

Summary
Ascertaining the dominant cell type driving an immunological disease is essential to understanding the causal pathology and, therefore, selecting or developing an effective treatment. Classifying immunological diseases in this way has led to successful treatment regimens for many monogenic diseases; however, when the dominant cell type is unclear and there is no obvious causal genetic mutation, then identifying the correct disease classification and appropriate therapy can be challenging. In this review we focus on pulmonary immunological diseases where an innate immune signature has been identified as a predominant aspect of the immunopathology. We describe the molecular pathology of ‘autoinflammatory diseases of the lung’ and propose that small molecule and biological therapies, including recombinant interleukin‐1 receptor antagonist, that target key innate immune pathways, are likely be beneficial in the control of pulmonary and systemic inflammation in these conditions. In addition, the successful use of macrolide antibiotics to treat lung infections in these conditions further confirms that the innate immune system is the key conductor of inflammation in these pulmonary diseases, as there is a strong body of evidence that macrolides are able to modulate the NLRP3 inflammasome and interleukin‐1β and interleukin‐18 secretion, both of which are central players in the innate immune response. Throughout this review we highlight the published evidence of autoinflammatory disease in chronic obstructive pulmonary disease, bronchiectasis, cystic fibrosis and rheumatoid lung disease and suggest that the fundamental pathology of these diseases places them towards the autoinflammatory pole of the immunological disease continuum.
Keywords: autoimmunity, bronchiectasis, COPD, cystic fibrosis, inflammation, panbronchiolitis
Introduction
The identification of an autoinflammatory basis for a significant proportion of human disease has significantly modified the nosology of inflammatory disorders over the past two decades.1, 2 The disease category, autoinflammation, was originally proposed to describe the underlying pathophysiology in a family of monogenic autosomal dominant periodic fever syndromes, but this classification has subsequently been applied to a broader range of disease entities, including polygenic autoinflammatory disease, such as Crohn's disease, as well as specific major histocompatibility complex class 1‐associated conditions, such as ankylosing spondylitis, psoriatic arthropathy and Behçet's disease.3, 4, 5 The term autoinflammation is based on central involvement of innate immune system activation, in association with a paucity of autoantibodies and autoantigen‐specific T and B cells.
Immunological diseases exist on a continuous spectrum, with autoimmune diseases, driven by the adaptive immune system, at one extreme, and autoinflammatory diseases, driven by the innate immune system, at the diametrically opposite end of that spectrum.3 The majority of immunological diseases are located somewhere in the interval between the autoimmune and autoinflammatory ends of the continuum, often with some degree of amalgamation of these two systems driving the underlying pathology.2 Diseases that are defined as wholly autoimmune or autoinflammatory in nature are most often the rare hereditary disorders associated with mutated genes/proteins in the underlying immunological innate or adaptive pathways. In that regard, the hereditary autoinflammatory diseases (HAIDs) constitute a set of conditions at the autoinflammatory end of the spectrum that have arisen due to mutations within genes involved in the innate immune system, and that lead to hyperresponsive or overactive innate immune responses.6 HAIDs usually present with periodic episodes or flares, which are often interleukin‐1 (IL‐1) ‐mediated and are particularly responsive to anakinra, a recombinant IL‐1 receptor antagonist (IL‐1Ra) molecule, or, indeed, to other forms of IL‐1 blockade, such as rilonacept and canakinumab. Rilonacept (IL‐1 Trap) is a decoy receptor for IL‐1, inhibiting both IL‐1α and IL‐1β signalling, whereas canakinumab is a humanized monoclonal antibody selectively binding to IL‐1β. HAIDs can be both monogenic and polygenic (Fig. 1); there is considerable overlap between polygenic autoinflammatory diseases and major histocompatibility complex class 1‐associated diseases.3 The periodic episodes associated with HAIDs involve systemic multi‐organ inflammation, fevers, arthritic joint pains, skin rashes, abdominal pain and pulmonary inflammation. Characteristic flares, associated with HAIDs, are often triggered by exposure to specific environmental conditions or agents; for example, low temperatures may precipitate an attack of familial cold urticaria, which is one of the conditions that falls under the umbrella term, cryopyrin‐associated periodic syndrome.
Figure 1.
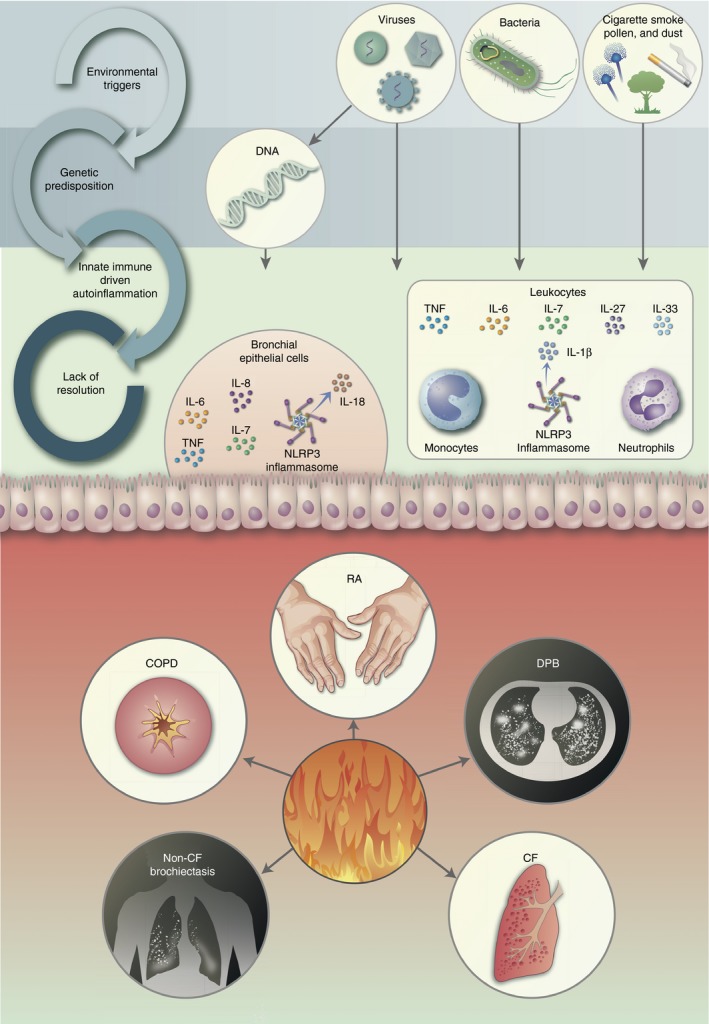
Non‐resolving cycle of autoinflammation in the lung and its related diseases. Chronic inflammation of the lungs results from the accumulation of a number of different factors, ranging from environmental insults (cigarette smoke, viral, bacterial, air pollution etc) to genetic predisposition; these cycles feed into each other resulting in innate immune driven autoinflammation, and ultimately results in chronic inflammation of the lungs, without resolution. A number of innate immune cells, such as neutrophils, monocytes and epithelial cells of the bronchi, play pivotal roles in the excessive response to stimuli and combine to secrete a range of inflammatory cytokines. DPB, diffuse panbronchiolitis; COPD, chronic obstructive pulmonary disorder; CF, cystic fibrosis; RA, rheumatoid arthritis.
