

Abstract
Histone deacetylase 8 (HDAC8) is a well-characterized member of the class I acetyl-lysine deacetylase (HDAC) family. Previous work has shown that the efficiency of HDAC8-catalyzed deacetylation of a methylcoumarin peptide varies depending on the identity of the divalent metal ion in the HDAC8 active site. Here we demonstrate that both HDAC8 activity and substrate selectivity for a diverse range of peptide substrates depends on the identity of the active site metal ion. Varied deacetylase activities of Fe(II)- and Zn(II)-HDAC8 toward an array of peptide substrates were identified using a high-throughput Self-Assembled Monolayers for MALDI-TOF Mass Spectrometry (SAMDI) mass spectrometry screen. Subsequently, the metal dependence of deacetylation of peptides of biological interest was measured using an in vitro peptide assay. While Fe(II)-HDAC8 is generally more active than Zn(II)-HDAC8, the ratio of Fe(II)/Zn(II) HDAC8 activity varies widely (2 to 150) among the peptides tested. These data provide support for the hypothesis that HDAC8 may undergo metal switching in vivo which, in turn, may regulate its activity. However future studies are needed to explore the identity of the metal ion bound to HDAC8 in cells under varied conditions.
Graphical Abstract
Protein lysine acetylation is an enzymatically reversible post translational modification. Acetylation is catalyzed by twenty lysine acetyl transferases (KATs) and hydrolysis of the acetyl moiety is catalyzed by eighteen acetyl-lysine deacetylases, including the metal-dependent histone deacetylases (HDACs) and the NAD(+)-dependent sirtuins (SIRTs). The balance of the enzymatic activities of HDACs and KATs regulates the acetylation state of the >3800 acetylated sites on thousands of proteins in the mammalian proteome involved in many cellular processes.2,3 With such a modest number of enzymes catalyzing the same chemistry on thousands of substrates, it is important to understand the mechanisms by which the selectivity of the enzymes is regulated.4–5 HDACs are medically relevant enzymes; aberrant HDAC activity is implicated in a number of disease states.5 Elucidating the determinants of HDAC substrate specificity will inform the engineering of selectivity into novel therapeutics targeting these enzymes.
HDAC8 is a biochemically well-characterized metal dependent HDAC. However, the protein substrates, cellular role, and regulation are still under investigation. This enzyme is expressed in both the nucleus and the cytoplasm with the highest expression levels in smooth muscle cells.6–7 Predicted HDAC8 substrates include the nuclear structural maintenance of chromosomes 3 (SMC3)8, histone proteins9, and several cytosolic substrates such as estrogen-related receptor alpha (ERRα).10–11 HDAC8 was originally described as a zinc-dependent enzyme because zinc co-purifies with the enzyme and this metal was visualized in the first HDAC8 crystal structure12–13, However several metal ions activate the enzyme. The kcat/KM (catalytic efficiency) trend, measured using a short methylcoumarin peptide substrate, for metal-substituted HDAC8 is: Co(II) > Fe(II) > Zn(II).1 Furthermore, the inhibition constant, Ki, for the T-cell lymphoma drug suberoylanilide hydroxamic acid (SAHA) is different for each metal-substituted HDAC8. The trend of inhibition constants is inverse that of kcat/KM, with Co(II)-HDAC8 binding SAHA most tightly.14 The fact that HDAC8 is activated by several divalent metal ions may suggest metal-dependent regulation of this enzyme in vivo.
Crystal structures of metal-substituted HDACs have not provided an explanation for the differential activation and inhibition. Structures of Fe(II)-, Co(II)-and Zn(II)-HDAC8 bound to the hydroxamic acid (metal chelating) inhibitor M344 demonstrate a common ligand coordination geometry for the three metal-substituted enzyme forms.14, The inhibitor-bound structures are a snapshot and cannot show the conformational changes or dynamic interactions that may occur when substrate binds to the enzyme and that may impact the activity and selectivity of HDAC8. Additionally, it is possible that the bound hydroxamic acid stabilizes a common metal coordination state regardless of the metal ion.
Protein metallation in cells is complicated, and not yet well-understood for many enzymes, but it depends on both cellular availability and, in some cases, metallochaperones.15 Either Zn(II)- or Fe(II)-HDAC8 are candidates for the in vivo enzyme form based on cellular metal concentrations and the affinity of HDAC8 for each metal. Exchangeable Zn(II) is present at an estimated 105-fold lower readily exchangeable concentration than Fe(II),16–20 but it has a 106-fold higher affinity for the HDAC8.1, 14 Fe(II)-HDAC8 is sensitive to oxidation and is not activated by Fe(III); acetyl-lysine deacetylase activity in both bacterial and mammalian cell lysates is oxygen sensitive, suggesting the presence of Fe(II)-dependent activity.1, 21 Moreover, immunopurified HDAC8 overexpressed in HeLa tissue culture cells demonstrates oxygen-sensitive activity as well.21 Taken together, the data to date suggest that iron may play a role in cellular HDAC8 activation and demonstrate the importance of determining which metal(s) activate and regulate HDAC8 in vivo.
Prompted by the difference in metal-dependent activity and the HDAC8 oxygen sensitivity, we further investigated the extent of the effect of metal ion identity on HDAC8 activity. Here we show, for the first time, that HDAC8 substrate selectivity depends on the identity of the metal ion at the active site; the selectivity toward peptide substrates changes in concert with the active site metal ion. This work suggests a new mechanism by which the specificity of HDAC8 may be regulated, with implications for cellular regulation of acetylation and deacetylase-targeting therapeutics.
METHODS
Metal free HEPES, NaCl, KCl and NaOH were purchased from Sigma. TCEP was purchased from GoldBio. All other reagents were purchased from Fisher unless otherwise specified.
