

Summary
We addressed the role of interleukin‐23 (IL‐23) in driving the intestinal T helper type 17 (Th17) response during obesity and metabolic syndrome progression induced by a high‐fat diet (HFD). Diet‐induced obese and lean mice received HFD or control diet (CTD), respectively, for 20 weeks. The nutritional, metabolic and immune parameters were examined at weeks 9 and 20. Gene and protein IL‐23p19 and IL‐23 receptor expression was increased in the ileum of obese wild‐type mice (WT) fed the HFD for 9 weeks. Mice lacking IL‐23 and fed the HFD exhibited greater weight gain, higher fat accumulation, adipocyte hypertrophy and hepatic steatosis. Notably, these mice had more glucose intolerance, insulin resistance and associated metabolic alterations, such as hyperinsulinaemia and hyperlipidaemia. IL‐23 deficiency also significantly reduced protein levels of IL‐17, CCL20 and neutrophil elastase in the ileum and reduced Th17 cell expansion in the mesenteric lymph nodes of the HFD mice. Of importance, IL‐23‐deficient mice exhibited increased gut permeability and blood bacterial translocation compared with WT mice fed HFD. Finally, metagenomics analysis of gut microbiota revealed a dramatic outgrowth of Bacteroidetes over Firmicutes phylum with the prevalence of Bacteroides genera in the faeces of IL‐23‐deficient mice after HFD. In summary, IL‐23 appears to maintain the Th17 response and neutrophil migration into the intestinal mucosa, minimizing the gut dysbiosis and protecting against obesity and metabolic disease development in mice.
Keywords: gut microbiota, inflammation, interleukin‐17‐producing T helper lymphocytes, interleulin‐23, metabolic disease, obesity
Abbreviations
- CTD
control diet
- DC
dendritic cells
- FITC
fluorescein isothiocyanate
- Foxp3
forkhead box P3
- GLUT‐4
glucose transporter type 4
- GTT
glucose tolerance test
- HFD
high‐fat diet
- IL‐23R
interleukin‐23 receptor
- ILC3
group 3 innate lymphoid cells
- IL
interleukin
- IRS‐1
insulin receptor substrate 1
- ITT
insulin tolerance test
- LPS
lipopolysaccharide
- MLN
mesenteric lymph nodes
- PBS
phosphate‐buffered saline
- T2D
type 2 diabetes
- Th17
T helper type 17 cells
- Treg
regulatory T
- VAT
visceral adipose tissue
- WT
wild‐type
Introduction
In addition to being considered one of the oldest metabolic disorders, obesity was recently described as the most important nutritional disorder in both developed and developing countries.1 According to the World Health Organization, the worldwide incidence of obesity has more than doubled since 1980 and in 2014 more than 1·9 billion adults were overweight.2 The causes of its increased incidence are diverse, but the nutritional transition within the last centuries, characterized by new eating habits, such as the so‐called western diet, is considered the main factor. The western diet is comprised of a high fat content (particularly of animal origin), refined foods (such as sugars and starches), and reduced complex carbohydrates and fibres. In Brazil, studies have shown that this transition in dietary patterns is related to demographic, socio‐economic and epidemiological changes over time, reflecting a gradual reduction of malnutrition and an increase in obesity.3
Metabolic syndrome is a common metabolic disorder that results from the increasing prevalence of obesity, which contributes to elevated risk of coronary heart disease, hypertension and stroke, certain types of cancer, diabetes, bladder disease, dyslipidaemia, osteoarthritis and gout.4 Although coronary diseases represent the major cause of deaths related to obesity, overweight people often develop other conditions that predispose to mortality, especially type 2 diabetes (T2D).5 Type 2 diabetes is considered a chronic inflammatory disease that is characterized by hyporesponsiveness to insulin and glucose intolerance, resulting in changes in β cell function, structure or both. The aetiology of T2D is multifactorial, being linked to genetic, environmental, dietary and metabolic factors. It is estimated that approximately 75% of the risk of developing T2D is associated with obesity.6 Recent studies have shown that adipose tissue in obese people provides an inflammatory environment, which leads to insulin resistance and T2D onset.6
Several types of immune cells are found in the adipose tissue and their recruitment and function are different in obese subjects.7 Macrophages and monocytes are the most studied immune cells that reside within the adipose tissue. Recently, other cells such as neutrophils, mast cells, eosinophils, dendritic cells (DCs), natural killer cells and even cells of the adaptive immune system, such as T helper type 1 (Th1), Th2, Th17 and B cells have been shown to be increased within the adipose tissue.8, 9 However, other cell subtypes, such as invariant natural killer T cells and regulatory T (Treg) cells are dramatically reduced in adipose tissue of obese individuals compared with lean controls.10, 11 Several studies have shown that interleukin‐23 (IL‐23) is directly related to terminal differentiation of Th17 cells, promoting their maintenance, expansion and consequent migration from the circulation into inflamed peripheral tissues.12 Furthermore, increased IL‐6‐dependent Th17 lymphocytes have been identified in the spleen of obese mice.13 More recently, it was reported that the prevalence of Th17 lymphocytes in the adipose tissue is associated with the progression of obesity in humans.14 However, another study found a decrease in the proportion of retinoic acid‐related orphan receptor γt‐expressing CD4+ T cells after 30 days of a high‐fat diet (HFD).15
During the last decade, some studies have highlighted the possible role of the gut microbiota in the development of obesity and related disorders.16 The intestinal microbiota is involved in energy production and storage through several metabolic functions, such as fermentation and absorption of complex carbohydrates.17 This microbial community contains bacteria, eukaryotes and viruses, which interact with each other and with the host, having a significant impact on their physiology and health.18 Alterations in the gut microbiota composition, named dysbiosis, and translocation have been proposed as a possible cause of systemic and intestinal inflammation related to obesity and metabolic complications, although the mechanisms involved are still not well understood.19 Furthermore, recent studies have found that Th17 cells can control the replication of commensal bacteria that have escaped the intestinal epithelial barrier, so preventing bacterial colonization and translocation to distal tissues.20
Based on this evidence, this study aimed to evaluate the role of IL‐23 in promoting intestinal Th17 response and its implication in the pathogenesis of obesity and metabolic syndrome. Using an experimental HFD‐induced obesity model, we found that protein and gene IL‐23 and its receptor (IL‐23R) are expressed in the ileum of obese mice at 9 weeks, declining at later periods. Inactivation of the gene encoding IL‐23, IL‐23p19, in mice fed the HFD led to a greater propensity to develop obesity, glucose intolerance and insulin resistance, as well as to increased intestinal permeability, dysbiosis and bacterial translocation. In addition, IL‐23‐deficient mice exhibited reduced Th17 cells in the mesenteric lymph nodes (MLNs), less IL‐17 cytokine and decreased CCL20 chemokine expression associated with lower neutrophilia in the intestinal mucosa. Our results identify IL‐23 as an immunoregulatory cytokine in obesity and metabolic syndrome development.
