

Abstract
The intrinsically disordered ATHP3 was studied at native conditions and in complex with DNA using single amino acid substitutions and high-resolution ion mobility spectrometry coupled to mass spectrometry (trapped IMS-MS). Results showed that ATHP3 can exist in multiple conformations at native conditions (at least 10 conformers were separated), with a variety of proline cis/trans orientations, side chain orientations and protonation sites. When in complex with AT rich DNA hairpins, the -RGRP- core is essential for stabilizing the ATHP3: DNA complex. In particular, the arginine in the sixth position plays an important role during binding to AT-rich regions of hairpin DNA, in good agreement with previous NMR and X-ray data. Mobility based correlation matrices are proposed as a way to reveal differences in structural motifs across the peptide mutants based on the conformational space and relative conformer abundance.
Introduction
The mammalian high mobility group protein (HMGA2, MW ~12 kDa), an intrinsically disordered protein (IDP) that aids in the regulation and expression of certain genes by influencing the remodeling of chromatin structure1–3, is also a known oncofetal antigen1,4, biomarker of cell transformation and metastasis5,6, and a target in cancer therapeutics7,8. In vivo studies have largely established the physiological role of the full length HMGA2 by monitoring the effects of overexpression and knockouts in mice models9. Specifically, HMGA2 gene amplification induces tumorigenesis, as observed in several cancers, including leukemia10, tongue cancer11, lung cancer12, breast cancer13,14 and pituitary adenomas15. A pygmy phenotype was observed in null mice6, whereas the truncated version of the protein is expressed in oversized mice16. More recently, in vitro analysis implicated HMGA2 in the process of aging17. HMGA2 contains three positively charged motifs, known as AT-hook peptides (ATHPs)18, that bind with high affinity AT-rich DNA duplex regions19, and a negatively charged c-terminal tail. HMGA2 can easily form homodimers20, an interaction most likely attributed to contacts forming between the acidic c-terminal tail of one protein with the basic residues of the counterpart protein, and interact with DNA and other proteins, making HMGA2 a protein with a large interaction network21. Despite progress made using in vivo and in vitro studies, there are no structural models for HMGA2, and only a few studies have contributed to the structural characterization (e.g., NMR and crystallography studies) of ATHP bound to duplex DNA22,23.
Ion mobility spectrometry and mass spectrometry (IMS-MS) have shown advantages for structural biology, and more specifically, for the study of biomolecules and biomolecular complexes without the influence of the solvent24–27. IMS-MS has addressed specific shortcomings of current conventional methods of structural analysis (i.e., NMR and X-ray crystallography) by reducing analysis time, sample consumption, sample purity requirements and restriction on biomolecule flexibility28,29. IMS-MS has gained great attention for the study of protein dynamics in their native, partially folded and unfolded forms24,30–32, and when complemented with molecular dynamic simulations, has allowed the identification of specific intramolecular interactions that stabilize conformational states (i.e.; proline and side-chain orientation, protonation site)33. IMS-MS can capture conformational interconversions and intermediate states for peptides, proteins, DNA and their complexes24,25,34. For example, in a recent study, the influence of the solvent composition on the ATHP3 conformational space and ATHP3 kinetically trapped intermediates were reported33. At the protein level, IMS-MS studies have shown that phosphorylation of the c-terminal tail can play a vital role during HMGA2 protein-protein and protein-DNA interactions35.
In the present study, the peptide sequence influence on the conformational space and DNA binding for the intrinsically disordered ATHP3 is studied using trapped ion mobility spectrometry with collision induced dissociation coupled to mass spectrometry (TIMS-CID-TOF MS) and single amino acid substitutions. A discussion on the conformational space as a function of the protonation sites, side chain and proline cis/trans orientation and the binding affinity to AT-rich DNA follows.
Methods
Sample preparation
Native AT-hook peptide 3 (Lys-Arg-Pro-Arg-Gly-Arg-Pro-Arg-Lys-Trp) and all amino acid substituted peptides were purchased from GenScript and used without further purification. An AT-rich DNA oligomer, denoted as FL876, sequence GGATATTGCCCCCGCAATATCC (C212H270N79O130P21, MW 6655.1561) was purchased from Eurofins Genomics (Luxembourg City, Luxembourg) and used as received. This 22 nucleotide DNA hairpin contains a 9 base pair stem comprised of a 5 base pair AT rich region in the middle of the stem. Solvents and ammonium acetate salts utilized in this study were analytical grade or better and purchased from Fisher Scientific (Pittsburgh, PA). A Tuning Mix calibration standard (G24221A) was obtained from Agilent Technologies (Santa Clara, CA) and used as received.
