

ABSTRACT
Vibrational spectroscopy provides non-destructively the molecular fingerprint of plant cells in the native state. In combination with microscopy, the chemical composition can be followed in context with the microstructure, and due to the non-destructive application, in-situ studies of changes during, e.g., degradation or mechanical load are possible. The two complementary vibrational microspectroscopic approaches, Fourier-Transform Infrared (FT-IR) Microspectroscopy and Confocal Raman spectroscopy, are based on different physical principles and the resulting different drawbacks and advantages in plant applications are reviewed. Examples for FT-IR and Raman microscopy applications on plant cell walls, including imaging as well as in-situ studies, are shown to have high potential to get a deeper understanding of structure–function relationships as well as biological processes and technical treatments. Both probe numerous different molecular vibrations of all components at once and thus result in spectra with many overlapping bands, a challenge for assignment and interpretation. With the help of multivariate unmixing methods (e.g., vertex components analysis), the most pure components can be revealed and their distribution mapped, even tiny layers and structures (250 nm). Instrumental as well as data analysis progresses make both microspectroscopic methods more and more promising tools in plant cell wall research.
KEYWORDS: Chemical imaging, confocal Raman microscopy, FT-IR microscopy, microspectroscopy, plant cell walls
1. Plant cell walls
Lignocellulosic biomass is the world's most abundant renewable material and represents mainly plant cell walls. It has a long tradition as an important source for building material, energy, textiles and paper production and plays a central role in human and animal nutrition. With fossil fuel reserves diminishing and their emission effects on global climate, there is an increasing interest and need for renewable carbon-neutral industrial feedstocks, like plant biomass.
The plant cell wall is the surface and product of the plant cell, which controls the composition of the wall according to specific needs: it is flexible during cell elongation (primary cell wall) and stiff in wood cells (secondary cell wall) (1). Cellulose synthase complexes produce crystalline cellulose ((1–4)-β-linked glucose) microfibrils as the basic structural elements, which are embedded in matrix polymers produced in Golgi compartments. In the thin primary cell wall (0.1–1 μm) built during early growth, the cellulose microfibril forms a network with mainly pectins and hemicelluloses (2). These thin walls give already shape and structure to plant cells, tissues, and ultimately organs and are sufficiently strong to prevent the cell from rupturing and yet flexible and plastic to accommodate growth (3). The secondary cell wall, a thick layer which is deposited after cells cease enlargement, is often lignified. Lignins are three-dimensional, amorphous heteropolymers that impart water impermeability and confer structural support and flexural stiffness to the plant cell wall (4, 5). The secondary cell wall is made of several layers, called S1, S2, S3, which differ in their thickness, chemical composition and cellulose microfibril angle (MFA, measured from the fiber longitudinal axis) (6). In particular, the alternation of high and low angles in the different layers of the secondary cell wall increases its rigidity and mechanical strength. From a mechanical point of view, it can indeed be regarded as a composite material, like reinforced concrete (7). With the movement of plants from the water to the land quite diverse body plans evolved with highly specialized tissues with distinct properties and functions (8, 9). To survive in the great variety of habitats many different functions had to be adapted and resulted in the high variability of plant cells and its wall composition and structure (10, 11).
For an optimized utilization or design of materials, basically four elements have to be understood and controlled: the structure and chemical composition, the synthesis and processing, the properties and the performance of a material. These four elements have been shown at the apices of a tetrahedron (Figure 1) to indicate the importance of the interrelationships of the elements, each with the other (12).
Figure 1.
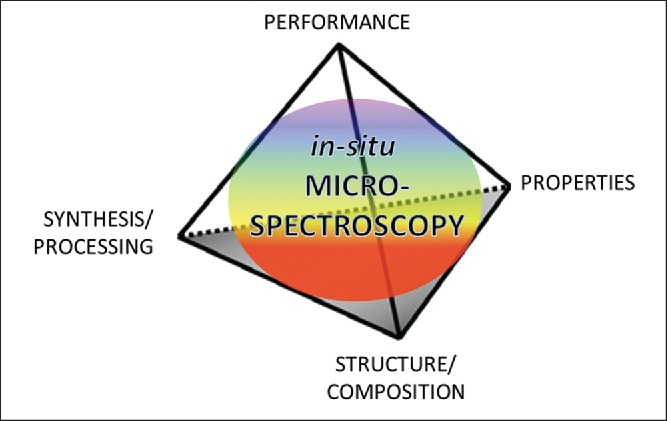
Fleming´s tetrahedron, illustrating the interrelationship of performance, process, structure and properties.
Like in other biological materials the structural arrangement of internal components encompasses several hierarchical levels, spanning the atomic and molecular, nano-, micro-, meso-, and macroscopic length scales. The wide range of length scales makes a comprehensive understanding of composition and structure a great challenge.
Vibrational spectroscopy gives access to the molecular structure of the material in the native state. In combination with microscopy, the chemical composition can be followed in context with the microstructure and furthermore due to the non-destructive application in-situ studies of performance (changes during degradation, mechanical load, temperature changes, etc.) are possible (Figure 1). In this work, the latest microscopic and in-situ performance studies using the two vibrational spectroscopic approaches, FT-IR and Raman spectroscopy, are reviewed including a general introduction into the principles of the methods. Three scientific disciplines (chemistry–physics–biology) come together as the chemical composition (molecular structure) is investigated with methods based on physical principles in context with the biological microstructure.
2. Vibrational microscopy
2.1. Raman and infrared (IR) spectroscopy: theory and principles
Both methods (IR and Raman spectroscopy) probe molecular vibrations, but the underlying physical mechanisms are different: absorption of light quanta and inelastic scattering of photons, respectively. Infrared absorption occurs, if the energy of an incident photon from a polychromatic light source matches the energy gap between the ground state of a molecule and an excited vibrational state (13). For simple vibrations within molecules, the matching frequency range of the spectrum is the mid-range infrared (400–4000 cm–1), corresponding to wavelengths of about 10 µm. In contrast in Raman spectroscopy, the scattering mechanism for exciting molecular vibrations requires monochromatic irradiation in the visible (VIS) light region (or ultraviolet (UV) or near-infrared (NIR) region) (Table 1). The Raman effect, that a very small portion of the incident photons is scattered inelastically (Stokes- and Anti-Stokes Lines) was for the first time experimentally proven in 1928 by C.V. Raman (14). The energy difference corresponds to the energy change of the molecule, which refers to the transition between two vibrational states. Nevertheless, most of the light is scattered without any interaction of the photons with the materials and is regarded as elastic scattering (Rayleigh scattering). The Raman signal is therefore a very weak signal and usually signal-to-noise ratio (S/N) is not as good as in Infrared spectroscopy. If absorption and electronic transitions occur undesirable fluorescence that masks the weaker Raman scattering signal or resonance enhancement of the Raman signal might be observed (15).
Table 1.
Comparison of the prinicipal characteristics of infrared and Raman microspectroscopy.
| Infrared | Raman | |
|---|---|---|
| Mechanism | Absorbance | Scattering |
| Excitation | Polychromatic | Monochromatic |
| -Source | Globar (or synchrotron source) | Laser |
| -Wavelength | IR | UV or VIS or NIR |
| Spectra | ||
| -Selection rule | Change in dipole moment (asymmetrical bonds, e.g., OH) | Change in polarizability (symmetrical bonds, e.g., C–C, backbone) |
| -frequency range | 4000–600 cm−1 | 4000–50 cm−1 (Stokes and Antistokes) |
| Resolution: | ||
| -Lateral | ∼5–20 µm | ∼0.3–2 µm |
| -Confocal/depth | not possible | ∼0.6–4 µm |
| Sample modes | Transmission, reflection, trans-flection; cassegrain or attenuated total reflectance (ATR) objective | Reflection (Scattering); immersion microscope objectives with high numerical aperture possible |
| Sample preparation | Optimal thickness (transmission mode), sample contact (ATR mode) | Plane surface for mapping/imaging, no contact, water as solvent or glass as container do not disturb |
| Problems | Strong absorption of glass, H2O and CO2 | Fluorescence and sample destruction (burning) |
Due to the different underlying mechanisms the sensitivity toward different types of chemical bonds (selection rules) differs between the two methods: IR is sensitive to the polarization (change in the dipole moment) of a bond, while Raman is sensitive to its polarizability (distortion of the electron cloud). Thus IR gives strong signals for polar functional groups, while Raman primarily shows vibrations of, e.g., the carbon backbone and vibrations of (nearly) symmetrical structures such as double bonds. Because of this fundamental difference, IR and Raman are often used as complimentary techniques, each highlighting different structures within the sample.
2.2. Microspectroscopy: spatial resolution and sample preparation
Resolution is the minimum distance at which two distinct points of a specimen can still be seen as separate objects. The spatial resolution of a microscope is ultimately determined by Abbe´s diffraction limit and given by r = 0.61 λ/NA, where λ is the wavelength of the light and NA the numerical aperture of the objective. NA is defined by the refractive index of the medium (n) in which the optics are immersed (e.g., 1.0 for air and up to 1.56 for oils) and the half-angle of the maximum cone of light that enters or exits the condenser or objective (θ) (NA = n sinθ). Two objects are completely resolved if they are separated by 2r and barely if they are separated by r (Rayleigh criterion of resolution) (16).
Considering the relation between r and the wavelength, it becomes clear that UV-excitation will achieve the highest spatial resolution, followed by VIS and NIR excitation and the lowest by IR-excitation. The need to use Cassegrain (Schwartzschild) objectives in IR-microscopy limits furthermore the spatial resolution as the largest achieved NA is approximately 0.6. Immersion optics are almost never used in IR because of the absorption of IR radiation by the oil, whereas in Raman microscopy the use of immersion objectives (e.g., oil with NA = 1.4) enhances the spatial resolution (16). Raman microscopy achieves a spatial resolution of ∼0.3 µm, which allows acquiring spectra selectively from the different cell wall layers and the cell corners (CC). For compositional changes of different cells or tissue types also IR-microscpectroscopy gives important information (10–20 µm). Enhanced spatial resolution (6–8 µm) in IR-microscopy can be achieved by using a so-called micro attenuated total reflection (ATR) objective with increased NA through the high refractive index of a Germanium crystal (17).
The use of a laser as excitation source in Raman microscopy allows performing confocal measurements and an axial resolution of about twice of the lateral resolution (18) (Table 1). Also if depth resolution is important, immersion objectives (oil, water) with high NA give the best results (19). In IR-microscopy, the use of ATR accessories with different angles of incidence provides an opportunity for depth profiling (17).