Methods
Animals and experimental groups
Female, 4‐ to 6‐week‐old, C57BL/6 mice deficient in IL‐23 and their littermate controls were used. IL‐23‐deficient mice were kindly provided by Dr Bernhard Ryffel from the Centre National de la Recherche Scientifique, Orleans (France). We are grateful to Noah W. Palm and Richard A. Flavell from the School of Medicine, Yale University, New Haven (CT, USA) for support with the metagenomic analysis. Mice were kept in the animal house of the Department of Biochemistry and Immunology, FMRP‐USP, where they were provided with filtered air and free access to water and food. Mice were reared under specific pathogen‐free conditions. The experiments were carried out in accordance with the National Council for Animal Experimentation Control (CONCEA) and were approved by the Ethics Committee on Animal Use (CEUA) of the University of Sao Paulo, Ribeirao Preto, Brazil (protocol number 144/2014). The animals were divided into group I, wild‐type (WT) mice fed a control diet (CTD‐AIN 93, comprising 9·7% fat, 77·1% carbohydrate and 13·4% protein); Group II, IL‐23‐deficient mice fed the CTD; group III, WT mice fed a high‐fat diet (HFD‐D12492, comprising 60% fat, 20% carbohydrate and 20% protein) and group IV, IL‐23‐deficient mice fed the HFD. C57BL/6 mice and IL‐23‐deficient mice were fed the CTD or HFD for 20 weeks. During this period, nutritional, metabolic and immunological parameters were analysed.
Nutritional parameters
The nutritional profile was determined by analysis of food intake, caloric intake, body weight, visceral (mesenteric), total fat mass and adiposity index. The body weight of animals was measured weekly, using a digital scale. The amount of total fat mass was determined by the sum of deposits of retroperitoneal and mesenteric fats. The adiposity index was calculated by dividing the total body fat by the final body weight, multiplied by 100.
Metabolic parameters
For the glucose tolerance test (GTT), mice were submitted to a 12‐hr fasting period. Blood samples were taken at baseline and after intraperitoneal administration of a solution containing 25% glucose (Sigma‐Aldrich, St Louis, MO) equivalent to 2·0 g/kg, being collected at 0, 15, 30, 60 and 120 min. The ACCU‐CHEK®Active equipment was used to read glucose levels. For the insulin tolerance test (ITT), mice were submitted to a 6‐hr fasting period. Blood samples were taken from mice at baseline and after intraperitoneal administration of regular insulin equivalent to 1·5 IU/kg, being collected at 0, 5, 10, 15, 20, 15 and 30 min.
Detection of total cholesterol, triglyceride and LPS levels
Mice were fasted for 12 hr and blood was collected from the tip of the tail vein. Haemolysis‐free serum was collected after centrifugation. Total cholesterol, triglyceride or lipopolysaccharide (LPS) concentrations were measured using kits from Labtest (St. Louis, MO) or Lonza (Basel, Switzerland).
Quantification of serum insulin levels
Insulin concentrations were determined using the Mouse Ultrasensitive Insulin kit (Alpco Diagnostics, Salem, NH) according to the manufacturer's instructions.
Quantification of cytokine, chemokine and elastase levels
Ileum or visceral adipose tissue (VAT) fragments were removed, weighed and placed in a tube containing 700 μl of Complete Protease Inhibitor Cocktail (Roche Diagnostics, Abbott Park, IL). The tissues were homogenized using a Polytron homogenizer (Thermo Fisher Scientific, Waltham, MA) and IL‐17, IL‐23, CCL20 and elastase levels were detected by enzyme‐linked immunosorbent assay using colorimetric kits according to the manufacturer's instructions (R&D Systems, Minneapolis, MN). The results are expressed as the mean ± SEM (nanograms per gram of tissue).
Intestinal permeability by FITC‐Dextran
After 12 hr of fasting, mice received fluorescein isothiocyanate (FITC)‐dextran by gavage (250 mg/kg) (Sigma‐Aldrich). After 4 hr, blood samples were collected from the tail vein. The blood was centrifuged at 4°, 1000 g for 3 min. The serum was diluted in the same volume of phosphate‐buffered saline (PBS; pH 7·4) for analysis of FITC‐dextran concentrations at excitation wavelength of 485 nm and emission wavelength of 535 nm.
Histopathology and immunohistochemistry analysis
Histopathological evaluations of the pancreas, VAT and liver were performed after staining with haematoxylin & eosin of fixed samples in PBS/10% formaldehyde. The material was sectioned, mounted on glass slides and kept in a dry oven at 60° for 1 hr. Then, the material was hydrated and deparaffinized using xylene, alcohol and water. Immunohistochemistry reactions were performed as previously described.21
Evaluation of bacterial translocation
To evaluate bacterial translocation in the blood and VAT, samples were aseptically collected. Subsequently, 50‐μl blood aliquots were spread with a sterile loop in brain–heart infusion medium‐containing agar plates and placed in an incubator at 37° for 48 hr to count the colony‐forming units.
RNA extraction and quantitative real‐time polymerase chain reaction
Total RNA was extracted from the ileum using Trizol (Life Technologies, Molecular Probes, Carlsbad, CA) following the manufacturer's instructions. cDNA was obtained using a High Capacity reverse transcription kit (Applied Biosystems, Foster City, CA) following DNase treatment (Life Technologies). The expression of IL‐23p19, IL‐23R and β‐actin was analysed by quantitative polymerase chain reaction (PCR) using the SYBR Green PCR Master Mix (Applied Biosystems). The following primers were used: β‐actin: forward: 5′‐AACGAGCGGTTCCGATG‐3′, reverse: 5′‐GGATTCCATACCCAACAAGGA‐3′, IL‐23p19 forward: 5′‐ATTGTGCCCCGTATCCAGTGT‐3′, reverse: 5′‐GGCTCCCCTTTGAAGATGTCA‐3′; IL‐23R forward: 5′‐GGCTTCTACTACATTTGGGACAT‐3′, reverse: 5′‐TACTTGTGATTCCTCCGTGACA‐3′. Specific mRNA expression levels were normalized relatively to β‐actin mRNA levels using the comparative 2ΔΔCt method.
Analysis of leucocytes by flow cytometry
Flow cytometry analysis was performed on samples with 1 × 106 cells/tube in 100 μl of PBS. First, cell suspensions were incubated with 5% normal rabbit serum for 30 min to block non‐specific binding. Next, antibodies against CD3, CD4, CD11b, Ly6G, Ly6C, CD103, CCR6, IL‐17, Foxp3 and their control isotypes (BD Pharmingen, San Diego, CA) were added and incubated for 30 min in the dark. IL‐17 production was evaluated after in vitro reactivation with phorbol myristate acetate (50 ng/ml) and ionomycin (500 ng/ml, Sigma‐Aldrich) and Golgi Stop (1,000X, BD Pharmingen), as previously described.25 The cells were analysed using a FACS Canto flow cytometer, and the data were analysed using flowjo (Tree Star, Ashland, OR) software.
Western blotting
Fifty micrograms of extracted proteins was loaded directly into sodium dodecyl sulphate sample buffer for 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis. After transferring the samples onto a nitrocellulose membrane (Trans‐Blot Transfer Medium; Bio‐Rad, Hercules, CA), the membranes were blocked with 5% milk in Tris buffer solution containing 0·1% Tween 20 for 1 hr and then incubated with antibodies against glucose transporter type 4 (GLUT‐4; Cell Signaling, Danvers, MA) or phosphorylated insulin receptor substrate (pIRS) in serine 1101 (Cell Signaling) overnight at 4°. Next, the cells were incubated with an enzyme horseradish peroxidase‐conjugated secondary antibody (Cell Signaling) for 1 hr at room temperature. After the membranes were rinsed, the immunocomplexes were developed using an enhanced peroxidase/luminol chemiluminescence reaction (ECL Western blotting detection reagents; Pierce Biotechnology, Waltham, MA) and exposed to X‐ray film with autoradiography (Carestream Health, Rochester, NY).
