new autoimmune manifestations (other than GvHD in transplanted patients) was significantly lower in patients surviving HSCT than under IS treatment (17% vs 51%, P = .001) showing that IS does not prevent disease progression. This latter conclusion is also supported by the DFS curve showing a progressive reduction of DFS probability during IS. On the contrary, DFS probability remains stable after the first 6 years posttransplant (Fig 5, B).
We did not observe any significant correlation between disease score or survival and the effect of the different mutations on the FOXP3 protein, as predicted by PolyPhen-2 or PROVEAN, for patients given either IS or HSCT (see Fig E3 in this article’s Online Repository at www.jacionline.org).
In the entire cohort from 28% (under IS) to 31% (after HSCT) of the patients experienced delayed neuromotor development or needed a support teacher, despite comparable rates of patients undergoing schooling/working activities adequate for age after either therapeutic approach. Of note, a significant percentage of children needed psychological support. Nutritional issues lead to a frequent and prolonged use of feeding support in both categories. In line with the outcome results, a significant percentage of patients surviving HSCT considered this therapy efficacious. In contrast, patients undergoing IS perceived an incomplete resolution of the disease and complained about chronic medications, side effects, and periodic follow-up (see Table E2 in this article’s Online Repository at www.jacionline.org). This confirms that patients surviving transplantation are less prone to disease evolution and complications over time.
DISCUSSION
The present retrospective study of IPEX patients provides an informative comparison of the currently available therapeutic options and their long-term outcome, together with a comprehensive and updated view of the disease and its initial presentation and progression. We demonstrate that IPEX patients have similar overall survival, regardless of whether they receive IS or HSCT, with a greater survival in the first years posttreatment for the non-HSCT group. However, the DFS of HSCT patients shows clear differences with stable resolution of autoimmunity as compared to the persistent disease progression in the nontransplanted IPEX patients. Therefore, the study highlights the therapeutic limitations of the current immunosuppressive regimens, although the use of rapamycin proved as the most beneficial IS therapy11,19,20 and appears to be superior to calcineurin inhibitors.13,21–24 Results from this study further indicate that a better survival outcome after HSCT is significantly affected by the patients’ pre-HSCT conditions, as defined by the OI score we established. IPEX patients with low OI score, either initially or after IS, had a survival advantage after HSCT. On the contrary, IPEX patients with severe organ impairment (high OI score) at HSCT had the lowest chance of survival even receiving a RIT, suggesting that the clinical status is more important than the conditioning regimens in the outcome of HSCT. Other variables (ie, type of donor, stem cells source, and chimerism) were not correlated with outcome, as was previously reported in a small cohort of patients.25 In contrast though with this other cohort,25 our data show that patients transplanted before 1 year of age tended to have a lower survival (although without statistical significance), which may reflect either a more severe disease status or an increased sensitivity to conditioning agents. Overall, these data point to the value of optimizing patient’s clinical condition prior to HSCT and considering HSCT before disease progression.
The majority of the patients in our study received a RIT,25–30 conditioning that is not always associated with mixed chimerism, frequently observed in IPEX transplanted patients.13,14,21,25,26,30 Mixed rather than full donor chimerism did not affect DFS probability, as has been the case for other primary immunodeficiencies.31 Importantly, in a small number of IPEX patients14,30 with mixed chimerism who achieved remission, all Treg cells were of donor origin, indicating that the presence of functional Treg cells may suffice to control autoimmunity and supporting the importance of evaluating lineage-specific chimerism.