Peptide nomenclature
The amino acid sequence of the ATHP3 and variants are presented in Table S1, along with the nomenclature followed throughout the text. Specifically, the variant peptides are referred to by the original amino acid residue followed by their position and the replacement amino acid in a single-code nomenclature.
Ion Mobility Spectrometry-Mass Spectrometry
Details regarding the trapped IMS (TIMS) operation and specifics compared to traditional IMS can be found elsewhere33,36–39. Briefly, a custom nESI-TIMS unit was coupled to a Maxis Impact Q-TOF mass spectrometer (Bruker, Billerica, MA). The TIMS unit is run by custom software in LabView (National Instruments) synchronized with the MS platform controls. Sample aliquots (10 μL) were loaded in a pulled-tip capillary biased at 700–1200 V to the MS inlet. The nitrogen bath gas flow is defined by the pressure differential between the entrance funnel (P1 = 2.6 mbar) and the exit funnel (P2 = 1.1 mbar) at ca. 294 K. A 880 kHz and 200 Vpp rf was applied. Deflector, capillary, entrance funnel, entrance and exit analyzer voltages were 60/−150, 50, 0, −200-0, and 60 V in positive mode (and −60/150, −50, 200-0, and −60 V in negative mode) to prevent ion heating prior to IMS separation. The reduced mobility, K, of an ion in a TIMS cell is described by:
| 1 |
where vg, E, Velution and Vout are the gas velocity, applied electric field, elution voltage and exit analyzer voltage, respectively. After thermalization, species elute from the TIMS cell by decreasing the electric field in stepwise decrements (referred to as the “ramp”) and can be described by a characteristic elution voltage (Velution). The mobility calibration constant A was determined using known reduced mobilities of Tuning Mix components (K0 of 1.013, 0.835, and 0.740 cm2/(V.s) for respective m/z 622, 922, and 1222). The scan rate (Sr = ΔVramp/tramp) was optimized for every experiment.
The measured mobilities were converted into CCS (Ω, Ų) using the Mason-Schamp equation:
| 2 |
where z is the charge of the ion, kB is the Boltzmann constant, N* is the number density of the bath gas and mi and mb refer to the masses of the ion and bath gas, respectively. TIMS-MS spectra were analyzed using Compass Data Analysis 5.0 (Bruker Daltonik GmbH) and TIMS Data Viewer 1.4.0.31397 (Bruker Daltonics Inc, Billerica). Heat maps of the corrected and normalized IMS profiles were built to facilitate the comparison between the experimental conditions explored.
The IMS corrected profiles were compared using the correlation coefficient function:
| 3 |
where and are the sample means average for IMS profile 1 and 2. Notice that in this approach, the entire profile is considered to obtain the correlation coefficient.
Correction of amino acid substituted collision cross sections
For direct comparison of the mobility profiles between the ATHP 3 and single amino acid substituted peptides (e.g., Arg to Ala, Pro to Ala and Trp to Ala), the CCS profiles were adjusted based on the method previously described in references26,40–42. To account for differences in CCS between N2 and He, the CCSN2 = 1.0857 (CCSHe) + 81.459 [Å2] conversion was used43,44. This resulted in Arg to Ala, Pro to Ala and Trp to Ala substitutions to be corrected by 17.43 Å2, 2.68 Å2 and 10.50 Å2 in N2, respectively.
Determination of binding affinities
Peptide-DNA binding affinities (Ka) were calculated using the general equation for an association reaction: peak area of the complex divided by the peak area of the unbound DNA and unbound peptide45:
| 4 |
A distance matrix was utilized to better evaluate relative changes associated with the single amino acid substitution using the equation:
| 5 |
Discussion
Three charge states in positive ion mode ([M + H]+ to [M + 3 H]3+) were observed for ATHP3 and the seven amino acid substituted peptides (see corrected CCS profiles and fingerprint in Fig. 1a,b) under native conditions (e.g., 10 mM ammonium acetate, pH ~6.8). A correlation matrix of the IMS fingerprints (Fig. 1c) is used for rapid comparison and identification of the single amino acid substitution that possess the smallest/largest influence in the ATHP3 mobility profile (first column and first row), as well as between the mutants. For example, following the color scale, P3A and R8A are the most and R2A is the least closely related mutants to ATHP3 under native conditions. That is, a quick assessment of the influence of peptide sequence (e.g., single amino acid substitution) on the peptide secondary structure can be perform.