IR-microscopy relies on the absorption process of the incident light and is therefore often performed in transmission mode, whereas Raman is based on the scattered light and has to be performed in reflection (Table 1). To work in transmission mode, a defined thickness of the sample is necessary and to work in reflectance mode a planar surface is required when scanning (imaging) of the samples is aimed at. Therefore, microspectroscopy methods often rely on proper sample preparations. For IR-and Raman microscopy fresh or embedded plant samples can be cut by a rotary or sliding microtome to a thickness between 1 and 20 µm. For the Raman imaging approach, cryo-cutting and Polyethylenglycol embedding have proven as the best approaches as high quality sections are achieved and no signal of the embedding medium has to be taken into account (20).
2.3. Instrumentation: first plant cell wall experiments & developments
Raman spectroscopy instrumentation advancements (e.g., laser technology that provide a highly monochromatic source, multilayer dielectric and holographic dichroic filters that effectively remove Rayleigh scattered laser light, imaging spectrometers that isolate wavelengths of interest, highly sensitive charge coupled device (CCD) imaging detectors, and powerful PCs having sufficient data storage capacities and processing speeds capable of handling large data) have led to a dramatic increase in use and development of Raman microscopy approaches (21). The first Raman studies on lignocellulosic materials were published in the 1980s (22–24). The problem of laser induced fluorescence that almost swamps the weaker Raman signal in many lignified samples was encountered and methods to deal with have been developed. One important step was to acquire the Raman spectra not from the dry sample, but under water and/or oxygen (22, 25). To choose an excitation wavelength where a sample and its impurities do not absorb (near IR region) was becoming possible through advances in instrumentation. Moving from the VIS to the NIR range, fluorescence virtually disappears as electronic absorption bands are unlikely. The use of Nd:YAG (neodymium-doped yttrium aluminum garnet) laser radiation at 1064 nm coupled with interferometers (involving Fourier transformations) led to so-called near-infrared Fourier Transform (NIR-FT) Raman spectrometers (26) and to more applications on plant cell wall e.g., (27–30). However, as the Raman scattering intensity is proportional to ν4,where ν is the frequency of the exciting laser radiation, longer collection times are required and also the spatial resolution is inferior (see 2.2). Other approaches to overcome fluorescence interference applied in the last years have been UV-resonance Raman spectroscopy and Kerr-gated Raman spectroscopy (31–33). A laser with wavelength in the VIS range (e.g., Ar+, He–Ne, Kr+, frequency doubled Nd:YAGlasers) is usually coupled with a dispersive spectrometer and a CCD for detection. These classical dispersive multichannel Raman spectrometers are nowadays often used in confocal microscope configurations with the advantage of superior rejection of fluorescence and depth resolution due to the pinhole (34). Together with in the beginning mentioned instrument improvements much shorter acquisition times (0.01–1 s per spectrum) and thus shorter exposure times to the laser light became possible (35) and opened up new fields for mapping and imaging. In point-by-point scanning, the sample is scanned with a laser beam using X, Y, Z scanning stages. At each position of the raster a Raman spectrum is acquired and out of these spectra an image generated. To speed up experiment time, instrument developments go toward line scanning (laser is elongated in a line with the help of a moving mirror of cylindrical optic devices) and wide-field imaging (whole sample field is illuminated and with different spectral channels images recorded) (21).
The application of FT-IR microscopy in plant sciences has become widespread, since the 90s, e.g., (36–42). In the last years FT-IR imaging technologies have benefited from developments in IR detector arrays leading to a marked growth of applications (43) as experiment time has been reduced drastically and signal-to-noise ratio increased. Focal plane array (FPA) imaging systems are built of an array of miniature detector elements that simultaneously record thousands of spectra over an area (16). Significant potential exists also in the applications of FT-IR spectroscopic imaging using ATR mode (17).
To reach higher signal intensities with high spatial resolution, the high brightness of IR synchrotron radiation (44, 45) has been successfully applied in some plant studies (46–50).
Several important reviews are available covering different FT-IR techniques used to study plant cell wall architecture and different plant tissues (36, 51–56). Also for the Raman studies on plant cell walls several reviews are recommended for reading (27, 29, 57–59). In this review, we will summarize which information can be gained from the plant cell wall spectra and then focus on recent imaging and in-situ performance studies on plant cell walls (24).
3. Raman and infrared spectra of plant cell walls
What kind of information do we get from plant cell wall spectra? What can we learn from it? The wall constituents, such as cellulose, hemicellulose, pectins, lignin and aromatic phenolics, have characteristic spectral features that can be used to identify and/or track these polymers in the native cell wall. Although it is not possible to assign each band in the complex cell wall spectra, the variation in intensity or absence of some bands reflect differences in polymer composition and structure (orientation) between cell walls. Many studies using conventional Raman and FT-IR spectroscopy on the isolated plant cell wall polymers and model compounds as well as different plant cell walls laid the groundwork for interpretation of plant cell wall spectra acquired in the microspectroscopic approaches. Yet still a lot of work needs to be done to establish more comprehensive spectral databases, band assignments as well as multivariate data analysis methods for extracting relevant data out of the acquired complex information.
3.1. Revealing composition and structure of plant cell wall polymers
The most studied plant cell wall polymer using vibrational spectroscopy is cellulose. The studies cover band assignment (60–64), identification of cellulose types (65–68) and determination of the degree of crystallinity (40, 41, 69–71). The relative ratio of cellulose Iα and Iß crystalline phases in developmental stages of wood cell wall formation was revealed by FT-IR microscopy and has led to a novel concept of on–off stressed crystallization, which may be responsible for the occurrence of both cellulose phases in wood formation (72). Very recently, Raman studies revealed that native plant cell walls lack crystalline cellulose domains and are formed during drying and processing (70). Crystalline cellulose bands are stronger and sharper than the bands deriving from other carbohydrate polymers (hemicellulose, pectin), which are present mostly in amorphous form. Because of the chemical similarity of the carbohydrate polymers the bands in the cell wall spectra partly overlap, but all the different hemicellluloses and pectins have characteristic band features and band characteristics and assignments are available (73–80). Spectra of lignin clearly differ from the carbohydrate spectra due to the aromatic structure. It is comprised of a variety of different types of covalent bonds derived from oxidative coupling of three different types of phenolic precursor units, p-coumaryl, coniferyl, and sinapyl alcohols and the heterogeneity of lignin structures in different species and tissue types is reflected in the vibrational spectra. Important work on lignin band assignment in infrared spectra was already published 1971 by Hergert (81) and later by Faix (82). For understanding, Raman spectra of lignin and deriving structural information important studies have been performed and provide a valuable source for interpretation of lignin structure based on Raman spectra, e.g., (83–89).
To deepen our understanding and knowledge gain of the acquired plant cell wall fingerprints a spectral database, including reference components as well as different plant cell walls is currently established and made available to the scientific community (http://bionami.at/spectra.html).
3.2. Alignment and association of polymers within the cell wall
Radiation polarized with its electric vector in one direction will only interact with similarly oriented molecular vibrations and therefore vibrational spectroscopy can also be used to elucidate molecular orientations. The change in the relative intensities of cellulose bands with alteration of the incident-beam polarization revealed new information concerning the ordering of molecular chains of cellulose within the cell walls of natural cellulose fibers, such as cotton or ramie (24). A few years later Raman microprobe evidence was reported that not only cellulose is arranged with a preferred orientation in the cell walls of woody tissue, but also lignin (90). Very recent, FT-IR microscopy studies have confirmed that lignin in hardwoods and softwoods are to some extent structurally ordered with a preferred direction following that of the cellulose microfibrils (91, 92). Also for the hemicelluloses in wood a high degree of association and orientation in parallel to the cellulose microfibrils has been revealed (92–95). Besides on cellulose fibers and wood, the polarization dependent response has also given important insights into the orientation of cell wall polymers in, e.g., pea stems (96), oat coleoptiles (97), dry and hydrated onion cells (55, 98) and elongating carrot cells (99).
3.3. Comparing FT-IR and Raman spectra of plant cell walls
The complementary nature of FT-IR and Raman spectroscopy and the sensitivity of the signals on the polarization direction of the incident light are shown exemplarily on spectra acquired from single spruce wood fibers (Figure 2). The spot size in Raman microscopy was ∼2 µm (inset Figure 2A), and the aperture of the FT-IR microscope was set to cover the whole width of the fiber (15 µm) and 100 µm along the length (inset Figure 2B). Due to the different selection rule (mechanism of inducing molecular vibration, see Table 1), different bands are observed in the spectra.
Figure 2.
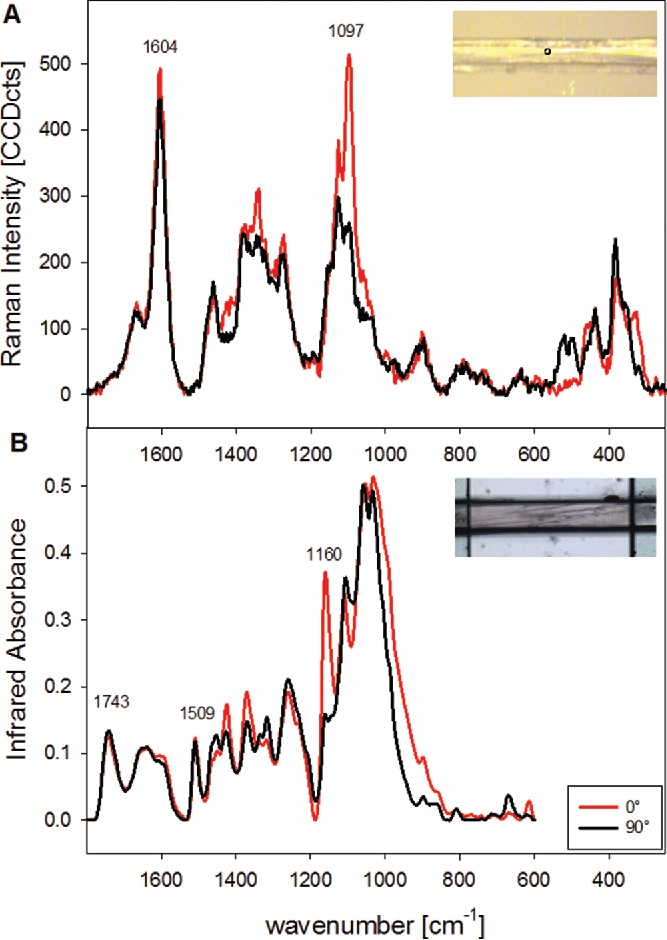
Baseline corrected Raman (A) and Infrared spectra (B) acquired on mechanically isolated single spruce wood fibers (microfibril angle < 10°) with the incident polarization direction parallel to the fiber (0° red spectra) and perpendicular to the fiber (90°, black spectra). (Experimental: A: Raman microscope (Hyperion 2000 BRUKER), 128scans, B: FT-IR microscope (WITEC), 785 nm laser, 10 s integration time, 10x objective (NA = 0.25).