Recombinant HDAC8 purification
HDAC8 was prepared using the following method, modified from (13). HDAC88-TEV-6His was transformed into BL21-DE3 Z-competent cells and grown in 2xYT media supplemented with 100 μg/mL ampicillin at 37°C until OD600 = 0.4–0.7. The temperature was decreased to 20°C for 45–60 minutes, followed by induction with isopropyl β-D-1-thiogalactopyranoside (IPTG) (0.5 mM) and addition of ZnSO4 (0.2 mM). Cells were harvested 15–16 hours post-induction by centrifugation (4000 × g, 15–20 minutes, 4°C) and resuspended in HDAC8 purification buffer (30 mM HEPES, pH 8, 150 mM NaCl, 1 mM imidazole, and 1 mM TCEP) supplemented with a complete protease inhibitor cocktail tablet (Roche). Cells were lysed using a microfluidizer (Microfluidics), followed by nucleic acid precipitation with polyethyleneimine (pH 7.9) and centrifugation (27,000 × g, 45 minutes, 4°C). The supernatant containing HDAC8 was loaded onto a Ni2+-charged chelating sepharose (GE Healthcare) gravity column and equilibrated with HDAC8 purification buffer. The column was washed with 20 mM imidazole purification buffer and HDAC8 was eluted using a linear gradient (25 – 250 mM imidazole). The 6His tag was cleaved using a 6His-tagged TEV protease during an overnight dialysis against HDAC8 purification buffer without imidazole. A second, stepwise-elution from a Ni2+-charged column was used to separate HDAC8 from TEV protease. HDAC8 was concentrated in 30k MWCO Amicon Ultra centrifugal concentrators and dialyzed against metal chelating buffer A (25 mM MOPS, pH 7.5, 1 mM TCEP, 5 mM KCl and 1 mM EDTA) followed by several, serial dialyses against metal free buffer B (25 mM MOPS pH 7.5, 1 mM TCEP and 5 mM KCl). For MALDI deacetylation assays, a PD-10 desalting column (GE Healthcare) in either PD-10 buffer A (25 mM HEPES pH 7.8, 150 mM NaCl, 3 mM KCl and 1 mM TCEP) or PD-10 buffer B (25 mM MOPS pH 7.5, 1 mM TCEP) was used to remove residual EDTA. HDAC8 was aliquoted, flash frozen in liquid nitrogen, and stored at −80°C. Concentration was measured by absorbance at 280 nm using the extinction coefficient of 52,120 M−1cm−1, which was determined previously.13 ICP-MS confirmed less than 10% Zn(II) present in the final enzyme sample.
High-throughput SAMDI mass spectrometry deacetylation assays
Selectivity screens were performed using Self-Assembled Monolayers for MALDI-TOF Mass Spectrometry (SAMDI). The SAMDI assays were performed as previously described.16 Peptides of varying sequence were transferred to a SAMDI array plate containing 384 gold spots, each with a monolayer presenting a maleimide group at a density of 10% against a background of tri-(ethylene glycol) groups. In this way, each peptide was immobilized to an individual spot through reaction of the thiol side chain of the terminal cysteine residue with a maleimide while the glycol groups prevent non-specific adsorption of proteins onto the monolayer. The peptide array was incubated with HDAC8, which had been reconstituted at 1:1 enzyme: metal ion, by dispensing 3 μL of a solution (0.5 μM enzyme, 25 mM Tris pH 8.0, 147 mM NaCl, 3 mM KCl) using a 12-channel pipette. Solutions were kept at 37°C for 30 minutes followed by stopping the reaction by rinsing the array plate with ethanol.
Enzyme-coupled assay for measuring deacetylation of non-methylcoumarin peptides
Peptides (Peptide 2.0 and/or Synthetic Biomolecules) were synthesized with an acetylated N-terminus and carboxyamide C-terminus. Zn(II)-HDAC8 and Fe(II)-HDAC8 were reconstituted as follows. Apo-HDAC8 (10 μM) was reconstituted with stoichiometric Zn(II) (Fluka) in peptide assay buffer (25 mM HEPES pH 8, 137 mM NaCl, 3 mM KCl) and incubated for 1 hour on ice. For Fe(II)-HDAC8, apo-HDAC8 was equilibrated in an anaerobic glove box (Coy Laboratory Products) for one hour prior to reconstitution. Solid FeCl2 (Sigma), L(+)-ascorbic acid (Fluka), and peptide assay buffer were equilibrated in the anaerobic chamber at least overnight. Fe(II) (100 μM) in 5 mM ascorbate and assay buffer was prepared daily. Fe(II)-HDAC8 (10 μM) was reconstituted anaerobically with 5-fold excess Fe(II) in assay buffer and 2.5 mM ascorbate for 1 hour in a 0 – 4°C CoolBox (Biocision) and maintained on ice until use. Assays with Fe(II)-HDAC8 were performed aerobically on ice within two hours, the effective working time for ascorbic acid to preserve Fe(II)-HDAC8 activity.22 The enzyme-coupled assay couples acetate production to the formation of NADH and was performed as described.19 Fe(II) - and Zn(II)-HDAC8 and peptides in assay buffer were individually equilibrated at 30°C for 15 minutes prior to reaction initiation. Reactions were initiated by the addition of enzyme (1 μM) to various concentrations of substrates (25 – 400 μM). Time points were quenched in 10% hydrochloric acid (HCl), flash frozen in liquid nitrogen and stored at −80°C. Assay workup was performed as described.19 Standards were prepared using acetic acid (Ricca Chemical Company). After thawing, time points were neutralized with 6% sodium bicarbonate (NaHCO3), centrifuged (16,000 × g, 1 minute), and added to an equilibrated coupled enzyme solution in a 96-well plate (Corning). The fluorescence of the resulting NADH was measured (ex. =340 nm, em. = 460 nm) and the initial rate was calculated from the time dependence of NADH production. The Michaelis-Menten (MM) equation or a line was fit to the concentration dependence of the initial rate to calculate kinetic parameters (kcat, KM and kcat/KM).