Figure 1.
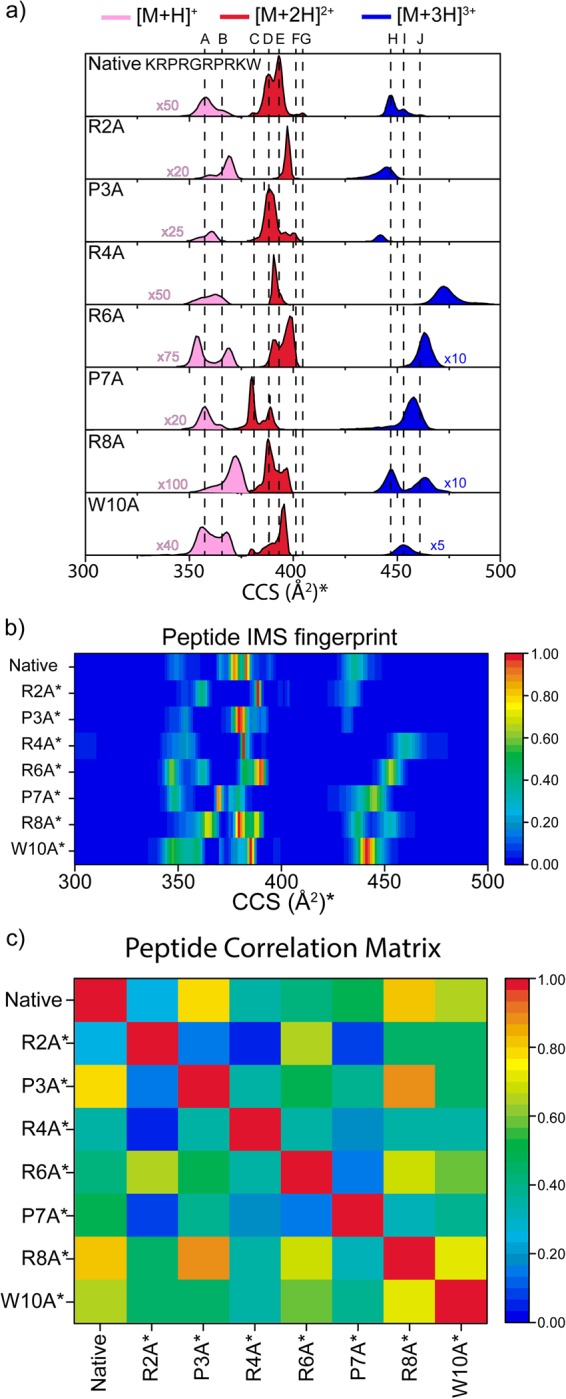
Typical, normalized ATHP3 and corrected variant (*) mobility profiles for the [M + H]+, [M + 2 H]2+ and [M + 3 H]3+ for intrinsic size parameters upon residue substitution to alanine (a). Peptide IMS fingerprint (b) and correlation matrix (c) are used for assessment of the amino acid sequence effect on the secondary structure.
