

Abstract
Aerosolized clinical grade azacitidine (Aza) was tested for aerosol quality in vitro and pharmacokinetics and systemic and pulmonary toxicity in vivo in mice. The aerodynamic size range of the aerosol droplets favored the deposit of the drug to the human lower airways and the aerosol process did not compromise the drug activity. No significant myelosuppression, pulmonary toxicity, hepatotoxicity, or nephrotoxicity were observed at a daily dose of 2.5 mg/m2 (0.83 mg/kg) 7 days. Reversible lung inflammation was found in mice treated with aerosolized Aza at dose 3-fold higher than the anticipated therapeutic dose (7.5 mg/m2 or 2.5 mg/kg daily for 7 days) at 3 weeks after the treatment, but not after 6 weeks. In a single dose pharmacokinetic study, aerosolized Aza was found to deposit mainly into the lung with very little drug detected in the circulation. The area under the curve (AUC) of drug in the lung was more than 22-fold greater than that of the peripheral blood: the peak concentration (Cmax) was 7.42 µg/g in lung as compared to 0.28 µg/ml in blood. The total clearance was only 0.010/h in lung vs 0.22/h for blood. In contrast, intravenously (IV) injected Aza resulted in a high Aza concentration in the peripheral blood (Cmax = 4.94 µg/ml), with only trace amounts of drug in the lung (Cmax = 0.35 µg/g). IV Aza was associated with bone marrow toxicity, as it caused significant myelosuppression in the mice. Our results indicate that aerosolized Aza has much less systemic toxicity than IV administered drug and that a safe starting dose of aerosolized Aza for clinical phase I trials in humans should be 2.5 mg/m2 (0.83 mg/kg) daily for 5 to 7 days.
INTRODUCTION
The majority of lung cancer occurs as a result of cumulative damage in the bronchial epithelium caused by inhaled carcinogens1–5. Additionally, damaged airway epithelium is prone to develop second primary malignancies during the lifespan of individuals with the diagnosis of primary lung cancer. Therefore, it is necessary to develop an early therapeutic or prevention method to specifically treat the abnormal airway epithelium. In theory this method should have definite advantages over systemic treatment.
The initial abnormalities in the airway epithelium consist of reversible epigenetic changes before the genetic changes occur. For example, the epigenetic-mediated silencing of tumor suppressor genes by CpG island hypermethylation in promoter regions appears dominant in transcriptional repression, and is tightly associated with the premalignant6 and malignant phenotypes1–5,7, 8, 9. Reversing aberrant DNA hypermethylation in the airway epithelium using an airway-targeted therapy is a promising strategy to prevent primary or secondary lung cancers, and inhibiting the growth of localized lung cancers.
Azacytidine (Aza) is an inhibitor of DNA methyltransferases (DNMT) and a prototypical demethylation agent and has been shown in vitro and in vivo to induce re-expression of genes silenced through promoter hypermethylation10–13. Aza has been approved to treat myelodysplastic syndrome (MDS). Clinical grade azacytidine (Vidaza) is delivered to patients by subcutaneous or intravenous administration (IV) and the primary toxicity of systemic delivery of this drug is myelosuppression. However, laboratory findings have revealed that the concentration of drug needed to induce gene demethylation and re-expression is much lower than that required to produce cytotoxicity14.
We have previously reported the therapeutic advantages of intratracheal and aerosol administration of Aza as compared to intravenous Aza in orthotopic human lung cancers in a mouse xenograft model10, 15. We have also demonstrated that aerosolized Aza can 1) inhibit human orthotopic lung cancers in mouse xenograft models, 2) reduce the promoter methylation of several tumor suppressor genes (TSGs) and increase their protein expression in xenograft models and 3) be better tolerated than IV administration. The use of aerosolized clinical grade azacytidine (Vidaza) and evaluation of its potential systemic and pulmonary toxicity has not been previously studied. In these studies, we examined the suitability of clinical grade azacitidine (Vidaza) for aerosol delivery with particular emphasis on lung deposition and pulmonary and systemic toxicity in mice.
MATERIALS AND METHODS
The drug
All azacitidine used in the toxicity and pharmacokinetic studies was clinical grade azacitidine (Vidaza; Celgene). Vidaza was purchased from the Department of Oncology pharmacy at Montefiore Medical Center as vials of lyophilized power containing 100 mg Aza and 100 mg mannitol in each vial for injection.
Aerosol administration
The aerosol equipment and the administration method were the same as described previously10. Briefly, the aerosol was generated with PARI’s personal compressor and LC Star nebulizer. The nose-only exposure system (CH Technologies, Inc.) linked with PARI’s aerosol system in a closed chemical hood was used for aerosol administration to mice (Figure 1c), and the aerosol time was strictly controlled. All aerosol doses used in the experiments reported in this paper are “lung deposited doses” calculated as previously reported by our group10.
Figure 1.


a and b. Aerodynamic size. The aerodynamic size of azacitidine (Vidaza) in suspension form (a) or solution form (b) were measured separately with extrusion-precipitation method using a 7-Stage Cascade Impactor linked to PARI’s personal compressor and LC star nebulizer system. Aerodynamic size and fraction of aerosol with a particular size range was measured and calculated as per manufacturer’s protocol. The data was mean ± standard deviation of the aerodynamic size based on weight (slant line bars, %Weight) and cumulative weight (empty bars, Accumulated) from 3 independent experiments. c. Aerosol administration system to treat mice. The system includes PARI’s personal compressor and LC Star nebulizer (red arrow) linked to the nose-only exposure system (green arrow) in a closed chemical hood.