Assay for measuring methylcoumarin-labeled peptides
Peptides containing a methylcoumarin-bound C-terminus were measured using the Fluor de Lys (FdL) assay (Enzo Life Sciences). Deacetylation of the substrates catalyzed by HDAC8 is followed by cleavage of the amide bond linking the C-terminal methylcoumarin to the peptide backbone catalyzed by trypsin, resulting in a fluorescence shift between the deacetylated product (ex. = 340 nm, em. = 450 nm) and the remaining substrate (ex. = 340 nm, em. = 380 nm). HDAC8 was reconstituted and equilibrated with either Fe(II) or Zn(II) as described previously. Reactions were initiated by the addition of enzyme (1 μM) to various concentrations of substrate (25 – 200 μM). Time points were quenched by the addition of trichostatin A (TSA) and trypsin developer. The initial rate was calculated from the time dependence of the changes in fluorescence with concentrations determined from a standard curve. The Michaelis-Menten (MM) equation or a line was fit to the concentration dependence of the initial rate to calculate kinetic parameters (kcat, KM and kcat/KM).
RESULTS
SAMDI deacetylation assays
As an initial screen to evaluate the substrate selectivity of HDAC8 reconstituted with either Zn(II) or Fe(II), we used SAMDI mass spectrometry to profile the reactivity of HDAC8 with a peptide array23 and found that even with short (6 amino acid) peptides the substrate selectivity of HDAC8 is metal ion-dependent. The peptides were of the form GXKAcZGC and were attached to a plate through reaction of the thiol side chain of the cysteine residue with a maleimide. The flanking residues X and Z were varied across nineteen amino acids (all natural residues excluding cysteine). In side-by-side experiments, we reconstituted apo-HDAC8 with either Fe(II) or Zn(II) and reacted each of these enzyme forms with the peptide array. After incubation, the monolayers were analyzed by matrix-assisted laser desorptionionization mass spectrometry using the SAMDI method to observe the masses of the substrate and product of the reaction. The extent of deacetylation for each peptide was determined by the ratio of the deacetylated peak area to the sum of the peak areas for the substrate and product.
Both Fe(II)- and Zn(II)-HDAC8 catalyze deacetylation of many of the acetylated peptides in the array (Figure 1A–B). For Zn(II)-HDAC8, 172 of the peptides resulted in no significant HDAC8 activity (< 3% deacetylation), 72 peptides showed moderate HDAC8 activity (3–15% conversion) and 117 peptides showed high activity (> 15% conversion). Similarly, Fe(II)-HDAC8 was inactive toward 139 of the peptides, moderately active toward 62 peptides, and highly active toward 160 peptides. These data are presented in heatmaps for each Me(II)-HDAC8 (Figure 1A, B).
Figure 1. Heat maps visualizing HDAC8 activity and selectivity.
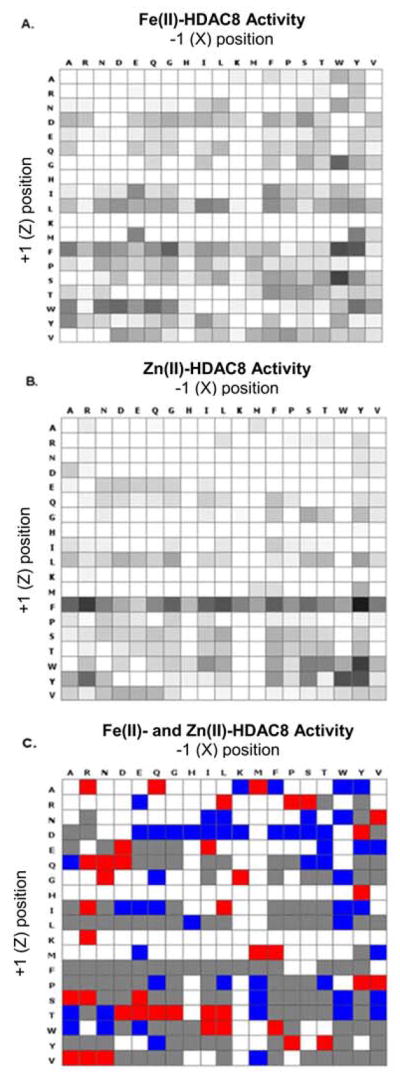
The selectivity of (A) Zn(II)- and (B) Fe(II)-bound HDAC8 was determined by applying each metal-substituted enzyme form to an array of 361 peptides of the sequence GXKAcZGC. The extent of deacetylation of each peptide is shown in grey scale on the heatmaps. (C) A metal-dependent peptide selectivity heat map was generated by taking the ratio of Zn(II)-HDAC8 to Fe(II)-HDAC8 product conversion for each peptide. Peptides with a greater than seven-fold preference for Zn(II)-HDAC8 are shown in red and Fe(II)-selective peptides are shown in blue. Peptides that were deacetylated similarly by both enzyme forms are shown in grey and peptides that were not deacetylated (<3%) by either enzyme form are shown in white.