In the Raman spectra (Figure 2A) also the polarization dependent and quite complex predominant skeletal bending modes vibrations of cellulose (60) below 600 cm−1 can be probed. The strong band at 1604 cm−1 is assigned to aryl stretching vibrations of lignin (75, 85). As this band has no overlap with the carbohydrate bands it can be used as a marker to image lignin distribution on the micron level (57, 100) (see 4.1.1). In the shoulder band contributions from coniferaldehyde units are expected at 1623 and 1660 cm−1, whereas coniferyl alcohol contributes at 1654 cm−1 as well as other chromophores (87). So, from this lignin derived band envelope also insights into lignin structure can be gained. Small changes in the lignin bands are observed, when the incident laser polarization direction is changed from 0° (parallel to fiber direction, red spectrum in Figure 2A) to 90° (perpendicular, black spectrum), which points to a preferred orientation, although only to a small extent. In the Infrared spectrum (Figure 2B) aromatic skeletal vibrations give a characteristic band at 1509 cm−1 and the 1607 cm−1 and 1664 cm−1 bands are overlapped with adsorbed water (1635 cm−1) (101, 102). Similar to the Raman spectra only small changes are observed in the lignin bands due to altered polarization direction. The most severe polarization dependent changes are with both techniques found for the bands attributed to the vibration of the cellulose (Figure 2A: Raman spectrum at 1097 cm−1; Figure 2B: FT-IR spectrum at 1160 cm−1). The orientation sensitivity of the 1097 cm−1 band can be used to probe changes in the cellulose microfibril orientation with the Raman imaging approach (103) (see 4.1.2).
In the infrared spectra, a clear band is observed at 1743 cm−1 assigned to C=O stretch of carboxylic acid of hemicelluloses(61) and C=O stretch of unconjugated ketones in Lignin(102), which is not clearly seen in the Raman spectra due to the different selection rules (Figure 2). Although the band assignment is of particular importance for the interpretation of IR and Raman microspectroscopy data, it is beyond the scope of this review article to provide a more extensive overview here. The most helpful papers for band assignments of the different polymers have been cited in the previous section and for wood spectra comprehensive band assignments are summarized for Raman by Agarwal and Ralph (75) and for FT-IR by Schwanninger et al. (101).
The spatial resolution of FT-IR microscopy is perfect for investigating changes on the wood fiber (cellular) or tissue level, e.g., for following changes in chemistry after different fiber isolation methods (104) or after enzymatic treatment (105) or as a high-throughput method to characterize cell wall mutant phenotypes (106). In the “whole fiber” or tissue characterization, the higher spatial resolution of Raman microscopy can be disadvantageous, but as soon as the different cell wall layers are of interest Raman imaging on micro-cut cross-sections is the method of choice as the plane cut surface gives more uniform scattering conditions than depth scans on fibers with changing fiber (cell) geometries.
4. Imaging chemical and structural changes on the sub-micron level
Based on thousands of position resolved spectra images are established either by univariate approaches (e.g., calculating the integral of marker bands, see 4.1) or by multivariate approaches (e.g., cluster analysis, principal component analysis (PCA), vertex component analysis (VCA), non-negative matrix factorization (NMF), see 4.2). The univariate analysis is always the beginning and can give a quick overview and based on selectively extracted average spectra also more detailed information (20).
The first univariate imaging studies on plant cell wall have been done 1998–1999 on flax stems (107, 108). FT-IR as well as NIR-FT-Raman microscopy were employed to map the distribution of the major components (cellulose, xylan, pectin and lignin) in the different anatomical cell types (107, 108). About 6 years later instrument advances in Confocal Raman microscopy are reflected in high resolution chemical images, in which the heterogeneity on the sub-micron level was followed in poplar (109, 110) and black spruce wood (111) cell walls. Around the same time FT-IR microscopy was introduced for imaging the chemical composition of, e.g., beech and poplar wood (112), transgenic aspen (113) and wheat grains (114, 115). In the following years, Raman imaging studies followed on different wood species (116–120), the model plants Arabidopsis (121, 122) and Brachypodium (123) and many other plant species, e.g., Newzealand flax (124), horsetail (125–127), bamboo (128–130), Miscanthus (131, 132) and tomato (133, 134). As Raman and FT-IR imaging gives complementary results with different spatial resolution in some studies also both methods have been applied, e.g., (135–137). Overall Raman imaging has been applied more often than FT-IR imaging, probably due to the fact that (1) only with a FPA detector imaging is achieved within reasonable measurement time and (2) the spatial resolution is limited by light diffraction 5–20 µm (138). Nevertheless FT-IR imaging is a promising tool in plant cell research (17, 50, 56, 112, 115, 139), especially on the plant tissue and cell level. For high resolution imaging, to elucidate, e.g., different cell wall layers and changes within the cell wall, Confocal Raman microscopy has shown high potential and is the focus of the next chapters.
4.1. Chemical and structural changes visualized by band integration
Raman imaging of plant organ cross-sections (stems, leaves or roots) reveals the chemical composition not only of different anatomical regions or tissues or cells, but in context with the microstructure. Cell layers and components are probed selectively on the sub-micron level as well as gradual changes are revealed. One major advantage of Raman imaging compared to staining and/or immunolabeling approaches is the fact that behind every pixel a full wavenumber spectrum is available for detailed information and final interpretation and/or verification of the imaging results. Therefore, always single spectra and/or average spectra based on marking regions of interests or band intensity thresholds should be extracted for verification and/or detailed analysis of molecular structure, e.g., the alignment of the cellulose microfibrils.
4.1.1. Following waterproofing and lignification of plant tissues
The advantage of Raman microscopy to investigate the sample in the wet native state enables to probe in-situ the water uptake (content) within the plant cells by integrating the broad water band (Figure 3A). In cross-sections of plant tissues, the highest intensity and pure water spectra are as expected observed within empty cell lumina (Figure 3A). In the shown poplar wood sample, an almost comparable high intensity is observed within the thick inner layers of the cells. This points to an enormous water uptake (swelling capacity) of this characteristic additional gelatinous layer (G-layer), which is laid down on the secondary cell wall layer in hardwoods to keep the stem and branches upright (140). A very small less hydrophilic layer was with the help of Raman imaging for the first time visualized as a lignified barrier on the G-layer toward the lumen (110). This tiny layer is not as highly lignified as the S-layer and not uniform, but with some accumulations preferably in the corners of the cells (Figure B). The occurrence of lignin in the G-layer during tension wood formation has long been debated (141). Some studies concluded that the G-layer mostly consists of cellulose and is totally free of lignin, e.g., (142, 143), whereas others reported lignin (144, 145). Raman imaging has given further evidence for lignification of the G-layer in maple (Acer spp.), beech (Fagus sylvatica) and oak (Quercus robur) (146). Lignin integration (Figure 3B) gives often a mirrored image of the water distribution (Figure 3A) as lignin is waterproofing the cell wall (122). Highest lignin intensity is often observed within the CC (Figure 3B), which are almost filled up with lignin to glue the wood fibers together.
Figure 3.
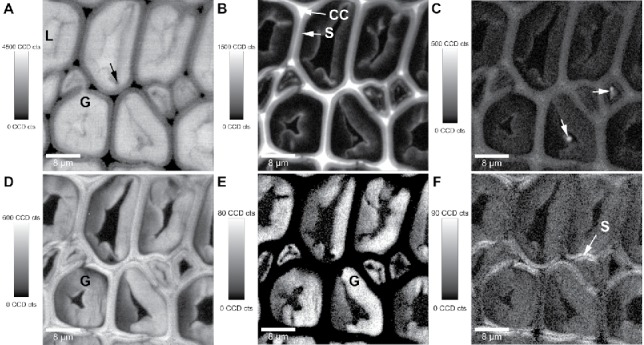
Raman images (30 µm × 30 µm) of poplar tension wood: Integrating the water band (A: 3111–3689 cm−1) shows the water content in the plant tissue, the vibrations mainly attributed to aromatic components (B: 1536–1724 cm−1) the lignification, while the 1460 cm−1 band visualizes deposits of lipid character. All the cell walls are highlighted by integration from 1076 to 1184 cm−1 (D, mainly carbohydrates), while the 1380 cm−1 band reveals selectively the G-layer (E: 1356 – 1404 cm−1) and the 1096 cm−1 the secondary cell wall in x-direction ( = laser polarization direction) (F: 1077–1115 cm−1). Experimental details see (110).
In this poplar tension wood sample other hydrophobic pointwise accumulations are observed within the lumen by integrating the 1460 cm−1 band (Figure 3C, arrows). The spectra at this position point toward a more lipid character (147) of these lumen deposits. The hydrophobic lipids in the lumen as well as the lignified layer toward the lumen are also visible in the water integration image as regions with less water content compared to the G-layer (Figure 3A).
Besides following changes in lignification and water distribution by Confocal Raman microscopy has also been successfully applied to reveal differences in wildtype and young transgenic aspen. Hand in hand with reduced lignin content a higher volume of water was found in the transgenic aspen, indicating an increase in the hydrophilicity of the cell wall after transgenic modification (120). Very recently, both μ-FTIR and confocal Raman spectroscopy have been shown to provide valuable information about the spatial distribution of adsorbed water in morphologically distinct cell wall regions of Ginkgo biloba (148)
4.1.2. Changes in cellulose and determination of cellulose microfibril angle
On a poplar wood cross-section integration from 1076 to 1138 cm−1 (mainly carbohydrates) visualizes all the cell wall material (Figure 3D), while the 1380 cm−1 band integration gives selectively the G-layer (Figure 3E). This is due to the fact that this band is only clearly separated in pure cellulosic cell walls, while it gets more hidden as a shoulder when lignin is present and a strong band arises at 1334 cm−1. The secondary cell wall (S) is lignified with medium intensity (Figure 3B) and the high signal of the 1097 cm−1 band (Figure 3F) only in the x-direction (laser polarization direction) points to high cellulose microfibril angle (MFA) with respect to the fiber axis. If the cellulose fibrils are aligned with a gentle helical slope (S-layer), the C–O–C bonding of the cellulose become more exposed on a cross-section and thus higher intensity of the corresponding 1097 cm−1 band in the polarization plane of the laser beam is observed (57, 109, 110). Therefore, changes in the cellulose bands due to changes in amount (all bands diminished and no change in laser orientation direction) have to be clearly distinguished from intensity changes due to changes in cellulose microfibril orientation (different band height ratios and laser polarization dependency). In other words, changes in amount cannot be probed by the band integration approach, if they go hand in hand with changes in cellulose microfibril orientation and/or crystallinity.