The aerodynamic size
Two formulations were tested separately; The suspension formulation was made by adding 4 ml of sterile water for injection, USP (Abbott Laboratories) to 100 mg Vidaza lyophilized powder for a final Aza concentration of 25 mg/ml. The solution formulation was made by adding 10 ml of sterile water to 100 mg Vidaza powder for a final Aza concentration of 10 mg/ml. The aerodynamic diameters of aerosol droplets were determined by extrusion-precipitation method using a seven-stage cascade impactor (In-Tox Products) linked to PARI’s aerosol system described above. The condensed aerosol samples were collected at three different periods, from 3.5 to 4 min, from 7.5 to 8 min, and from 11.5 to 12 min. Aerodynamic size and fraction of aerosol with a particular size range were measured and calculated as per manufacturer’s protocol. The mean and standard deviation were obtained from 3 independent experiments.
Aerosolized Aza activity
Clinical grade azacitidine (Vidaza) was aerosolized with Pari’s aerosol system for more than 1 hour, and the aerosol fog from 58 min to 62 min was condensed into a sterile tube (Aero Vidaza). Aerosolized Mannitol (Sigma) using the same procedure (Aero Mannitol) and non-aerosolized Vidaza (Vidaza) at the same concentration were used as negative and positive controls. The condensed liquids were used to treat the human NSCLC cell line H226 at 0.5 µM for 10 days; the medium and drug were changed every other day. Ten days later, the nuclear proteins of the cells were extracted. EpiQuik DNA methyltransferase (DNMT) activity assay kit (Epigentek, Farmingdale NY) was used to measure DNMT activity quantitatively as per manufacturer’s instructions.
Animals
Male and female ICR mice, 6–8 weeks old (Harlan) were housed in the animal facility at Albert Einstein College of Medicine. All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol (Number: 20130312) was approved by the Institutional Animal Care and Use Committee (IACUC) of Albert Einstein College of Medicine. During the studies, animals were observed daily. Humane endpoints were used during this study: moribund animals were euthanized by CO2 in an inhalation chamber followed by exsanguination. We used two criteria to identify the moribund animals based on our animal use protocol: 1) mouse had difficulty breathing, eating, or drinking; 2) a mouse lost ≥15% body weight over a period of 4 days. Anesthesia (isoflurane) was used to minimize discomfort during blood sampling.
Pharmacokinetics of the aerosolized Aza in mice
Twelve ICR mice (male and female) were divided into 2 groups and given a single dose of either aerosolized or IV Aza (Vidaza) at 2.5 mg/m2 using the method described previously. At 20 min, 2 h, 6 h and 24 h, three mice in each group were euthanized and blood was taken from the abdominal vena cava. The mice were then perfused with saline via the right ventricle and the lungs were excised. Azacytidine in the blood and the lungs was extracted and quantitatively measured by a previously reported method using LC-MS system16. The quantitative detection was performed by Millis Scientific Inc. The sensitivity was 1 ng Aza/ml of sample. The Aza concentration in blood/tissue vs. time data were analyzed and simulated with the best fit (the highest R2 value). The equation of the simulated curves C = f(t) were used to calculated AUC from 0 to 24 hours as AUC = ∫0–24 f(t) dt. Peaking time (T) and peak concentration were obtained directly from observation of the curves.
White blood cell count
ICR mice were distributed into 3 groups of 12 mice (6 male and 6 female) each. Each group was treated once daily for 7 days with either aerosolized Aza at 2.5 mg/m2 (0.83 mg/kg) or 7.5 mg/m2 (2.5 mg/kg), or tail vein injection of Aza at 7.5 mg/m2. The aerosol dose was calculated and controlled as described previously10, 17. Blood samples (50 µl) were obtained from the facial vein18 under isoflurane anesthesia at 1 week prior to and 1, 3, and 6 weeks after the treatment. Red blood cells were removed from the blood samples using RBC lysis buffer (eBioscience, Inc., San Diego, CA) and white blood cells (WBCs) were collected as per the manufacturer’s protocol and counted using a hemocytometer.
Comprehensive blood chemistry
The potential therapeutic dose of aerosol Aza (in our case 2.5 mg/m2) was selected for a more detailed evaluation of the blood counts and chemistries. The blood samples (~400µl) were taken from retro-orbital sinus at the same time point mentioned above. Antech Diagnostics (Lake Success, NY) performed the complete blood count with differential as well as comprehensive blood chemistries.
Pulmonary toxicity hepatoxicity, nephrotoxicity, and myelotoxicity
At week 3 and 6 after the final Aza administration, mice were humanely euthanized and the lungs, kidneys, liver, and sternum were removed and fixed in 10% neutral buffered formalin and routinely processed to paraffin, sectioned to 5 µm, stained with hematoxylin and eosin at the Histology and Comparative Pathology facility at the Albert Einstein College of Medicine (AECOM). All samples were evaluated histologically for evidence of pulmonary toxicity, hepatoxicity, nephrotoxicity, and myelotoxicity, The pulmonary toxicity was determined by pathological evaluation using previously described method17.
