

 A fluorescent τ-probe was demonstrated to quantitate ultra-trace endogenous hydrogen sulfide (H2Sn) within cells and zebrafish through changes of fluorescence lifetime.
A fluorescent τ-probe was demonstrated to quantitate ultra-trace endogenous hydrogen sulfide (H2Sn) within cells and zebrafish through changes of fluorescence lifetime.
Abstract
Hydrogen sulfide (H2S) has been recognized as an important endogenous gasotransmitter associated with biological signaling transduction. However, recent biological studies implied that the H2S-related cellular signaling might actually be mediated by hydrogen polysulfides (H2Sn, n > 1), not H2S itself. Unraveling such a mystery strongly demanded the quantification of endogenous H2Sn in living systems. However, endogenous H2Sn has been undetectable thus far, due to its extremely low concentration within cells. Herein, we demonstrated a strategy to detect ultra-trace endogenous H2Snvia a fluorescent τ-probe, through changes of fluorescence lifetime instead of fluorescence intensity. This τ-probe exhibited an ultrasensitive response to H2Sn, bringing about the lowest value of the detection limit (2 nM) and a lower limit of quantification (10 nM) to date. With such merits, we quantified and mapped endogenous H2Sn within cells and zebrafish. The quantitative information about endogenous H2Sn in cells and in vivo may have a significant implication for future research on the role of H2Sn in biology. The methodology of the τ-probe established here might provide a general insight into the design and application of any fluorescent probes, beyond the limit of utilizing fluorescence intensity.
Introduction
Reactive sulfur species (RSS) are a family of endogenous sulfur-containing molecules in biological systems related to a series of signaling transduction processes.1–3 Among RSS, hydrogen sulfide (H2S) has been studied for many years as the third most important endogenous gasotransmitter after CO and NO and has been supposed to be involved in regulating neuromodulation,4 cytoprotection,5 vascular tone regulation,6 oxygen sensing,7 inflammatory regulation,8 and cell growth.9 However, recent studies have implied that hydrogen polysulfides (H2Sn, n > 1) might act as the real signaling mediator instead of H2S.10–12 For example, the conversion of cysteine residues (–SH) to persulfides (–S–SH) through S-sulfhydration was originally ascribed to H2S, but H2S itself can hardly induce S-sulfhydration.13–16 Also, studies have shown that H2Sn can compensate for physiological functions that H2S cannot achieve.17–19 There are still many unaddressed arguments associated with the biological role of H2Sn.
A good start to clarify such a conundrum might begin with the quantification of endogenous H2Sn in living systems. Such a task seems straightforward, but actually remains a great challenge. The H2S level in biological systems has been reported from 17 nM to 3 μM.20 As a downstream product of H2S, endogenous H2Sn was conjectured to be <100 nM or even less. Unfortunately, it is still difficult to detect H2Sn at such low concentration. UV-vis spectroscopy and mass spectrometry (MS) have been applied to investigate H2Sn,21,22 but they were limited by poor sensitivity and insufficient operability in organisms. Fluorescent probes were feasible for H2Sn detection in living systems, owing to their high sensitivity and compatibility with bioimaging facilities.23,24 In 2014, the Xian group reported the first H2Sn-specific probes (DSP) exploiting a 2-fluoro-5-nitrobenzoic ester template,25 and several subsequent H2Sn fluorescent probes were designed afterwards.26–33 The DSP-type probes showed satisfactory sensitivity and selectivity for H2Sn;32 however, they would also react with biothiols, resulting in a slow response and low yield. To settle this issue, several other recognition groups were developed, including nucleophilic ring-opening reaction,34 α-substituted acrylate ester,35 and phenyl 2-(benzoylthio)benzoate formation.36–38 Studies have also been performed to improve the signal-to-noise ratio through the red shift of the wavelength,30,32,38,39 two-photon excitation31,40 or ratio detection,29,41,42 and also to achieve simultaneous detection of H2Sn together with H2S,37 glutathione,43 or superoxide anions.44 Although many efforts have been done, all these probes can still detect H2Sn only through external stimulation28,32,35 or H2Sn addition,36,45 but not endogenous H2Sn in native cells. In 2015, the Xian group reported a new H2Sn-probe (PSP), showing the lowest detection limit extrapolated from theory (3 nM), and an actual working range from 0.25 μM to 20 μM.36 Although the lower limit of quantification was as low as 0.25 μM, endogenous H2Sn in native cells still cannot be detected. It was expected that the lower limit of quantification must reach tens of nM, and the detection limit must reach a few nM, so as to detect native endogenous H2Sn. Such a goal seemed hard to achieve by using intensity-based fluorescent probes, either from theoretical deduction or from the best result reported so far. In addition to fluorescence intensity, fluorescence lifetime is another fundamental photophysical parameter associated with any fluorescence emission. Different from fluorescence intensity that depends on emitter numbers, fluorescence lifetime mainly depends on the instinct photophysical characteristic of each emitter.46 Fluorescence lifetime imaging has been commonly applied as a powerful tool of biology studies.47 Current time-resolved techniques are able to distinguish lifetime changes as low as ps–fs levels.48
In spite of the important progress made in studying H2Sn, the quantification of endogenous H2Sn in native biological systems still remain unsolved. The most sensitive fluorescence intensity probes are currently unable to achieve such a low detection limit. In this work, we developed an alternative strategy of using a fluorescent τ-probe to detect H2Sn through changes of fluorescent lifetime rather than fluorescence intensity. The detection limit of the τ-probe was as low as 2 nM, and the actual working range was from 10 nM to 5 μM, which allows us to detect ultra-trace endogenous H2Sn in cells and in zebrafish. The concentration of endogenous H2Sn in a HeLa cell was distributed between ∼0 and 120 nM, with an averaged value about ∼60–70 nM. The concentration distribution in zebrafish slightly broadened to ∼0–160 nM, but still has a comparable averaged concentration of ∼70–80 nM with the value of cells. To the best of our knowledge, this is the first time to detect endogenous H2Sn in a native biological system. These results provided fundamental quantitative information about endogenous H2Sn in cells and in vivo and have a significant implication for deeply understanding the role of H2Sn in biology. Meanwhile, the methodology of the τ-probe established here might provide a general insight into the design and application of any analytical strategy exploiting fluorescent probes, to remedy the disadvantage of fluorescence intensity probes that are insensitive at extremely low concentration.