Figure 2.

Innate immune frontier. The lung is one of the main organs where the environment comes into direct contact with the innate immune system, termed here as the innate immune frontier. The autoinflammatory lung diseases CF, COPD, panbronchiolitis, bronchiectasis and RA lung disease all have a similar innate immune frontier response, which results in a chronic inflammatory state, with cytokines, such as IL‐18 and TNF, and fever inducing IL‐1β being released in excessive amounts into the lung parenchyma and stroma. Anti‐IL‐1 treatments are effective in reducing this exaggerated response.
This review will propose that many immunological diseases exhibiting pulmonary manifestations are driven by innate immune cells and can be situated towards the autoinflammatory pole of the immunological disease continuum (IDC) (Fig. 1). We will also explore the molecular mechanisms of pulmonary flares in HAIDs and discuss the similarities in autoinflammatory pathology that are shared by many pulmonary immunological diseases at the molecular level.
The pulmonary innate immune system
The pulmonary system is an integral part of the innate immune system, by providing crucial barrier function between the environment and the circulation, and by employing various mechanisms to prevent foreign bodies entering the body.7, 8 The innate immune system's main purpose is to provide the initial outposts, in the form of toll‐like receptor (TLR), expressed on sentinel cells, such as macrophages and dendritic cells, to detect and respond to invading pathogens; this is achieved by the recognition of a broad range of structurally conserved molecules derived from microbes, termed pathogen‐associated molecular patterns. In addition, damage‐associated molecular patterns are endogenous molecules, released by the host's dead or dying cells, which are also recognized by TLRs with triggering of innate immune responses in the lung's microenvironment.8
The innate immune system of the lung is diverse in nature and includes itinerant leucocytes such as monocytes, neutrophils and macrophages, as well as structural cells, such as epithelial cells and fibroblasts. Dendritic cells and mast cells are of haematopoietic origin, but may be found in the lung and combine to orchestrate immune responses in that organ. A wide variety of microbiocidal soluble factors are secreted by cells of the innate immune system to counter invading pathogens. However, the pulmonary innate immune system does not just rely on myeloid and haematopoietic immune cells for defence, as pulmonary epithelial cells are also a vital cell type in detection and prevention of spread by invasive foreign pathogens.8 These epithelial cells are often targeted by both bacteria and viruses, which conspire to evade the immune system; however, the pulmonary epithelium is able to orchestrate the degree and magnitude of the inflammatory response as they express high levels of TLRs capable of detecting a broad range of pathogen‐associated molecular patterns. Epithelial cells also undergo shedding,9, 10, 11 a process whereby they can mediate cell death and are subsequently replaced by a new layer of epithelial cells. This process reduces the spread of foreign organisms throughout the epithelial layers and, in the process, exposes intracellular pathogens to specialized phagocytic cells, such as macrophages and dendritic cells.12
The initiation of an innate immune response is mediated by a key set of cytokines. The IL‐1 cytokine family is primarily comprised of innate pro‐inflammatory cytokines and chemokines plus their antagonists and receptors. The most comprehensively studied IL‐1 cytokines are IL‐1β and IL‐18, both of which are inflammasome mediated.13 The inflammasomes are key intracellular innate immune macromolecular protein complexes that require two signals to become primed and activated. Once activated, the inflammasomes bring inactive pro‐caspase‐1 and inactive zymogens, pro‐IL‐1β and pro‐IL‐18, into close proximity. The pro‐caspases then self‐cleave in an autocatalytic reaction that culminates in the cleavage and activation of pro‐IL‐1β and pro‐IL‐18 into their active forms. The IL‐1β and IL‐18 serve different pro‐inflammatory purposes in fine tuning the innate immune response.14 Haematopoietic innate immune cells predominantly secrete IL‐1β, a highly biologically active pro‐inflammatory cytokine that provokes a systemic inflammatory state by inducing fever and activation, with subsequent recruitment of other immune cells. Epithelial cells are reported to preferentially secrete IL‐18 over IL‐1β (Fig. 1). IL‐18 is responsible for recruiting neutrophils to sites of inflammation as well as differentiating T cells towards T helper type 17 and T helper type 1 phenotypes, and activating natural killer cells.15
The air we breathe contains damaging foreign bodies in abundance, all capable of initiating an innate immune response. The successful resolution of such pro‐inflammatory responses is equally important to their initiation. Uncontrollable or excessive pulmonary inflammation is highly damaging, and conditions such as sepsis or chronic obstructive pulmonary disease (COPD) may arise from local inflammation that is not efficiently resolved (Fig. 1). The inherent capacity of a host to initiate and resolve lung inflammation has further implications than merely containing specific lung conditions. Many chronic infections, in addition to cancers,16 heart disease17 and immunological diseases, are thought to begin in the lung, either due to inadequate control of local infection or potent carcinogens, resulting in DNA mutation or the development of autoantigens capable of breaking tolerance. As the lung is on the frontline in protecting against environmental injury, innate immune responses, including those of epithelial origin, are of particular importance in this regard;7 if resolution or activation of these pivotal responses goes awry, then immunological disease may ensue. This review will explore the molecular mechanisms involved in chronic innate immune‐mediated inflammation in the lung and will also examine the particular aspects of such autoinflammation that enable immunological disease progression.
Autoinflammatory diseases
Respiratory manifestations may occur in many cases of autoinflammatory disease (Fig. 1). This is in part due to the systemic nature of autoinflammation.18 However, the innate immune response within HAIDs is one that is primed and hyperresponsive and when innate immune cells come into contact with antigens entering the lung, the response will often turn out to be inappropriate and prolonged. Recurrent and severe respiratory infections often coincide with the periodic flares associated with autoinflammatory disease.19 Recurrent respiratory tract infections, often pneumonia, as well as restrictive lung disease and interstitial fibrosis occur in spondyloenchondrodysplasia with immune dysregulation,20 stimulator of interferon genes‐associated vasculopathy with onset in infancy,21 and acute febrile neutrophilic dermatosis (Sweet syndrome).