16S rRNA gene sequencing
16S rRNA gene sequencing was performed, as previously described.22 The Ribosomal Database Project classifier and the May 2013 Greengenes taxonomy were used to assign taxonomy to representative operational taxonomic units and the Linear Discriminant Analysis Effect Size Galaxy module was used for additional statistical analyses.23
Statistical analysis
The data are expressed as mean ± SEM. The differences observed among the several experimental groups were analysed by one‐way analysis of variance followed by the parametric Tukey test for comparing multiple groups. All analyses were performed using prism 5·0 software (GraphPad Software, San Diego, CA). Statistical significance was set at P < 0·05.
Results
Up‐regulation of IL‐23 and IL‐23 receptor gene and protein expression in the ileum of obese mice
Initially, we analysed the protein and gene expression of IL‐23p19 and IL‐23R in the ileum of diet‐induced obese mice after 9 and 20 weeks of the HFD. Increased gene IL‐23p19 expression was found in the ileum of obese mice at 9 weeks of HFD when compared with lean mice fed the CTD. Gene IL‐23p19 expression was decreased after 20 weeks of the HFD (Fig. 1a). Accordingly, gene IL‐23R expression in the ileum increased significantly after 9 weeks of HFD (Fig. 1b). In addition, IL‐23 protein expression was significantly increased in the ileum of HFD‐fed obese mice at 9 weeks, but reduced at 20 weeks (Fig. 1c).
Figure 1.
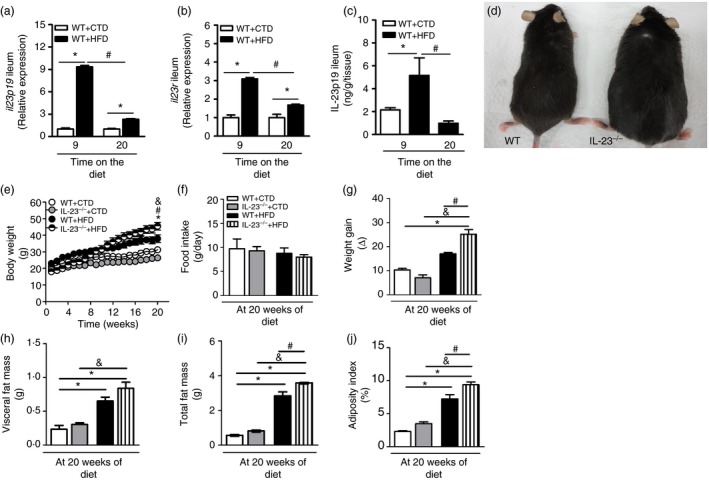
Time–course of interleukin‐23 (IL‐23) p19 and interleukin‐23 receptor (IL‐23R) gene and protein expression in the ileum and nutritional parameters of wild‐type (WT) and IL‐23p19–/– mice fed a control diet (CTD) or a high‐fat diet (HFD). Relative gene IL‐23p19 (a) or IL‐23R (b) expression in the ileum was analysed by RT‐PCR. IL‐23p19 protein expression was detected in the ileum by enzyme‐linked immunosorbent assay (c) of mice fed an HFD or a CTD. Representative images of IL‐23p19–/– mice (right) and WT mice (left) fed an HFD for 20 weeks (d). Body weight (e), food intake (f), weight gain (g), visceral adipose tissue (VAT) mass (h), total fat mass (i) and adiposity index (j) were determined in IL‐23p19–/– and WT mice after 20 weeks on HFD or CTD. Asterisks represent statistically significant differences (*P < 0·05) compared with WT on CTD; (# P < 0·05) compared with WT on HFD; (& P < 0·05) compared with IL‐23p19–/– mice on CTD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐23 activation elicits protection to obesity in the diet‐induced obesity model
The nutritional profile is based on the analysis of factors that characterize the physical state of mice, such as body weight, food intake, caloric intake, fat accumulation and adiposity index. All of these parameters were assessed in WT and IL‐23‐deficient (IL‐23p19–/–) mice fed either the HFD or the CTD. As shown in Fig. 1, WT mice fed the HFD exhibited a higher weight gain compared to lean mice fed the CTD. IL‐23p19–/– mice were more prone to HFD‐induced obesity. In fact, these mice exhibited increased weight gain starting from 12 weeks of the HFD (Fig. 1e), which was significantly increased at 20 weeks compared with WT mice (Fig. 1d,g). However, no differences in food intake were observed between the several experimental groups (Fig. 1f). Although IL‐23p19–/– mice showed a trend towards increased VAT accumulation (Fig. 1h), they only had a significant increase in total fat accumulation and adiposity index compared with HFD‐fed WT mice (Fig. 1i,j). These data indicate that IL‐23 plays a protective role during diet‐induced obesity development.
IL‐23 activation impairs adipocyte hypertrophy and hepatic steatosis induced by obesity
By analysing the VAT, IL‐23p19–/– mice fed a CTD displayed a small increase in adipocyte size and a decrease in adipocyte number compared with WT mice on the same diet (Fig. 2a,b,m,n). However, IL‐23p19–/– mice exhibited a greater increase in adipocyte size, characterized as hypertrophy, and decreased adipocyte number compared with WT mice fed the HFD (Fig. 2c,d,m,n). Also, we noted slow fat deposition in hepatic tissues of IL‐23p19–/– mice compared with WT mice fed the CTD (Fig. 2e,f). However, IL‐23p19–/– mice fed the HFD exhibited much more fat accumulation in the liver compared with WT mice on the HFD (Fig. 2g, h). In parallel, we detected that IL‐23 deficiency did not change the morphology of the pancreatic islets compared with WT mice on the CTD (Fig. 2i,j). On the other hand, HFD‐fed IL‐23p19–/– mice appear to have increased pancreatic islet size compared with HFD‐fed WT mice (Fig. 2k,l), but the morphometry of the insulin‐positive pancreatic islets, determined by immunohistochemistry, did not confirm this difference between the experimental groups (Fig. 2o). Hence, our findings suggest that IL‐23 prevents adipocyte hypertrophy and hepatic steatosis in HFD‐induced obesity.
Figure 2.