Results and discussion
Concept and design of the fluorescent τ-probe
The design of the τ-probe was inspired by Xian group's PSP-3 probe,36 but its structure was modified to maximize the sensitivity. Detailed synthesis protocols and structural characterization are provided in the ESI (Scheme S1, Fig. S1–S3†). Products from the reaction between the τ-probe and H2Sn are shown in Fig. S4.† Compared to the PSP-3 probe with two recognition groups that would consume two H2Sn per probe, our τ-probe has only one recognition group, which consumes one H2Sn per probe. As shown in Fig. 1a, this probe initially showed a “dark” state with a weak fluorescence emission (Fig. 1b) and a short lifetime τdark = 0.75 ns (Fig. 1d). When reacted with H2Sn (Scheme S2†), fluorescein was released to a “bright” state with a strong fluorescence emission (Fig. 1c) and a long lifetime τbright = 3.47 ns (Fig. 1e). This probe is a typical “turn-on”-type fluorescent probe. Here we presented that, in addition to fluorescence intensity, the fluorescence lifetime could also be utilized for quantitative analysis.
Fig. 1. Concept and design of the τ-probe. (a) Reaction with H2Sn switched the τ-probe from “dark” to “bright” states with distinct fluorescence characteristics. (b–e) Fluorescence intensity and lifetime of the “dark” state τ-probe (b, d) and “bright” state τ-probe (c, e), respectively.

To prove the concept of the τ-probe, we studied the response of fluorescence intensity and lifetime to the concentration of H2Sn. Theoretically, for an emitter with unchanged fluorescence emission characteristics, the fluorescence intensity depends on the numbers of emitters, and the fluorescence lifetime depends on the instinct photophysical properties of each emitter. However, for systems mixed with two emitters with distinct fluorescence emission characteristics, the total value of fluorescence intensity and lifetime in such mixed systems was a result contributed by the two emitters. Particularly, for our τ-probe that has “dark” and “bright” states, the total weighted value of steady-state fluorescence intensity (Itotal) consisted of “bright” state fluorescence intensity (Ibright) and “dark” state fluorescence intensity (Idark), and then Itotal could be calculated by:
| Itotal = Ibright + Idark | 1 |
Since fluorescence intensity is proportional to the number of fluorescent molecules (N) and their emission cross section (σ), the fluorescence intensity (I) can also be expressed as:
| I = fcVNaσ | 2 |
where f is a proportional constant related to the incident photon flux. cbright and cdark are the concentrations of “bright” and “dark” state τ-probes, V is the volume of the solution, and Na is the Avogadro constant. Combined with eqn (2), Itotal could be expressed as:
| Itotal = fcbrightVNaσbright + fcdarkVNaσdark | 3 |
Because the parameters f, V and Na are constants for a given system, they could be totally represented by another constant γ. Then eqn (3) could be described as:
| Itotal = γcbrightσbright + γcdarkσdark | 4 |
When the emitters were excited by a pulsed light source, for instance in fluorescence lifetime measurement, the transient fluorescence intensity at time t (Itotal(t)) could be calculated by:
 |
5 |
where A and B are the compensation coefficients of fluorescence intensity at the bright state and dark state, respectively. τbright and τdark are the fluorescence lifetime of “bright” and “dark” state τ-probes. Therefore, the total weighted mean of fluorescence lifetime (τtotal) could be calculated by:
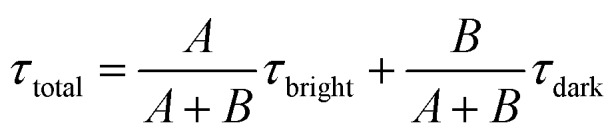 |
6 |
A and B could be described as:
| A ∝ cbrightσbright | 7 |
| B ∝ cdarkσdark | 8 |
Then the total fluorescence lifetime (τtotal) could be described as:
 |
9 |
According to eqn (4) and (9), we find that I(total) and τ(total) are only related to the concentration of fluorescent molecules cbright and cdark, owing to the fact that σbright and σdark are fixed values in a given system (σbright ≫ σdark). Consequently, if we use cB0 and cD0 which represent the original concentration of “bright” and “dark” state τ-probes, the concentration (c) of H2Sn reacted with the τ-probe could be calculated from the total fluorescence intensity (Itotal(c)) or fluorescence lifetime (τtotal(c)):
| Itotal(c) = γ(cB0 + c)σbright + γ(cD0 – c)σdark | 10 |
 |
11 |
Fluorescence response to H2Sn: intensity versus lifetime