One of the advantages of using single point residue mutations in combination with IMS-MS is the possibility to identify the conformational state of the prolines (i.e., cis/trans orientation), the side chain orientations and charge (proton) location by comparison of the corrected IMS profiles26. Prolines in the trans position require to overcome a ~13 kcal mol−1 energy barrier in order to convert to the cis position (the trans state is ~0.5 kcal mol−1 lower in energy than the cis form)46. However, alanine residues (not bound to proline) are energetically more favorable in the trans position (the trans state is ~2.5 kcal mol−1 lower in energy with a trans to cis ~20 kcal mol−1 barrier46). This means that the determination of the ATHP3 trans or cis state can be accomplished by looking at the conservation (proline in trans) or absence (proline in cis) of each of IMS band of the native ATHP3 peptide26. That is, comparison of the corrected profiles for native, P3A and P7A permits the assignment of the proline bands in the native ATHP3 (results summarized in Table 1). Analogous, we can suggest that a single point mutation of a basic residue (e.g., R) allows for the assignment of the proton location (or charge) in the native ATHP3, since the loss of a charged basic residue (R to A mutation) very likely promotes the disruption of proton-assisted, intramolecular interactions that stabilizes a conformational state. Specifically, the determination of the ATHP3 charge location can be accomplished by looking at the conservation (N) or absence (+1) of the IMS band after the mutation of a basic residue with a neutral residue (e.g., R to A mutation). If an IMS band is present in the ATHP3 and the ATHP3 RxA modified peptides profiles, there is likely no proton-assisted intramolecular interaction at that position. In the case of the ATHP3, up to seven protons can be attached; however, during native conditions (pH ~ 6.7), a maximum of three protons were observed (i.e., +1– +3 charge states). Comparison of the ATHP3 profile with those of R2A, R4A, R6A and R8A permitted the evaluation of intramolecular interactions involving a proton in the basic residue of the native, intrinsically disordered ATHP3 (see Table 1). Knowing the total peptide charge and likelihood of the basic residues to be protonated, the protonation probability of the N-term and the lysine at position one was estimated (i.e., difference from the total charge state and number of protonated arginines). In addition to this information, the W10A substitution provides insight in the role of the tryptophan on the overall conformational space (i.e., is it coordinating or non-coordinating with other residues). Overall, the high number of IMS bands can be attributed to conformations with different locations of the protons (i.e., protomers), potential constraints generated from the proline configuration and orientations of the side chains.
Table 1.
Summary of charge location, proline cis/trans configuration and side chain orientation (W10) of the Native ATHP3 per IMS band (A–J).
| [M + H]+ | [M + 2 H]2+ | [M + 3 H]3+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | J | |
| Residue | ||||||||||
| R2 | +1* | N | +1* | +1* | +1 | +1* | +1* | N | +1* | +1 |
| P3 | trans | cis | trans | trans | trans | trans | cis | cis | cis | cis |
| R4 | N | N | +1* | +1* | +1 | +1* | +1* | +1 | +1* | +1 |
| R6 | N | N | +1* | +1* | N | N | +1* | +1 | +1* | N |
| P7 | trans | trans | trans | trans | cis | cis | cis | cis | trans | trans |
| R8 | +1* | N | N | N | N | +1* | +1* | N | +1* | N |
| W10 | NC | NC | NC | NC | NC | C | C | C | NC | NC |
| N-ter/K | N | +1 | N | N | N | N | N | +1 | N | +1 |
N = neutral. C = coordinated bond. NC = non-coordinated bond. *Denotes cases where charge assignment between basic residues cannot be distinguished.
Inspection of Table 1 shows that ATHP 3 [M + H]+ conformers (i.e., A and B bands) differ in the orientation of P3 (trans vs. cis) and the location of the charge (R2/R8 vs. N-term/K). In the case of ATHP 3 [M + 2 H]+2, differences between the five bands (C-G) are also associated to the orientation of the prolines P3 and P7, the protonation of the basic residues R2, R4, R6 and R8, and the coordination of W10. In the case of ATHP [M + 3 H]+3, differences between the three bands (H–J) are associated to the proline P7, the protonation of the basic residues R2, R4, R6 and R8, and the coordination of W10. While in some cases the charge residue that is protonated can be uniquely assigned (bands B, E and J), for the other bands we cannot discriminate (denoted with a * in Table 1); that is, different protonation patterns can share the same IMS band. In summary, the data presented in Table 1 shows that the native, intrinsically disordered ATHP3 can exist in a variety of conformations (at least 10), with different proline configurations, protonation sites and side chain orientations.
Peptide-DNA complexes were formed in solution and observed in the gas-phase at [M + 4 H]4+ and [M + 5 H]5+ charge states under native conditions for all considered peptide mutants (Fig. 2a). Beside the peptide-DNA complexes, unbound DNA and peptides were also observed at [M + 3 H]3+ – [M + 4 H]4+ and at [M + H]+ – [M + 3 H]3+ charge states, respectively. IMS profiles of the peptide-DNA complexes (Fig. 2b,c) can be characterized by a single, broad mobility band. Inspection of the peptide-DNA plots, generated from the corrected IMS profiles, allows for a quick description of the influence of the single amino acid substitution on the structure of the peptide-DNA complex (Fig. 2c). Further information can be extracted from the correlation matrix (Fig. 2d). If the single amino acid substitution does not dramatically affect the corrected IMS profiles (similar fingerprint) or has a high correlation value, then most likely this amino acid has a lower role during the peptide -DNA complex formation and is not an active participant in the binding site. For example, comparison of the ATHP3 and the R2A/R4A/R8A/W10A peptides shows higher similarities and correlation values, which suggest that these amino acids are less likely to regulate and participate in the peptide-DNA complex interaction. Moreover, P3A/R6A/P7A showed larger differences and lower correlation values, which suggest that there are most likely involved in the peptide-DNA complex interaction. These observations are in good agreement with previous NMR results that suggest that the -PRGRP- core most likely interacts with the DNA22. A crystal structure of the ATHP3 in complex with duplex B-form DNA revealed that the central core arginine residues protrude inward creating hydrogen bonds from the amino acid NH group to the thymine oxygen atom23. Nevertheless, it should be pointed out that multiple conformations can lead to a similar CCS (or IMS bands) and in the case of the peptide-DNA complexes, the largest contribution to the total CCS is from the DNA.