RESULTS
Aerodynamic size of clinical grade azacitidine
Aerodynamic size is an important characteristic for predicting the deposition of aerosol droplets and estimate aerosol inhalation efficiency in clinical use. It has been suggested that aerosol droplets < 5 µm, particularly < 3 µm in diameter, deposit most frequently in the lower airways and are therefore appropriate for pharmaceutical inhalation of aerosolized preparations in humans19,20. In order to make a high quality aerosol formulation for further clinical trials, we modified the commercial Aza formulation to have an optimal aerosol droplet size range of < 3~5 µm to optimize lung distribution. Vidaza is a lyophilized power formulation containing 50% of Aza and 50% of mannitol. The manufacturer suggests reconstituting the formulation with sterile water into a 25 mg/ml suspension for subcutaneous injection and less than 10 mg/ml for intravenous administration to patients. With the objective of optimizing aerosol efficiency, we also made a Vidaza solution with the highest possible Aza concentration: 10 mg/ml in water. We compared the aerodynamic size of these two formulations under our experimental conditions. We found that about 66% of the droplets from the 25 mg/ml suspension formulation were ≤ 5 µm and only 45% of these droplets were ≤ 3 µm (Fig 1a); However, about 80% of the droplets from the 10 mg/ml solution formulation were <5 µm and about 70% of them were from 0.01 to 3 µm (Fig 1b). Because the 10 mg/ml solution achieved a better size distribution for optimal lung distribution than the 25 mg/ml suspension, all further studies were performed using the 10 mg/ml solution to generate the aerosol formulation.
The activity of aerosolized Aza
Aza is an unstable compound. The estimated t1/10 in water at room temperature is less than 2 h (the t1/10 = 40 min at 40°C)21. Unlike bolus injection, aerosol administration can take more time: in our studies, aerosol administration took up to ~ 50 min per group of mice. In addition, the aerosol shearing power may reduce the activity of the drug. Therefore, we evaluated whether the aerosolization process would reduce the activity of Aza.. We selected the DNMT activity as the parameter of activity, as Aza inhibits DNMT activity. Using the longest possible time (1 h) to aerosolize Vidaza, aerosol fog was collected into a sterile tube and the condensed liquid was used to treat the human NSCLC cell line H226 in vitro. The DNMT activity was measured with an ELISA method using EpiQuik DNMT assay kit, and the non-aerosolized Vidaza at the same concentration was used as the control comparator. We found no statistically significant difference in the activity between the aerosolized and non-aerosolized Vidaza (Fig 2), thus indicating that the basic epigenetic activity of Aza was preserved after one-hour aerosolization.
Figure 2.

The activity of aerosolized drug. The condensed (from 58 min to 62 min) aerosolized Aza solution (Aero Vidaza) were used to treat the human NSCLC cell line H226. Aerosolized Mannitol (Aero Mannitol) and non-aerosolized Vidaza (Vidaza) were used as negative and positive controls, separately. The columns and the error bars are the mean and standard deviation from 3 independent experiments.
Aerosolized Vidaza mainly deposits in lung and much less in peripheral blood
In order to know the aerosol delivery efficiency and measure the amount of the drug deposited in the lungs of mice, we measured the lung deposition and pharmacokinetics of the aerosolized Aza in mice. Intravenously injected Aza at the same dose was used as a comparator.
Our results demonstrated that in aerosol administration, a large portion of the drug deposited into the lung, with a low drug distribution into the peripheral blood. The peak concentration of Aza in the lung was 10.6 µg/g of lung at 20 min after a single 2.5 mg/m2 (0.83 mg/kg) aerosol dose of Aza. At this time point the amount of Aza per each lung was 1.55 µg, representing a recovery of 7.4% of the total Aza administered [1.55 µg ÷ (2.5/3 µg/g × 25)]. The Aza concentration in the blood increased over time and was highest at 24 hours post-administration (0.28 ± 0.16 µg/ml). The slow rise in blood concentrations of Aza after aerosol administration is interpreted to be the result of drug clearance from the lungs. The area under the concentration vs. time curve (AUC0~24h) for Aza in lung (66.5 µg/g × h) was 19-fold higher than that of blood (3.55 µg/ml × h) (p = 0.003)
Intravenously injected Aza had a peak blood concentration of 9.69 µg/ml at 20 min, which was about 81% of the initial dose (basing peripheral blood as 7% of body weight); the blood drug concentration initially declined rapidly, which was followed by a slower phase of drug concentration decrease. With IV administration, there was a trace amount of drug in the lung peaking at 0.38 µg/g, which was < 0.3% of the initial drug dose. The blood AUC was 12-fold higher (42.5/3.55) and the lung AUC was 62-fold lower (1.07/66.5) than those obtained from mice treated with aerosol Aza (Table 1).
Table 1.
Pharmacokinetic parameters
| Aerosol | IV | |
|---|---|---|
| Lung AUC (h·µg/g) | 66.5 | 1.07 |
| Blood AUC (h·µg/g) | 3.55 | 10.7 |
| AUC ratio (Lung/Blood) | 18.8 | 0.01 |
| Lung AUC ratio (Aero/IV) | 62.0 | |
| Blood AUC ratio (IV/Aero) | 3.0 | |
| Lung Cmax (µg/g) | 10.6 | 0.38 |
| Blood Cmax (µg/g) | 0.28 | 5.09 |
| t1/2.lung (h) | 2.3 | 2.0 |
| t1/2.blood (h) | N/A | 0.8 |
Clearance of Aza given via aerosol showed a very different pattern compared to Aza delivered intravenously. Aza delivered by aerosol had a 62-fold higher AUC in the lung than IV-administered Aza. The clearance of Aza from the lung was also much slower when delivered by aerosol (t1/2 = 2.3 h), indicating the aerosolized drug not only efficiently deposited but also had prolonged retention in the lungs without notable systemic drug concentrations. Our results indicate that aerosolized Aza is a much more efficient method to deliver drug to the lung or airway epithelium than IV administration.