Theoretical response curves of Itotal(c) and τtotal(c) to H2Sn concentration (c) could be obtained by substituting experimental data into eqn (10) and (11): cB0 = 0 μM, cD0 = 10 μM, σbright = 20σdark, τbright = 3.47 ns, and τdark = 0.75 ns. Giving consideration to both accuracy and sensitivity, the concentration of the τ-probe was chosen as 10 μM in the following experiments (Fig. S5†). Experimental response curves were obtained by measuring changes of fluorescence intensity and lifetime by the addition of 0–25 μM Na2S4 (commercial H2Sn donor), as adopted previously.31,35,41 Theoretical curves matched experimental data well for either fluorescence intensity (Fig. 2a) or lifetime (Fig. 2b). The response features of Itotal(c) and τtotal(c) were totally different. Itotal(c) exhibited a nearly linear response in a wide range of concentration (c). However, τtotal(c) showed a non-linear response, which is much more sensitive at low concentration. For instance, when the concentration of Na2S4 adds to 0.2 μM, τtotal(c) increased 0.93 ns, and the rate of change was about 34.19% compared with the value of the “bright” state. At the same condition, Itotal(c) is almost unchanged and the variation is as low as 0.09% (Fig. 2c). According to the temporal resolution and noise level of the instrument (∼10 ps), the detection limit of the τ-probe was about ∼2 nM, and the experimental working range was from ∼10 nM to 5 μM. Compared to H2Sn probes reported previously (Fig. 2d), our τ-probe reached the lowest value of the detection limit and a lower limit of quantification to date. Note that even though τtotal(c) is sensitive at low concentration, it does not have as broad a response range as Itotal(c) does. Therefore, these two methods with high sensitivity or wide response range would complement each other, rather than replace each other.
Fig. 2. Response characteristics: intensity versus lifetime. (a and b) Theoretical and experimental response curves of the fluorescence intensity (a) and lifetime (b) of the τ-probe to Na2S4 (as a H2Sn donor). (c) Response of fluorescence intensity and lifetime at extremely low concentration. (d) Comparison of the detection limit and working range between the current τ-probe and some intensity-based H2Sn probes reported previously: 1-current work, 2-(ref. 36), 3-(ref. 39), 4-(ref. 37), 5-(ref. 43), 6-(ref. 25), 7-(ref. 35), and 8-(ref. 42). All experiments were repeated at least three times. Error bars represent standard deviation.

Time-dependent response and photostability of the τ-probe to Na2S4 are shown in Fig. S6 and S7.† Fluorescence intensity reached almost maximum within 5 min, and thus a reaction time of 20 min was employed subsequently to ensure complete reactions. The effects of pH were investigated; the τ-probe worked effectively in the pH range of 7–8 (Fig. S8†). To obtain a working curve, varying concentrations of Na2S4 (0.01–25 μM) were added to the τ-probe (10 μM). Fluorescence intensity almost linearly correlated with Na2S4 in a broad range of 0.5–10 μM (Fig. S9†), but changes at 0.5 μM and below were hard to distinguish. However, fluorescence lifetime still has a sensitive response in the range of 10 nM to 0.2 μM (Fig. S10†). The correlation of fluorescence lifetime (τ) with the concentration of H2Sn (c) in the range of 0–0.2 μM could be well fitted to a linear equation (R2 = 0.99267):
| τ = 0.77 + 4.60c (0 < c < 0.2 μM) | 12 |
Test of selectivity and specificity
To test the selectivity of the τ-probe to H2Sn, the τ-probe was treated with a series of reactive sulfur species (RSS) including glutathione (GSH), cysteine (Cys), homocysteine (Hcy), glutathione disulfide (GSSG), H2S, S2O32–, SO32–, SO42–, and CH3SSSCH3. A negligible change of fluorescence lifetime was observed for any of these compounds (Fig. 3a). Only Na2S4 generated a significant change (column blank). The interference of these RSS with the τ-probe upon addition of Na2S4 was tested and minute interference was found (column red). We also validated whether the τ-probe could sense H2Sn formation from H2S and reactive oxygen species (ROS) in situ. The τ-probe did not show a response (Fig. 3b) to H2S (in the form of Na2S) in the presence of common ROS including hydrogen peroxide (H2O2), superoxide (O2–), hydroxyl radical (OH), and singlet oxygen (1O2). Only the ClO–/H2S system showed an obvious response. This result was consistent with previous reports that H2Sn can be derived from H2S and ClO–, but not other ROS.36 All these results indicated the high sensitivity and high selectivity of the τ-probe to H2Sn, suggesting that the τ-probe could be used to detect endogenous H2Sn in native biological systems.
Fig. 3. Selectivity and specificity of the τ-probe to H2Sn. (a) Fluorescence lifetime changes caused by various reactive sulfur species, with or without Na2S4 (as a H2Sn donor). (b) Fluorescence lifetime changes caused by various reactive oxygen species, with or without Na2S (as a H2S donor). See experimental details in the Experimental section. All experiments were repeated at least three times. Error bars represent standard deviation.