To quantify differences in reactivity of the metal-substituted enzymes, we calculated the ratio of the Zn(II)-HDAC8 to Fe(II)-HDAC8 product conversion and we generated a specificity heat map where the peptides are binned into four categories: little preference (gray, 122 peptides), higher reactivity with Zn(II)-HDAC8 (red, 40 peptides), higher reactivity with Fe(II)-HDAC8 (blue, 54 peptides) and little reactivity with HDAC8 (gray, 145 peptides) (Figure 1C). This heat map demonstrates that the substrate selectivity of HDAC8 depends on the identity of the active site metal ion. In the screen, approximately 15% of the peptides were better substrates for Fe(II)-HDAC8 and 11% were better substrates for Zn(II)-HDAC8 while 34% of the peptides had comparable reactivity with both enzyme forms, and 40% of the peptides resulted in negligible deacetylation for at least one form of the enzyme.
In addition to differences in selectivity based on the active site metal ion, the arrays suggest general peptide sequence selectivity trends for HDAC8. For example, both HDAC8 metalloforms display a preference for peptides containing an aromatic side chain (F, W or Y) on the C-terminal side of the acetyl-lysine residue, while methionine in the Z position was largely unfavorable to both enzyme forms.
Kinetic assays quantify metal-dependent substrate selectivity for Fe(II)- and Zn(II)-HDAC8
To further test the dependence of substrate selectivity on the identity of the active site metal ion, we next probed metal-dependent selectivity using in vitro kinetic assays in solution. Concurrent work to identify HDAC substrates has suggested interesting putative protein substrates, and we selected peptides from these proteins to enhance the biological relevance of the results. We assayed deacetylation of peptides (listed in Table 1) taken from acetylated proteins that have been proposed as in vivo substrates of HDAC8 using proteomic and computational methods.24–25 The peptide length was varied to enhance solubility.
Table 1.
Fe(II)/Zn(II)-HDAC8 reactivity ratios for deacetylation of various peptide substrates. Metal selectivity and reactivity varied significantly with both the identity of the active site divalent metal ion and the residues at the +1 and −1 positions of the acetyl lysine.
| Protein | Sequence | Fe(II) kcat/KM (M−1s−1) | Zn(II) kcat/KM (M−1s−1) | Fe(II)/Zn(II) Ratio |
|---|---|---|---|---|
| H3K9 13mer | TKQTARK(ac)STGGKA | 290 ± 20 | 50 ± 5 | 6 ± 1 |
| La-related protein 1 | LGK(ac)FRR | 1080 ± 180 | 7 ± 1 | 154 ± 33 |
| SMC3 9mer | RVIGAKK(ac)DQ | 240 ± 80 | 14 ± 2 | 17 ± 6 |
| SMC3 10mer | RVIGAKK(ac)DQY | 220 ± 40 | 36 ± 2 | 6 ± 1 |
| CREB94 | CKDLK(ac)RLFS | 8 ± 0.5 | 2 ± 0.2 | 4 ± 0.5 |
| THRAP3 | LGDGK(ac)MKS | 20 ± 8 | 10 ± 1 | 2 ± 0.8 |
| CSRP2BP | STPVK(ac)FISR | 660 ± 120 | 40 ± 11 | 17 ± 5 |
| HDAC8 Fluor-de-Lys | RHK(ac)K(ac)-coumarin | 2300 ± 160A | 800 ± 50A | 3 ± 0.2A |
| SIRT1 Fluor-de-Lys | RHKK(ac)-coumarin | 6150 ± 870 | 460 ± 35 | 13 ± 2 |
| Boc-K(ac)- Fluor-de-Lys | Boc-K(ac)-coumarin | 975 ± 120 | 255 ± 25 | 4 ± 0.6 |
Data for the HDAC8 Fluor-de-Lys peptide was adapted from (1).
Deacetylation was quantified using an acetate-NADH coupled assay that measures the conversion of acetate product to NADH by fluorescence.26–27 The quenched assay was performed using Zn(II)- or Fe(II)-HDAC8 under multiple turnover conditions. The dependence of the initial rates of peptide deacetylation on the peptide concentration was used to calculate kcat/KM values (see Figure 2). We also measured the reactivity and metal selectivity of several commercially available coumarin-labeled peptides using changes in fluorescence upon deacetylation.
Figure 2. Representative peptide assay data.
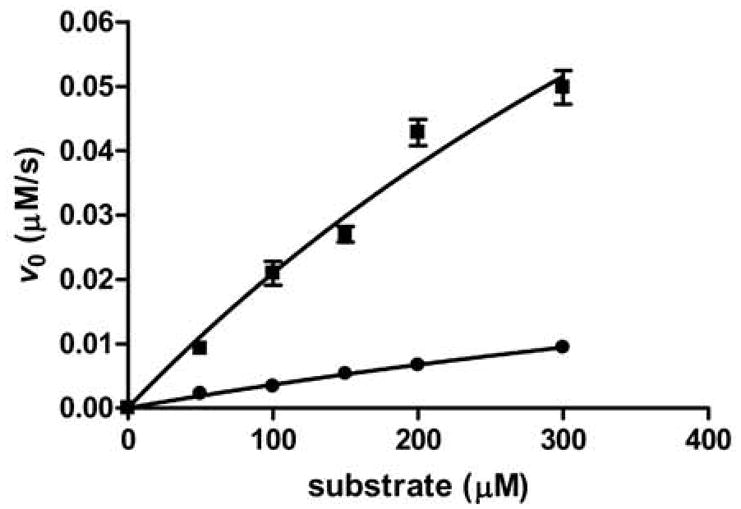
The dependence of the initial rate on substrate concentration for Fe(II)-(■) and Zn(II)- (●) HDAC8-catalyzed deacetylation of SMC3 10-mer peptide measured using the acetate assay. A hyperbola was fit to the data to calculate the Michaelis-Menten parameters
The catalytic efficiency for deacetylation of these peptides ranged from 2 to 800 M−1s−1 for Zn(II)-HDAC8 and 8 to 6000 M−1 s−1 for Fe(II)-HDAC8. The Fe(II)-HDAC8 enzyme was faster for all of the peptides tested in solution. The ratio of kcat/KM values varied widely for the two metal enzyme forms, as predicted by the HDAC8 selectivity in the initial screen. The Fe(II)-to Zn(II)-HDAC8 ratio of the kcat/KM values ranged from 2 to 154 (Figure 3, Table 1). Variations in the metal-dependent activity (ratios of 3 – 13) are also observed for the commercially available coumarin-labeled peptides, despite the higher values of kcat/KM. In general, an increase in the value of kcat/KM for Fe(II)-HDAC8 correlates with an increase in the Fe(II)/Zn(II) selectivity ratio.