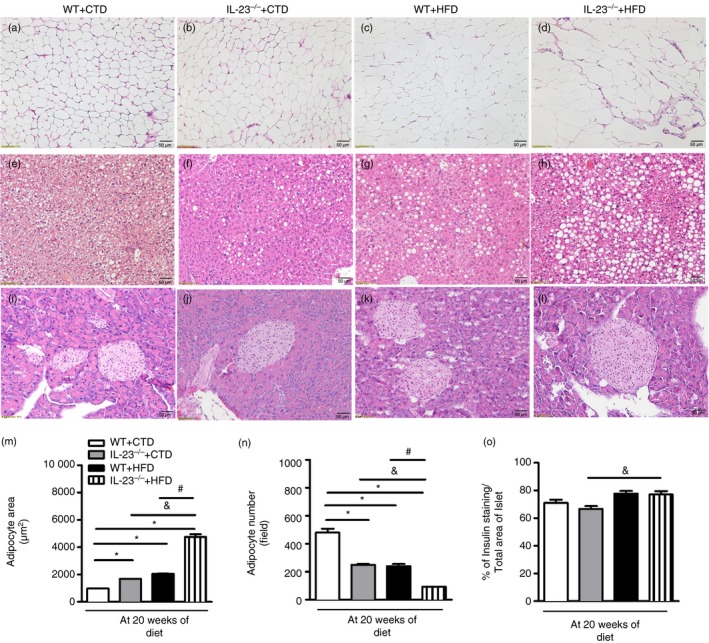
Histopathological analysis of the visceral adipose tissue (VAT), liver and pancreatic tissues of wild‐type (WT) and interleukin‐23 p19‐deficient (IL‐23p19–/–) mice fed a control diet (CTD) or a high‐fat diet (HFD) for 20 weeks. Adipocyte hypertrophy was assessed in VAT of mice on HFD or CTD (a–d). Hepatic steatosis was assessed in liver of mice on HFD or CTD (e–h). Histological analysis of inflammatory infiltrate into pancreatic islets stained with haematoxylin & eosin (i–l). Morphometric quantification of adipocyte size (m) or numbers was performed in adipose tissue (n). A quantitative analysis of insulin‐producing β cell expression was performed into pancreatic islets immunostained with insulin‐specific antibody (o). The images are representative of a single experiment repeated three times (original magnification 200×). Asterisks represent statistically significant differences (*P < 0·05) compared with WT on CTD; (# P < 0·05) compared with WT on HFD; (& P < 0·05) compared with IL‐23p19–/– mice on CTD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐23 activation ameliorates the metabolic syndrome induced by obesity
Next, the impact of IL‐23 deficiency on glucose metabolism was addressed by measuring fasting glucose levels (Fig. 3a) or by the GTT (Fig. 3b,d). Interestingly, IL‐23p19–/– mice had increased blood glucose levels and became more glucose intolerant after the HFD (Fig. 3a,b,d). In agreement, these mice exhibited greater insulin resistance, as shown by the ITT, compared with WT after the HFD (Fig. 3c,e). Moreover, IL‐23p19 deficiency in the presence of the HFD significantly increased fasting insulin levels (Fig. 3f) and triglyceride levels (Fig. 3g) without affecting total cholesterol levels (Fig. 3h) compared with WT mice. In parallel, protein expression of the GLUT‐4 and IRS‐1 in serine 1101 (Ser1101), molecules involved in glucose uptake and insulin signalling inhibition, respectively, was analysed in skeletal muscle of mice fed the CTD and the HFD. Surprisingly, GLUT‐4 expression was increased in HFD‐fed IL‐23p19–/– mice compared with the other experimental groups (Fig. 3i). The IL‐23 deficiency also increased Ser1101 phosphorylated IRS‐1 expression when compared with HFD‐fed WT mice, suggesting that these mice developed more insulin resistance (Fig. 3i). These results emphasize that IL‐23 attenuates obesity‐associated metabolic alterations, including glucose intolerance, insulin resistance and hyperlipidaemia, induced by an HFD.
Figure 3.
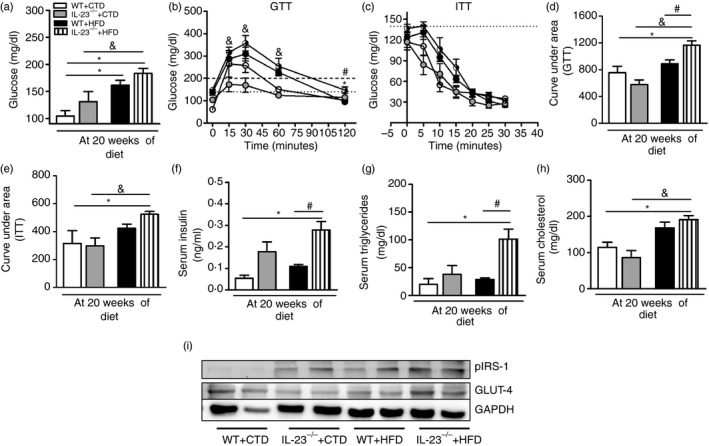
Metabolic parameters and total glucose transporter type 4 (GLUT‐4) and phosphorylated insulin receptor substrate 1 (IRS‐1) expression in the muscle of wild‐type (WT) and interleukin‐23 p19‐deficient (IL‐23p19–/– mice fed a control diet (CTD) or high‐fat diet (HFD) for 20 weeks. Fasting blood glucose levels (a) and glucose levels after glucose tolerance test (GTT) (b) or insulin tolerance test (ITT) (c). Area under the curve for the GTT (d) or ITT (e) were also calculated. Concentrations of insulin (f), triglycerides (g) and cholesterol (h) were determined in the serum. Total GLUT4 and Ser1101 phosphorylated IRS‐1 expression in skeletal muscle was determined by Western blot (i). Asterisks represent statistically significant differences (*P < 0·05) compared with WT on CTD; (# P < 0·05) compared with WT on HFD; (& P < 0·05) compared with IL‐23p19–/– mice on CTD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐23 induces intestinal IL‐17 production and neutrophil migration in obese mice
Subsequently, protein expression of IL‐17 cytokine, CCL20 chemokine and elastase was determined in the small intestine (ileum) and serum of mice fed the CTD or HFD. Although not statistically significant, HFD‐fed WT mice displayed increased IL‐17 levels in the ileum compared with WT mice on the CTD. However, IL‐17 levels significantly decreased in HFD‐fed IL‐23p19–/– mice compared with WT mice on the HFD (Fig. 4a). HFD‐fed IL‐23p19–/– mice also exhibited a significant reduction of CCL20 levels in the ileum compared with WT mice on the HFD (Fig. 4b). In addition, a significant decrease in elastase levels, which is a serine protease expressed by neutrophils, was observed in the ileum of IL‐23p19–/– mice compared with WT mice on the HFD (Fig. 4c). In accordance, elastase levels were significantly decreased in the serum of HFD‐fed IL‐23p19–/– mice compared with WT mice on the HFD (Fig. 4d). Similarly, these mice had a significant increase in neutrophil population in the MLNs (Fig. 4e,f,i). However, no significant alterations in the frequency or absolute numbers of inflammatory monocyte populations in the MLNs were observed in these mice (Fig. 4g,h). Taken together, our findings imply that IL‐23 contributes to IL‐17 production and neutrophil migration to the intestinal mucosa during diet‐induced obesity.
Figure 4.