Aerosolized Aza does not cause myelosuppression in mice
Myelosuppression is the dose limiting toxicity in patients receiving Aza by IV administration22. In our previous preclinical study, we found marked myelosuppression in mice after IV injection of Aza but not in mice receiving aerosolized Aza10. Mice treated with IV Aza (non clinical grade) at a dose of 75 mg/m2 (25 mg/kg) daily (a clinical therapeutic dose) had an average reduction in the WBC of 70% at day 510, 15. In contrast, intratracheal administration of Aza at the same dose did not result in myelosuppression in mice15. Our present study was designed to identify potential toxicities of aerosolized clinical grade Aza at potentially effective therapeutic doses in mice. Healthy mice were treated with two different doses of aerosol Vidaza, 2.5 or 7.5 mg/m2 daily (0.83 or 2.5 mg/kg) for 7 days. There was no significant reduction in the WBCs in mice treated with aerosol Vidaza at ether dose (Fig 4a). The WBCs at all time points from week 1 to week 6 were within the normal range (5000~10000 cells/µl). Aerosol administration of Aza at these doses also did not have evidence of bone marrow toxicity histologically (Fig 4b~m. 12 sternum pictures, b~g: wk3; h~m: wk6). In contrast, IV administered Vidaza at 7.5 mg/m2 (2.5 mg/kg), a dose 10-fold lower than clinical therapeutic dose, caused significant myelosuppression (Fig 4a): the WBC decreased to 30~50% below the normal range (p = 0.00009), confirming the myelosuppressive effects of IV administered Aza.
Figure 4.
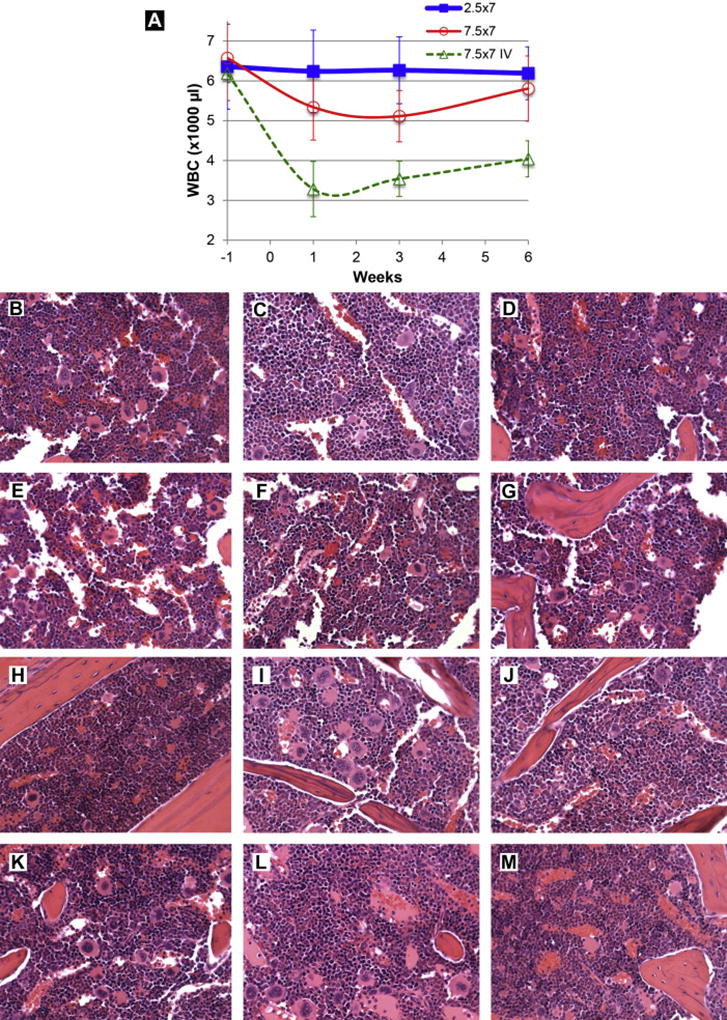
a. White blood cell count. Three groups of mice were treated once daily for 7 days by either aerosolized Aza at 2.5 mg/m2 (0.83 mg/kg; square) or 7.5 mg/m2 (2.5 mg/kg; round), or tail vein injection of Aza at 7.5 mg/m2 (2.5 mg/kg; triangle, dash line). Fifty microliter blood samples from each mouse were obtained under isoflurane anesthesia from the facial vein18 at 1 week prior and 1, 3, and 6 weeks after the treatment. White blood cells (WBCs) were collected and counted with a hemocytometer under a microscope after removing red blood cells. The data was mean and standard deviation from 6 to 12 mice per time point.
b~m. Pathological evaluation of the myelosuppression. Representative images of bone marrow (from sternum) 3 weeks (b~g) or 6 weeks (h~m) after the final administration of aerosolized Aza at 2.5 mg/m2 (0.83 mg/kg) daily × 7 days. In all cases, the bone marrow was histologically normal. The pictures were taken from hematoxylin and eosin stained slides and the objective magnification was 40×.