Quantifying endogenous H2Sn within cells and zebrafish
We then demonstrated that our τ-probe could quantify H2Sn in cells by using a suspension of HeLa cells. For positive control cells incubated with 20 μM Na2S4 (as a H2Sn donor), the fluorescence lifetime increased dramatically (Δτ = 2.70 ns). For negative control cells incubated with 100 μM N-ethylmaleimide (NEM) to eliminate the endogenous H2Sn, the fluorescence lifetime remained almost unchanged (Δτ = 0.05 ns) compared to the “dark” state τ-probe (Fig. 4a). With the positive and negative controls, endogenous H2Sn in native HeLa cells was obtained by substituting fluorescence lifetime into the working curve (eqn (3)). The averaged H2Sn concentration in native HeLa cells is about ∼0.065 μM (65 nM). We then further confirmed this by adding 0.065 μM (1×) and 0.130 μM (2×) additional Na2S4 to cell samples, and the fluorescence lifetime changed linearly according to the working curve. Cell viability assay demonstrated that the τ-probe has nearly no cytotoxicity to cells (Fig. S11†).
Fig. 4. Quantifying endogenous H2Sn in a HeLa cell suspension. (a) Fluorescence lifetime change of the τ-probe in negative control cells (H2Sn(–)) incubated with N-ethylmaleimide (NEM) to eliminate H2Sn (left column), native cells (H2Sn-cell) without any treatment (middle column) and positive control cells (H2Sn(+)) incubated with Na2S4 (right column). (b) Fluorescence lifetime change of the τ-probe in native HeLa cells (H2Sn = 65 nM) and cells incubated with 0.065 μM (1×) or 0.13 μM (2×) additional Na2S4. All experiments were repeated at least three times. Error bars represent standard deviation.

With results in the cell suspension, we then quantified endogenous H2Sn within single HeLa cells, utilizing both fluorescence intensity and lifetime. Fluorescence imaging was performed on a confocal microscope. As reported previously,35,36 fluorescence was observed only in positive control cells (+20 μM Na2S4), but not in native HeLa cells (Fig. S12†). Fluorescence lifetime imaging was carried out on a time-gated transient microscope (see details in the Method and Scheme S3 in the ESI†). Dark field imaging was utilized to locate cells (Fig. 5a and b). As expected, fluorescence lifetime changes were observed in native HeLa cells (Fig. 5c and d). Then the map and histogram of H2Sn within a HeLa cell were obtained by applying the working curve to fluorescence lifetime imaging (Fig. 5e and f). The H2Sn level in three different HeLa cells was determined to be about 60–70 nM (Fig. 5g), which is comparable with the value of the cell suspension (∼65 nM). It is interesting to note that even the intracellular concentration of H2Sn covers a wide range of 0–120 nM (Fig. 5f), and the averaged values of different individual cells were quite close to each other (Fig. 5g). This result is sort of out of expectation since studies have shown heterogeneity between individual cells in the concentration of biomolecules.49 Further in-depth studies are definitely needed to get a better understanding about the role of H2Sn in cells. Afterwards, we mapped H2Sn distribution in zebrafish (Fig. 6). The endogenous H2Sn within zebrafish was mainly localized in its abdominal region, and the distribution covers a wide range of 0–160 nM, with an average value of about ∼70–80 nM. It was also found that endogenous H2Sn within zebrafish was hard to observe in the fluorescence intensity imaging (Fig. S13†). These results demonstrated that the τ-probe could be an ultrasensitive tool for studying H2Sn in living systems.
Fig. 5. Quantitative mapping of H2Sn in single HeLa cells. (a and b) Dark field imaging of a native HeLa cell (a) and HeLa cells treated with Na2S4 (b). (c and d) Fluorescence lifetime imaging of a native HeLa cell (c) and HeLa cells treated with Na2S4 (d). (e) Map of H2Sn concentration in a native HeLa cell. (f) Histogram of H2Sn concentration in the native HeLa cell showing in (e). (g) Averaged H2Sn concentration within three different native HeLa cells. Scale bars: 10 μm.

Fig. 6. Quantitative mapping of H2Sn in zebrafish. (a and b) Dark field imaging of untreated zebrafish (a) and zebrafish treated with Na2S4 (b). (c and d) Fluorescence lifetime imaging of untreated zebrafish (c) and zebrafish treated with Na2S4 (d). (e) Map of H2Sn concentration in untreated zebrafish. (f) Histogram of H2Sn concentration in the untreated zebrafish showing in (e). Scale bars: 150 μm.

Conclusions
In summary, we developed an alternative strategy to detect H2Sn through a fluorescence lifetime probe (termed: τ-probe) rather than fluorescence intensity. The detection limit of the τ-probe was as low as 2 nM, and the actual working range was from 10 nM to 5 μM, which allows us to detect ultra-trace endogenous H2Sn in cells and in zebrafish. The endogenous H2Sn in native HeLa cells was distributed between ∼0 and 120 nM, with an average value of about ∼60–70 nM. The value in zebrafish slightly broadened to ∼0–160 nM, but still has a comparable average level ∼70–80 nM. To the best of our knowledge, this is the first time to detect endogenous H2Sn in a native biological system. This information has a significant implication for deeply understanding the role of H2Sn in biology. The concept of the τ-probe provided a general insight into the application of fluorescent probes, beyond the limit of fluorescence intensity.