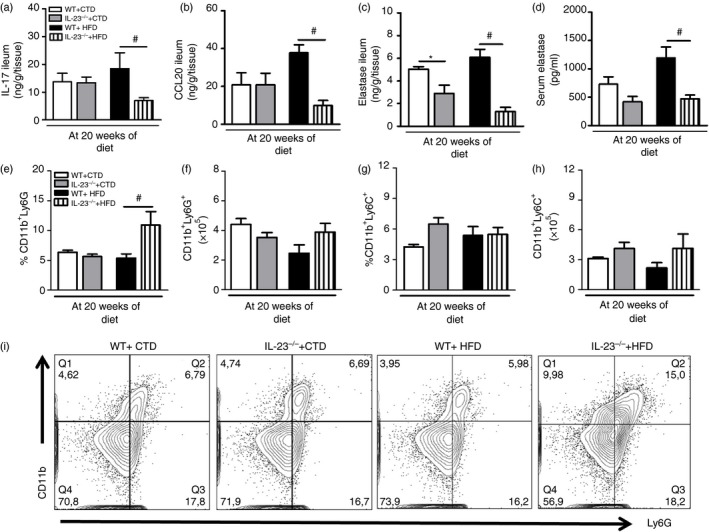
Expression of T helper type 17 (Th17) profile‐related molecules in the ileum and mesenteric lymph nodes (MLNs) of wild‐type (WT) and interleukin‐23 p19‐deficient (IL‐23p19–/–) mice on control diet (CTD) or high‐fat diet (HFD) for 20 weeks. Levels of interleukin‐17 (IL‐17) (a), CCL20 (b) and elastase (c) in the ileum and in the serum (d) were quantified by enzyme‐linked immunosorbent assay. Percentage and absolute numbers of neutrophils CD11b+ Ly6G+ (e, f) and monocytes CD11b+ Ly6C+ (g, h) were determined in the MLNs by flow cytometry. Percentages of neutrophils are shown in representative dot plots (i). Asterisks represent statistically significant differences (*P < 0·05) compared with WT on CTD; (# P < 0·05) compared with WT on HFD; (& P < 0·05) compared with IL‐23p19–/– mice on CTD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐23 promotes Th17 cell expansion in the mesenteric lymph nodes in obese mice
We also investigated whether reduced IL‐17 production in IL‐23p19–/– mice correlates with lower population of Th17 cells (CD3+ CD4+ IL‐17+) and IL‐17‐producing group 3 innate lymphoid cells (ILC3) (CD3– CCR6+ IL‐17+) in the MLNs. In fact, the percentage and absolute numbers of Th17 cells were significantly decreased in the MLNs of HFD‐fed IL‐23p19–/– mice compared with WT mice on the HFD (Fig. 5a,b,i). However, the percentage and absolute numbers of IL‐17‐producing ILC3 cells were not significantly reduced in the MLNs of these mice compared with WT mice on HFD (Fig. 5c,d). Conversely, there was a significant increase in the percentage of Treg cells (CD4+ Foxp3+) in the MLNs of HFD‐fed IL‐23p19–/– mice compared with WT mice on HFD (Fig. 5e), but there was no statistical difference in the absolute numbers of Treg cell population between the experimental groups (Fig. 5f). Also, the percentage of tolerogenic DCs (CD11c+ CD103+), but not the absolute numbers, was significantly increased in the MLNs of HFD‐fed IL‐23p19–/– mice compared with WT mice on HFD (Fig. 5g,h). Overall, our findings indicate that IL‐23 induces Th17 cell expansion in the MLNs and migration to intestinal mucosa during diet‐induced obesity.
Figure 5.
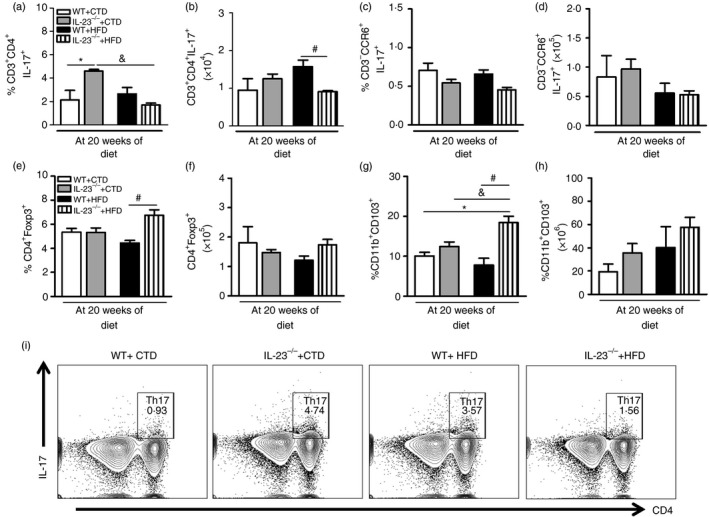
T helper type 17 (Th17), group 3 innate lymphoid cell (ILC3), regulatory T (Treg) cell and tolerogenic dendritic cell (DC) numbers in the mesenteric lymph nodes (MLNs) of wild‐type (WT) and interleukin‐23 p19‐deficient (IL‐23p19–/–) mice on control diet (CTD) or high‐fat diet (HFD) for 20 weeks. Percentage and absolute numbers of Th17 CD3+ CD4+ IL‐17+ (a, b) and ILC3 CD3– CCR6+ IL‐17+ (c, d) were determined in MLNs by flow cytometry. Percentage and absolute numbers of Treg CD4+ Foxp3+ (e, f) and tolerogenic DCs CD11b+ CD103+ (g, h) were determined in MLNs by flow cytometry. Percentages of Th17 cells are shown in representative dot plots (i). Asterisks represent statistically significant differences (*P < 0·05) compared with WT on CTD; (# P < 0·05) compared with WT on HFD; (& P < 0·05) compared with IL‐23p19–/– mice on CTD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐23 controls the gut microbiota dysbiosis and bacterial translocation in obesity
Considering the importance of Th17 cells in maintaining both the composition of the gut microbiota and the integrity of the gut epithelial barrier, we hypothesized that in the deficiency of IL‐23, which plays a crucial role in the induction of Th17 cells, the gut microbiota exhibits dysbiosis. As shown, several differences were found at the phylum (Fig. 6a), family (Fig. 6b) and genus (Fig. 6c) levels in IL‐23‐deficient HFD‐fed mice. HFD‐fed IL‐23p19–/– mice had an increase in Bacteroidetes and Verrucomicrobia phyla when compared with all other groups (Fig. 6a). Interestingly, at the family level, CTD‐fed IL‐23p19–/– mice presented several differences when compared with WT mice. More specifically, a decrease in Erysipelotrichaceae and an increase in both Clostridiaceae and Ruminococcaceae families were found in CTD‐fed IL‐23p19–/– mice, but not in their WT counterparts fed either the CTD or HFD (Fig. 6b). Of note, HFD‐fed WT mice had an increase in Lactobacillaeceae and S24‐7 and a decrease in Lachnospiraceae (Fig. 6b). Interestingly, these differences were also noted in HFD‐fed IL‐23p19–/– mice compared with CTD‐fed IL‐23p19–/– mice. Finally, at the genus level, both CTD‐fed and HFD‐fed IL‐23p19–/– mice presented several changes when compared with their WT counterparts, such as an increase in Akkermansia and Bacteroides genera (Fig. 6c). We have also determined possible biomarkers of each group by using the linear discriminant analysis effect size.24 Several biomarkers for each group were found (Fig. 6d). More specifically, the genus Turicibacter in CTD‐fed WT mice, the genus Jeotgalicoccus in HFD‐fed WT mice, the genus Clostridium in CTD‐fed IL‐23p19–/– mice and the genera Adlecreutzia, Bacteroides, Ruminococcus and Oscillospira in HFD‐fed IL‐23p19–/– mice were found to be significantly more expressed, and hence were considered as biomarkers of each phenotype. Considering that gut dysbiosis is intimately related to bacterial translocation and gut permeability,25 we analysed whether the dysbiosis induced by IL‐23 deficiency had any effect on the gut epithelial barrier. As expected, we observed increased gut permeability, measured by FITC‐Dextran, in HFD‐fed IL‐23p19–/– mice compared with WT mice (Fig. 6e) and a trend to increased LPS levels (Fig. 6f). In accordance, we detected increased colony‐forming units count in the blood (Fig. 6g), but not in the VAT (Fig. 6h) of these mice. Overall, IL‐23 has a crucial role in limiting gut microbiota dysbiosis and maintaining the gut epithelial barrier integrity, resulting in lower bacterial translocation induced by obesity.