Other autoinflammatory diseases may also develop acute respiratory distress syndrome, a condition in which high levels of autoinflammation increase the alveolocapillary space, thereby impairing oxygen gas exchange with consequent reduced blood oxygenation. Acute respiratory distress syndrome has been observed in adult‐onset Still's disease,22, 23 familial haemophagocytic lymphohistiocytosis,24 and NLRC4‐related macrophage activation‐like syndrome,25, 26, 27, 28 resulting in pulmonary fibrosis. Autoinflammation and PLCG2‐associated antibody deficiency and immune dysregulation has been described as manifesting with respiratory bronchiolitis and recurrent sinopulmonary infections, driven by innate immune cellular infiltrations (neutrophils, eosinophils, histiocytes and lymphocytes (Fig. 1),29 with reduced IgA and IgM levels and memory B‐cells.29
The fact that autoinflammatory diseases present with pulmonary inflammation confirms that the lung is a site that is vulnerable to chronic, unresolved innate immune‐mediated damage (Fig. 1). Therefore, immunological diseases, where the lung is the primary site of chronic inflammation, could be expected to develop a predominantly innate immune signature.30 Where pulmonary innate immune cells respond inappropriately, excessively and without proper resolution, this can be thought of as autoinflammatory disease of the lung. As described above, autoinflammatory diseases, although self‐perpetuating, require a specific trigger(s) to develop into a characteristic systemic flare; indeed, this is also the case for many pulmonary immunological diseases.
Macrolides
Rapamycin is a macrolide antibiotic with potent immunosuppressor activity. The drug is widely used in vitro as an inhibitor of NLRP3 inflammasome. The introduction of other macrolides including erythromycin, clarithromycin and azithromycin appears to have similar anti‐inflammatory and immunomodulatory properties.31, 32They reduce IL‐1β and IL‐6 responses to challenge with lipopolysaccharide and decrease bacterial burden, lung inflammation and, in the mouse model, enhance bacterial clearance of Burkholderia cepacia complex through induction of autophagy.31, 32, 33, 34 The macrolide azithromycin has also been shown to have anti‐inflammatory effects in bronchiectasis, as it enhances the clearance of apoptotic cells, such as neutrophils, by improved macrophage phagocytic function.35, 36 Non‐antibiotic macrolide derivatives have also been shown to inhibit lipopolysaccharide‐induced mucus production, neutrophil infiltration and the production of inflammatory cytokines and suppression of IL‐1β induced nuclear factor‐κB activation in airway epithelial cells.37
Low‐dose macrolide therapy can reduce pulmonary exacerbation rates in patients suffering from various lung diseases including cystic fibrosis (CF), non‐CF bronchiectasis, COPD, asthma, bronchiolitis obliterans syndrome, diffuse panbronchiolitis and chronic rhinosinusitis.38, 39, 40, 41, 42 This class of drug has both anti‐inflammatory and immunomodulatory effects, which are independent of antimicrobial activity. Low doses of macrolide inhibit the innate immune response, as well as altering the lung microbiota and bacterial quorum sensing.43, 44 Individual response to low‐dose macrolide therapy in conditions such as COPD and bronchiectasis is variable and may reflect differences in aetiology as well as the balance between infection and autoinflammation.
Diffuse panbronchiolitis
Several prospective clinical trials of macrolides in CF have shown variable improvements in lung function, weight, quality of life and a reduction in pulmonary exacerbations.45 These studies were prompted by the successful use of erythromycin in diffuse panbronchiolitis, a disease of chronic airway inflammation and sinobronchial infection.37, 46 The aetiology of diffuse panbronchiolitis remains unclear although there are both environmental and genetic predisposing factors, with most cases occurring in East Asia.37, 47, 48, 49, 50, 51 This condition shares some features with CF and, if left untreated, may result in disease progression, bronchiectasis and end‐stage lung disease. Like CF, diffuse panbronchiolitis is associated with endobronchial neutrophilic inflammation, with elevation of IL‐1β and IL‐8 levels in the lungs.52, 53, 54 In the respiratory bronchioles, lymphocytic inflammation appears to predominate, with peribronchial infiltration by lymphocytes, plasma cells and histiocytes.55 Inhibition of this inflammatory response by low‐dose macrolides supports a significant autoinflammatory component to the disease.
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease is a leading cause of death worldwide and the condition is characterized by chronic bronchitis, airway obstruction and emphysema. Smoking is the primary cause of COPD and triggers an autoinflammatory response through induction of reactive oxygen species (ROS), acquired dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, impaired autophagy, endoplasmic reticulum stress and activation of the unfolded protein response.56, 57, 58 This inflammatory process is unable to resolve effectively and is ultimately highly destructive, manifesting in chronic bronchitis and emphysema.59, 60, 61 There is evidence for an autoinflammatory signature in individuals with COPD, suggesting that there may be a genetic predisposition beyond α‐1‐antitrypsin deficiency, which, when combined with chronic exposure to external stimuli, may progress to COPD.62 The various clinical subtypes of COPD may reflect variation in the balance between inflammation and infection, as well as disease aetiology.
The external stimuli that trigger COPD are numerous and comprehensive, and embody both infectious organisms and noxious chemicals, such as those found in cigarette smoke (Fig. 1). This feature of COPD's pathogenesis is shared with many HAIDs. In addition to similarities in disease onset, the type of inflammation in COPD also bears many parallels with HAIDs. A strong IL‐1β, IL‐6, IL‐18, IL‐27 and IL‐33 signature has been found in the lungs of individuals with COPD, as well as macrophage and neutrophilic infiltrations.63, 64, 65 A recent study by Faner et al.,64 described how the NLRP3 inflammasome is primed in COPD patients and that during exacerbations the pre‐primed NLRP3 inflammasome releases an excess of IL‐1β family cytokines into the surrounding tissues. Further evidence for IL‐1β‐driven inflammation in COPD has been demonstrated in a COPD mouse model, whereby tobacco smoke inhalation over 10 months was used to induce COPD in NLRP3 −/− and wild‐type mice.66 NLRP3 −/− mice developed no pulmonary lung damage or IL‐1β secretion related to the smoke inhalation. In addition, the levels of innate immune cellular infiltration were significantly higher in COPD mice compared with NLRP3 −/− mice. This suggests that NLRP3‐mediated IL‐1β drives COPD pathology and that chronic NLRP3 priming prevents resolution of COPD lung inflammation. Another study found no significant increase in NLRP3 or IL‐1β cytokines in lung samples from COPD patients; however, the IL‐7 level was elevated in COPD.65 In the lung, IL‐7 is secreted by epithelial cells, which then drives monocytic and T‐cell‐mediated inflammation. This suggests that IL‐7 may be recruiting inflammatory cells into the COPD lung and thereby supporting and prolonging the inflammation. Trials of low‐dose macrolides have demonstrated clinical benefit with a reduction in pulmonary exacerbations, increased time to next exacerbation and improved quality of life. Patients who are not actively smoking appear to gain greater benefit, possibly reflecting persistence of autoinflammation in the absence of a primary trigger.67 , 68 These data support the notion that COPD is towards the autoinflammatory end of the IDC and that established COPD has an intrinsic priming of the innate IL‐1β cytokine pathway. A positive response to low‐dose macrolide therapy may reflect a subgroup of individuals where autoinflammation is driving disease progression. Despite evidence for overexpression of NLRP3 in the lung of stable COPD patients, treatment with anti‐IL‐1β, anti‐IL‐1R1 and anti‐IL‐18 monoclonal antibodies have not proved beneficial.