Experimental section
Reagents
Fluorescein, methyl iodide (MeI), potassium carbonate (K2CO3), dimethyl formamide (DMF), sodium hydroxide (NaOH), methyl alcohol (MeOH), hydrochloric acid (HCl), dichloromethane (CH2Cl2), thiosalicylic acid, benzoic acid, 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC), 4-dimethylaminopyridine (DMAP), dimethyl sulfoxide (DMSO), glutathione (GSH), cysteine (Cys), homocysteine (Hcy), glutathione disulfide (GSSG), Na2S, Na2S2O3, Na2SO3, Na2SO4, CH3SSSCH3, hydrogen peroxide (H2O2), sodium hypochlorite (NaClO), KO2, FeSO4, and ethyl 3-aminobenzoate methanesulfonate were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). High glucose Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), and trypsin were purchased from Invitrogen (Carlsbad, U.S.A.). The cell cytotoxicity assay kit, human cervical cancer (HeLa) cells, and phosphate buffer solution (PBS, 10 mM, pH = 7.4) were purchased from KeyGEN Biotech. Co (Nanjing, China). Zebrafish were purchased from the Model Animal Research Center of Nanjing University. Ultra-pure water from a Millipore Milli-Q (18 MΩ cm–1) was used in the experiments. All other reagents were of analytical reagent grade.
Apparatus
UV-vis absorption spectra were recorded using a UV-vis spectrophotometer (Nanodrop-2000C, Nanodrop, USA). The fluorescence emission spectra were obtained on a Shimadzu fluorescence S-3 spectrophotometer (RF-5301PC, Shimadzu Co., Japan). The fluorescence lifetime spectra were obtained on a FLS 980 spectrophotometer (Edinburgh instruments, U.K.). Confocal fluorescence images of cells were acquired with TCS SP8 confocal microscopy (Leica, Germany). The cell viability assay was performed using a Thermo Scientific Varioskan Flash (Thermo Fisher Scientific, American). The τ-Probe was purified by Ultimate 3000 high performance liquid chromatography (Thermo Fisher Scientific, American). Fluorescence lifetime imaging was achieved by using instruments built in our laboratory.
Synthesis of the τ-probe
The synthesis procedure of the τ-probe is shown in Scheme S1.† CH2Cl2 (20 mL) was added to a mixture of 3-O-methylfluorescein50 (69.2 mg, 0.2 mmol), 2-(benzoylthio)benzoic acid37,51 (78 mg, 0.3 mmol), EDC (58 mg, 0.3 mmol) and DMAP (12.2 mg, 0.1 mmol) at room temperature and the mixture was stirred for 5 h. Then the solvent was evaporated under reduced pressure and the resulting residue was subjected to high performance liquid chromatography for purification. The τ-probe was obtained as a yellow solid. 1H-NMR and 13C-NMR spectra of τ-probe are shown in Fig. S1 and S2,† respectively. 1H NMR (400 MHz, CD3Cl) δ 8.23 (dd, J = 1.6, 8.0 Hz, 1H), 8.03–8.01 (m, 3H), 7.72–7.57 (m, 6H), 7.49–7.47 (m, 2H), 7.18 (d, J = 2.4 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 6.88 (dd, J = 2.4, 8.8 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 2.4 Hz, 1H), 6.69 (d, J = 8.8 Hz, 1H), 6.62 (dd, J = 2.4, 8.8 Hz, 1H), 3.82 (s, 3H). 13C NMR (100 MHz, CD3Cl) δ 189.44, 169.80, 164.27, 161.51, 153.05, 152.28, 151.98, 151.92, 137.33, 136.36, 135.26, 133.90, 133.47, 132.81, 131.66, 129.92, 129.72, 129.11, 129.00, 128.93, 128.83, 127.59, 126.41, 125.16, 124.05, 117.53, 116.74, 111.99, 110.72, 110.49, 100.90, 82.97, 55.62. LC-MS (ESI+): m/z C35H23O7S+, calcd 587.1168, found [M+] 587.1169.
Fluorescence lifetime measurement
The fluorescence lifetime measurements were performed on a FLS 980 spectrophotometer (Edinburgh instruments, U.K.). The instrument is based on time-correlated single photon counting (TCSPC). Photons are counted in a time window which sweeps across the full time range following each excitation pulse, creating a histogram of counts versus time. The data quality of the resulting histogram is improved by adding the data of repeated sweeps. A 488 nm picosecond laser (∼3 ps, 5.5 MHz) was used to pump the fluorescence, and the collection time window is 0–50 ns in the wavelength range of 500–650 nm, with a minimum time per channel = 305 fs. The time jitter in the delay generation system is about ∼10 ps.