Figure 6.
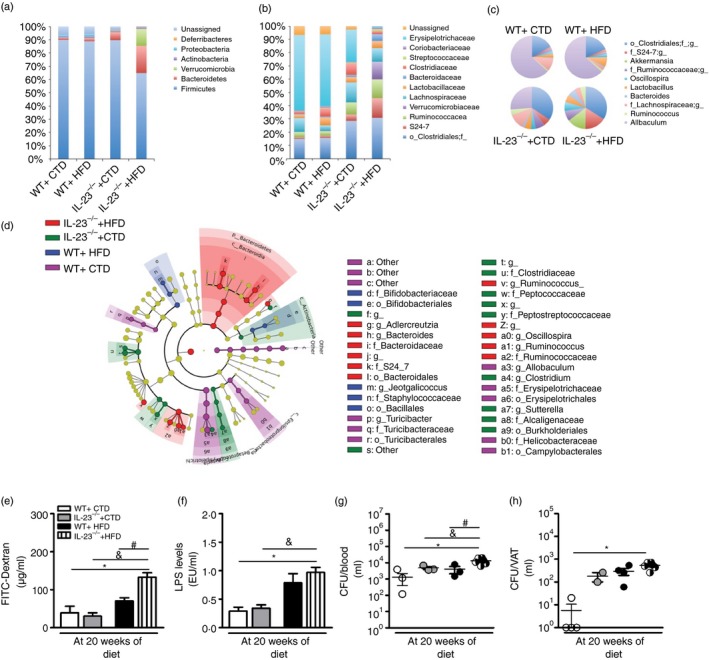
Gut dysbiosis, intestinal permeability and bacterial translocation in wild‐type (WT) and interleukin‐23 p19‐deficient (IL‐23p19–/–) mice on control diet (CTD) or high‐fat diet (HFD) for 20 weeks. Relative abundance of faecal bacterial phylum (a), family (b) and genus (c) were evaluated by 16S rRNA gene sequencing. Taxonomic cladogram comprising all detected taxa (d). Bacterial taxa that were significantly different among all pairwise comparisons were used as inputs for the Linear Discriminant Analysis Effect Size software. The rings of the cladogram stand for phylum (innermost), class, order, family and genus (outermost), respectively. Enlarged coloured circles are the differentially abundant taxa identified to be metagenomic biomarkers. Fluorescein isothiocyanate (FITC) ‐Dextran assay (e), serum lipopolysaccharide (LPS) levels (f) and colony‐forming unit (CFU) numbers in the blood (g) or visceral adipose tissue (VAT) (h). Asterisks represent statistically significant differences (*P < 0·05) compared with WT on CTD; (# P < 0·05) compared with WT on HFD; (& P < 0·05) compared with IL‐23p19–/– mice on CTD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
Discussion
Obesity is an epidemic disease whose prevalence worldwide has increased dramatically over the past few decades. In specific countries, like the USA, obesity continues to expand at alarming rates with an increase of 75% since 1980.26 The development of obesity is a complex process involving genetic predisposition and environmental factors. The excessive accumulation of fat in the body is the main cause of metabolic syndrome. The metabolic syndrome, which includes glucose intolerance, insulin resistance, dyslipidaemia, associated with abdominal obesity, can induce T2D onset. In this context, evidence of a close relationship between obesity and gut microbiota dysbiosis, which leads to increased circulating LPS and insulin signalling abnormalities in metabolic tissues, has also been reported.27
Obesity is an inflammatory disease where both resident cells in the adipose tissue as well as adipocytes and innate immune cells are activated and secrete cytokines in response to metabolic products, such as fatty acids, or microbial products from the gut microbiota. The IL‐23 cytokine is derived from antigen‐presenting cells, which are activated after the recognition of pathogen‐related molecular patterns or damage‐related molecular patterns.28, 29 IL‐23R is expressed in both innate and adaptive immune cell types, including T cells and ILC3 cells. IL‐23 has an important role in later stages of Th17 differentiation and also promotes Th17 cell maintenance in inflammatory sites.12 In this context, a study demonstrated that immature adipose tissue DCs express higher levels of IL‐6, transforming growth factor‐β and IL‐23, resulting in the enhancement of Th17 cell response in a diet‐induced obesity model.30 In accordance, we observed that gene and protein IL‐23p19 and IL‐23R expression was significantly increased in the ileum of obese mice after 9 weeks of HFD, decreasing after 20 weeks of the HFD. These results indicate that obesity promotes IL‐23 production in the small intestine at an earlier phase of the disease.
Interleukin‐23 deficiency upon an HFD led to excessive weight gain, increased VAT accumulation and adiposity index. In agreement, HFD‐fed IL‐23p19–/– mice exhibited increased adipocyte size, named hypertrophic obesity, and hepatic fat deposition. On the other hand, a study showed that IL‐23a–/– mice are protected from HFD‐induced weight gain and excess adiposity.31 Non‐alcoholic fatty liver disease or hepatic steatosis is a chronic disease characterized by the accumulation of triglycerides in more than 5% of hepatocytes, which is associated with insulin resistance.32 In particular, the increase of free fatty acids in the circulation and their accumulation in the liver can lead to insulin resistance.33 In turn, insulin resistance reduces the suppression of lipolysis in the adipose tissue by increasing the levels of free fatty acids and triglycerides.34 Notably, HFD‐fed mice lacking IL‐23 also have more hepatic steatosis and exhibited a significant increase in triglyceride levels when compared with WT mice. In this context, a study demonstrated a correlation between increased levels of Th17 cell‐related cytokines, including IL‐6, IL‐17 and IL‐23, in hepatic tissue and significant pathological and biochemical changes in the liver.35 Overall, these data indicate that IL‐23 deficiency predisposes to obesity and exacerbates the deposition of hepatic fat and dyslipidaemia.
During an insulin‐resistant pre‐diabetic state, the insulin‐producing β‐cell mass expands functionally through intense proliferation and differentiation to compensate the insulin resistance, producing large amounts of this hormone and inducing a hyperinsulinaemic state. In fact, we noted that IL‐23‐deficient mice on HFD exhibit increased glucose intolerance, hyperglycaemia and hyperinsulinaemia. Insulin resistance and overproduction can lead to dysfunction/exhaustion of β cells because the greater mass of these cells is reduced in humans with diabetes or mice with T2D.36, 37 Although the deficiency of IL‐23 induces a state of insulin resistance; IL‐23p19–/– mice on HFD did not exhibit profound alterations in pancreatic islet morphology, suggesting that IL‐23 improves glucose homeostasis, possibly due to restoration of insulin sensitivity. In accordance, other results showed that conditional inactivation of the gene encoding the IL‐6Rα chain of the receptor for IL‐6 in myeloid cells promotes glucose intolerance and obesity‐associated resistance to insulin.38 However, another study showed that neutralizing IL‐23 in vitro reduces pancreatic oxidative stress and improves insulin biosynthesis in obesity.39 In summary, our data suggest that IL‐23 plays a protective role in glucose intolerance and insulin resistance, which precedes obesity‐induced T2D onset.