Figure 2.

Typical mass spectra (a) and ‘corrected’ IMS profiles (b) of the native and substituted ATHP3: DNA complexes. The IMS profile fingerprint (c) of the complexes was used to generate a correlation matrix (d). Peptide: DNA: complex ratios are reported in the inset in (a).
A complementary way to evaluate the influence of each amino acid on the ATHP3: DNA complex is through the inspection of the dissociation curve as a function of the collision energy (Fig. 3a). The assumption is that the rupture of the intramolecular interactions that stabilizes the peptide-DNA complex using single amino acid substitutions will result in different collision energies required to disrupt the complex. Inspection of Fig. 3a shows that the R6A peptide presents the largest change in dissociation profiles, which suggest that the arginine in the sixth position is essential for the stabilization of the ATHP3: DNA complex in the -RGR- core, in good agreement with previous NMR observations22,47.
Figure 3.
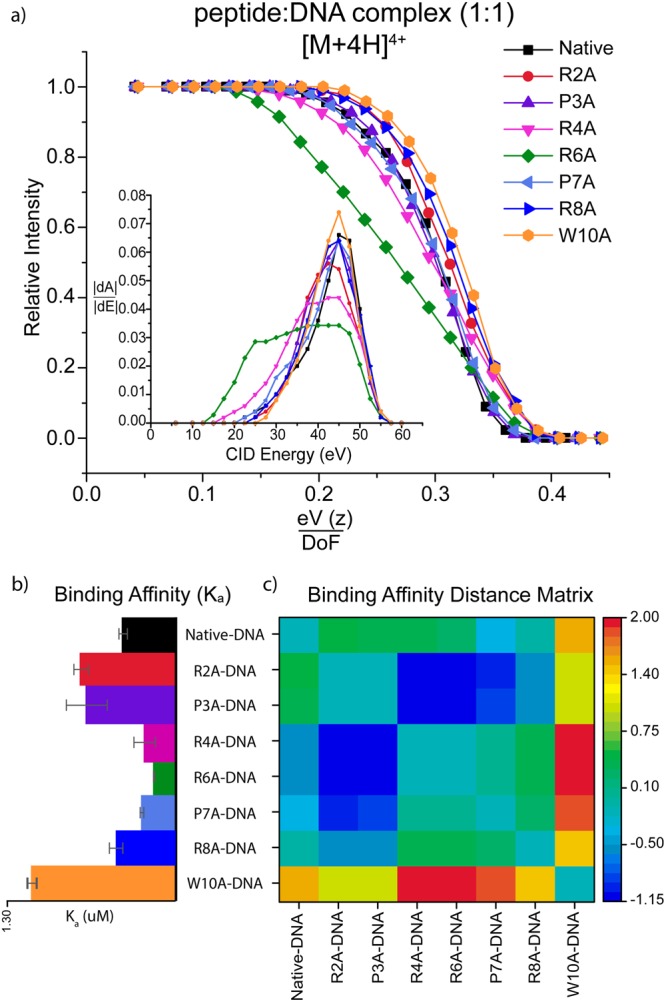
Typical CID profiles (a) showing the decrease in intensity of the [M + 4 H]4+ peptide: DNA complexes as a function of collision energy (eV), charge (z) and degrees of freedom (DoF). The dissociation threshold (inset) shows the absolute derivative of each CID profiles as a function of the collision energy. The binding affinity (b) and distance matrix (c) illustrates the influence of the single amino acid substitution on the peptide-DNA binding affinity.