General procedure for spectral measurements
A stock solution of the τ-probe was prepared at 1 mM in DMSO. A PBS (10 mM, pH 7.4) solution with DMSO as the cosolvent (DMSO/PBS = 1 : 9, v/v) was used for performing all spectroscopic measurements. The fluorescence signal was recorded with the excitation wavelength at 488 nm and the emission wavelength range from 500 to 650 nm. The maximum fluorescence emission was obtained at 515 nm. Glutathione (GSH, 5 mM), cysteine (Cys, 1 mM), homocysteine (Hcy, 100 μM), glutathione disulfide (GSSG, 100 μM), Na2S (200 μM), Na2S2O3 (100 μM), Na2SO3 (100 μM), Na2SO4 (100 μM), CH3SSSCH3 (100 μM), hydrogen peroxide (H2O2, 200 μM), hypochlorite (ClO–, 50 μM), superoxide (O2–, 50 μM), hydroxyl radical (˙OH, 50 μM), singlet oxygen (1O2, 50 μM), H2O2 (200 μM) + Na2S (50 μM), H2O2 (200 μM) + Na2S (100 μM), ClO– (50 μM) + Na2S (100 μM), O2– (50 μM) + Na2S (100 μM), ˙OH (50 μM) + Na2S (100 μM), and 1O2 (50 μM) + Na2S (100 μM) were all dissolved in prepared solutions for various testing species. The preparation of reactive oxygen species was according to the literature.36 Na2S4 (commercial H2Sn donor) was dissolved in PBS solution, to prepare a sample solution (1 mM). A volume of 10 μL of τ-probe stock solution was added to 1 mL volumes of various analyte buffer/DMSO solutions (H2O/DMSO = 9 : 1, v/v), and 20 μL of Na2S4 stock solution was employed in some of the experiments. After vigorous shaking, the resulting solutions were kept at room temperature for 30 min, and their fluorescence signals were collected.
Cell culture
HeLa cells were cultured in high glucose DMEM containing 10% fetal bovine serum (FBS), 100 IU mL–1 penicillin and 100 IU mL–1 streptomycin at 37 °C in a 5% CO2–95% air incubator MCO-15AC (Sanyo, Tokyo, Japan).
Cell viability assay
Firstly, HeLa cells were seeded in 96-well plates (3000 cells per well) and maintained at 37 °C in a humidified atmosphere (95% air and 5% CO2) for 24 h. After 24 h incubation, the cells were washed with PBS and then cultured in a DMSO/PBS solution (pH 7.4, 1 : 9 v/v) containing the τ-probe with various concentrations. After 30 min, the cells were washed with PBS and incubated in a fresh medium for an additional 24 h. Then, according to the manufacturer's instructions, 50 μL MTT solution was added to each well and incubated for 4 h at 37 °C. After removing the medium, 150 μL DMSO was added to solubilize the blue-colored tetrazolium, the plates were gently shaken for 5 min, and the optical density values at 550 nm were monitored using a Thermo Scientific Varioskan Flash. The cell viability was set as 100% in control cells.
Intracellular H2Sn detection by using a fluorescence spectrometer
Cells were subjected to different treatments: the first group was incubated with 100 μM N-ethylmaleimide (NEM) for 15 min, the second group was untreated, the third group was incubated with 20 μM Na2S4 for 30 min, and the forth group was incubated with 0.065 μM Na2S4 for 30 min, while the fifth group was incubated with 0.13 μM Na2S4 for 30 min. After washing with PBS, the cells were cultured in a DMSO/PBS solution (pH 7.4, 1 : 9 v/v) containing a 10 μM τ-probe for 30 min. Then fluorescence lifetime was acquired after washing with PBS (pH = 7.4). The cells were excited at 488 nm and the emission was collected at 515 nm.
Confocal fluorescence images of HeLa cells upon incubation
HeLa cells were digested and seeded on confocal dishes for 24 h at 37 °C. The cells were subjected to different treatments. The first group was incubated with 100 μM N-ethylmaleimide (NEM) for 15 min, and the second group was untreated, while the third group was incubated with 20 μM Na2S4 for 30 min. After washing with PBS, the cells were cultured in a DMSO/PBS solution (pH 7.4, 1 : 9 v/v) containing a 10 μM τ-probe for 30 min. Then confocal images were acquired after washing with PBS (pH = 7.4). The cells were excited at 488 nm and the emission was collected from 505 to 550 nm.
Optical configuration of fluorescence lifetime microscopy
The homemade fluorescence lifetime microscopy system was made up of a 488 nm nanosecond pulse laser, an inverted microscope and a delay generator. The pulse laser (20 Hz, ∼5 ns, Quanta-Ray INDI, spectra-physics, USA) acted as the excitation light source and was focused on the sample through a beam expander. The fluorescence intensity of the sample was collected by using a 20× objective and recorded with an ICCD camera (iStar CCD 334, Andor Technology Ltd., UK), with an optical gate width of ∼2 ns. To avoid the influence from the excitation light source, a 500 nm long-pass filter was added before the ICCD camera. In addition, the recorded delay was provided by a delay generator (DG645, Stanford Research Systems, USA).
Fluorescence lifetime images of HeLa cells
HeLa cells were digested and seeded on confocal dishes for 24 h at 37 °C. The cells were washed with PBS and then cultured in a DMSO/PBS solution (pH 7.4, 1 : 9 v/v) containing a 10 μM τ-probe for 30 min, and then washed with PBS and subjected to different treatments. The first group was untreated, while the second group was incubated with 20 μM Na2S4 for 30 min. Then fluorescence lifetime images were acquired after washing with PBS (pH = 7.4). The cells were excited by using a pulse laser (∼5 ns) with a wavelength of 488 nm and the fluorescence emission was collected by using an ICCD camera through a 500 nm long-pass filter. Every group of fluorescence decaying images were captured after a delay time of 0, 2, …, 26 ns. The map of fluorescence lifetime was obtained through fitting the emission intensities of each pixel at different delay times. The time baseline was corrected using a FLS 980 spectrophotometer by measuring the same sample, and the response curves of the laser pulse and instrument were deconvoluted from the decay curve of fluorescence emission to obtain the final lifetime. Finally, the intracellular concentration distribution of H2Sn was obtained by substituting the fluorescence lifetime into the working equation.