Aerosolized Aza does not cause kidney or liver toxicity in mice, but does cause mild reversible lung toxicity at a high dose
Blood chemistries and liver, kidney and lung histology in mice were evaluated for evidence of systemic toxicity after aerosolized Aza treatment. Blood chemistries, in particular the liver and kidney function parameters, were unaffected in mice treated with aerosol Aza at 2.5 mg/m2 (0.83 mg/kg) daily × 7. Only this does was evaluated, as it is the anticipated therapeutic dose based on the antitumor efficacy in mice in previous studies10. All blood chemistry data are shown in Supplementary Table 1. Lung, liver, and kidneys of mice treated with aerosolized Aza at 2.5 and 7.5 mg/m2 (0.83 and 2.5 mg/kg) daily for consecutive 7 days were evaluated histologically at 2 time points: 3 and 6 weeks after the completion of aerosol Aza administration. There were no treatment-related findings in the kidneys or livers of mice treated mice at either time point (data not shown). Mice treated with the low dose (2.5 mg/m2 or 0.83 mg/kg) of Aza did not have any lung toxicity at either the 3 or 6 week time point. However, in the high dose group (7.5 mg/m2 or 2.5 mg/kg) slight but reversible pulmonary toxicity was evident at week 3 in 4 of the 6 mice treated: 2 had minimal, 1 had moderate, and 1 had median pneumonitis (Fig 5 d~i). The lung lesions were typical of pneumonitis: fibrin accumulation along alveolar walls with increased infiltrates of alveolar macrophages and perivascular lymphoplasmacytic cuffing. The lesions were typically multifocal and of limited severity. No mice had treatment-related lesions at the 6 week time point (Fig 5 j~o), indicating that the pulmonary toxicity associated with aerosol Aza at 7.5 mg/m2 (2.5 mg/kg) × 7 days was reversible.
Figure 5.
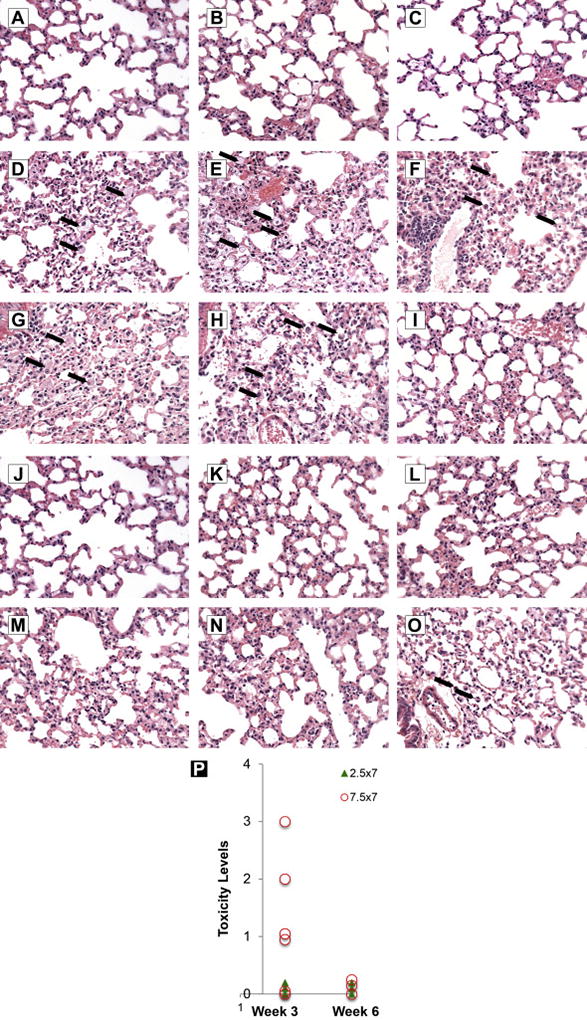
Pathological evaluation of the lungs from mice treated with aerosolized vehicle or Vidaza at 3 and 6 weeks after final treatment. a~c: Lungs from mice treated with aerosolized vehicle (10 mg/ml mannitol in sterile water). The lungs are normal. d~i: Lungs from mice treated with 7.5 mg/m2 (2.5 mg/kg) of aerosolized Vidazla three weeks after the final dose. There are low numbers of scattered foamy macrophages (arrows) within the alveoli in 5 of 6 mice. j~o: Lungs from mice treated with 7.5 mg/m2 (2.5 mg/kg) of aerosolized Vidazla six weeks after the final dose. One of the 6 mice had small alveolar macrophages (arrows), which was incidental and not a toxic lesion (i.e. occasionally present in vehicle-treated animals). The pictures were taken from hematoxylin and eosin stained slides and the objective magnification was 40×. p: The lung toxicity score: The toxicity of each lung were evaluated pathologically and characterized as 5 different grades: 0 = no toxicity/normal, 1 = minimal, 2 = moderate, 3 = median, 4 = severe. The circles represent high dose (7.5 mg/m2 or 2.5 mg/kg daily ×7, the H & E pictures presented above d~o), the solid triangles represent low dose (2.5 mg/m2 or 0.83 mg/kg daily ×7).
DISCUSSION
The common conclusions of the clinical trials of aerosolized chemo- or immune-therapeutic agents previously reported23,24,25,26,27,28,29,30 are definite local efficacy and significantly altered toxicity profiles with no systemic toxicity and very low local toxicity compared with their systemic administration. Because the chronic inhalation of carcinogens damages the airway epithelium along the entire respiratory tree1–5, the ideal preventative strategy should involve treating the entire airway epithelium in order to prevent the initiation and progression of lung cancer. Aerosol administration directly delivers the agent/drug to the entire airway epithelium and therefore is the ideal method of drug administration for diseases of the bronchial epithelium.
In this study we modified a clinical grade Aza to a high quality aerosol formulation: the aerodynamic size was optimal for clinical application of inhalation treatment and we showed that the drug’s activity is preserved under the aerosol conditions. Our pharmacokinetics study confirmed the aerosol delivery efficiency in mice: the majority of the drug was delivered into the lungs with only a small amount of drug released into the peripheral blood. When compared to IV administration, aerosolized Aza, had an AUC 60-fold greater in the lung and 3-fold lower in the peripheral blood demonstrating the superior toxicologic profile of local drug delivery..