From spectra extracted from the cell walls mapped parallel to the laser polarization direction, the MFA within the different cell wall layers can be calculated based on mathematical models (103). These models are based on a detailed study on the change of Raman spectra with respect to the angle between cellulose fiber (molecule) and laser orientation direction. By acquiring spectra from a single spruce fiber (MFA <10°) at one position during changing the laser polarization direction in 3° steps (Figure 4A), the orientation dependent changes of the spectra were recorded (Figure 4B) and described by quadratic linear regression (band height ratios) and partial least square regressions (selected wavenumber regions), respectively (103).
Figure 4.
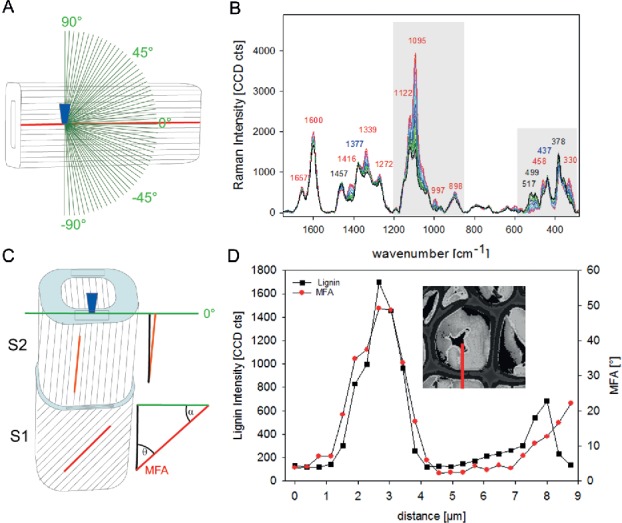
Experimental set up (A) for investigating the dependence of the Raman intensity of the cellulose bands on microfibril angle and laser polarization direction. The laser beam is focused through a microscopic objective (dark blue) within the tangential S2 layer of a single latewood fiber, which cellulose fibril orientation is known parallel with respect to the fiber axis. Spectra are acquired from one position while changing the laser polarization direction in 3° steps (green lines) to investigate the dependency between cellulose fibril orientation and laser polarization direction (B). In the imaging approach, the laser beam (microscopic objective, dark blue) is focused on a cross-sectional area (light blue) of wood samples with the laser polarization direction (green line) constant parallel to the tangential cell wall. By extracting spectra from cell wall areas parallel to the laser polarization direction (black rectangle, C) the angle between cellulose orientation and laser polarization direction (α) can be predicted from the spectral characteristics of the cell wall layers to finally calculate the microfibril angle (MFA, θ = 90-α) (details, see(103)). Change in lignin amount (integrated intensity of the aromatic lignin peak) and microfibril orientation (calculated based on models shown in Gierlinger et al. (103)) across a tension wood cell (red line marked in the insert) (D).
For the prediction of unknown samples with different and changing lignin content, the models based on two spectral regions (selected to include most of the orientation dependent changes and almost no lignin contribution) work out best (Figure 4B, grey parts). The prerequisite of exact geometric sample and laser alignment has to be taken into account for prediction of microfibil angle based on spectra of cross-section Raman images (Figure 4C). Only those parts of the cells aligned completely parallel to the laser polarization direction (Figure 4C, rectangular drawing in the cell wall layers) can be used for spectra extraction to predict the MFA. In the image scan of Figure 3, this constraint is fulfilled only in the radial walls of the cell in the left corner. On this position, a cross-section can be laid (Figure 4D, inset) and the microfibril calculated across the cell wall based on the extracted spectra. The highest MFA are calculated for the CML and S2 (Figure 4D). The CML is usually smaller than 0.6 µm, and thus the highest detected value of 50° will be underestimated as the S2 will contribute to the signal. Along the S2-CML-S2 an average of 34° is calculated for the secondary cell wall, which coincides with the value determined with x-ray after enzymatic removal of the G-layer (149). Within the G-layer 2–3° are calculated and an increase toward the lignified lumen-side is observed (Figure 4D). Along the investigated cell wall layers of tension wood the change in MFA goes hand in hand with a change in the lignin amount (Figure 4D).
Getting knowledge on cellulose microfibril orientation and cell wall polymer composition on the tissue and micro-level helps to understand plant cell wall design and its optimization for different functions. Raman imaging studies gave, therefore, important insights in understanding the structure–function relationships of stress and movement generation in reaction wood tissues (110, 146, 149, 150). Analogously to poplar tension wood, a thick almost pure cellulosic layer on a thin lignified secondary cell wall layer with higher MFA was identified by the Raman imaging approach in roots of red clover. The same cell wall design like in tension wood was revealed and concluded to be responsible for moving the foliage buds deeper into the soil (151).
Collecting the whole Raman images with different degrees of laser polarization direction revealed detail on cellulose orientation within dislocations in hemp (Cannabis sativa (L.)) fibers. While the chemical composition did not change, it was found that cellulose microfibrils within dislocations have a different orientation as well as the transition zones. Furthermore, cellulose orientation seemed to be less uniform within dislocations than in the surrounding cell walls (152).
Very recently, a similar to the above described polarized Raman microspectroscopic method was applied on rice plant cell walls, whereby differences between wild type rice plants and the rice brittle culm mutant with a more disordered cellulose microfibril arrangement have been detected (153). Also, a polarized light microscopy approach was introduced to determine the cellulose MFA and applied on cells from an Araucaria branch, in a wheat (Triticum turgidum) awn's hygroscopically active region, and a storkbill's (Erodium gruinum) coiling awn (154).
In any of these studies on cellulose orientation and mechanical properties great care has to be taken in sample preparation (fiber cell alignment and cutting axis) as even small misalignment by a few degrees can result in different mechanical properties assessed with nanoindentation as well as a change in the measured cellulose MFA (155).
4.1.3. Minerals and carbohydrates
Minerals and inorganic substances in plants are normally determined as the residue remaining after all the combustible material has been burned off and therefore no detailed information about the kind of inorganic is gained. Raman microscopy studies allow the identification, characterization and spatial localization of inorganic substances in fossil e.g (156) and ancient plants (125–127, 157, 158) as well as in modified (mineralized) wood (159, 160).
Imaging can be applied directly on plant tissues (125–127) or also on isolated phytoliths to reveal organic matter within them (161). One of the minerals of great interest in plants is silicon (Si). It is one of the most abundant elements on earth and although not considered essential for the growth and development of higher plants, it is known to increase vigor and to play protective roles. A lot of research has been done on Equisetaceae, one of the oldest living plant families, known for high silica accumulations (162) and having a stem with a special lightweight design (163) of interest for biomimetic approaches (164).
With the help of Raman imaging the silica distribution was imaged in horsetail on a stem cross-sections (126) (Figure 5) and through the native outer stem surface with the help of a depth scan (127). All imaging results showed that the highest silica accumulation was in the typical knobs (126, 127). Compared to the Raman image based on CH-stretching (Figure 5A), which shows all the organic cell wall structures, integration over the 976 cm−1 band, assigned to silanol groups, highlights just the outer part of the knob (Figure 5B). In the spectrum extracted from this knob region (Figure 5G, red spectrum), a broad band below 500 cm−1, attributed to amorphous silica, is seen additionally to the band at 976 cm−1, and smaller contributions at 800 cm−1 and 1088 cm−1. Comparing the spectrum with silica gel (Figure 5H, red spectrum), it can be concluded that these very outer part of the protrusions were almost pure amorphous, hydrated silica. Integrating the broad amorphous silica band around 500 cm−1, still high silica accumulation is suggested in the lower part of the knob and decreasing amounts in the epidermal layer and below (Figure 5C). Extracting an average spectrum from the lower part of the knob (Figure 5G, pink spectrum) shows beside the broad silica band clear carbohydrate bands (e.g., 1092 cm−1). All different carbohydrates present in the cell walls (cellulose, hemicelluloses, pectin) give rise to the Raman signature of plant cell wall spectra with partly overlapping bands, but also some marker bands exist (20). In Figure 5C, the typical spectra for glucomannan, pectin and cellulose are listed, and it is clearly seen that many bands are overlapping and the band integration approach is coming to its limit of application. Based on these spectra, the integration of the 900 cm−1 band was chosen to follow hemicelluloses (glucans) as it is very prominent in glucomannan, while in cellulose it results in a rather weak band. For pectin, the 855 cm−1 band works as an excellent marker band, as this is neither present in cellulose, nor in glucomannan. For cellulose again many prominent bands are overlapping with hemicellulose and therefore the small 380 cm−1 band was chosen, although it might be masked by the broad silica band within the Equisetum sample. Based on these three images it is suggested that (1) hemicellulose (a glucan) is found in high amounts in the knob, but also in the epidermis and the sterome fibers below, (2) pectin shows high amount in the epidermal layer and is deposited in the sterome fibers as a glue between the fiber and that (3) the 380 cm−1 band is especially high in the inner sterome fibers. Extracted average spectra confirm that the epidermal layer is rich in pectin (Figure 5G, green spectrum), while below in the sterome fibers cellulose contributions are more prominent (Figure 5G, blue spectrum). The throughout high 1260 cm−1 and 900 cm−1 bands in all three parts (knob, epidermis sterome fibers) suggest that a glucan plays an important role in all parts, especially also in the silica deposition in the knob as here the highest intensity is observed (Figure 5G, pink spectrum).
Figure 5.