Some studies showed that IL‐23 is produced in response to colonization of segmented filamentous bacteria or systemic dissemination of microbial products followed by rupture of the intestinal barrier. The main function of IL‐23 is to maintain Th17 cell differentiation and proliferation.20 Several findings have reported the importance of Th17 cells in host defence against bacterial and fungal pathogens, particularly those found on the mucosal surfaces.40 In addition, Th17 cells are fundamental to control the replication of commensal bacteria inside the intestinal mucosa and avoid translocation of bacteria to distal tissues. A study reported that intestinal tissues from obese mice exhibit significant reductions in the proportion of Th17 cells relative to non‐obese mice.15 Likewise, our data show that IL‐23 deficiency in HFD‐fed mice significantly reduces Th17 cells in the MLNs and decreases IL‐17 expression in the ileum when compared with WT mice on HFD. The CCR6 chemokine receptor is expressed on activated Th17 cells, which is involved in the recruitment of these cells to inflammatory sites through CCL19 and CCL20 chemokines.41 As mice lacking IL‐23 on HFD also have reduced CCL20 expression in the ileum, it is plausible that these mice produce less IL‐17 in the ileum as a consequence of defective migration of Th17 cells into the intestinal mucosa.
Interleukin‐17A regulates granulopoiesis, neutrophil recruitment and the production of antimicrobial peptides.42 In addition, IL‐17A has been shown to delay the apoptosis of neutrophils and mononuclear phagocytes through stimulation of cytokines like granulocyte colony‐stimulating factor and granulocyte–macrophage colony‐stimulating factor in homeostatic conditions.43 HFD‐fed IL‐23p19–/– mice also exhibited a significant decrease in the expression of elastase, a proteolytic enzyme produced by neutrophils, in the ileum and serum. Other studies already demonstrated that serum neutrophil activity is increased in experimental models and in patients with obesity, suggesting a deleterious role of neutrophils in inflammation and insulin resistance.44 However, neutrophil migration has a protective role in the intestinal lumen, resulting in the formation of organized intraluminal structures that encapsulate commensals and limiting their contact with the epithelium during gut dysbiosis.45 Taken together, these data suggest that IL‐23 sustains Th17 cell induction and neutrophil migration into intestinal mucosa during HFD‐induced obesity in order to control intestinal homeostasis.
Patients with T2D exhibit an imbalance in Th1 over Treg cell populations. Treg cells are immunoregulatory cells important for prevention of body weight gain, adipocyte hypertrophy and insulin resistance.46 Surprisingly, we verified a significant increase in the percentage of Treg cells in the MLNs of HFD‐fed mice lacking IL‐23. Similarly, these mice display an increase of tolerogenic DCs, which are important to induce the differentiation of intestinal Treg cells.47 Our findings are unexpected because the worsening of obesity and insulin resistance in mice lacking IL‐23 correlates with a higher immunoregulatory profile in the intestine. A possible explanation is that Treg cell and DC expansion is a consequence of the intense inflammatory response and/or bacterial stimuli in the gut of these mice. ILC3 are a subpopulation of cells of the immune system, which are important to induce organogenesis and intestinal homeostasis. These cells are differentiated in the presence of the cytokines IL‐2, IL‐7 and IL‐23 and produce IL‐17 and IL‐22.48 Recent evidence showed the importance of the interaction of ILC3 cells with the commensal microbiota in the intestine.49 However, we did not detect alterations in the ILC3 population in the MLNs of IL‐23‐deficient mice on HFD. Hence, these data suggest that IL‐23 counter‐regulates Treg cell and tolerogenic DC responses in MLNs during HFD‐induced obesity.
In humans, obesity and metabolic syndrome are associated with a higher intestinal Firmicutes/Bacteroidetes ratio in comparison with lean individuals.50, 51 Despite the absence of differences in the phylum levels in obese WT mice, HFD‐fed IL‐23‐deficient mice exhibited an increased abundance of Verrucomicrobia and Bacteroidetes phyla. At the genus level, these mice revealed greater abundance of Akkermansia and Bacteroides compared with WT mice on HFD. Several gut bacterial strains, including Bacteroides and Escherichia coli strains, increase adiposity when introduced as monocultures into germ‐free mice fed a low‐fat, polysaccharide‐rich diet. The most pronounced effect on adiposity was verified after associating germ‐free mice with either Parabacteroides distasonis or Bacteroides vulgatus.52 In contrast, Akkermansia muciniphila is a bacterium colonizing the intestinal mucosa and its abundance is negatively correlated with body weight in humans.53, 54 It was found that the abundance of A. muciniphila decreases in obese mice and the administration of this bacterium improves metabolic changes induced by diet such as metabolic endotoxaemia, intestinal permeability and insulin resistance.55 In agreement, a study reported that the adoptive transfer of gut‐tropic Th17 cells to obese mice reduces metabolic defects and leads to expansion of commensal microbes associated with leanness.15 Overall, our data suggest that IL‐23 is involved in the establishment of the intestinal Th17 response, control of the gut dysbiosis and protection against obesity‐associated metabolic syndrome in a murine model.
Experimental models suggest that obesity is associated with alterations of the composition and function of gut microbiota,56 which can lead to intestinal inflammation and increased permeability.49 In this context, Cani et al. demonstrated that after 4 weeks of high‐fat feeding, mice exhibited an obese phenotype accompanied by a change in gut microbiota composition and an increase in circulating LPS levels.57 Consistently, HFD‐fed mice have reduced expression of epithelial tight‐junction proteins like occludin and zona occludens proteins, but increased intestinal permeability and LPS levels.58 In fact, we observed increased intestinal permeability associated with a trend to augmented serum LPS levels in HFD‐fed WT mice, and these alterations are potentiated in IL‐23‐deficient mice on HFD. HFD‐fed IL‐23p19–/– mice also exhibited more bacterial translocation in the blood compared with WT mice. Our findings demonstrate that the IL‐23 production in the gut maintains intestinal barrier integrity, thereby minimizing bacterial translocation in an obesity‐induced metabolic syndrome model. Therefore, IL‐23 appears to be indispensable for inducing an intestinal Th17 response and neutrophil migration, limiting gut microbiota dysbiosis, maintaining epithelial integrity and preventing metabolic syndrome associated with obesity. These findings identify IL‐23 as a novel immunotherapy target for the treatment of human metabolic diseases.
Author contributions
LMSM performed experiments and analysed the results; MMP, CAP, FRCC and MSD contributed to the analysis and helped with in vivo experiments; SGR supported us with histology and imaging data. MZ helped with the metagenomic analyses. BR genotyped the IL‐23‐deficient mice and provided scientific assistance. RCT and JSS edited the manuscript, provided scientific assistance and revised it critically. DC provided intellectual support in addition to directing and supervising the study.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
This study was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP ‐ Process: 2014/15462‐2). We are grateful to Elaine Medeiros Floriano from the Laboratory of Pathology of the Ribeirão Preto Medical School, University of São Paulo, for helping with the histological analysis.
Contributor Information
João S. Silva, Email: jsdsilva@fmrp.usp.br
Daniela Carlos, Email: danicar@usp.br.
References
- 1. Dyer RG. Traditional treatment of obesity: does it work? Baillieres Clin Endocrinol Metab 1994; 8:661–88. [DOI] [PubMed] [Google Scholar]
- 2. WHO . Obesity and Overweight. Geneva: World Health Organization, 2015. [Google Scholar]
- 3. Monteiro CA, Mondini L, Souza ALM, Popkin BM. Da desnutrição para a obesidade: a transição nutricional no Brasil. Velhos e novos males da saúde no Brasil: a evolução do país e de suas doenças São Paulo: Hucitec 1995:247–55.