Clearly the risk of potential lung toxicity is a concern with any drug delivered by aerosol. Azacitidine’s main toxicity when administered by IV is myelsuppression and has not been reported to cause pulmonary toxicity. It was therefore essential for us to understand any potential pulmonary toxicity associated with inhaled Aza prior to the initiation of any clinical trials. In our previous studies using intratracheal delivery of Aza at 2.5 mg/m2 (0.83 mg/kg), 7.5 mg/m2 (2.5 mg/kg), and 22.5 mg/m2 (7.5 mg/kg) daily for 7 days, only the highest dose resulted in severe pulmonary toxicity and death. In the studies reported here, we did not observe any lung toxicity at 2.5 mg/m2 (0.83 mg/kg) but we did observe slight but reversible lung toxicity in 4/6 mice at 7.5 mg/m2 (2.5 mg/kg) three weeks after completing treatment. By week 6, no treatment-related lesions were evident. From these results, we concluded that we couldn’t exclude the possibility of lung toxicity in humans as increasing doses of inhaled Aza are given. Therefore, we decided to select 2.5 mg/m2 (0.83 mg/kg) as the safe starting dose for the clinical trial and incorporate close monitoring of pulmonary function in the patients enrolled in the trial, as pulmonary toxicity may well be the limiting toxicity of inhaled Aza.
Although we did not observe clinically significant myelosuppression with inhaled Aza at 7.5 mg/m2 (2.5 mg/kg) or 2.5 mg/m2 (0.83 mg/kg) in mice, the possibility of myelosuppression cannot be ruled out in humans, and patients in clinical trials should be followed with regular complete blood counts. However, based on the pharmacokinetic, blood, and histological data, we do not expect myelosuppression to be a significant toxicity or dose limiting toxicity of inhaled Aza.
Aza is currently the best available demethylating agent used for cancer treatment. It is a non-specific DNA methyl transferase inhibitor. We used Aza as a prototype drug to prove the concept of using aerosolized epigenetic agents for lung cancer therapy and/or prevention. Once a better drug with a more specific demethylation function on TSGs is developed, we plan to use it to replace Aza.
Aza has shown antitumor activity against human leukemia31–33 and lung cancer in preclinical and clinical models10,15,34,35,36. In addition, its demethylating effect was found to be associated with its antitumor activity. While it may also demethylate and thus activate promoters for oncogenes is possible, this effect may not have a significant impact. Typically, the activation of oncogenes in cancer occurs as a result of other more permanent mechanisms such as gene mutation, gene amplification, and chromosome rearrangements37,38,39. Therefore, the possibility that Aza may further, activate them or increase their expression remains unlikely. On the other hand, in cancer, the majority of TSGs are silenced and their reactivation can have a beneficial impact on cell cycle repression and apoptosis and exert therapeutic effects40,41,42. Because it has been demonstrated that hypermethylation of promoter regions of TSGs is one of the major epigenetic alterations in lung cancer development43,41,42, treating the respiratory epithelium with demethylating agents would help tip the scales toward tumor suppression. Further detailed investigations however, should be pursued to uncover whether Aza can hypomethylate the promoter of oncogenes in cancers and its impact on potential cancer development.
In conclusion, inhalation delivery changes the toxicity profile of Aza. Based on previous studies inhalation of Aza is also an effective way to treat the entire airway epithelium. We plan to expand our strategy to other compounds that may be useful for the prevention of lung cancer. The Phase I clinical study of inhaled Aza has been recently initiated at Montefiore Medical Center/Albert Einstein College of Medicine. Three patients have been enrolled at the first dose level with no apparent side effects. This study will be key in guiding us in the development of other aerosolized prevention strategies of primary or/and second primary lung cancer, as strongly indicated by the field cancerization theory.
Supplementary Material
Figure 3.

Pharmacokinetics of aerosolized Aza. After aerosol (a) or intravenous (b) administration of clinical grade Aza in mice, the Aza levels in peripheral blood (triangle) and lung tissue (round dots) of mice were measured with a LC-MS method and plotted against the sampling time. The data in each figure was mean and standard deviation from 3 mice/time point.
CLINICAL PRACTICE POINTS.
Azacitidine has been used systemically in humans to treat myelodysplastic syndromes and, in combination with HDAC inhibitors, lung cancer. Systemic administration of azacytidine causes myelosuppression in humans and animals, which is dose-limiting. The use of aerosolized azacytidine for localized delivery of the drug to the lungs was highly effective in an orthotopic animal model of lung. This method of delivery was associated with longer survival, less toxicity, and less lung cancer burden in those animals as compared to those treated with the drug systemically.
The toxicity and pharmacokinetic profiles of aerosolized clinical grade azacitidine were determined. It was found that there was no pulmonary or systemic toxicity at a predicted therapeutic dose, and there was reversible lung toxicity at a dose 3-fold the anticipated therapeutic dose. An appropriate dose equivalent to the starting dose for a clinical phase I trial was determined.
Acknowledgments
The research was supported by a NIH (NCI) grant 5R01CA154755-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE
All authors have stated that they have no conflict of interest.
A supplemental table is included.