Figure 3. Fe(II)/Zn(II) reactivity ratios of various peptide substrates with HDAC8.
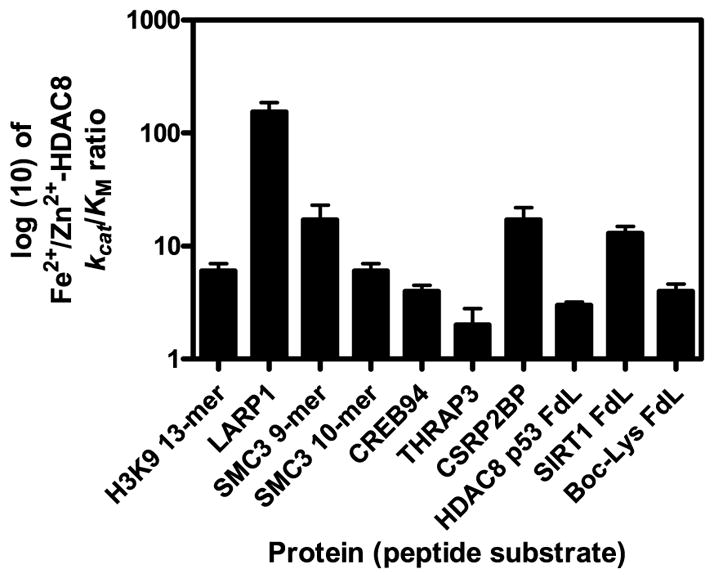
Metal reactivity and selectivity varied significantly with the sequence of the peptide. Commercially available methylcoumarin bound substrates were used as controls. HDAC8 p53 FdL data are reported in (1).
The lack of peptides with higher kcat/KM values for Zn(II)-HDAC8 compared to Fe(II)-HDAC8 is a major difference between the SAMDI screen and the solution measurements. This difference likely reflects alterations in the peptide structures and the assay conditions. First, the Fe(II)-HDAC8 activity in the SAMDI assay may be underreported as the enzyme was reconstituted with stoichiometric iron and was thus likely not saturated with Fe(II). Additionally, the SAMDI screen uses maleimide-modified peptides that are attached to a PEG-derivatized gold surface compared to the solution conditions of the peptide assays and the SAMDI screen may measure values for kcat rather than kcat/KM Nonetheless, both assay methods clearly demonstrate that the Fe(II)/Zn(II) selectivity ratio varies significantly for different peptides.
DISCUSSION
Our in-solution peptide assays provide insight into several determinants of HDAC8 selectivity. First, while we did not set out to analyze the role of peptide length, our data suggest that this is not a primary determinant of selectivity, consistent with previous work.28 The peptide with the largest Fe(II)/Zn(II) specificity ratio was 6 amino acids long while peptides with ratios between 2 and 20 were 8 to 9 amino acids long.
Second, these data suggest that the peptide sequence is an important determinant of metal-dependent selectivity. For example, the two SMC3 peptides differ only by the presence or absence of a tyrosine at the +3 position, which leads to a significant difference in metal-dependent selectivity (17 ± 6 for the 9-mer and 6 ± 1 for the 10-mer). In this case, the change in the ratio results from a 2.5-fold decrease in the Zn(II)-dependent activity upon removal of the +3 Tyr residue. However, an aromatic side chain at the +3 position is not sufficient to lead to high catalytic activity or a large Fe(II)/Zn(II) activity ratio. CREB94 has a Phe at the +3 position, but is a poor substrate for both metal-bound HDAC8 forms. The activity of Zn(II)-HDAC8 and Fe(II)-HDAC8 toward the CREB94 peptide is about 2 M−1 s−1 and 8 M−1 s−1, respectively, making the selectivity ratio about 4.
The SAMDI screen predicts that HDAC8 has selectivity for an aromatic side chain at the +1 position, consistent with previous data. 23 Consistent with this, both of the peptides containing a Phe side chain at the +1 position (La-related protein 1 and CSRP2BP) have the highest kcat/KM values for Fe(II)-HDAC8 and are both Fe(II)-selective peptides with Fe(II)/Zn(II) ratios of 154 and 17, respectively (Table 1). These two peptides share the following sequences: +1 Phe, −3 aliphatic, and +3/4 arginine. Nonetheless, the remaining alterations in sequence lead to the almost 10-fold difference in the Fe(II)/Zn(II) ratio. This alteration is due to an almost 2-fold decrease in activity of Fe(II)-HDAC8 toward CSRP2BP compared to La-related protein 1 combined with a 5-fold increase in the Zn(II)-HDAC8 activity toward deacetylation of CSRP2BP compared to La-related protein 1.