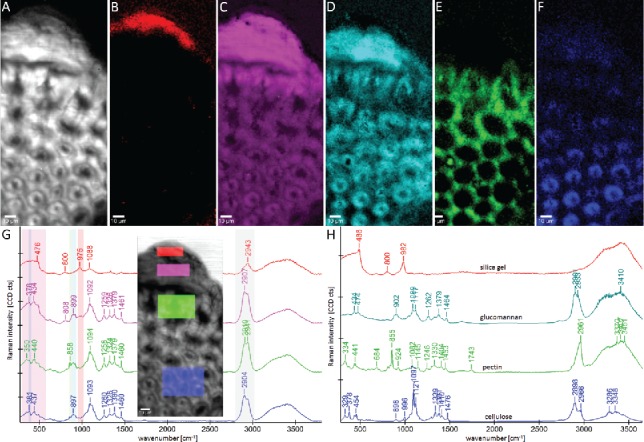
Raman images (95 µm × 200 µm) of horsetail (Equisetum hyemale): Integrating the CH-stretching band (A: 2825–3035 cm−1) shows all organic cell wall structure. On the outer part of the knob almost pure amorphous hydrated silica is visualized by integrating the 974 cm−1 band (B: 934–1005 cm−1), while the whole knob and in lower intensity the epidermal layer and sterome fibers are seen by integrating the broad silica band from 330 to 600 cm−1(C). The changes in carbohydrate composition are visualized by integrating the 900 cm−1 band (D, hemicellulose glucan and cellulose), the marker band for pectin at 858 cm−1 (E) and the OH-vibration of cellulose at 380 cm−1 (F). Average spectra have been extracted from the chemically distinct regions (G, colored regions within the picture) and compared with reference spectra of silica gel and carbohydrates (H). The spectral ranges used for establishing the Raman images (A–F) are marked in the shown average spectra (G). Sample and experimental details see in (126).
The role of carbohydrates in silica deposition is highly debated. While Perry and Lu (1992) (165) reported that the addition of insoluble cellulose to an aqueous solution of octahedral silicon complex favored the formation of ordered aggregates (with the shape of sheets) and a role of cellulose was suggested. Later on mixed-linkage glucans, have been proposed to act as template for silicification in Equisetum (166), an observation recently corroborated in rice by Kido et al. (2015)(167). But also callose was proofed to play role in Equisetum (168) and in the fern Adiantum raddianum pectic arabinan was suggested (169). Based on these Raman studies, a role of a glucan seems very reasonable, since its marker bands are clearly present in the highly silicified knobs.
Beside minerals also other plant accessory compounds have been studied (e.g., carotenoids and polyacetylenes (170, 171)).
4.2. Better image quality and more information by multivariate data analysis?
Multivariate data analysis takes at every pixel the whole wavenumber spectrum into account and reduces the redundant information substantially to finally come to a better understanding of the reduced terms. The most wide used method is PCA, which decomposes a data table into a bilinear model of latent variables, the so-called principal components. Although the “score maps” often are reminiscent of the real distribution maps, the “loading spectra” have negative bands and therefore lack any chemical meaning (172). Therefore, other Image segmentation methods (unsupervised and supervised) as well as image resolution methods are more suitable for chemical imaging. With the help of segmentation methods (e.g., cluster analysis) similar pixels ( = similar spectra) are identified and therefore regions with similar chemical composition and properties visualized. The goal of image resolution methods is to know the spatial (distribution map) and chemical (pure spectrum) information about each particular image constituent. Resolution methods decompose the original raw image into the Beer–Lambert bilinear model, D = CST +E, where D is the raw image, St the matrix of pure spectra, C are the stretched concentration profiles and E the error. Many different methods exist (for details, see e.g., (172–177)) and only three promising methods, cluster analysis, VCA and NMF, are shown here on examples.
4.2.1. Segmentation of chemically or structurally different regions by cluster analysis
Cluster analysis sorts the tens of thousands of spectra in a data set according their spectral similarities and by this the variation of chemical composition along the scanned surface is visualized (178). As mathematical indicator distances, like Euclidean distance, are used to quantify the similarity among pixel spectra Hierarchical methods begin by considering each pixel as an individual class, after which the two most similar pixels are linked to constitute a new class. On the before shown example of poplar tension wood (Figure 3), 4 main clusters corresponding to CC and compound middle lamella (CML+CC, red), S2 (blue) and two parts of the G-layer (green: main part, turquois: lumen-side border) are distinguished (Figure 6A). Small parts within the G-layer are displaced as blue spots on the lumen-side and thus resemble more the spectral characteristics of the S2. From a first sight it becomes clear that the main differences between the calculated clusters are due to changes in the lignin bands (Figure 6B, e.g., 1601 cm−1 and 1331 cm−1). It is evident that typical lignin bands are found in the G-layer, especially in the innermost layer toward the lumen (cluster 4: Gin in turquois).
Figure 6.
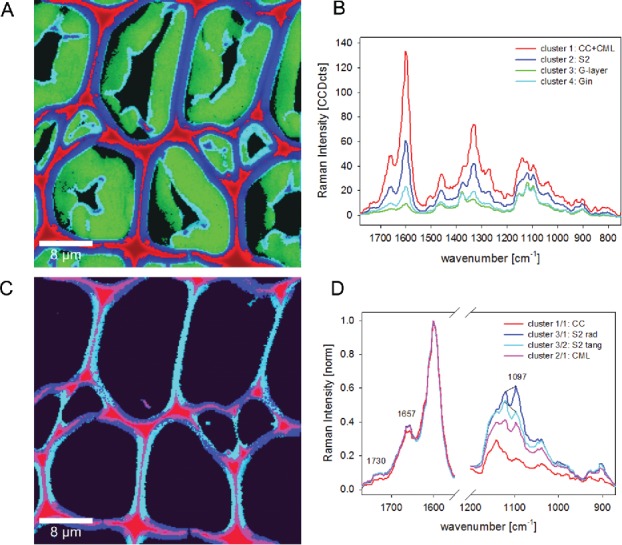
Raman images (30 µm × 30 µm) of poplar tension wood based on hierarchical cluster analysis: the four main clusters separate cell corner (CC) together with compound middle lamella (CML) from secondary cell wall (S2) and two parts of the gelatinous layer (G-layer and Gin (A). The corresponding cluster average spectra show that differences are mainly due to changes in lignin content (B). Cluster analysis within cluster 1 enables to separate CC and CML and within cluster 2 the radial and tangential S2 layer (C). Average spectra reveal differences in lignin composition and amount between CC and CML and the effect of high microfibril angle within the S2 with a pronounced 1097 cm−1 band in x-direction (radial walls) (D).
Continuing cluster analysis within the CC and CML (red) and S-layer (blue) can reveal more subtle differentiation (Figure 6C). Within the first cluster, the CC (red) can be separated from the CML (pink), and within the second S-layer cluster the radial walls (blue) are clearly different from the tangential walls (turquois) (Figure 6C). Normalizing the corresponding spectra on the lignin band and zooming into the lignin and cellulose region (Figure 6D) reveals that (1) CC spectra represent almost pure lignin (1097 cm−1 and 1022 cm−1 band marginal) and (2) CC lignin has a slightly different composition than the cell wall (Figure 6D: 1657 cm−1 and 1732 cm−1 band are slightly reduced and shoulder around 1624 cm−1), (3) the radial and tangential S2 spectra are separated based on the typical change of the 1097 cm−1 band in relation to the 1122 cm−1 band, if the microfibrils are aligned with a slope (high MFA).
So, by cluster analysis the band integration results on changes in lignin amount and MFA have been proofed and furthermore a change in lignin composition within the CC revealed. By extracting average spectra of each of the clusters detailed information on the molecular composition can be gained. These average spectra are often more valuable compared to average spectra extracted based on band integration images, as the regions have not to be set by visual inspection or selecting a band intensity threshold.
4.2.2. Image resolution methods to reveal more details and pure components
One of the promising image resolution methods is VCA, which projects the data to the identified orthogonal subspace in an interactive way and finds the endmember by repeated iteration (179). The result are distribution maps of the endmembers and the corresponding endmember spectra, which represent the most pure component spectra found in the analyzed data set. As the cell wall represents a polymer blend the pure components might not be present at any single pixel of the Raman map. The aim is therefore to differentiate first of all cell wall regions different in molecular structure and then get new insights into composition of these defined areas based on the analysis of the endmember spectra. In spruce wood indeed only one pure component spectrum (lignin from the CC) was extracted, while all other endmember spectra represented mixtures characteristic for the different resolved spatial areas. VCA revealed for the first time that the lumen sided S3 layer has a similar molecular composition as the pit membrane, both revealing a clear change in lignin composition compared to all other cell wall regions. Within the S2 layer, a lamellar structure was visualized, which was elucidated to derive also from slight changes in lignin composition and content and might be due to successive but not uniform lignification during growth (180).
Also in the current running investigations on pits (the connecting valves between tracheids), new insights will be gained with the help of this analysis method as subtle chemical differences are revealed in context with structure with an excellent image resolution (Figure 7). Beside the cell wall (Figure 7A), CC and middle lamella (Figure 7B), the tiny pit membrane (Figure 7C, arrow) is visualized with different composition in one endmember together with spots at the torus and a tiny outer surface of the cell walls with exceedingly high spatial resolution. The final endmember represents the water around (Figure 7D).
Figure 7.
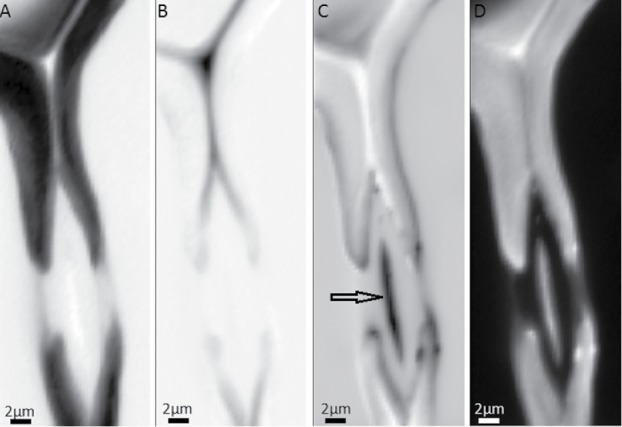
Raman images of spruce tracheid pits based on vertex component analysis (VCA) using four endmembers: abundance maps visualize the secondary cell wall (A), cell corner and compound middle lamella (B) and even details of the pit structure (C) within the water filled lumen(D).
VCA was also successfully applied on Arabidopsis to reveal details on the waterproofing in lignified tissues and the outer cuticle. Differences in lignin composition in vessels and fibers as well as a change in the aromatic composition within the lipidic cuticle of a trichome have been elucidated with the help of VCA (122).
Recently, an approach based on NMF combined with a clustering algorithm was introduced for analysis of Raman maps of tissues from carrot root (Daucus carota L. subsp. Sativus). The output of the NMF analysis was used as a starting point for two clustering algorithms, k-means clustering and hierarchical clustering. Both methods converged with similar results providing precise spatial separation of spectral data according to the most predominant component (pectins, cellulose and lignins) in specific areas of the studied tissues (181).