In addition to the analysis of outcomes, results from the present data collection strengthen the notion that the disease onset is usually early, with half of the patients presenting within the first month of life. We show that enteropathy, T1D, and eczema are the initial symptoms, especially in neonates, while in the later onset patients, failure to thrive becomes a predominant symptom at presentation. With the increasing number of IPEX patients diagnosed over time, a group of asymptomatic patients or patients initially presenting with only T1D have been identified. With disease progression, multiple other autoimmune symptoms arise with blood, kidney, and liver frequently targeted. Unexpectedly, neurological impairment, respiratory involvement, and cardiac complications have been often observed, though their autoimmune origin rather than toxic or infectious pathogenesis remains unclear and should be investigated in a prospective study. As has been previously suggested,9,32 it is difficult to correlate the site of mutation with disease course or outcome. For example, mutations in the FKH domain, required for FOXP3 nuclear localization and DNA binding, correlated with the earliest onset, while there was no association between type of mutation and outcome. Furthermore, the same genotype can present with variable phenotypes. Indeed, asymptomatic children carrying the same mutation of their affected siblings have been recently published15 and herein reported (siblings carrying the c.1190 G>A mutation). Future prospective studies should be considered to better understand the functional effects of each FOXP3 mutation, at disease onset and during disease course. Similarly, future studies should focus on evaluating the effect of different immunosuppressive regimens on the regulation of FOXP3 expression and its epigenetic modification such as demethylation at the Treg cell–specific demethylated region, which is essential to maintain Treg cells’ identity.33,34
Overall, this retrospective study instructs that rapamycin should be the preferred choice as IS treatment, while HSCT should be considered in patients with low OI score and stable clinical conditions. In conclusion, our comprehensive description of the natural history of IPEX and comparison of the effects of IS and HSCT may assist in determining therapeutic choices for these complex patients.
Supplementary Material
Key messages.
Pretreatment organ impairment score in IPEX best predicts overall survival after HSCT.
HSCT and IS recipients experience similar overall survival, but those receiving HSCT demonstrate higher rates of disease-free survival.
Acknowledgments
This work has been supported by Telethon (Tele10-A4 to R.B.), the Jeffrey Modell Foundation Travel Award to F.B., and a generous gift to the Stanford Center for Genetic Immune Diseases.
E. Gambineri serves as a consultant for Baxalta/Shire. M. Cavazzana receives grant support from the Institut National de la Santé et de la Recherche Médicale/Assistance Publique-Hôpitaux de Paris–European Research Council (ERC) Advanced Grant. W. Qasim receives grant support from the National Institute of Health Research, Cellectis SA, Cellmedica, Bellicum and Autolus; serves as a consultant for Autolus Ltd and Servier; receives royalties from Orchard; owns stock options for Autolus. M. H. Albert serves as a consultant for GlaxoSmithKline; receives grant support from GlaxoSmithKline; receives payment for lectures from MSD and Jazz; holds stock options for Amgen, Bristol-Myers S, and Juno; and receives travel support from Jazz and Medac. F. Haerynck receives grant support from Jeffery Modell Foundation (JMF) Diagnostic and Research Centre. C. Dhooge receives payment for lectures for nurses; travel support for Society for industrial and organizational psychology (SIOP), European Society for Blood and Marrow Transplantation, and European Musculo-Skeletal Oncology Society meetings. R. G. Bredius. E. Haddad serves as a consultant for Leadiant; and receives grant support from CSL Behring. S.-Y. Pai receives grant support from the Boston Children’s Hospital. F. Goldman serves as a consultant for Jazz Pharmaceuticals; receives grant support from the Department of Defense; and payment for lectures from Jazz Pharmaceuticals. M. J. Cowan receives grant support from the National Institutes of Health–National Institute of Allergy and Infectious Diseases and California Institute of Regenerative Medicine; serves on the board for Bluebird Bio, Exogen Bio, and Homology Medicine; holds a patent for lentiviral vector; and has stock options with Homology Medicine and Exogen Bio.
We are grateful to Robertson Parkman, for his thoughtful comments and revisions. We thank Ken Weinberg for critical revision and Rajni Agarwal-Hashmi and Maria Ester Bernardo for helping with transplanted patients’ stratification. We thank Alessandro Ambrosi for his contribution in the initial statistical analysis and the Italian Network for Primary Immunodeficiencies (IPINET) for support. We sincerely thank all the patients and families for their participation and trust.
Abbreviations
- DFS
Disease-free survival
- FKH
Forkhead domain
- GvHD
Graft-versus-host disease
- HSCT
Hematopoietic stem cell transplantation
- IPEX
Immunodysregulation polyendocrinopathy enteropathy x-linked
- IS
Immunosuppression
- OI
Organ involvement
- RIT
Reduced intensity transplant
- T1D
Type 1 diabetes
- Treg
Regulatory T
Footnotes
Disclosure of potential conflict of interest:
The rest of the authors declare that they have no relevant conflicts of interest.