Non‐cystic fibrosis bronchiectasis
Bronchiectasis is a complex heterogeneous group of disorders with different underlying aetiologies, presenting with varied prevalence across geography and ethnicity, indicating both environmental and genetic links to disease susceptibility.69 The term bronchiectasis refers to the permanent dilatation of the airways due to airway injury and remodelling, as a consequence of infection, inflammation and autoimmune disease.69, 70 Chronic inflammation remains a key component of bronchiectasis with autoinflammation often driving disease progression even in the absence of active infection. Inflammation occurs in the bronchial wall, mainly of the smaller airways, with predominantly macrophages and lymphocytes (mainly T cells) migrating into the cell wall,71, 72, 73 with the neutrophils being the most prominent cell type occupying the bronchial lumen.71, 73 Once neutrophils have migrated to sites of infection in the lungs, they move along a chemoattractant gradient (e.g. IL‐8, leukotriene B4, tumour necrosis factor and IL‐1β) and switch to their antimicrobial function.74 By contrast, elevation in IL‐13 reflects a more eosinophilic phenotype and an exaggerated IL‐17 response occurs in primary immunodeficiency.75, 76, 77 The increased number of apoptotic neutrophils in the airways indicates the failure of phagocytic cells such as macrophages to clear the apoptotic cells, leading to increased inflammation and airway damage, through the uncontrolled release of the neutrophils’ granular contents.78, 79
Bronchial epithelial cells excessively secrete pro‐inflammatory cytokines and express adhesion molecules such as intercellular adhesion molecule 1 when stimulated with a bacterial trigger.80, 81, 82 This results in the recruitment of neutrophils to the site of infection, exacerbating inflammation.
Neutrophilic airway inflammation can persist in the absence of infection, and the vicious circle of host‐mediated autoinflammation can be further exacerbated by the presence of chronic bronchial sepsis.83
Cystic fibrosis
Cystic fibrosis is one of the most common life threatening genetically inherited conditions affecting Caucasians. The disease is caused by an absence or defect in the CFTR protein, which is expressed throughout the body. In the lung defective CFTR function results in abnormal ion transport, dehydrated airway surface liquid and abnormal mucociliary clearance. These changes lead to recurrent infections, hypoxia, anaerobic biofilm formation, innate immune cell infiltration, excessive inflammation and bronchiectasis. Epithelial cells do not exclusively express the CFTR, with strong evidence that fibroblasts, lymphomas, leukaemia cells, lymphocytes, neutrophils, monocytes and alveolar macrophages also express the CFTR protein. Reduced CFTR expression and Cl− flux have also been shown in CF monocytes. Therefore, with both epithelial cells and innate immune cells being affected by the CFTR mutation and with a clinical presentation of disproportionate pulmonary inflammation, CF is firmly located towards the autoinflammatory end of the IDC.
Evidence for autoinflammatory disease in the CF lung is supported by a recent study showing that IL‐1β secretion and NLRP3 inflammasome activation are exaggerated in Pseudomonas aeruginosa infection in murine CF.84 Data suggesting that NLRP3 inflammasome‐dependent secretion of IL‐1β is elevated in cftr −/− mice also indicate that elevated IL‐1β secretion in CF is intensified by insufficient NLRC4‐mediated IL‐1Ra production. This study proposes that further genetic deficiency within NLRC4 or IL1RN (IL‐1Ra gene) would exacerbate and predispose to severe autoinflammatory lung disease in CF. The highlights of this elegant study include reduced bacterial colonization of cftr −/− mice after anakinra treatment, which was corroborated by reduced inflammation in both cftr −/− mice and human epithelial cells treated with anakinra. The authors advocate the use of anakinra therapy in CF, as their data show an anakinra‐dependent reduction in NLRP3 inflammasome activation, by not merely assuaging IL‐1β production but also by inducing autophagy and so NLRP3‐inflammasome degradation.
In addition to an excessive response to bacterial infection, the intrinsic defect in CF predisposes innate immune cells towards a pro‐inflammatory phenotype, with a decrease in alternatively activated, anti‐inflammatory M2 phenotype macrophages.85 This is a hallmark of autoinflammatory diseases such as macrophage activation‐like syndrome25, 26, 28 and deficiency of adenosine deaminase 2, an inherited cause of vasculitis.86, 87
Data suggesting that mitochondrial calcium (Ca2+) and the mitochondrial Ca2+ uniporter have a role in supporting NLRP3 inflammasome signalling in CF support the idea that inflammation in CF is autoinflammation‐based.88 Dysfunctional or mutated CFTR perturbs intracellular Ca2+ signalling, in combination with P. aeruginosa infection, and decreases mitochondrial membrane potential, increases mitochondrial fragmentation and induces mitochondrial ROS (mROS) production. This study clearly outlines the role exerted by P. aeruginosa infection, and specifically flagellin/TLR5/Myd88 signalling, on the integrity and function of the mitochondria and how this mitochondrial damage induces exaggerated inflammatory responses in CF. The authors focused on NLRP3‐mediated inflammation, as CF lung disease is often characterized by IL‐1β accumulation. By using sophisticated silencing experiments, they described mitochondrial perturbation as being upstream of NLRP3 inflammasome activation and that P. aeruginosa flagellin amplified this activation via a Ca2+‐dependent mechanism. The mitochondrial dysfunction caused by loss of CFTR function is driven by P. aeruginosa infection and associated NLRP3 activation, leading to susceptibility to the pathogen. The mitochondrial dysfunction was dependent on mitochondrial calcium uniporter (MCU) expression as the channel facilitated the influx of calcium into the mitochondria. The fact that mitochondrial dysfunction and mROS production activated NLRP3 supplements the evidence for an autoinflammatory disease process in the CF lung.