For unmixing methods, high-resolution scans are beneficial to enhance the chance to collect pure component pixels. On the already beforehand shown example of poplar tension wood (Figure 3 and Figure 6), a pure lignin spectrum is resolved (red) with predominant abundance in the CC and very thin resolved CML, whereas a pure cellulose spectrum is extracted (green) from the G-layer area. The secondary cell wall is expressed by a mixture of components and the abundance of this endmember in the lumen sided G-layer confirms again the lignified inner wall of this tension wood (Figure 8). Comparing the spectra of the same measurement with the spectra based on cluster analysis (Figure 6), the pure nature of the cellulose and lignin spectra becomes clear.
Figure 8.
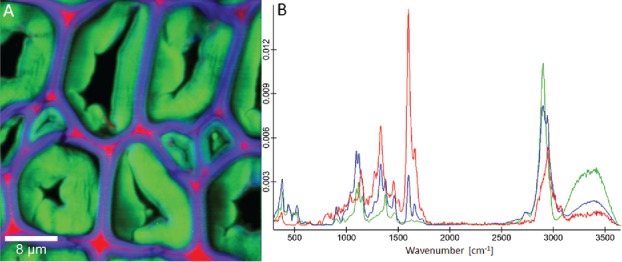
Raman images (30 µm × 30 µm) of poplar tension wood based on non-negative matrix factorization (NMF) based on 4 endmembers: Beside the lumen (not shown, black area) again the three chemically most different areas, cell corner and middle lamella (red), secondary cell wall (blue) and G-layer (green) are visualized (A). The corresponding endmember spectra present pure lignin (red) and cellulose (green), while the third one (blue) represents a composite spectrum of the secondary cell wall.
As a third unmixing method for biological materials, a protocol for multivariate curve resolution-alternating least squares (MCR-ALS) analysis has been published. The analysis considers also the full spectral profile to identify the chemical compounds and to visualize their distribution across the sample to categorize chemically distinct areas (182). Very recently, the aromatic lignin ring as well as the alcohol and aldehyde groups have been mapped using hyperspectral stimulated Raman microscopy together with MCR analysis (183).
5. In-situ studies: probing plant cell wall changes
5.1. Molecular changes during tensile load
Micromechanical studies combined with Raman spectroscopy for evaluating microstructural and molecular changes that occur in samples subjected to stress and strain have been performed on wood tissues and paper (184), cellulose composites (185–190), natural and regenerated cellulose fibers (191–193) as well as cotton, flax (194), hemp (195, 196), coir and celery (197) and single wood fibers (198). In all these studies, a shift of the 1095 cm−1 cellulose band (corresponding to C–O and C–O–C stretching, Figure 11C) was observed toward a lower wavenumber due to molecular deformation. The magnitude of the induced wavenumber shift and the recovery after cyclic deformation depends on the molecular structure and microstructure of the sample (197).
Figure 11.
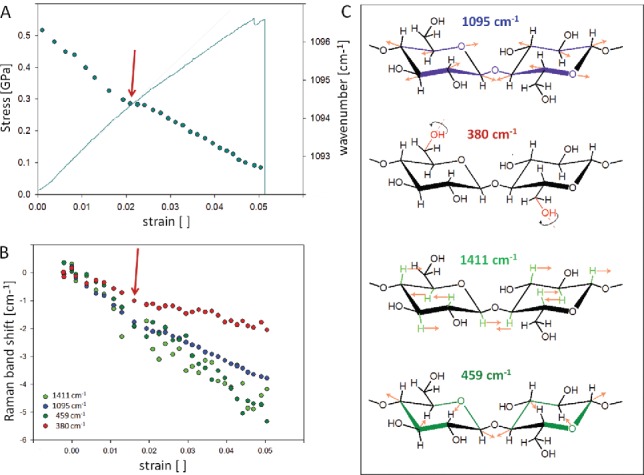
Molecular changes during tensile testing of a single Ramie fiber (almost pure cellulose, microfibrils aligned parallel to the fiber). The shift of the 1097 cm−1 band is plotted together with the stress–strain curve (A) and also compared to the shift of other cellulose bands (B). The involved vibrations are assigned according to Wiley and Atalla (60) and Barsberg (200) and visualized in (C). Experimental details see in Gierlinger et al. (198).
Comparing the mechanical behavior of wood tissues and mechanically isolated single wood fibers (Figure 9A and B) together with their molecular behavior, clearly revealed that the cellulose in a single wood fiber “feels” double of the tensing load with a peak shift from 1097 cm−1 to 1091 cm−1 compared to wood tissues (1097 cm−1 to 1095 cm−1) (Figure 9C and D). The rate of the peak shift of both samples falls together with the applied stress level (Figure 9C, all points on one line), while the correlation with strain results in two lines (Figure 9D). This fact is explained by the fiber composite design of the wood tissues, which makes the interface the first weak point and enables higher strain levels without having stress on the cell wall of the fiber. Thus working on the single fiber level reveals more the excellent properties of the wooden cell wall, whereas the wood tissue shows the secure design by having the interfaces between the fibers to allow higher strain levels without coming to a load to rupture the cell wall material itself.
Figure 9.
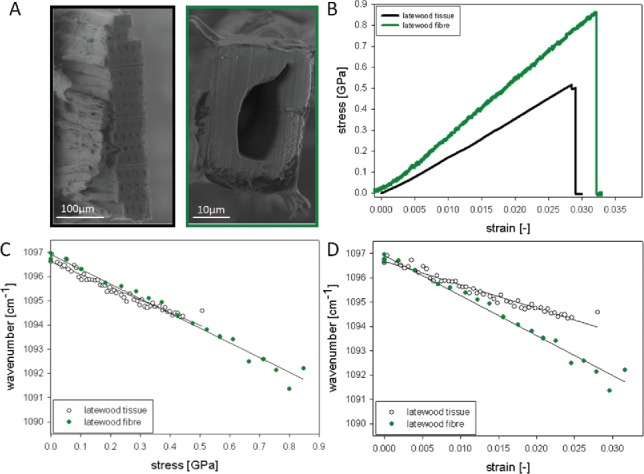
Following molecular changes during tensile tests of wood tissues and single wood fibers: Cross-sectional areas of the tested samples are shown (A) and their different mechanical behavior (B). Plotting the shift of the 1097 cm−1 band in dependence of stress (C) and strain (D) shows clearly that lower stress levels in the tissue are reflected in a shift of only 3 cm−1, whereas the single fibers show double of the shift (6 cm−1). A clear relationship between applied stress and wavenumber shift (points are on one line) is seen, whereas the strain levels result in two lines. Experimental details see in Gierlinger et al. (109).
When comparing single fibers with low (5°) and medium MFA (15°) both show a very high peak shift (8-9 cm−1), although the lower MFA fibers slightly more (Figure 10). Plotting the peak shifts with stress and strain shows once again the strong relation of the band shift with applied stress (Fig. 10A) and different strain responses in dependence of the MFA (Figure 10B). In this case, the reason for higher strain levels in the fibers with higher microfibrils might be explained by a possible realignment of the cellulose microfibrils during tensing and more gliding in the matrix as the microfibrils are loaded with an angle.
Figure 10.
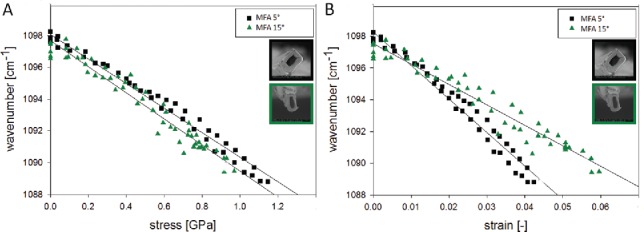
Following molecular changes during tensile tests of single wood fibers with different microfibril angle: The shift of the 1097 cm−1 band is quite high in the normal wood fibers with low microfibril angle (MFA∼ 5°) as well as in the juvenile wood fibers (MFA∼ 15°). A clear relationship between applied stress and wavenumber shift is seen, whereas the strain levels result in two lines as the high MFA fibers are able to align and strain more. Experimental details see in Gierlinger et al. (198).
While wood fibers have many broad overlapping bands of all wood polymers (cellulose, hemicellulose and lignin) and usually less signal due to higher fluorescence background of lignin the molecular deformation studies on pure cellulose fibers allow a more detailed study of the cellulose deformation. In regenerated cellulose II fibers, a non-linear shift following the shape of the stress–strain curve could be related to the breakdown of hydrogen bonding and the onset of relative molecular motion. In this study beside the 1095 cm−1 band also the 1414 cm−1 band was investigated in detail as reported to correspond to side groups (C–O–H) along the chain and to be influenced by main-chain hydrogen bonding. The non-linear shift toward a lower wavenumber and the reaching of a plateau at the yield point is interpreted as a breakdown of the hydrogen network (199). Investigating a native cellulose fiber (Ramie, almost pure cellulose with microfibril aligned parallel to the fiber axis) in more detail, showed that even in case of only a slight non-linear tendency in the stress–strain curve (Figure 11A, red arrow) changes on the molecular level are revealed. Plotting the shift of the 1095 cm−1 band shows clearly this transition: after three stable experimental points the shift goes toward lower wavenumbers with a lower rate (Figure 11A, circles). Plotting additionally other cellulose bands it becomes clear that the shift rate changes at this point not in the same manner for the shown bands (Figure 11B). While the 380 cm−1 band is shifted with the lowest rate, followed by the 1095 cm−1 band and with almost the same rate the band at 1411 and 459 cm−1. Based on the density functional theory (DFT) band assignments the 380 cm−1 is assigned to OH-vibrations (200, 201), while the band at 1425 cm−1 involves OH but also CH wagging (60, 200), as illustrated in Figure 11C. Thus the lower shift of the 380 cm−1 band might reflect a loosening of the hydrogen network at some places, which also slightly affects the load on the main axis reflected by the 1095 cm−1 band (Figure 11B-C). The load on the 1411 cm−1 and 459 cm−1 is shifting with the same rate as vibrations involved are on the glucose ring and not so much involved in connecting molecules and fibrils (Figure 11C). Another explanation might be that the cellulose molecule is from this point on not loaded exactly in fiber (molecule) direction due to a defect on one side, and thus the vibrations along the fiber axis are less affected than the ones involving also the vibrations on the ring. This measurement is shown as an example how sensitive the changes in the different involved vibrations can be tracked and reveals the molecular mechanisms behind to explain different observed stress–strain curves.