The CrossMark symbol notifies online readers when updates have been made to the article such as errata or minor corrections
References
- 1.Powell BR, Buist NR, Stenzel P. An x-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–7. doi: 10.1016/s0022-3476(82)80573-8. [DOI] [PubMed] [Google Scholar]
- 2.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 3.Wildin RS, Ramsdell F, Peake J, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 4.Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211. doi: 10.3389/fimmu.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bacchetta R, Barzaghi F, Roncarolo Mg. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. 2016 Feb 25; doi: 10.1111/nyas.13011. [E-pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6.Bacchetta R, Passerini L, Gambineri E, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006;116:1713–22. doi: 10.1172/JCI25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinnunen T, Chamberlain N, Morbach H, Morbach H, Choi J, Kim S, Craft J, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood. 2013;121:1595–603. doi: 10.1182/blood-2012-09-457465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Passerini L, Olek S, Di Nunzio S, Barzaghi F, Hambleton S, Abinun M, et al. Forkhead box protein 3 (FOXP3) mutations lead to increased th17 cell numbers and regulatory T-cell instability. J Allergy Clin Immunol. 2011;128:1376–9.e1. doi: 10.1016/j.jaci.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Bin Dhuban K, Piccirillo CA. The immunological and genetic basis of immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome. Curr Opin Allergy Clin Immunol. 2015;15:525–32. doi: 10.1097/ACI.0000000000000214. [DOI] [PubMed] [Google Scholar]
- 10.Sheikine Y, Woda CB, Lee PY, Chatila TA, Keles S, Charbonnier LM, et al. Renal involvement in the immunodysregulation, polyendocrinopathy, enteropathy, x-linked (IPEX) disorder. Pediatr Nephrol. 2015;30:1197–202. doi: 10.1007/s00467-015-3102-x. [DOI] [PubMed] [Google Scholar]
- 11.Zama D, Cocchi I, Masetti R, Specchia F, Alvisi P, Gambineri E, et al. Late-onset of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (IPEX) with intractable diarrhea. Ital J Pediatr. 2014;40:68. doi: 10.1186/s13052-014-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Benedetti F, Insalaco A, Diamanti A, Cortis E, Muratori F, Lamioni A, et al. Mechanistic associations of a mild phenotype of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome. Clin Gastroenterol Hepatol. 2006;4:653–9. doi: 10.1016/j.cgh.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Mazzolari E, Forino C, Fontana M, D’Ippolito C, Lanfranchi A, Gambineri E, et al. A new case of IPEX receiving bone marrow transplantation. Bone Marrow Transplant. 2005;35:1033–4. doi: 10.1038/sj.bmt.1704954. [DOI] [PubMed] [Google Scholar]
- 14.Seidel Mg, Fritsch G, Lion T, Jurgens B, Heitger A, Bacchetta R, et al. Selective engraftment of donor CD4125high FOXP3-positive T cells in IPEX syndrome after nonmyeloablative hematopoietic stem cell transplantation. Blood. 2009;113:5689–91. doi: 10.1182/blood-2009-02-206359. [DOI] [PubMed] [Google Scholar]
- 15.Seidel Mg, Boztug K, Haas Oa. Immune dysregulation syndromes (IPEX, CD27 deficiency, and others): always doomed from the start? J Clin Immunol. 2016;36:6–7. doi: 10.1007/s10875-015-0218-5. [DOI] [PubMed] [Google Scholar]
- 16.Veys P. Reduced intensity transplantation for primary immunodeficiency disorders. Pediatr Rep. 2011;3(suppl 2):E11. doi: 10.4081/pr.2011.s2.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Apperley J, Carreras D, Gluckman E, Masszi T, editors. The EBMT handbook on haematopoietic stem cell transplantation. 6th. Leiden, The Netherlands: EBMT; 2012. [Google Scholar]
- 18.Shin CR, Kim MO, Li D, Bleesing JJ, Harris R, Mehta P, et al. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012;47:1428–35. doi: 10.1038/bmt.2012.31. [DOI] [PubMed] [Google Scholar]