A key aspect of immunological diseases is that the underlying inflammation is sterile, despite infection being one of the triggers. The above studies both used infection models to elucidate the extent to which CF is an IL‐1‐mediated disease. However, the fact that there is an IL‐1 signature does not automatically assign CF to a place among the autoinflammatory diseases. A feature of CF is periodic pulmonary infection and, therefore, it is important to establish whether the intrinsic CFTR defect is the root cause of the inflammation, rather than the recurrent infections. Animal model studies have demonstrated that CF does produce sterile inflammation in the lung when animals are housed in germ‐free environments. These animals develop lung and gut inflammation despite the absence of microorganisms. This is due to the fact that colonization of the lung and gut after birth leads to life‐long periodic lung infections, so providing a constant trigger for CFTRmut‐dependent autoinflammation.
There is also strong evidence for the presence of elevated oxidative stress in CF.89, 90, 91 Oxidative stress with associated ROS production is a known activator of innate immune signalling and aberrant NLRP3‐inflammasome activation.
Due to the nature of the disease, misfolded CFTR protein is often present in many genetic classes of CF. Misfolded proteins drive many autoinflammatory diseases, such as tumour necrosis factor‐receptor‐associated periodic fever syndrome (TRAPS) and familial Mediterranean fever and generate intrinsic endoplasmic reticulum stress that serves to prime and initiate pro‐inflammatory signalling pathways via the unfolded protein response. X‐box binding protein 1 (XBP1), a transcription factor spliced and activated by Inositol‐requiring enzyme 1 alpha (IRE1α), is a major arm of the unfolded protein response, and induces pro‐inflammatory cytokine signalling; CF and TRAPS have this pathophysiology in common. To summarize, CF fits the characteristics of an HAID, due to the combined effects of several aberrant molecular pathways being adversely affected by loss of CFTR function and the presence of defective protein integrity, which results in the autoinflammatory phenotype of CF.
Rheumatoid arthritis lung disease
Rheumatoid arthritis (RA) is a systemic inflammatory disorder, affecting about 0·7–1·0% of adults of Western European ancestry, which is characterized by synovial inflammation and swelling that may ultimately lead to erosive destructive changes in cartilage and bone.92 The disease is usually associated with the presence of autoantibodies, including rheumatoid factor and antibodies to citrullinated protein antigens, in over 60% of patients.93 Patients with RA frequently have extra‐articular manifestations, including vasculitis, inflammatory eye disease and lung disease. There is considerable debate about when and where the inflammation begins in RA; in this regard, a number of initiating sites of inflammation have been proposed for the immune‐mediated injury in RA.94 These include oral bacteria95 as well as gut microbiota,96 and a number of studies have suggested that the systemic inflammation originates within the lungs, with cigarette smoking being a potent inducer of RA,97, 98 for poorly understood mechanisms.
Although RA is often considered as an autoimmune disease, the pathophysiology has many innate immune signatures and it may be better placed along the IDC, rather than at the autoimmune end of the spectrum. The recent definition of immunologically defined disease subsets of RA, using immunohistochemistry and gene expression data, from both synovial tissues99, 100 and blood (TACERA – Towards A Cure for Early Rheumatoid Arthritis), has led to an improved understanding of the complex pathobiology of RA. Different synovial phenotypes in RA have been correlated with response to biological therapeutics, with the myeloid (innate immune‐mediated) and lymphoid (predominantly adaptive immune‐mediated) phenotypes being associated with differential clinical responses; the myeloid subtype responds primarily to anti‐tumour necrosis factor, and the adaptive subtype responds better to anti‐IL6R therapy, especially in later RA.99 These studies have created a paradigm shift in the classification of RA, with the autoimmune subtype being associated with the presence of autoantibodies and the myeloid subtype more likely to be driven by innate immune mechanisms.
A wide range of lung diseases may be associated with RA, including bronchiectasis, pulmonary parenchymal disease (interstitial lung disease) (Fig. 1), bronchiectasis, bronchiolitis (predominantly obliterative in nature), and inflammation of the pleura (pleural thickening and effusions), and diseases of the pulmonary vasculature (vasculitis and pulmonary hypertension). These changes may reflect increased susceptibility to infection (often related to medications), chronic immune activation, or toxicity from disease‐modifying or biological therapies.
A recent study by Lasithiotaki et al. investigated the role of the NLRP3 inflammasome in rheumatoid lung disease, both idiopathic pulmonary fibrosis (IPF) and RA–usual interstitial pneumonia (RA‐UIP) by using in vitro stimulation studies of patient bronchoalveolar lavage fluid (BALF) samples.101 There were distinct NLRP3 inflammasome activation profiles between patients with IPF and RA‐UIP. Both IL‐1β and IL‐18 levels were elevated in RA‐UIP BALF, and also in BALF macrophages before and after stimulation in RA‐UIP, suggesting pre‐existing NLRP3 inflammasome activation in these patients. These observations were further supported by the elevated IL‐18 levels, in particular, being decreased by caspase‐1 inhibition in RA‐UIP but not in IPF, as caspase‐1 maturation and release is mediated by NLRP3 inflammasome activation.102 Therefore, this study provides evidence for RA‐UIP being essentially an innate immune‐driven autoinflammatory condition, despite the authors’ conclusion that the disease is autoimmune. Furthermore, there was failure of NLRP3 inflammasome activation in alveolar macrophages in BALF samples from the patients with IPF, and it was proposed that this impaired activation may be a key mechanism in generating the autoinflammatory fibrotic phenotype in IPF.
There is an emerging body of work regarding the role of the microbiome in lung disease and the potential for gut or oral microbiota to contribute to the pathogenesis of these conditions and also, indeed, to beneficially modulate the innate immune response.103 Furthermore, there is evidence for neural regulation of innate immunity, as a coordinated host response to pathogens.104 However, these studies are still in their infancy and the tools to decipher the complexity of the relationship between the microbiome, the central nervous system and immune regulation of disease are not yet available. We can expect these to be areas of intensive research effort in the near future.
Conclusions
Autoinflammation is an emerging component of a growing number of diseases and understanding their immunopathogenesis is an important endeavour that will lead to greater personalization of therapies, with targeting of pathways and cytokines that are relevant to disease pathogenesis. This process involves reassessing already‐characterized diseases to elucidate disease subsets that may be identifiable after more detailed probing of the immune signatures involved. Many lung diseases have not yet been recognized as part of the immunological disease spectrum, and characterizing them in this way may reveal new pathogenic pathways that are targetable by existing small molecules and/or biologicals.
The pathologies of many chronic respiratory diseases involve a combination of genetic and environmental factors that, working in concert, prime and activate innate immune‐associated inflammation. This scenario is typical of autoinflammatory diseases, particularly the HAIDs. This review has highlighted aspects of COPD, bronchiectasis, CF and RA lung disease that are quintessentially autoinflammatory in nature and has proposed that the core pathologies of these diseases place them towards the autoinflammatory pole of the IDC.