We attempted to predict the metal selectivity based solely on the +1 (Z) and −1 (X) positions using the data from the SAMDI screen. This was successful for the two SMC3 peptides (X= K, Z=D), predicting peptides that would react more rapidly with Fe(II)-HDAC8. However, the predictions for the other peptides were incorrect. The H3K9- and CREB-derived peptides were predicted as zinc-selective substrates, however our kinetic analysis shows that these peptides react more rapidly with Fe(II)-HDAC8. Finally, the SAMDI screen suggests that peptides containing the +1 (Z) and −1 (X) amino acids observed in the THRAP3- and CSRP2BP-derived peptides should be inactive. This lack of correlation between the predictions from the SAMDI screen and the peptide kinetic data demonstrate that although the amino acids at the +1 and −1 positions contribute to reactivity and metal-dependent selectivity, the residues at the other sites in the sequence are also important determinants of HDAC8 selectivity. Consistent with this, previous studies investigating the correlation between peptide sequence and HDAC8 activity using kinetic and computational studies have demonstrated that the 3 amino acids on both sides of the acetyl-lysine contribute to reactivity.25, 23 These data in our current work highlight the nuance of HDAC8 peptide sequence selectivity and demonstrate the complexity that the metal ion identity adds to substrate recognition.
Regulation of HDAC8 activity by metal-switching
The data presented here reveal novel insights into HDAC8 substrate selectivity and highlight the importance of addressing HDAC8 metal selectivity in both in vitro and in vivo studies, a facet of HDAC biochemistry that has been neglected. The activity data demonstrate that Fe(II)-HDAC8 catalyzes deacetylation of peptides comparable to or faster than Zn(II)-HDAC8. Although both protein structure and long-range HDAC8-substrate interactions contribute to substrate selectivity28–29, peptides mimic short-range substrate interactions22 and are a useful predictor of possible cellular substrates. Thus, these peptide data suggest that either metal-bound form of HDAC8 could be a relevant in vivo deacetylase, and that manipulating the metal-substituted HDAC8 identity in the cell could alter the pool of recognized substrates and thereby influence downstream cellular effects.
We showed previously that Fe(II)-HDAC8 had a higher kcat/KM value than Zn(II)-HDAC8 for the one commercial peptide tested.1 Here we broaden the scope of peptide substrates and show that even among short 6-mer peptides, which interact only with the active site and substrate binding surface directly near the active site, there are significant differences in selectivity based on the identity of the metal ion. If the Fe(II)/Zn(II) activity ratio was constant, this would suggest intrinsic differences in the reactivity of the active site metal ion. However, the peptide-dependent variation in the Fe(II)/Zn(II) activity ratio suggests a more complicated mechanism of regulation involving both metals. In this case, variation of the active site metal ion alters both HDAC8 reactivity and selectivity. The structural basis for the metal-dependent substrate selectivity is unclear. Crystal structures of HDAC8 with bound ligands indicate that the substrate binding site is primarily composed of flexible loops that accommodate a range of substrates but also influence the enzyme’s specificity10, 14, 30–35. Furthermore, the residues that coordinate the active site metal ion (His 180, Asp267 and Asp 178) are positioned by these loops.14 Intrinsic properties of the metal ion, including Lewis acidity and size, could influence the structure and dynamics of the loop regions and alter the binding interface presented to substrates. Additionally, altering the active site metal ion coordination is expected to propagate structural changes to the peptide binding site via the residues in the hydrophobic shell around the metal ligands.36 However, alterations in the geometry of the metal polyhedron have not yet been observed; structures of Fe(II)-, Co(II)- and Zn(II)-HDAC8 bound to the hydroxamic acid inhibitor M344 demonstrate a common ligand coordination geometry for the three metal-substituted enzyme forms. 14 The bound hydroxamic acid may stabilize the metal coordination geometry in the active site of HDAC8.
These results suggest that metal-dependent selectivity of HDAC may be important for regulating deacetylation in the cell. The readily exchangeable concentrations of Zn(II) and Fe(II) in cells is estimated to be in the pM and μM ranges, respectively, under normal conditions.16–20 Therefore, HDAC8 could bind either Zn(II) or Fe(II) in cells despite the 106-fold higher affinity for Zn(II).1, 14 Furthermore, the relative concentrations of readily exchangeable Zn(II) and Fe(II) increase and decrease depending on the cellular context, suggesting the possibility of metal switching in response to cellular conditions.37–38 Zn(II) concentrations can change by several orders of magnitude, from picomolar to nanomolar levels.39 For example, cellular zinc is generally tightly buffered but the concentration of exchangeable Zn(II) increases under redox stress as protein thiol groups that coordinate zinc are oxidized, releasing Zn(II). 39–40 The dependence of Fe(II) concentration on the redox state of the cell is unclear, although it is reasonable to assume that it might decrease under oxidative stress.41
Metal-dependant substrate selectivity and inhibitor selectivity have been observed in bacterial methionine aminopeptidases, as well as other enzymes. Trypanosoma brucei methionine aminopeptidases 1, while likely a zinc enzyme in vivo, is activated by multiple metal ions (Co(II), Zn(II), Ni(II), Mn(II), Fe(II) and displays metal-dependent substrate selectivity for short peptide substrates.42 E. coli methionine aminopeptidase demonstrates metal-dependent inhibitor selectivity (Fe(II), Co(II), and Mn(II)).43 Klebsiela oxytoce acire-ductone dioxygenase is an example of a known cambialistic enzyme; if Fe(II) is bound it catalyzes one reaction, and if Ni(II) or Co(II) is bound it catalyzes a different chemical reaction.44 The metal-dependent bacterial deacetylase LpxC from E. coli has been shown to bind either Zn(II) or Fe(II) based on the relative abundance of these metals in the cell.45 In this case, Fe(II)-LpxC has 8-fold higher activity than the zinc-bound enzyme. We would expect a similar model for metal-dependent HDACs; Fe(II)-HDAC8 (KD 0.2 – 1 μM)14, 46 and/or apo-HDAC8 may exist when the Zn(II) concentration is low. However, upon an increase in the exchangeable Zn(II) concentration HDAC8 could exchange the active site metal ion to form Zn(II)-HDAC14,46, maintaining active HDAC8 but altering the activity level and substrate selectivity.