Beside Raman microscopy also FT-IR spectroscopy has been used to monitor molecular straining in wood. Similarly in Raman (198), only groups assigned clearly to cellulose showed a deformation based on a band shift and confirm the major role of cellulose during tensing along the fiber axis (202). In the native epidermal cell walls of onion a distinct reorientation of the cellulose and pectin molecule has been observed during mechanical stress and with the help of two-dimensional infrared spectroscopy evidence that cellulose and xyloglucan associate, while pectin creates an independent network (55). In another system, the Chara corallina cell wall, FT-IR microspectroscopy revealed the cellulose microfibrils aligned predominantly transverse to the long axis of the cell and becoming orientated increasingly transversely as longitudinal strain increased, while the pectic polysaccharide matrix underwent molecular orientation parallel to the direction of strain (203).
Currently, all studies rely on single point measurements and thus present only a part of the fiber or tissue. Combining with the Raman imaging approach is more challenging as the focal plane is often changing during tensing and/or relaxation of the fiber and the danger of sample damage by longer measurement time, but would have the great potential to study stress distribution with respect to the microstructure.
5.2. Following plant cell wall treatments
Treatment of lignocellulosic plant cell walls involves on the one hand deconstruction to fermentable sugars by thermochemical and/or biological means and on the other hand improving the properties by modifying the native plant cell walls. Vibrational microspectroscopy has high potential not only to follow the changes of the treatment per se, but also resolve accessibility and different reactions on the micro level. To follow the treatment step by step consecutive measurements are necessary, which is sometimes difficult with Raman microscopy in the visible laser range, due to the fact that repeating measurements have to be taken with great care and very low laser power not to induce changes in the sample due to the repeated laser irradiation instead of the treatment. On the other hand, Raman microscopy has the much better spatial resolution on the cell wall level and therefore is very suitable for a detailed comparison of samples with and without treatment, e.g., in wood modification.
The enzymatic degradation of poplar wood was investigated in-situ in a step wise manner using FT-IR microscopy. Cross-sections of poplar wood were treated with cellulase Onozuka RS within a custom-built fluidic cell (Figure 12A) and at the same time light-optical images and FT-IR spectra acquired. Light-optical micrographs showed almost complete removal of the gelatinous (G) layer in tension wood fibers (Figure 12B, 2nd row at room temperature, 3rd row at 50 °C), while no structural and spectral changes were observed in the lignified normal wood fibers (Figure 12B first row, C) within the same section and experiment. The removal of the G-layer was also clearly shown by the FT-IR spectra as mainly the cellulose bands have been reduced (Figure 12D). At room temperature, spectral changes have been observed over 40 hours, whereas at 50° an enormous enhancement of the hydrolysis has been observed in the very beginning and the experiment time could be reduced to a third (204). The accessibility of cellulose within the lignified cell wall was found to be the main limiting factor, whereas the depletion of the enzyme due to lignin adsorption could be ruled out as comparative measurements have always been taken within one experiment on one cross-section. The fast, selective hydrolysis of the cellulose in the G-layer, even at room temperature, has been explained by the gel-like structure and the highly porous surface. Young plantation grown hardwood trees with a high proportion of G-fibers were thus suggested as an interesting resource for bioconversion to fermentable sugars in the process to bioethanol. Based on these experiments, it is furthermore clear that the occurrence of tension wood, which is quite common in young trees, will influence tremendously the chemical composition when determined by wet chemistry, but also experiments on sugar release.
Figure 12.
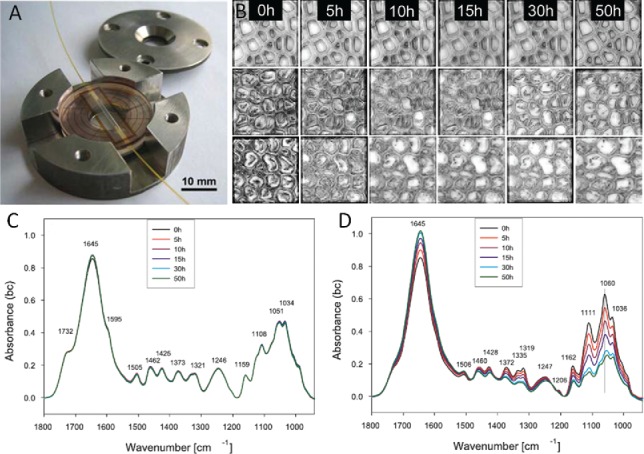
Custom built fluid cells enabling temperature controlled FT-IR microscopy in water and enzyme solution (A). Lightmicroscopic images (B) have been acquired on normal wood (first row) and tension wood (2nd and 3rd row) during Cellulase Onozuka treatment at room temperature (2nd row) and 50 °C (3rd row). During the treatment FT-IR spectra have been acquired from the shown region of normal (C) and tension wood (D). Experimental details see in Gierlinger et al. (204).
Confocal Raman microscopy has been used to monitor the structural and chemical changes upon NaOH treatment of longitudinal and transversal sections of Miscanthus x giganteus. A globular structure, proposed to be predominantly of hemicellulose and lignin was revealed on the interior cell wall, which was preferentially removed by the NaOH treatment, while the cellulose was largely undisturbed (131, 205). Nevertheless, the suggested lignocellulose complex is questioned based on the presented spectra of the globules. The strong band at 478 cm−1 as well as two characteristic bands between 800 and 1000 cm−1 are typical for starch (bionami.at/spectra), which is known to occur as granules in parenchyma cells.
Understanding the natural decay was the aim of studying waterlogged archaeological Norway spruce [Picea abies (L.) Karst] poles by means of confocal Raman imaging and attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) analysis. The waterlogged approximately 400 year old poles were submerged under anoxic conditions and solely decayed by erosion bacteria. A residual material with heterogeneous lignin distribution and a strong depletion of carbohydrates was found in the decayed tracheids, indicating that erosion bacteria are able to utilize the carbohydrate fraction of the cell wall effectively. Regions free from decay showed evidence of a loss of acetyl groups in glucomannan, loss of un-conjugated ester linkages in the lignin–carbohydrate complexes between xylan and lignin, and minor oxidation of the lignin polymer compared to recent reference material (206).
Besides deconstructing the cell wall for industrial purposes there are also efforts to avoid the natural destruction and/or improve dimension stability and fire retardancy by wood modification. In this case, it is of interest whether the modification is uniform and/or if only the lumen is affected or the entire cell wall modified. One of the first studies to follow wood modification with Raman microscopy was on tracking the melamin–formaldehyde resin in the secondary cell walls (207). During the last years, the Raman imaging approach has been very successfully used to follow the insertion of hydrophobic polymers (208–210) and minerals (159, 160) into the cell wall.
6. Outlook
FT-IR and Raman microscopy have shown to have high potential to get a deeper understanding of structure–function relationships as well as biological processes and technical treatments as it is possible to reveal plant cell wall chemistry and structure non-destructively in context with the microstructure. Both probe molecular vibrations of all present molecules at once and result in many overlapping bands, wherefore understanding and interpreting the acquired spectra is still a challenge. The combination with microscopy in mapping mode allows position resolved acquisition, and in combination with multivariate unmixing methods to resolve more pure spectra (components) and their distribution. Still progress in band assignment is needed, but on a good way as a solid databases with reference spectra is currently established (http://bionami.at/spectra.html) and with experience and every sample measured new knowledge is gained. As discussed in this paper, the two methods are complementary and each has its advantages and drawbacks. Depending on the research question the best method should be chosen, but they should also be more often be applied together to ease spectral data interpretation and derive a maximum of information. Furthermore, the combination with other methods, especially Atomic force microscopy, has high potential to get an even more complete picture of cell wall design by revealing at the same position nanostructure and nanomechanics and in the future probably also nanochemistry by Tip-enhanced Raman spectroscopy or nano-IR (211, 212).
Funding Statement
Austrian Science Fund Y-728-316; H2020 European Research Council ERC consolidator grant "SCATAPNUT" No681885
Acknowledgments
The research presented was done over quite a long time and at many different places. For the support, I thank Prof. Peter Fratzl (Max-Planck-Institute for Colloids and Interfaces, Department Biomaterials), Prof. Ingo Burgert (ETH Zürich), Prof. Sabine Hild (JKU Linz) and Prof. José Toca-Herrera (BOKU, Vienna). I acknowledge funding by the Austrian Science Fund (START-project SURFINPLANT Y-728-316) and the European community (ERC-consolidator grant SCATAPNUT 681885).