Fluorescence lifetime images of zebrafish
Zebrafish were anesthetized in a PBS (pH = 7.4) solution containing 0.125% ethyl 3-aminobenzoate methanesulfonate. Firstly, the experimental group was incubated with 20 μM Na2S4 for 30 min, and then washed with PBS (pH = 7.4). Afterwards, the experimental group and the control group were both incubated with the τ-probe (10 μM) for 30 min in DMSO/PBS (pH 7.4, 1 : 9 v/v) solution. Then fluorescence lifetime images were acquired after washing with PBS (pH = 7.4). The fluorescence lifetime of the τ-probe and intracellular concentration distribution of H2Sn in zebrafish were obtained by the same procedure as that of the cells. The fish was too big to fit in the view field of the microscope, so the image was made up of three pieces. The experimental conditions for these three images were exactly the same. All animal work was approved by the Animal Care Committee of Nanjing University in accordance with Institutional Animal Care and Use Committee guidelines.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21327902, 21535003, and 21675081), State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM1715) and program B for Outstanding PhD candidate of Nanjing University (201702B052).
Footnotes
†Electronic supplementary information (ESI) available: Detailed synthesis procedure and design strategies of the τ-probe, schematic diagram of the fluorescence lifetime microscope, characterization of the τ-probe, and additional figures. See DOI: 10.1039/c8sc01879k
References
- Fukuto J. M., Carrington S. J., Tantillo D. J., Harrison J. G., Ignarro L. J., Freeman B. A., Chen A., Wink D. A. Chem. Res. Toxicol. 2012;25:769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiol. Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wang X., Sun J., Zhang W., Ma X., Lv J., Tang B. Chem. Sci. 2013;4:2551–2556. [Google Scholar]
- Austgen J. R., Hermann G. E., Dantzler H. A., Rogers R. C., Kline D. D. J. Neurophysiol. 2011;106:1822–1832. doi: 10.1152/jn.00463.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H., Shibuya N., Kimura Y. Antioxid. Redox Signaling. 2012;17:45–57. doi: 10.1089/ars.2011.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert J. W., Jha S., Gundewar S., Elrod J. W., Ramachandran A., Pattillo C. B., Kevil C. G., Lefer D. J. Circ. Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson K. R., Whitfield N. L. Antioxid. Redox Signaling. 2010;12:1219–1234. doi: 10.1089/ars.2009.2921. [DOI] [PubMed] [Google Scholar]
- Li L., Bhatia M., Zhu Y. Z., Zhu Y. C., Ramnath R. D., Wang Z. J., Anuar F. B. M., Whiteman M., Salto-Tellez M., Moore P. K. FASEB J. 2005;19:1196–1198. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- Baskar R., Bian J. Eur. J. Pharmacol. 2011;656:5–9. doi: 10.1016/j.ejphar.2011.01.052. [DOI] [PubMed] [Google Scholar]
- Kimura H. Nitric Oxide. 2015;47:S6. doi: 10.1016/j.niox.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Jacob C., Burkholz T., Griffin S., Faulstich L., Estevam E. C. Curr. Org. Chem. 2016;20:211–217. [Google Scholar]
- Ono K., Akaike T., Sawa T., Kumagai Y., Wink D. A., Tantillo D. J., Hobbs A. J., Nagy P., Xian M., Lin J., Fukuto J. M. Free Radical Biol. Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishanina T. V., Libiad M., Banerjee R. Nat. Chem. Biol. 2015;11:457. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav P. K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M. R., Banerjee R. J. Am. Chem. Soc. 2016;138:289–299. doi: 10.1021/jacs.5b10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N., Fu C., Pappin D. J., Tonks N. K. Sci. Signaling. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Proc. Jpn. Acad., Ser. B. 2015;91:131–159. doi: 10.2183/pjab.91.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen C. E., Carroll K. S. Chem. Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Nitric Oxide. 2014;41:4–10. doi: 10.1016/j.niox.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Kimura H. Neurochem. Int. 2013;63:492–497. doi: 10.1016/j.neuint.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Hideo K. Antioxid. Redox Signaling. 2017;27:619–621. doi: 10.1089/ars.2017.7076. [DOI] [PubMed] [Google Scholar]
- Greiner R., Pálinkás Z., Bäsell K., Becher D., Antelmann H., Nagy P., Dick T. P. Antioxid. Redox Signaling. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y., Mikami Y., Osumi K., Tsugane M., Oka J.-i., Kimura H. Nitric Oxide. 2014;39:S39. [Google Scholar]