The trend of greater catalytic efficiency for the iron-bound enzyme among more physiological peptides is consistent with the hypothesis that HDAC8 is activated, at least in part, by Fe(II) in the cell. Interestingly, Fe(II)-HDAC8 activity toward these peptides shows a correlation with the Fe/Zn specificity ratio. It is possible that a high ratio of iron to zinc activity may be indicative of HDAC8 substrates in vivo. This is supported by the fact that the three highest Fe(II)/Zn(II) ratios correspond to proteins recently identified as potential HDAC8 substrates in a proteomics screen.24 Conversely, despite in vitro activity, evidence is beginning to accumulate suggesting that histones may not be principal targets of HDAC8 in vivo24, 47–48 and this is consistent with the H3K9 13-mer peptide having a Fe(II)/Zn(II) ratio less than 10.
This study is the first to demonstrate that the peptide sequence selectivity of HDAC8 varies with the identity of the active site metal ion. The SAMDI peptide screen enabled a broad survey of enzyme selectivity, and the enzyme assays in solution demonstrate varied Fe/Zn selectivities toward substrates of likely physiological relevance. In vivo evidence consistent with the hypothesis of metal switching regulation is still needed, however the data presented here are consistent with the possibility that alteration in cellular conditions that alter metal ion concentrations are coupled to modulation of deacetylation of target proteins by dictating the identity of the active site metal ion.
Acknowledgments
Funding Sources
This work was funded by National Institutes of Health (NIH) Grant 5-R01-GM-040602 (CAF), F31-GM-116619 (JEL), the University of Michigan Chemistry-Biology Interface (CBI) training program NIH grant 5T32GM008597 (CAC), and Rackham Graduate School (CAC, JEL). SAMDI screens were funded by the NTU-NU Institute for NanoMedicine located at the International Institute for Nanotechnology, Northwestern University, USA and the Nanyang Technological University, Singapore; Agmt 10/20/14
Footnotes
ASSOCIATED CONTENT
Full details of experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.”
Notes
The authors declare no competing financial interests
References
- 1.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45(19):6170–8. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 2.Norris KL, Lee J-Y, Yao TP. Acetylation Goes Global: The Emergence of Acetylation Biology. Science Signaling. 2009;2(97):76. doi: 10.1126/scisignal.297pe76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Scientific reports. 2011:1. doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith KT, Workman JL. Introducing the acetylome. Nature biotechnology. 2009;27(10):917–9. doi: 10.1038/nbt1009-917. [DOI] [PubMed] [Google Scholar]
- 5.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waltregny D, de Leval L, Glénisson W, Ly Tran S, North BJ, Bellahcène A, Weidle U, Verdin E, Castronovo V. Expression of Histone Deacetylase 8, a Class I Histone Deacetylase, Is Restricted to Cells Showing Smooth Muscle Differentiation in Normal Human Tissues. The American Journal of Pathology. 2004;165(2):553–564. doi: 10.1016/S0002-9440(10)63320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu E, Chen Z, Fredrickson T, Zhu Y, Kirkpatrick R, Zhang GF, Johanson K, Sung CM, Liu R, Winkler J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. The Journal of biological chemistry. 2000;275(20):15254–64. doi: 10.1074/jbc.M908988199. [DOI] [PubMed] [Google Scholar]
- 8.Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, Gil-Rodríguez C, Arnedo M, Loeys B, Kline AD, Wilson M, Lillquist K, Siu V, Ramos FJ, Musio A, Jackson LS, Dorsett D, Krantz ID. Mutations in Cohesin Complex Members SMC3 and SMC1A Cause a Mild Variant of Cornelia de Lange Syndrome with Predominant Mental Retardation. The American Journal of Human Genetics. 2007;80(3):485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buggy JJ, Sideris ML, Mak P, Lorimer DD, McIntosh B, Clark JM. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem J. 2000;350(Pt 1):199–205. [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: Histones and beyond. Biopolymers. 2013;99(2):112–126. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguere V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Molecular endocrinology. 2010;24(7):1349–58. doi: 10.1210/me.2009-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401(6749):188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 13.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(42):15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling DP, Gattis SG, Fierke CA, Christianson DW. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry. 2010;49(24):5048–56. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster AW, Osman D, Robinson NJ. Metal preferences and metallation. J Biol Chem. 2014;289(41):28095–103. doi: 10.1074/jbc.R114.588145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bozym RA, Thompson RB, Stoddard AK, Fierke CA. Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem Biol. 2006;1(2):103–11. doi: 10.1021/cb500043a. [DOI] [PubMed] [Google Scholar]
- 17.Vinkenborg JL, Nicolson TJ, Bellomo EA, Koay MS, Rutter GA, Merkx M. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat Methods. 2009;6(10):737–40. doi: 10.1038/nmeth.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10(6):997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin Y, Dittmer PJ, Park JG, Jansen KB, Palmer AE. Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc Natl Acad Sci USA. 2011;108(18):7351–6. doi: 10.1073/pnas.1015686108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik I. Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem. 1997;248(1):31–40. doi: 10.1006/abio.1997.2126. [DOI] [PubMed] [Google Scholar]
- 21.Gattis SG. Dissertation. University of Michigan; 2010. Mechanism and metal specificity of zinc-depdendent deacetylases. [Google Scholar]