Disclosures
The authors declare no conflict of interest.
Acknowledgements
The authors would like to thank Dr Chi Wong, Dr Heledd Jarosz‐Griffiths and Samuel Lara Reyna for critical reading of the manuscript. The authors are supported by a grant (SRC009) from the Cystic Fibrosis Trust.
References
- 1. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M et al Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–44. [DOI] [PubMed] [Google Scholar]
- 2. Wekell P, Berg S, Karlsson A, Fasth A. Toward an inclusive, congruent, and precise definition of autoinflammatory diseases. Front Immunol 2017; 8:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med 2006; 3:e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kastner DL, Aksentijevich I, Goldbach‐Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell 2010; 140:784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stoffels M, Kastner DL. Old dogs, new tricks: monogenic autoinflammatory disease unleashed. Annu Rev Genomics Hum Genet 2016; 17:245–72. [DOI] [PubMed] [Google Scholar]
- 6. Gabay C, Lamacchia C, Palmer G. IL‐1 pathways in inflammation and human diseases. Nat Rev Rheumatol 2010; 6:232–41. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki T, Chow CW, Downey GP. Role of innate immune cells and their products in lung immunopathology. Int J Biochem Cell Biol 2008; 40:1348–61. [DOI] [PubMed] [Google Scholar]
- 8. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol 2015; 16:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hato T, El‐Achkar TM, Dagher PC. Sisters in arms: myeloid and tubular epithelial cells shape renal innate immunity. Am J Physiol Renal Physiol 2013; 304:F1243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weitnauer M, Mijosek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol 2016; 9:287–98. [DOI] [PubMed] [Google Scholar]
- 11. Puchelle E, Zahm JM, Tournier JM, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3:726–33. [DOI] [PubMed] [Google Scholar]
- 12. Liesman RM, Buchholz UJ, Luongo CL, Yang L, Proia AD, DeVincenzo JP et al RSV‐encoded NS2 promotes epithelial cell shedding and distal airway obstruction. J Clin Invest 2014; 124:2219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)‐1 cytokine family – balance between agonists and antagonists in inflammatory diseases. Cytokine 2015; 76:25–37. [DOI] [PubMed] [Google Scholar]
- 14. Martinon F, Gaide O, Petrilli V, Mayor A, Tschopp J. NALP inflammasomes: a central role in innate immunity. Semin Immunopathol 2007; 29:213–29. [DOI] [PubMed] [Google Scholar]
- 15. Dinarello CA. IL‐18: a TH1‐inducing, proinflammatory cytokine and new member of the IL‐1 family. J Allergy Clin Immunol 1999; 103:11–24. [DOI] [PubMed] [Google Scholar]
- 16. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. Effect of interleukin‐1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double‐blind, placebo‐controlled trial. Lancet 2017; 390:1833–42. [DOI] [PubMed] [Google Scholar]
- 17. Harrington RA. Targeting inflammation in coronary artery disease. N Engl J Med 2017; 377:1197–8. [DOI] [PubMed] [Google Scholar]
- 18. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH et al Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med 2008; 14:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tarantino G, Esposito S, Andreozzi L, Bracci B, D'Errico F, Rigante D. Lung involvement in children with hereditary autoinflammatory disorders. Int J Mol Sci 2016; 17:2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader‐Meunier B et al Tartrate resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet 2011; 43:127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Canna SW, Goldbach‐Mansky R. New monogenic autoinflammatory diseases – a clinical overview. Semin Immunopathol 2015; 37:387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dua AB, Manadan AM, Case JP. Adult onset Still's disease presenting with acute respiratory distress syndrome: case report and review of the literature. Open Rheumatol J 2013; 7:125–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suleiman M, Wolfovitz E, Boulman N, Levy Y. Adult onset Still's disease as a cause of ARDS and acute respiratory failure. Scand J Rheumatol 2009; 31:181–3. [PubMed] [Google Scholar]
- 24. Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol 2011; 163:271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manappallil RG. A case of macrophage activation syndrome with acute respiratory distress syndrome. J Clin Diagn Res 2016; 10:OD11–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fischbach KE, Coe B, Radwan M, Rodriguez J, Carter J, Rumbak MJ. Macrophage activation syndrome in a patient with adult Still's disease following rituximab. IU Director 2013; 4:248–51. [Google Scholar]
- 27. Kovach MA, Stringer KA, Bunting R, Wu X, San Mateo L, Newstead MW et al Microarray analysis identifies IL‐1 receptor type 2 as a novel candidate biomarker in patients with acute respiratory distress syndrome. Respir Res 2015; 16:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loh NK, Lucas M, Fernandez S, Prentice D. Successful treatment of macrophage activation syndrome complicating adult Still disease with anakinra. Intern Med J 2012; 42:1358–62. [DOI] [PubMed] [Google Scholar]
- 29. Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A et al A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet 2012; 91:713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peckham D, Scambler T, Savic S, McDermott MF. The burgeoning field of innate immune‐mediated disease and autoinflammation. J Pathol 2017; 241:123–39. [DOI] [PubMed] [Google Scholar]
- 31. Gualdoni GA, Lingscheid T, Schmetterer KG, Hennig A, Steinberger P, Zlabinger GJ. Azithromycin inhibits IL‐1 secretion and non‐canonical inflammasome activation. Sci Rep 2015; 5:12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lendermon EA, Coon TA, Bednash JS, Weathington NM, McDyer JF, Mallampalli RK. Azithromycin decreases NALP3 mRNA stability in monocytes to limit inflammasome‐dependent inflammation. Respir Res 2017; 18:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH et al Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011; 7:1359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Balloy V, Deveaux A, Lebeaux D, Tabary O, le Rouzic P, Ghigo JM et al Azithromycin analogue CSY0073 attenuates lung inflammation induced by LPS challenge. Br J Pharmacol 2014; 171:1783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hodge S, Hodge G, Jersmann H, Matthews G, Ahern J, Holmes M et al Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 178:139–48. [DOI] [PubMed] [Google Scholar]