The probability of peptide: DNA complex formation (or binding affinity) can also be evaluated by the differences in the relative abundance of the product (i.e., complex) with respect to the initial reactants (i.e., peptide and DNA) using equations 4 and 5 (Fig. 3b,c). Inspection of Fig. 3b,c shows that the substitutions R2A/P3A/W10A has higher binding affinity (Ka) relative to the ATHP3 peptide, while the substitutions R4A/R6A/P7A has lower binding affinity compared to the ATHP3. These results suggest that R4A/R6A/P7A are directly related to the ATHP3: DNA complex formation.
Conclusion
The intrinsically disordered ATHP3 peptide in complex with DNA was studied using single amino acid substitutions and trapped IMS-MS at native conditions. Seven amino acid substitutions were considered and changes in the peptide and peptide-DNA complexes conformational space as well as their relative abundance and dissociation kinetics were used to establish the amino acid positions that stabilize the ATHP3:DNA complex formation in solution. The high resolution of the trapped IMS (TIMS) analyzer permitted the observation of multiple IMS bands for the intrinsically disordered ATHP3 peptide (at least 10 conformers). Using the corrected IMS profiles, the proline orientations (cis-trans), protonation site (i.e., basic residues and N-terminal), and side chain orientation (W10) per IMS band of the native ATHP3 were determined, providing a detailed description of the intramolecular interactions that drive the native ATHP3 conformational space. That is, these studies provided significant insight on the critical role of cis and trans proline configurations, charge location, and W10 coordination in the populations of ATHP3 structures that are observed in native conditions.
The study of peptide-DNA complexes suggests that the -RGRP- core is essential for stabilizing ATHP3: DNA complexes at native conditions. Moreover, results showed that the weakest peptide: DNA complex is formed with R6A, suggesting that this basic residue plays the most important role during binding to AT-rich regions of the DNA, in good agreement with previous NMR and X-ray data22,23.
It should be noted that the trapped IMS-MS workflow permits the study of multiple kinetically trapped conformations simultaneously, in contrast to the most abundant (or energetically favored) typically described using NMR and X-Ray measurements. This makes trapped IMS-MS a powerful platform for the study of intrinsically disordered biomolecules and their kinetically trapped conformers. This work highlights the potential of trapped IMS-MS combined with single amino acid substitutions for the evaluation of the peptide sequence influence on the secondary structure of peptides at native conditions. When combined with CID-MS, this workflow can be translated to the study of peptide/protein -ligand complexes. A potential limitation of the workflow is the need for higher mobility resolution as the size and complexity of the biological system increases. With the high resolving power of the TIMS analyzer (R up to 400), we estimate that this workflow can be easily extended to peptides with 35–40 amino acid residues.
Electronic supplementary material
Acknowledgements
The authors acknowledge the financial support from the National Science Foundation Division of Chemistry, under CAREER award CHE-1654274, with co-funding from the Division of Molecular and Cellular Biosciences to FFL. AG will like to acknowledge the 2017–2018 Dissertation Year Fellowship from Florida International University.
Author Contributions
F.F.L. designed and oversaw the entire project along with writing and reviewing the manuscript. A.G. conducted all the key experiments, analyzed the data and wrote the manuscript. A.B. assisted during sample preparation, data processing and contributed to the writing of the manuscript. F.L. made equal contributions to the experimental design and to the writing and reviewing of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-28956-z.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 2.Zhou X, Chada K. HMGI family proteins: architectural transcription factors in mammalian development and cancer. Keio J.Med. 1998;47:73–77. doi: 10.2302/kjm.47.73. [DOI] [PubMed] [Google Scholar]