- 22.Kim BC. Probing the determinants of the molecular recognition in metal-dependent deacetylase. University of Michigan; 2014. [Google Scholar]
- 23.Gurard-Levin ZA, Kim J, Mrksich M. Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. Chembiochem: a European journal of chemical biology. 2009;10(13):2159–61. doi: 10.1002/cbic.200900417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, Svinkina T, Natoli T, Lu X, Paulk J, McCarren P, Wagner FF, Barker D, Howe E, Lazzaro F, Gale JP, Zhang Y-L, Subramanian A, Fierke CA, Carr SA, Holson EB. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS chemical biology. 2014;9(10):2210–6. doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alam N, Zimmerman L, Wolfson Noah A, Joseph Caleb G, Fierke Carol A, Schueler-Furman O. Structure-Based Identification of HDAC8 Non-histone Substrates. Structure. 2016;24(3):458–468. doi: 10.1016/j.str.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfson NA, Pitcairn CA, Sullivan ED, Joseph CG, Fierke CA. An enzyme-coupled assay measuring acetate production for profiling histone deacetylase specificity. Analytical Biochemistry. 2014;456(0):61–69. doi: 10.1016/j.ab.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baumann M, Stürmer R, Bornscheuer UT. A High-Throughput-Screening Method for the Identification of Active and Enantioselective Hydrolases. Angewandte Chemie International Edition. 2001;40(22):4201–4204. doi: 10.1002/1521-3773(20011119)40:22<4201::AID-ANIE4201>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 28.Castaneda CA, Wolfson NA, Leng KR, Kuo Y-M, Leng KR, Andrews AJ, Fierke CA. Histone deacetylase 8 substrate specificity for full-length histone substrates is determined by long- and short-range interactions. doi: 10.1074/jbc.M117.811026. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gurard-Levin ZA, Mrksich M. The Activity of HDAC8 Depends on Local and Distal Sequences of Its Peptide Substrates. Biochemistry. 2008;47:6242–6250. doi: 10.1021/bi800053v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47(51):13554–63. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8(9):879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12(7):1325–34. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 33.Decroos C, Clausen DJ, Haines BE, Wiest O, Williams RM, Christianson DW. Variable Active Site Loop Conformations Accommodate the Binding of Macrocyclic Largazole Analogues to HDAC8. Biochemistry. 2015;54(12):2126–35. doi: 10.1021/acs.biochem.5b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(42):15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. Journal of the American Chemical Society. 2011;133(32):12474–7. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim B, Pitcairn CA, Lopez JE, Fierke CA. Second Shell Residues Modulate the Reactivity and Metal Selectivity of Histone Deacetylase. :8. In preparation. [Google Scholar]
- 37.Kim AM, Vogt S, O’Halloran TV, Woodruff TK. Zinc availability regulates exit from meiosis in maturing mammalian oocytes. Nat Chem Biol. 2010;6(9):674–81. doi: 10.1038/nchembio.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim AM, Bernhardt ML, Kong BY, Ahn RW, Vogt S, Woodruff TK, O’Halloran TV. Zinc sparks are triggered by fertilization and facilitate cell cycle resumption in mammalian eggs. ACS chemical biology. 2011;6(7):716–23. doi: 10.1021/cb200084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maret W, Krężel A. Cellular Zinc and Redox Buffering Capacity of Metallothionein/Thionein in Health and Disease. Molecular Medicine. 2007;13(7–8):371–375. doi: 10.2119/2007-00036.Maret. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kröncke K-D. Cellular stress and intracellular zinc dyshomeostasis. Archives of Biochemistry and Biophysics. 2007;463(2):183–187. doi: 10.1016/j.abb.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxidants & redox signaling. 2006;8(9–10):1419–1441. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- 42.Marschner A, Klein CD. Metal promiscuity and metal-dependent substrate preferences of Trypanosoma brucei methionine aminopeptidase 1. Biochimie. 2015;115:35–43. doi: 10.1016/j.biochi.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 43.Huguet F, Melet A, Alves de Sousa R, Lieutaud A, Chevalier J, Maigre L, Deschamps P, Tomas A, Leulliot N, Pages JM, Artaud I. Hydroxamic acids as potent inhibitors of Fe(II) and Mn(II) E. coli methionine aminopeptidase: biological activities and X-ray structures of oxazole hydroxamate-EcMetAP-Mn complexes. ChemMedChem. 2012;7(6):1020–30. doi: 10.1002/cmdc.201200076. [DOI] [PubMed] [Google Scholar]
- 44.Dai Y, Wensink PC, Abeles RH. One Protein, Two Enzymes. Journal of Biological Chemistry. 1999;274(3):1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 45.Gattis SG, Hernick M, Fierke CA. Active Site Metal Ion in UDP-3-O-((R)-3-Hydroxymyristoyl)-N-acetylglucosamine Deacetylase (LpxC) Switches between Fe(II) and Zn(II) Depending on Cellular Conditions. Journal of Biological Chemistry. 2010;285(44):33788–33796. doi: 10.1074/jbc.M110.147173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim B, Pithadia AS, Fierke CA. Kinetics and Thermodynamics of Metal-binding to Histone Deacetylase 8. Protein Science. 2015;24(3):11. doi: 10.1002/pro.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T, Ota Y, Ri M, Bando M, Gotoh A, Itoh Y, Tsumoto H, Tatum PR, Mizukami T, Nakagawa H, Iida S, Ueda R, Shirahige K, Miyata N. Rapid Discovery of Highly Potent and Selective Inhibitors of Histone Deacetylase 8 Using Click Chemistry to Generate Candidate Libraries. Journal of Medicinal Chemistry. 2012;55(22):9562–9575. doi: 10.1021/jm300837y. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki T, Muto N, Bando M, Itoh Y, Masaki A, Ri M, Ota Y, Nakagawa H, Iida S, Shirahige K, Miyata N. Design, Synthesis, and Biological Activity of NCC149 Derivatives as Histone Deacetylase 8-Selective Inhibitors. ChemMedChem. 2014;9(3):657–664. doi: 10.1002/cmdc.201300414. [DOI] [PubMed] [Google Scholar]