- 36. Wong C, Jayaram L, Karalus N, Eaton T, Tong C, Hockey H et al Azithromycin for prevention of exacerbations in non‐cystic fibrosis bronchiectasis (EMBRACE): a randomised, double‐blind, placebo‐controlled trial. Lancet 2012; 380:660–7. [DOI] [PubMed] [Google Scholar]
- 37. Shimizu T, Suzaki H. Past, present and future of macrolide therapy for chronic rhinosinusitis in Japan. Auris Nasus Larynx 2016; 43:131–6. [DOI] [PubMed] [Google Scholar]
- 38. Zhuo GY, He Q, Xiang‐Lian L, Ya‐Nan Y, Si‐Te F. Prolonged treatment with macrolides in adult patients with non‐cystic fibrosis bronchiectasis: meta‐analysis of randomized controlled trials. Pulm Pharmacol Ther 2014; 29:80–8. [DOI] [PubMed] [Google Scholar]
- 39. Wu Q, Shen W, Cheng H, Zhou X. Long‐term macrolides for non‐cystic fibrosis bronchiectasis: a systematic review and meta‐analysis. Respirology 2014; 19:321–9. [DOI] [PubMed] [Google Scholar]
- 40. Tong X, Guo T, Liu S, Peng S, Yan Z, Yang X et al Macrolide antibiotics for treatment of asthma in adults: a meta‐analysis of 18 randomized controlled clinical studies. Pulm Pharmacol Ther 2015; 31:99–108. [DOI] [PubMed] [Google Scholar]
- 41. Saiman L, Marshall BC, Mayer‐Hamblett N, Burns JL, Quittner AL, Cibene DA et al Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2003; 290:1749–56. [DOI] [PubMed] [Google Scholar]
- 42. Carr RR, Nahata MC. Azithromycin for improving pulmonary function in cystic fibrosis. Ann Pharmacother 2004; 38:1520–4. [DOI] [PubMed] [Google Scholar]
- 43. Burr LD, Rogers GB, Chen AC, Hamilton BR, Pool GF, Taylor SL et al Macrolide treatment inhibits Pseudomonas aeruginosa quorum sensing in non‐cystic fibrosis bronchiectasis. An analysis from the bronchiectasis and low‐dose Erythromycin Study Trial. Ann Am Thorac Soc 2016; 13:1697–703. [DOI] [PubMed] [Google Scholar]
- 44. Essilfie AT, Horvat JC, Kim RY, Mayall JR, Pinkerton JW, Beckett EL et al Macrolide therapy suppresses key features of experimental steroid‐sensitive and steroid‐insensitive asthma. Thorax 2015; 70:458–67. [DOI] [PubMed] [Google Scholar]
- 45. Southern KW, Barker PM. Azithromycin for cystic fibrosis. Eur Respir J 2004; 24:834–8. [DOI] [PubMed] [Google Scholar]
- 46. Hoiby N. Diffuse panbronchiolitis and cystic fibrosis: east meets west. Thorax 1994; 49:531–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin X. Macrolides for diffuse panbronchiolitis. Cochrane Database Syst Rev 2015; 25:CD007716. [DOI] [PubMed] [Google Scholar]
- 48. Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H, Zhou Y, Fan F, Zhang Y, Li X, Yu H et al Effect of azithromycin on patients with diffuse panbronchiolitis: retrospective study of 51 cases. Intern Med 2011; 50:1663–9. [DOI] [PubMed] [Google Scholar]
- 50. Kudoh S. Applying lessons learned in the treatment of diffuse panbronchiolitis to other chronic inflammatory diseases. Am J Med 2004; 117(Suppl. 9A):12s–9s. [DOI] [PubMed] [Google Scholar]
- 51. Keicho N, Hijikata M. Genetic predisposition to diffuse panbronchiolitis. Respirology 2011; 16:581–8. [DOI] [PubMed] [Google Scholar]
- 52. Kadota J, Matsubara Y, Ishimatsu Y, Ashida M, Abe K, Shirai R et al Significance of IL‐1β and IL‐1 receptor antagonist (IL‐1Ra) in bronchoalveolar lavage fluid (BALF) in patients with diffuse panbronchiolitis (DPB). Clin Exp Immunol 1996; 103:461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sakito O, Kadota J, Kohno S, Abe K, Shirai R, Hara K. Interleukin 1β, tumor necrosis factor α, and interleukin 8 in bronchoalveolar lavage fluid of patients with diffuse panbronchiolitis: a potential mechanism of macrolide therapy. Respiration 1996; 63:42–8. [DOI] [PubMed] [Google Scholar]
- 54. Hiratsuka T, Mukae H, Iiboshi H, Ashitani J, Nabeshima K, Minematsu T et al Increased concentrations of human β‐defensins in plasma and bronchoalveolar lavage fluid of patients with diffuse panbronchiolitis. Thorax 2003; 58:425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matsuura H, Yoshida Y, Yamaji Y. Diffuse panbronchiolitis. QJM 2017; 110:253. [DOI] [PubMed] [Google Scholar]
- 56. Vij N, Chandramani P, Westphal CV, Hole R, Bodas M. Cigarette smoke induced autophagy‐impairment accelerates lung aging, COPD‐emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol 2016; 12:e0182420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao H. Measuring the impact of cigarette smoke on the UPR. Methods Enzymol 2011; 489:147–64. [DOI] [PubMed] [Google Scholar]
- 58. Tagawa Y, Hiramatsu N, Kasai A, Hayakawa K, Okamura M, Yao J et al Induction of apoptosis by cigarette smoke via ROS‐dependent endoplasmic reticulum stress and CCAAT/enhancer‐binding protein‐homologous protein (CHOP). Free Radic Biol Med 2008; 45:50–9. [DOI] [PubMed] [Google Scholar]
- 59. Duijts L, Reiss IK, Brusselle G, de Jongste JC. Early origins of chronic obstructive lung diseases across the life course. Eur J Epidemiol 2014; 29:871–85. [DOI] [PubMed] [Google Scholar]
- 60. Narang I, Bush A. Early origins of chronic obstructive pulmonary disease. Semin Fetal Neonatal Med 2012; 17:112–8. [DOI] [PubMed] [Google Scholar]
- 61. Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet 2015; 385:899–909. [DOI] [PubMed] [Google Scholar]
- 62. Molfino NA, Coyle AJ. Gene–environment interactions in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2008; 3:491–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N et al Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin‐33‐dependent response to infection. Immunity 2015; 42:566–79. [DOI] [PubMed] [Google Scholar]
- 64. Faner R, Sobradillo P, Noguera A, Gomez C, Cruz T, López‐Giraldo A et al The inflammasome pathway in stable COPD and acute exacerbations. ERJ Open Res 2016; 2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Di Stefano A, Caramori G, Barczyk A, Vicari C, Brun P, Zanini A et al Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax 2014; 69:516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang W, Ni H, Wang H, Gu H. NLRP3 inflammasome is essential for the development of chronic obstructive pulmonary disease. Int J Clin Exp Pathol 2015; 8:13209–16. [PMC free article] [PubMed] [Google Scholar]
- 67. Han MK, Tayob N, Murray S, Dransfield MT, Washko G, Scanlon PD et al Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014; 189:1503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Colarusso C, Terlizzi M, Molino A, Pinto A, Sorrentino R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget 2017; 8:81813–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Boyton RJ, Altmann DM. Bronchiectasis: current concepts in pathogenesis, immunology, and microbiology. Annu Rev Pathol 2016; 11:523–54. [DOI] [PubMed] [Google Scholar]