References
- 1.Leng S, Liu Y, Weissfeld JL, et al. 15q12 Variants, Sputum Gene Promoter Hypermethylation, and Lung Cancer Risk: A GWAS in Smokers. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsay JC, Li Z, Yie TA, et al. Molecular characterization of the peripheral airway field of cancerization in lung adenocarcinoma. PLoS One. 2015;10:e0118132. doi: 10.1371/journal.pone.0118132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadara H, Fujimoto J, Yoo SY, et al. Transcriptomic architecture of the adjacent airway field cancerization in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:dju004. doi: 10.1093/jnci/dju004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu CT, Lin MW, Hsieh MS, Kuo SW, Chang YL. New aspects of the clinicopathology and genetic profile of metachronous multiple lung cancers. Ann Surg. 2014;259:1018–1024. doi: 10.1097/SLA.0000000000000385. [DOI] [PubMed] [Google Scholar]
- 5.Nakachi I, Rice JL, Coldren CD, et al. Application of SNP microarrays to the genome-wide analysis of chromosomal instability in premalignant airway lesions. Cancer Prev Res (Phila) 2014;7:255–265. doi: 10.1158/1940-6207.CAPR-12-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phillips JM, Goodman JI. Inhalation of cigarette smoke induces regions of altered DNA methylation (RAMs) in SENCAR mouse lung. Toxicology. 2009;260:7–15. doi: 10.1016/j.tox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Pattani KM, Zhang Z, Demokan S, et al. Endothelin receptor type B gene promoter hypermethylation in salivary rinses is independently associated with risk of oral cavity cancer and premalignancy. Cancer Prev Res (Phila) 2010;3:1093–1103. doi: 10.1158/1940-6207.CAPR-10-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang S, Mahadevan S, Qadir F, et al. Identification of FOXM1-induced epigenetic markers for head and neck squamous cell carcinomas. Cancer. 2013;119:4249–4258. doi: 10.1002/cncr.28354. [DOI] [PubMed] [Google Scholar]
- 9.Wang JL, Lu FZ, Shen XY, Wu Y, Zhao LT. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem Biophys Res Commun. 2014;455:229–233. doi: 10.1016/j.bbrc.2014.10.153. [DOI] [PubMed] [Google Scholar]
- 10.Qiu X, Liang Y, Sellers RS, Perez-Soler R, Zou Y. Aerosol azacytidine inhibits orthotopic lung cancers in mice through Its DNA demethylation and gene reactivation effects. PLoS One. 2014;9:e109874. doi: 10.1371/journal.pone.0109874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soncini M, Santoro F, Gutierrez A, et al. The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim Biophys Acta. 2013;1832:114–120. doi: 10.1016/j.bbadis.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Thepot S, Lainey E, Cluzeau T, et al. Hypomethylating agents reactivate FOXO3A in acute myeloid leukemia. Cell Cycle. 2011;10:2323–2330. doi: 10.4161/cc.10.14.16399. [DOI] [PubMed] [Google Scholar]
- 13.Polakova K, Bandzuchova E, Sabty FA, Mistrik M, Demitrovicova L, Russ G. Activation of HLA-G expression by 5-aza-2 - deoxycytidine in malignant hematopoetic cells isolated from leukemia patients. Neoplasma. 2009;56:514–520. doi: 10.4149/neo_2009_06_514. [DOI] [PubMed] [Google Scholar]
- 14.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 15.Mahesh S, Saxena A, Qiu X, Perez-Soler R, Zou Y. Intratracheally administered 5-azacytidine is effective against orthotopic human lung cancer xenograft models and devoid of important systemic toxicity. Clin Lung Cancer. 2010;11:405–411. doi: 10.3816/CLC.2010.n.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao M, Rudek MA, He P, et al. Quantification of 5-azacytidine in plasma by electrospray tandem mass spectrometry coupled with high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;813:81–88. doi: 10.1016/j.jchromb.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Zou Y, Tornos C, Qiu X, Lia M, Perez-Soler R. p53 aerosol formulation with low toxicity and high efficiency for early lung cancer treatment. Clin Cancer Res. 2007;13:4900–4908. doi: 10.1158/1078-0432.CCR-07-0395. [DOI] [PubMed] [Google Scholar]
- 18.Golde WT, Gollobin P, Rodriguez LL. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim (NY) 2005;34:39–43. doi: 10.1038/laban1005-39. [DOI] [PubMed] [Google Scholar]
- 19.Bates DV, Fish BR, Hatch TF, Mercer TT, Morrow PE. Deposition and retention models for internal dosimetry of the human respiratory tract. Task group on lung dynamics. Health Phys. 1966;12:173–207. [PubMed] [Google Scholar]
- 20.Davies DS. Pharmacokinetics of inhaled substances. Scand J Respir Dis Suppl. 1979;103:44–49. [PubMed] [Google Scholar]
- 21.Israili ZH, Vogler WR, Mingioli ES, Pirkle JL, Smithwick RW, Goldstein JH. The disposition and pharmacokinetics in humans of 5-azacytidine administered intravenously as a bolus or by continuous infusion. Cancer Res. 1976;36:1453–1461. [PubMed] [Google Scholar]
- 22.Weiss AJ, Stambaugh JE, Mastrangelo MJ, Laucius JF, Bellet RE. Phase I study of 5-azacytidine (NSC-102816) Cancer Chemother Rep. 1972;56:413–419. [PubMed] [Google Scholar]