- 4. WHO . Obesity: Preventing and Managing the Global Epidemic. Geneva: World Health Organization, 2000. [PubMed] [Google Scholar]
- 5. Jung R. Obesity as a disease. Br Med Bull 1997; 53:307–21. [DOI] [PubMed] [Google Scholar]
- 6. Henriksen EJ, Diamond‐Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med 2011; 51:993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cildir G, Akincilar SC, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med 2013; 19:487–500. [DOI] [PubMed] [Google Scholar]
- 8. Guzmán‐Flores JM, López‐Briones S. Células de la inmunidad innata y adaptativa en la diabetes mellitus tipo 2 y obesidad. Gaceta Médica de México 2012; 148:381–9. [PubMed] [Google Scholar]
- 9. Sundara Rajan S, Longhi MP. Dendritic cells and adipose tissue. Immunology 2016; 149:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lynch L. Adipose invariant natural killer T cells. Immunology 2014; 142:337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cipolletta D. Adipose tissue‐resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology 2014; 142:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chung Y, Dong C. Don't leave home without it: the IL‐23 visa to TH‐17 cells. Nat Immunol 2009; 10:236–8. [DOI] [PubMed] [Google Scholar]
- 13. Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, et al Obesity predisposes to Th17 bias. Eur J Immunol 2009; 39:2629–35. [DOI] [PubMed] [Google Scholar]
- 14. Eljaafari A, Robert M, Chehimi M, Chanon S, Durand C, Vial G, et al Adipose tissue‐derived stem cells from obese subjects contribute to inflammation and reduced insulin response in adipocytes through differential regulation of the Th1/Th17 balance and monocyte activation. Diabetes 2015; 64:2477–88. [DOI] [PubMed] [Google Scholar]
- 15. Hong CP, Park A, Yang BG, Yun CH, Kwak MJ, Lee GW, et al Gut‐specific delivery of T‐helper 17 cells reduces obesity and insulin resistance in mice. Gastroenterology 2017; 152:1998–2010. [DOI] [PubMed] [Google Scholar]
- 16. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low‐grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012; 3:279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al Metagenomic analysis of the human distal gut microbiome. Science 2006; 312:1355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 2012; 148:1258–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol 2013; 10:637–44. [DOI] [PubMed] [Google Scholar]
- 20. Shih VF, Cox J, Kljavin NM, Dengler HS, Reichelt M, Kumar P, et al Homeostatic IL‐23 receptor signaling limits Th17 response through IL‐22‐mediated containment of commensal microbiota. Proc Natl Acad Sci U S A 2014; 111:13942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carlos D, Yaochite JN, Rocha FA, Toso VD, Malmegrim KC, Ramos SG, et al Mast cells control insulitis and increase Treg cells to confer protection against STZ‐induced type 1 diabetes in mice. Eur J Immunol 2015; 45:2873–85. [DOI] [PubMed] [Google Scholar]
- 22. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014; 158:1000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012; 37:158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med 2016; 213:1223–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999–2000. JAMA 2002; 288:1723–7. [DOI] [PubMed] [Google Scholar]
- 27. Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell 2013; 152:673–84. [DOI] [PubMed] [Google Scholar]
- 28. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin‐12/interleukin‐23 production and the T‐helper 17 response in humans. Immunol Rev 2008; 226:112–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y, Tian J, Tian X, Tang X, Rui K, Tong J, et al Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS ONE 2014; 9:e92450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Upadhyay V, Poroyko V, Kim TJ, Devkota S, Fu S, Liu D, et al Lymphotoxin regulates commensal responses to enable diet‐induced obesity. Nat Immunol 2012; 13:947–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003; 37:917–23. [DOI] [PubMed] [Google Scholar]
- 33. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012; 148:852–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Avramoglu RK, Basciano H, Adeli K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin Chim Acta 2006; 368:1–19. [DOI] [PubMed] [Google Scholar]
- 35. He B, Wu L, Xie W, Shao Y, Jiang J, Zhao Z, et al The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol 2017; 18:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011; 11:98–107. [DOI] [PubMed] [Google Scholar]
- 37. Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell 2012; 148:1160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al Signaling by IL‐6 promotes alternative activation of macrophages to limit endotoxemia and obesity‐associated resistance to insulin. Nat Immunol 2014; 15:423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, et al Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med 2014; 20:1417–26. [DOI] [PubMed] [Google Scholar]
- 40. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al Specific microbiota direct the differentiation of IL‐17‐producing T‐helper cells in the mucosa of the small intestine. Cell Host Microbe 2008; 4:337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chabaud M, Page G, Miossec P. Enhancing effect of IL‐1, IL‐17, and TNF‐α on macrophage inflammatory protein‐3α production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol 2001; 167:6015–20. [DOI] [PubMed] [Google Scholar]
- 42. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL‐17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 2007; 25:821–52. [DOI] [PubMed] [Google Scholar]
- 43. Hergott CB, Roche AM, Tamashiro E, Clarke TB, Bailey AG, Laughlin A, et al Peptidoglycan from the gut microbiota governs the lifespan of circulating phagocytes at homeostasis. Blood 2016; 127:2460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mansuy‐Aubert V, Zhou QL, Xie X, Gong Z, Huang JY, Khan AR, et al Imbalance between neutrophil elastase and its inhibitor α1‐antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab 2013; 17:534–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Molloy MJ, Grainger JR, Bouladoux N, Hand TW, Koo LY, Naik S, et al Intraluminal containment of commensal outgrowth in the gut during infection‐induced dysbiosis. Cell Host Microbe 2013; 14:318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeng C, Shi X, Zhang B, Liu H, Zhang L, Ding W, et al The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med (Berl) 2012; 90:175–86. [DOI] [PubMed] [Google Scholar]
- 47. Varol C, Vallon‐Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, et al Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 2009; 31:502–12. [DOI] [PubMed] [Google Scholar]
- 48. Tait Wojno ED, Artis D. Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell Host Microbe 2012; 12:445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cupedo T. Human lymph node development: an inflammatory interaction. Immunol Lett 2011; 138:4–6. [DOI] [PubMed] [Google Scholar]
- 50. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444:1022–3. [DOI] [PubMed] [Google Scholar]
- 51. Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al Differential adaptation of human gut microbiota to bariatric surgery‐induced weight loss: links with metabolic and low‐grade inflammation markers. Diabetes 2010; 59:3049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe‐host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 2014; 6:220ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karlsson CL, Onnerfalt J, Xu J, Molin G, Ahrne S, Thorngren‐Jerneck K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity (Silver Spring) 2012; 20:2257–61. [DOI] [PubMed] [Google Scholar]
- 54. Santacruz A, Collado MC, Garcia‐Valdes L, Segura MT, Martin‐Lagos JA, Anjos T, et al Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr 2010; 104:83–92. [DOI] [PubMed] [Google Scholar]
- 55. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al Cross‐talk between Akkermansia muciniphila and intestinal epithelium controls diet‐induced obesity. Proc Natl Acad Sci U S A 2013; 110:9066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005; 102:11070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56:1761–72. [DOI] [PubMed] [Google Scholar]
- 58. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP‐2‐driven improvement of gut permeability. Gut 2009; 58:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]