- 70. King PT. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis 2009; 4:411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eller J, Lapa e Silva JR, Poulter LW, Lode H, Cole PJ. Cells and cytokines in chronic bronchial infection. Ann N Y Acad Sci 1994; 725:331–45. [DOI] [PubMed] [Google Scholar]
- 72. Lapa e Silva JR, Guerreiro D, Noble B, Poulter LW, Cole PJ. Immunopathology of experimental bronchiectasis. Am J Respir Cell Mol Biol 1989; 1:297–304. [DOI] [PubMed] [Google Scholar]
- 73. Gaga M, Bentley AM, Humbert M, Barkans J, O'Brien F, Wathen CG et al Increases in CD4+ T lymphocytes, macrophages, neutrophils and interleukin 8 positive cells in the airways of patients with bronchiectasis. Thorax 1998; 53:685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shockley RA, Shaw J, Hill SL, Burnett D. Neutrophil chemotaxis in bronchiectasis: a study of peripheral cells and lung secretions. Clin Sci (Lond) 1988; 74:645–50. [DOI] [PubMed] [Google Scholar]
- 75. Dente FL, Bilotta M, Bartoli ML, Bacci E, Cianchetti S, Latorre M et al Neutrophilic bronchial inflammation correlates with clinical and functional findings in patients with noncystic fibrosis bronchiectasis. Mediators Inflamm 2015; 2015:642503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Saleh AD. The heterogeneity of systemic inflammation in bronchiectasis. Respir Med 2017; 127:33–9. [DOI] [PubMed] [Google Scholar]
- 77. Tsikrika S. The role of non‐invasive modalities for assessing inflammation in patients with non‐cystic fibrosis bronchiectasis. Cytokine 2017; 99:281–6. [DOI] [PubMed] [Google Scholar]
- 78. Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med 1999; 160:S5–11. [DOI] [PubMed] [Google Scholar]
- 79. Watt A, Brown V, Courtney J, Kelly M, Garske L, Elborn J et al Neutrophil apoptosis, proinflammatory mediators and cell counts in bronchiectasis. Thorax 2004; 59:231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Humlicek AL, Pang L, Look DC. Modulation of airway inflammation and bacterial clearance by epithelial cell ICAM‐1. Am J Physiol Lung Cell Mol Physiol 2004; 287:L598–607. [DOI] [PubMed] [Google Scholar]
- 81. Devalia JL, Davies RJ. Airway epithelial cells and mediators of inflammation. Respir Med 1993; 87:405–8. [DOI] [PubMed] [Google Scholar]
- 82. Hill S, Mitchell J, Burnett D, Stockley R. IgG subclasses in the serum and sputum from patients with bronchiectasis. Thorax 1998; 53:463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Angrill J, Agusti C, De Celis R, Filella X, Rano A, Elena M et al Bronchial inflammation and colonization in patients with clinically stable bronchiectasis. Am J Respir Crit Care Med 2001; 164:1628–32. [DOI] [PubMed] [Google Scholar]
- 84. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M et al IL‐1 receptor antagonist ameliorates inflammasome‐dependent inflammation in murine and human cystic fibrosis. Nat Commun 2016; 7:10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tarique AA, Sly PD, Holt PG, Bosco A, Ware RS, Logan J et al CFTR‐dependent defect in alternatively activated macrophages in cystic fibrosis. J Cyst Fibros 2017; 16:475–82. [DOI] [PubMed] [Google Scholar]
- 86. Hashem H, Kelly SJ, Ganson NJ, Hershfield MS. Deficiency of adenosine deaminase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders. Curr Rheumatol Rep 2017; 19:70. [DOI] [PubMed] [Google Scholar]
- 87. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL et al Early‐onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014; 370:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+‐dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa‐driven inflammatory response in cystic fibrosis. Nat Commun 2015; 6:6201. [DOI] [PubMed] [Google Scholar]
- 89. Wood LG, Fitzgerald DA, Lee AK, Garg ML. Improved antioxidant and fatty acid status of patients with cystic fibrosis after antioxidant supplementation is linked to improved lung function. Am J Clin Nutr 2003; 77:150–9. [DOI] [PubMed] [Google Scholar]
- 90. Ntimbane T, Comte B, Mailhot G, Berthiaume Y, Poitout V, Prentki M et al Cystic fibrosis‐related diabetes: from CFTR dysfunction to oxidative stress. Clin Biochem Rev 2009; 30:153–77. [PMC free article] [PubMed] [Google Scholar]
- 91. van der Vliet A, Eiserich JP, Marelich GP, Halliwell B, Cross CE. Oxidative stress in cystic fibrosis: does it occur and does it matter? Adv Pharmacol 1997; 38:491–513. [DOI] [PubMed] [Google Scholar]
- 92. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet 2016; 388:2023–38. [DOI] [PubMed] [Google Scholar]
- 93. Niewold TB, Harrison MJ, Paget SA. Anti‐CCP antibody testing as a diagnostic and prognostic tool in rheumatoid arthritis. QJM 2007; 100:193–201. [DOI] [PubMed] [Google Scholar]
- 94. Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr Opin Rheumatol 2014; 26:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Loyola‐Rodriguez JP, Martinez‐Martinez RE, Abud‐Mendoza C, Patino‐Marin N, Seymour GJ. Rheumatoid arthritis and the role of oral bacteria. J Oral Microbiol 2010; 2:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wu X, He B, Liu J, Feng H, Ma Y, Li D et al Molecular insight into gut microbiota and rheumatoid arthritis. Int J Mol Sci 2016; 17:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J et al A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA‐DR (shared epitope)‐restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006; 54:38–46. [DOI] [PubMed] [Google Scholar]
- 98. Stolt P, Kallberg H, Lundberg I, Sjogren B, Klareskog L, Alfredsson L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis 2005; 64:582–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dennis G Jr, Holweg CT, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA et al Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther 2014; 16:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol 2013; 25:334–44. [DOI] [PubMed] [Google Scholar]
- 101. Lasithiotaki I, Giannarakis I, Tsitoura E, Samara KD, Margaritopoulos GA, Choulaki C et al NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur Respir J 2016; 47:910–8. [DOI] [PubMed] [Google Scholar]
- 102. Laliberte RE, Eggler J, Gabel CA. ATP treatment of human monocytes promotes caspase‐1 maturation and externalization. J Biol Chem 1999; 274:36944–51. [DOI] [PubMed] [Google Scholar]
- 103. Dickson RP, Erb‐Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med 2013; 7:245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 2006; 6:318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]