- 3.Baldassarre G, et al. Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI-C gene by the chronic stimulation of the IL-2 and IL-15 pathway. Proc. Natl. Acad. Sci. USA. 2001;98:7970–7975. doi: 10.1073/pnas.141224998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sgarra R, et al. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004;574:1–8. doi: 10.1016/j.febslet.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Morishita A, et al. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013;73:4289–4299. doi: 10.1158/0008-5472.CAN-12-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Benson KF, Ashar HR, Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995;376:771–774. doi: 10.1038/376771a0. [DOI] [PubMed] [Google Scholar]
- 7.Fedele M, Fusco A. HMGA and cancer. Biochim. Biophys. Acta. 2010;1799:48–54. doi: 10.1016/j.bbagrm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Rajamani D, Bhasin MK. Identification of key regulators of pancreatic cancer progression through multidimensional systems-level analysis. Genome Med. 2016;8:38. doi: 10.1186/s13073-016-0282-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashar HR, Chouinard RA, Jr., Dokur M, Chada K. In vivo modulation of HMGA2 expression. Biochim. Biophys. Acta. 2010;1799:55–61. doi: 10.1016/j.bbagrm.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 10.Efanov A, et al. Human HMGA2 protein overexpressed in mice induces precursor T-cell lymphoblastic leukemia. Blood Cancer J. 2014;4:e227. doi: 10.1038/bcj.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X-P, et al. Overexpression of HMGA2 promotes tongue cancer metastasis through EMT pathway. J. Transl. Med. 2016;14:26. doi: 10.1186/s12967-016-0777-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Cello F, et al. HMGA2 Participates in Transformation in Human Lung Cancer. Mol. Cancer Res. MCR. 2008;6:743–750. doi: 10.1158/1541-7786.MCR-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rogalla P, et al. Expression of HMGI-C, a member of the high mobility group protein family, in a subset of breast cancers: relationship to histologic grade. Mol. Carcinog. 1997;19:153–156. doi: 10.1002/(SICI)1098-2744(199707)19:3<153::AID-MC2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 14.Sun M, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA. 2013;110:9920–9925. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fedele M, Pierantoni GM, Visone R, Fusco A. Critical role of the HMGA2 gene in pituitary adenomas. Cell cycle (Georgetown, Tex.) 2006;5:2045–2048. doi: 10.4161/cc.5.18.3211. [DOI] [PubMed] [Google Scholar]
- 16.Battista S, et al. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999;59:4793–4797. [PubMed] [Google Scholar]
- 17.Yu KR, et al. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell res. 2013;10:156–165. doi: 10.1016/j.scr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Manfioletti G, et al. cDNA cloning of the HMGI-C phosphoprotein, a nuclear protein associated with neoplastic and undifferentiated phenotypes. Nucleic Acids Res. 1991;19:6793–6797. doi: 10.1093/nar/19.24.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson KR, Lehn DA, Reeves R. Alternative processing of mRNAs encoding mammalian chromosomal high-mobility-group proteins HMG-I and HMG-Y. Mol. Cell. Bio. 1989;9:2114–2123. doi: 10.1128/MCB.9.5.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frost L, et al. The Dimerization State of the Mammalian High Mobility Group Protein AT-Hook 2 (HMGA2) PLoS ONne. 2015;10:e0130478. doi: 10.1371/journal.pone.0130478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sgarra R, et al. HMGA molecular network: From transcriptional regulation to chromatin remodeling. Biochim. Biophys. Acta. 2010;1799:37–47. doi: 10.1016/j.bbagrm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Huth JR, et al. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat. Struct. Bio. 1997;4:657–665. doi: 10.1038/nsb0897-657. [DOI] [PubMed] [Google Scholar]
- 23.Fonfria-Subiros E, et al. Crystal structure of a complex of DNA with one AT-hook of HMGA1. PLoS One. 2012;7:e37120. doi: 10.1371/journal.pone.0037120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molano-Arevalo JC, et al. Characterization of Intramolecular Interactions of Cytochrome c Using Hydrogen–Deuterium Exchange-Trapped Ion Mobility Spectrometry–Mass Spectrometry and Molecular Dynamics. Anal. Chem. 2017;89:8757–8765. doi: 10.1021/acs.analchem.7b00844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schenk ER, et al. Kinetic Intermediates of Holo- and Apo-Myoglobin Studied Using HDX-TIMS-MS and Molecular Dynamic Simulations. J. Am. Soc. Mass Spectrom. 2015;26:555–563. doi: 10.1007/s13361-014-1067-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierson NA, Chen L, Russell DH, Clemmer DE. Cis–Trans Isomerizations of Proline Residues Are Key to Bradykinin Conformations. J. Am. Chem. Soc. 2013;135:3186–3192. doi: 10.1021/ja3114505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierson NA, Chen L, Valentine SJ, Russell DH, Clemmer DE. Number of Solution States of Bradykinin from Ion Mobility and Mass Spectrometry Measurements. J. Am. Chem. Soc. 2011;133:13810–13813. doi: 10.1021/ja203895j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJR. Ion mobility mass spectrometry of proteins and protein assemblies. Chem. Soc. Rev. 2010;39:1633–1655. doi: 10.1039/B914002F. [DOI] [PubMed] [Google Scholar]