- Carter K. P., Young A. M., Palmer A. E. Chem. Rev. 2014;114:4564–4601. doi: 10.1021/cr400546e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Gao X., Shi W., Ma H. Chem. Rev. 2014;114:590–659. doi: 10.1021/cr300508p. [DOI] [PubMed] [Google Scholar]
- Liu C., Chen W., Shi W., Peng B., Zhao Y., Ma H., Xian M. J. Am. Chem. Soc. 2014;136:7257–7260. doi: 10.1021/ja502968x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso A., Bruno E., Parise M., Pepe M. Analyst. 2015;140:3766–3772. [Google Scholar]
- Gao M., Yu F., Chen H., Chen L. Anal. Chem. 2015;2015:3631–3638. doi: 10.1021/ac5044237. [DOI] [PubMed] [Google Scholar]
- Zeng L., Chen S., Xia T., Hu W., Li C., Liu Z. Anal. Chem. 2015;87:3004–3010. doi: 10.1021/acs.analchem.5b00172. [DOI] [PubMed] [Google Scholar]
- Han Q., Mou Z., Wang H., Tang X., Dong Z., Wang L., Dong X., Liu W. Anal. Chem. 2016;88:7206–7212. doi: 10.1021/acs.analchem.6b01391. [DOI] [PubMed] [Google Scholar]
- Huang Y., Yu F., Wang J., Chen L. Anal. Chem. 2016;88:4122–4129. doi: 10.1021/acs.analchem.6b00458. [DOI] [PubMed] [Google Scholar]
- Zhang J., Zhu X.-Y., Hu X.-X., Liu H.-W., Li J., Feng L. L., Yin X., Zhang X.-B., Tan W. Anal. Chem. 2016;88:11892–11899. doi: 10.1021/acs.analchem.6b03702. [DOI] [PubMed] [Google Scholar]
- Ma J., Fan J., Li H., Yao Q., Xu F., Wang J., Peng X. J. Mater. Chem. B. 2017;5:2574–2579. doi: 10.1039/c7tb00098g. [DOI] [PubMed] [Google Scholar]
- Zhang J., Hao X., Sang W., Yan Q. Small. 2017;13:1701601. doi: 10.1002/smll.201701601. [DOI] [PubMed] [Google Scholar]
- Chen W., Rosser E. W., Zhang D., Shi W., Li Y., Dong W.-J., Ma H., Hu D., Xian M. Org. Lett. 2015;17:2776–2779. doi: 10.1021/acs.orglett.5b01194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Yang S., Guo C., Zeng Q., Qing Z., Cao Z., Li J., Yang R. Anal. Chem. 2018;90:881–887. doi: 10.1021/acs.analchem.7b03755. [DOI] [PubMed] [Google Scholar]
- Chen W., Rosser E. W., Tetsuro M., Armo P., Takaaki A., Xian M. Angew. Chem. 2015;54:13961–13965. doi: 10.1002/anie.201506887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Pacheco A., Takano Y., Day J. J., Hanaoka K., Xian M. Angew. Chem., Int. Ed. 2016;55:9993–9996. doi: 10.1002/anie.201604892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Chen W., Shi W., Li H., Xian M., Ma H. Chem. Commun. 2017;53:8759–8762. doi: 10.1039/c7cc04093h. [DOI] [PubMed] [Google Scholar]
- Li K.-B., Chen F.-Z., Yin Q.-H., Zhang S., Shi W., Han D.-M. Sens. Actuators, B. 2018;254:222–226. [Google Scholar]
- Shang H., Chen H., Tang Y., Guo R., Lin W. Sens. Actuators, B. 2016;230:773–778. [Google Scholar]
- Takano Y., Hanaoka K., Shimamoto K., Miyamoto R., Komatsu T., Ueno T., Terai T., Kimura H., Nagano T., Urano Y. Chem. Commun. 2017;53:1064–1067. doi: 10.1039/c6cc08372b. [DOI] [PubMed] [Google Scholar]
- Kawagoe R., Takashima I., Uchinomiya S., Ojida A. Chem. Sci. 2017;8:1134–1140. doi: 10.1039/c6sc03856e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Yue X., Zhang H., Li W., Zhang L., Xiao Q., Huang C., Sheng J., Song X. Anal. Chem. 2017;89:12984–12991. doi: 10.1021/acs.analchem.7b04033. [DOI] [PubMed] [Google Scholar]
- Yu F., Gao M., Li M., Chen L. Biomaterials. 2015;63:93–101. doi: 10.1016/j.biomaterials.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Chen W., Yue X., Sheng J., Li W., Zhang L., Su W., Huang C., Song X. Sens. Actuators, B. 2018;258:125–132. [Google Scholar]
- Valeur B. and Berberan-Santos M. N., Molecular Fluorescence: Principles and Applications, Wiley, 2012. [Google Scholar]
- Gryczynski Z., Gryczynski I. and Lakowicz J. R., in Molecular Imaging, ed. R. N. Day, American Physiological Society, San Diego, 2005, pp. 21–56. [Google Scholar]
- Ammasi Periasamy R. M. C., FLIM Microscopy in Biology and Medicine, Chapman and Hall/CRC, New York, 2009. [Google Scholar]
- Pan R., Xu M., Jiang D., Burgess J. D., Chen H.-Y. Proc. Natl. Acad. Sci. U. S. A. 2016;113:11436–11440. doi: 10.1073/pnas.1609618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugherli L., Burchak O. N., Chatelain F., Balakirev M. Y. Bioorg. Med. Chem. Lett. 2006;16:4488–4491. doi: 10.1016/j.bmcl.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Bahde R. J., Appella D. H., Trenkle W. C. Tetrahedron Lett. 2011;52:4103–4105. doi: 10.1016/j.tetlet.2011.05.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.