- 19.Bindl L, Torgerson T, Perroni L, Youssef N, Ochs HD, Goulet O, et al. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome) J Pediatr. 2005;147:256–9. doi: 10.1016/j.jpeds.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 20.Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol. 2008;28:581–7. doi: 10.1007/s10875-008-9196-1. [DOI] [PubMed] [Google Scholar]
- 21.Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, et al. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med. 2001;344:1758–62. doi: 10.1056/NEJM200106073442304. [DOI] [PubMed] [Google Scholar]
- 22.Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, x linked (IPEX) syndrome. J Med Genet. 2002;39:537–45. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taddio A, Faleschini E, Valencic E, Granzotto M, Tommasini A, Lepore L, et al. Medium-term survival without haematopoietic stem cell transplantation in a case of IPEX: insights into nutritional and immunosuppressive therapy. Eur J Pediatr. 2007;166:1195–7. doi: 10.1007/s00431-006-0395-6. [DOI] [PubMed] [Google Scholar]
- 24.Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. 2008;122:1105–12.e1. doi: 10.1016/j.jaci.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 25.Kucuk ZY, Bleesing JJ, Marsh R, Zhang K, Davies S, Filipovich AH. A challenging undertaking: stem cell transplantation for immune dysregulation, polyendocrinopathy, enteropathy, x-linked (IPEX) syndrome. J Allergy Clinical Immunol. 2016;137:953–5.e4. doi: 10.1016/j.jaci.2015.09.030. [DOI] [PubMed] [Google Scholar]
- 26.Nademi Z, Slatter M, Gambineri E, Mannurita SC, Barge D, Hodges S, et al. Single centre experience of haematopoietic SCT for patients with immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome. Bone Marrow Transplant. 2014;49:310–2. doi: 10.1038/bmt.2013.181. [DOI] [PubMed] [Google Scholar]
- 27.Lucas Kg, Ungar D, Comito M, Bayerl M, Groh B. Submyeloablative cord blood transplantation corrects clinical defects seen in IPEX syndrome. Bone Marrow Transplant. 2007;39:55–6. doi: 10.1038/sj.bmt.1705542. [DOI] [PubMed] [Google Scholar]
- 28.Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007;109:383–5. doi: 10.1182/blood-2006-05-025072. [DOI] [PubMed] [Google Scholar]
- 29.Burroughs LM, Torgerson TR, Storb R, Carpenter PA, Rawlings DJ, Sanders J, et al. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome. J Allergy Clin Immunol. 2010;126:1000–5. doi: 10.1016/j.jaci.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasow KA, Morales-Tirado VM, Wichlan D, Shurtleff SA, Abraham A, Persons DA, et al. Therapeutic in vivo selection of thymic-derived natural t regulatory cells following non-myeloablative hematopoietic stem cell transplant for IPEX. Clin Immunol. 2011;141:169–76. doi: 10.1016/j.clim.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slatter MA, Boztug H, Potschger U, Sykora KW, Lankester A, Yaniv I, et al. Treosulfan-based conditioning regimens for allogeneic haematopoietic stem cell transplantation in children with non-malignant diseases. Bone Marrow Transplant. 2015;50:1536–41. doi: 10.1038/bmt.2015.171. [DOI] [PubMed] [Google Scholar]
- 32.d’Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo CA. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, x linked (IPEX) syndrome. J Med Genet. 2012;49:291–302. doi: 10.1136/jmedgenet-2012-100759. [DOI] [PubMed] [Google Scholar]
- 33.Barzaghi F, Passerini L, Gambineri E, Ciullini Mannurita S, Cornu T, Kang ES, et al. Demethylation analysis of the FOXP3 locus shows quantitative defects of regulatory T cells in IPEX-like syndrome. J Autoimmun. 2012;38:49–58. doi: 10.1016/j.jaut.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iizuka-Koga M, Nakatsukasa H, Ito M, Akanuma T, Lu Q, Yoshimura A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J Autoimmun. 2017;83:113–21. doi: 10.1016/j.jaut.2017.07.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.