- 29.Robinson CV, Sali A, Baumeister W. The molecular sociology of the cell. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- 30.Benigni P, et al. Towards the analysis of high molecular weight proteins and protein complexes using TIMS-MS. Int. J. Ion Mobil. Spectrom. 2016;19:95–104. doi: 10.1007/s12127-016-0201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruotolo BT, et al. Ion mobility-mass spectrometry reveals long-lived, unfolded intermediates in the dissociation of protein complexes. A Angew. Chem. Int. Ed. 2007;46:8001–8004. doi: 10.1002/anie.200702161. [DOI] [PubMed] [Google Scholar]
- 32.Scott D, Layfield R, Oldham NJ. Ion mobility-mass spectrometry reveals conformational flexibility in the deubiquitinating enzyme USP5. Proteomics. 2015;15:2835–2841. doi: 10.1002/pmic.201400457. [DOI] [PubMed] [Google Scholar]
- 33.Schenk ER, Ridgeway ME, Park MA, Leng F, Fernandez-Lima FA. Isomerization Kinetics of AT Hook Decapeptide Solution Structures. Anal. Chem. 2014;86:1210–1214. doi: 10.1021/ac403386q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garabedian A, et al. Structures of the kinetically trapped i-motif DNA intermediates. Phys. Chem. Chem. Phys. 2016;18:26691–26702. doi: 10.1039/C6CP04418B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurizio E, et al. Conformational Role for the C-Terminal Tail of the Intrinsically Disordered High Mobility Group A (HMGA) Chromatin Factors. J. Proteome Res. 2011;10:3283–3291. doi: 10.1021/pr200116w. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Lima, F. A., Kaplan, D. A. & Park, M. A. Note: Integration of trapped ion mobility spectrometry with mass spectrometry. R Rev. Sci. Instrum. 82 (2011). [DOI] [PMC free article] [PubMed]
- 37.Fernandez-Lima F, Kaplan D, Suetering J, Park M. Gas-phase separation using a trapped ion mobility spectrometer. Int. J. Ion Mobil. Spectrom. 2011;14:93–98. doi: 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez DR, et al. Ion dynamics in a trapped ion mobility spectrometer. Analyst. 2014;139:1913–1921. doi: 10.1039/C3AN02174B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schenk ER, et al. Direct Observation of Differences of Carotenoid Polyene Chain cis/trans Isomers Resulting from Structural Topology. Anal. Chem. 2014;86:2019–2024. doi: 10.1021/ac403153m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valentine SJ, Counterman AE, Clemmer DE. A database of 660 peptide ion cross sections: use of intrinsic size parameters for bona fide predictions of cross sections. J. Am. Soc. Mass Spectrom. 1999;10:1188–1211. doi: 10.1016/S1044-0305(99)00079-3. [DOI] [PubMed] [Google Scholar]
- 41.Srebalus Barnes CA, Clemmer DE. Assessing Intrinsic Side Chain Interactions between i and i + 4 Residues in Solvent-Free Peptides: A Combinatorial Gas-Phase Approach. The J.Phys. Chem A. 2003;107:10566–10579. doi: 10.1021/jp030519s. [DOI] [Google Scholar]
- 42.Dilger JM, Valentine SJ, Glover MS, Ewing MA, Clemmer DE. A database of alkali metal-containing peptide cross sections: Influence of metals on size parameters for specific amino acids. Int. J. Mass Spectrom. 2012;330-332:35–45. doi: 10.1016/j.ijms.2012.05.001. [DOI] [Google Scholar]
- 43.Bush MF, Campuzano IDG, Robinson CV. Ion Mobility Mass Spectrometry of Peptide Ions: Effects of Drift Gas and Calibration Strategies. Anal. Chem. 2012;84:7124–7130. doi: 10.1021/ac3014498. [DOI] [PubMed] [Google Scholar]
- 44.Bush MF, et al. Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, Kitova EN, Klassen JS. Influence of Solution and Gas Phase Processes on Protein−Carbohydrate Binding Affinities Determined by Nanoelectrospray Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2003;75:4945–4955. doi: 10.1021/ac034300l. [DOI] [PubMed] [Google Scholar]
- 46.Stewart DE, Sarkar A, Wampler JE. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 1990;214:253–260. doi: 10.1016/0022-2836(90)90159-J. [DOI] [PubMed] [Google Scholar]
- 47.Reeves R, Nissen MS. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 1990;265:8573–8582. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.