- 23.Lorenz J, Wilhelm K, Kessler M, et al. Phase I trial of inhaled natural interleukin 2 for treatment of pulmonary malignancy: toxicity, pharmacokinetics, and biological effects. Clin Cancer Res. 1996;2:1115–1122. [PubMed] [Google Scholar]
- 24.Anderson PM, Markovic SN, Sloan JA, et al. Aerosol granulocyte macrophage-colony stimulating factor: a low toxicity, lung-specific biological therapy in patients with lung metastases. Clin Cancer Res. 1999;5:2316–2323. [PubMed] [Google Scholar]
- 25.Skubitz KM, Anderson PM. Inhalational interleukin-2 liposomes for pulmonary metastases: a phase I clinical trial. Anticancer Drugs. 2000;11:555–563. doi: 10.1097/00001813-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Verschraegen CF, Gilbert BE, Loyer E, et al. Clinical evaluation of the delivery and safety of aerosolized liposomal 9-nitro-20(s)-camptothecin in patients with advanced pulmonary malignancies. Clin Cancer Res. 2004;10:2319–2326. doi: 10.1158/1078-0432.ccr-0929-3. [DOI] [PubMed] [Google Scholar]
- 27.Otterson GA, Villalona-Calero MA, Sharma S, et al. Phase I study of inhaled Doxorubicin for patients with metastatic tumors to the lungs. Clin Cancer Res. 2007;13:1246–1252. doi: 10.1158/1078-0432.CCR-06-1096. [DOI] [PubMed] [Google Scholar]
- 28.Wittgen BP, Kunst PW, van der Born K, et al. Phase I study of aerosolized SLIT cisplatin in the treatment of patients with carcinoma of the lung. Clin Cancer Res. 2007;13:2414–2421. doi: 10.1158/1078-0432.CCR-06-1480. [DOI] [PubMed] [Google Scholar]
- 29.Lemarie E, Vecellio L, Hureaux J, et al. Aerosolized gemcitabine in patients with carcinoma of the lung: feasibility and safety study. J Aerosol Med Pulm Drug Deliv. 2011;24:261–270. doi: 10.1089/jamp.2010.0872. [DOI] [PubMed] [Google Scholar]
- 30.Zarogoulidis P, Eleftheriadou E, Sapardanis I, et al. Feasibility and effectiveness of inhaled carboplatin in NSCLC patients. Invest New Drugs. 2012;30:1628–1640. doi: 10.1007/s10637-011-9714-5. [DOI] [PubMed] [Google Scholar]
- 31.Griffin PT, Komrokji RS, De Castro CM, et al. A multicenter, phase II study of maintenance azacitidine in older patients with acute myeloid leukemia in complete remission after induction chemotherapy. Am J Hematol. 2015;90:796–799. doi: 10.1002/ajh.24087. [DOI] [PubMed] [Google Scholar]
- 32.Fozza C, Corda G, Barraqueddu F, et al. Azacitidine improves the T-cell repertoire in patients with myelodysplastic syndromes and acute myeloid leukemia with multilineage dysplasia. Leuk Res. 2015;39:957–963. doi: 10.1016/j.leukres.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Abou Zahr A, Saad Aldin E, Barbarotta L, Podoltsev N, Zeidan AM. The clinical use of DNA methyltransferase inhibitors in myelodysplastic syndromes. Expert Rev Anticancer Ther. 2015;15:1019–1036. doi: 10.1586/14737140.2015.1061936. [DOI] [PubMed] [Google Scholar]
- 34.Wrangle J, Wang W, Koch A, et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget. 2013;4:2067–2079. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed MD, Tellez CS, Grimes MJ, et al. Aerosolised 5-azacytidine suppresses tumour growth and reprogrammes the epigenome in an orthotopic lung cancer model. Br J Cancer. 2013;109:1775–1781. doi: 10.1038/bjc.2013.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu BF, Karpenko MJ, Liu Z, et al. Phase I study of 5-aza-2'-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother Pharmacol. 2013;71:115–121. doi: 10.1007/s00280-012-1986-8. [DOI] [PubMed] [Google Scholar]
- 37.Gene "Switch" Stops Colon Cancer in Mice. Cancer Discov. 2015;5:789–790. doi: 10.1158/2159-8290.CD-NB2015-096. [DOI] [PubMed] [Google Scholar]
- 38.Stella GM, Luisetti M, Pozzi E, Comoglio PM. Oncogenes in non-small-cell lung cancer: emerging connections and novel therapeutic dynamics. Lancet Respir Med. 2013;1:251–261. doi: 10.1016/S2213-2600(13)70009-2. [DOI] [PubMed] [Google Scholar]
- 39.Anderson MW, Reynolds SH, You M, Maronpot RM. Role of proto-oncogene activation in carcinogenesis. Environ Health Perspect. 1992;98:13–24. doi: 10.1289/ehp.929813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teschendorff AE, Yang Z, Wong A, et al. Correlation of Smoking-Associated DNA Methylation Changes in Buccal Cells With DNA Methylation Changes in Epithelial Cancer. JAMA Oncol. 2015;1:476–485. doi: 10.1001/jamaoncol.2015.1053. [DOI] [PubMed] [Google Scholar]
- 41.Li W, Deng J, Wang SS, et al. Association of methylation of the RAR-beta gene with cigarette smoking in non-small cell lung cancer with Southern-Central Chinese population. Asian Pac J Cancer Prev. 2014;15:10937–10941. doi: 10.7314/apjcp.2014.15.24.10937. [DOI] [PubMed] [Google Scholar]
- 42.Tan Q, Wang G, Huang J, et al. Epigenomic analysis of lung adenocarcinoma reveals novel DNA methylation patterns associated with smoking. Onco Targets Ther. 2013;6:1471–1479. doi: 10.2147/OTT.S51041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han F, Liu W, Jiang X, et al. SOX30, a novel epigenetic silenced tumor suppressor, promotes tumor cell apoptosis by transcriptional activating p53 in lung cancer. Oncogene. 2015;34:4391–4402. doi: 10.1038/onc.2014.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.