References
- 1. Emons A. M. C., and Fagerstedt K. V. (2010) The plant cell surface. J. Integr. Plant Biol. 52: 126–130. [DOI] [PubMed] [Google Scholar]
- 2. O ´Neill M. A., and York W. S. (2003) The composition and structure of plant primary cell walls In The plant cell wall – annual plant reviews, Rose J., Ed., pp. 1–44. [Google Scholar]
- 3. Cosgrove D. J. (2005) Growth of the plant cell wall. Nature Rev. Mol. Cell Biol. 6: 850–861. [DOI] [PubMed] [Google Scholar]
- 4. Boerjan W., Ralph J., and Baucher M. (2003) Lignin biosynthesis. Annu. Rev. Plant Biol. 54: 519–543. [DOI] [PubMed] [Google Scholar]
- 5. Vanholme R., Demedts B., Morreel K., Ralph J., and Boerjan W. (2010) Lignin biosynthesis and structure. Plant Physiol. 153: 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dejardin A., Laurans F., Arnaud D., Breton C., Pilate G., and Leple J. C. (2010) Wood formation in Angiosperms. Cr. Biol. 333: 325–334. [DOI] [PubMed] [Google Scholar]
- 7. Kerstens S., Decraemer W. F., and Verbelen J.-P. (2001) Cell walls at the plant surface behave mechanically like fiber-reinforced composite materials. Plant Physiol. October2001, 127: 381–385. [PMC free article] [PubMed] [Google Scholar]
- 8. Graham L. E., Cook M. E., and Busse J. S. (2000) The origin of plants: Body plan changes contributing to a major evolutionary radiation. Proc. Natl. Acad. Sci. USA. 97: 4535–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niklas K. J. (2000) The evolution of plant body plans – a biomechanical perspective. Annal. Botany 85: 411–438. [Google Scholar]
- 10. Sarkar P., Bosneaga E., and Auer M. (2009) Plant cell walls throughout evolution: Towards a molecular understanding of their design principles. J. Exp. Botany 60: 3615–3635. [DOI] [PubMed] [Google Scholar]
- 11. Burton R. A., Gidley M. J., and Fincher G. B. (2010) Heterogeneity in the chemistry, structure and function of plant cell walls. Nature Chem. Biol. 6: 724–732. [DOI] [PubMed] [Google Scholar]
- 12. Flemings M. C. (1999) What next for departments of materials science and engineering? Annu. Rev. Mater. Sci. 29: 1–23. [Google Scholar]
- 13. Siebert F., and Hildebrandt P. (2008) Vibrational spectroscopy in life science, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. [Google Scholar]
- 14. Raman C. V., and Krishnan K. S. (1928) A new type of secondary radiation. Nature 121: 501–502. [Google Scholar]
- 15. Smith E., and Dent G. (2005) Modern raman spectroscopy – a practical approach, John Wiley & Sons Ltd. [Google Scholar]
- 16. Griffith P. R. (2009) Infrared and Raman instrumentation for mapping and imaging In Infrared and Raman Spectroscopic Imaging, Salzer R. and Siesler H. W., Eds., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, pp. 3–64. [Google Scholar]
- 17. Kazarian S. G., and Chan K. L. A. (2010) Micro- and macro-attenuated total reflection fourier transform infrared spectroscopic imaging. Appl. Spectrosc. 64: 135A–152A. [DOI] [PubMed] [Google Scholar]
- 18. Bruneel J. L., Lassegues J. C., and Sourisseau C. (2002) In-depth analyses by confocal Raman microspectrometry: Experimental features and modeling of the refraction effects. J. Raman Spectrosc. 33: 815–828. [Google Scholar]
- 19. Everall N., Lapham J., Adar F., Whitley A., Lee E., and Mamedov S. (2007) Optimizing depth resolution in Confocal Raman microscopy: A comparison of metallurgical, dry corrected, and oil immersion objectives. Appl. Spectrosc. 61: 251–259. [DOI] [PubMed] [Google Scholar]
- 20. Gierlinger N., Keplinger T., and Harrington M. (2012) Imaging of plant cell walls by confocal Raman microscopy. Nat. Protocols 7: 1694–1708. [DOI] [PubMed] [Google Scholar]
- 21. Nelson M. P., and Treado P. J. (2010) Raman imaging instrumentation In Raman, infrared, and near-infrared chemical imaging, Sasic S. and Ozaki Y., Eds., John Wiley & Sons, Inc., Hoboken, New Jersey, pp. 23–55. [Google Scholar]
- 22. Atalla R. H., and Agarwal U. P. (1986) Recording Raman-spectra from plant cell walls. J. Raman Spectrosc. 17: 229–231. [Google Scholar]
- 23. Atalla R. H. (1987) Raman-spectroscopy and the Raman microprobe – Valuable new tools for characterizing wood and wood pulp fibers. J. Wood Chem. Technol. 7: 115–131. [Google Scholar]
- 24. Atalla R. H., Whitmore R. E., and Heimbach C. J. (1980) Raman spectral evidence for molecular orientation in native cellulosic. Fibers Macromolecules 13: 1717–1719. [Google Scholar]
- 25. Agarwal U. P., and Atalla R. H. (1986) In-situ raman microprobe studies of plant cell walls – Macromolecular organization and compositional variability in the secondary wall of Picea mariana (Mill). Bsp Planta 169: 325–332. [DOI] [PubMed] [Google Scholar]
- 26. Hirschfeld T., and Chase B. (1986) FT-Raman Spectroscopy – development and justification. Appl. Spectrosc. 40: 133–137. [Google Scholar]
- 27. Agarwal U. P. (1999) An overview of Raman spectroscopy as applied to lignocellulosic materials In Advances in lignocellulosics characterization, Argyropoulos D. S., Ed., TAPPI Press, Atlanta, GA, pp. 209–225. [Google Scholar]
- 28. Séné C. F. B., McCann M. C., Wilson R. H., and Crinter R. (1994) Fourier-transform Raman and fourier-transform infrared spectroscopy. An lnvestigation of five higher plant cell walls and their components. Plant Physiol. 106: 1623–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schrader B., Klump H. H., Schenzel K., and Schulz H. (1999) Non-destructive NIR FT Raman analysis of plants. J. Mol. Struct. 509: 201–212. [Google Scholar]
- 30. Agarwal U. P. (2014) 1064nm FT-Raman spectroscopy for investigations of plant cell walls and other biomass materials. Front Plant Sci. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barsberg S., Matousek P., Towrie M., Jorgensen H., and Felby C. (2006) Lignin radicals in the plant cell wall probed by Kerr-gated resonance Raman spectroscopy. Biophys. J. 90: 2978–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saariaho A. M., Jaaskelainen A. S., Matousek P., Towrie M., Parker A. W., and Vuorinen T. (2004) Resonance Raman spectroscopy of highly fluorescing lignin containing chemical pulps: Suppression of fluorescence with an optical Kerr gate. Holzforschung 58: 82–90. [Google Scholar]
- 33. Yakovlev V. V. (2007) Time-gated confocal Ramon microscopy. Spectroscopy 22:34–41. [Google Scholar]
- 34. Das R. S., and Agrawal Y. K. (2011) Raman spectroscopy: Recent advancements, techniques and applications. Vib. Spectrosc. 57: 163–176. [Google Scholar]
- 35. Hollricher O. (2010) Raman Instrumentation for confocal raman microscopy In Confocal Raman microscopy, Diening T., Hollricher O., and Toporski J., Eds., Springer-Verlag, Berlin Heidelberg, pp. 43–60. [Google Scholar]
- 36. McCann M. C., Chen L., Roberts K., Kemsley E. K., Sene C., Carpita N. C., Stacey N. J., and Wilson R. H. (1997) Infrared microspectroscopy: Sampling heterogeneity in plant cell wall composition and architecture. Physiol. Plant. 100: 729–738. [Google Scholar]
- 37. McCann M. C., Hammouri M., Wilson R. H., Belton P., and Roberts K. (1992) Fourier transform infrared microspectroscopy is a new way to look at plant cell walls. Plant Physiol. 100: 1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stewart D. (1996) Fourier transform infrared microspectroscopy of plant tissues. Appl. Spectrosc. 50: 357–365. [Google Scholar]
- 39. Stewart D., Mcdougall G. J., and Baty A. (1995) Fourier-transform infrared microspectroscopy of anatomically different cells of flax (Linum-Usitatissimum) stems during development. J. Agric. Food Chem. 43: 1853–1858. [Google Scholar]
- 40. Kataoka Y., and Kondo T. (1996) Changing cellulose crystalline structure in forming wood cell walls. Macromolecules 29: 6356–6358. [Google Scholar]
- 41. Kataoka Y., and Kondo T. (1998) FT-IR microscopic analysis of changing cellulose crystalline structure during wood cell wall formation. Macromolecules 31: 760–764. [Google Scholar]
- 42. Séné C. F. B., McCann M. C., Wilson R. H., and Crinter R. (1994) Fourier-Transform Raman and fourier-transform lnfrared spectroscopy. An investigation of five higher plant cell walls and their components. Plant Physiol. 106: 1623–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sellors J., Hoult R. A., Crocombe R. A., and Wright N. A. (2010) FT-IR imaging hardware In Raman, Infrared, and near-infrared chemical imaging, Sasic S. and Ozaki Y., Eds., John Wiley & Sons, Inc., Hoboken, New Jersey, pp. 55–73. [Google Scholar]
- 44. Carr G. L. (1999) High-resolution microspectroscopy and sub-nanosecond time-resolved spectroscopy with the synchrotron infrared source. Vib. Spectrosc. 19: 53–60. [Google Scholar]
- 45. Dumas P., Carr G. L., and Williams G. P. (2000) Enhancing the lateral resolution in infrared microspectrometry by using synchrotron radiation: Applications and perspectives. Analusis, 28: 68–74. [Google Scholar]
- 46. Raab T. K., and Vogel J. P. (2004) Ecological and agricultural applications of synchrotron IR microscopy. Infrared Phys. Technol. 45: 393–402. [Google Scholar]
- 47. Schmidt M., Gierlinger N., Schade U., Rogge T., and Grunze M. (2006) Polarized infrared microspectroscopy of single spruce fibers: Hydrogen bonding in wood polymers. Biopolymers 83: 546–555. [DOI] [PubMed] [Google Scholar]
- 48. Wetzel D. L., Eilert A. J., Pietrzak L. N., Miller S. S., and Sweat J. A. (1998) Ultraspatially-resolved synchrotron infrared microspectroscopy of plant tissue in situ. Cell. Mol. Biol. 44: 145–168. [PubMed] [Google Scholar]
- 49. Gou J. Y., Park S., Yu X. H., Miller L. M., and Liu C. J. (2008) Compositional characterization and imaging of “wall-bound” acylesters of Populus trichocarpa reveal differential accumulation of acyl molecules in normal and reactive woods. Planta 229: 15–24. [DOI] [PubMed] [Google Scholar]
- 50. Dokken K. M., and Davis L. C. (2007) Infrared imaging of sunflower and maize root anatomy. J. Agric. Food Chem. 55: 10517–10530. [DOI] [PubMed] [Google Scholar]
- 51. Moore A. K., and Owen N. L. (2001) Infrared spectroscopic studies of solid wood. Appl. Spectrosc. Rev. 36: 65–86. [Google Scholar]
- 52. Jarvis M. C., and McCann M. C. (2000) Macromolecular biophysics of the plant cell wall: Concepts and methodology. Plant Physiol. Biochem. 38: 1–13. [Google Scholar]
- 53. McCann M. C., Bush M., Milionia D., Sadoa P., Stacey N. J., Catchpole G., Defernez M., Carpita N. C., Hoft H., Ulvskov P., Wilson R. H., and Roberts K. (2001) Approaches to understanding the functional architecture of the plant cell wall. Phytochemistry 57: 811–821. [DOI] [PubMed] [Google Scholar]
- 54. Dokken K. M., Davis L. C., and Marinkovic N. S. (2005) Use of infrared microspectroscopy in plant growth and development. Appl. Spectrosc. Rev. 40: 301–326. [Google Scholar]
- 55. Wilson R. H., Smith A. C., Kacurakova M., Saunders P. K., Wellner N., and Waldron K. W. (2000) The mechanical properties and molecular dynamics of cell wall polysacharides studied by fourier-transform infrared spectroscopy. Plant Physiol. 124: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kumar S., Lahlali R., Liu X., and Karunakaran C. (2016) Infrared spectroscopy combined with imaging: A new developing analytical tool in health and plant science. Appl. Spectrosc. Rev. 51: 466–483. [Google Scholar]
- 57. Gierlinger N., and Schwanninger M. (2007) The potential of Raman microscopy and Raman imaging in plant research – review. Spectroscopy 21: 69–89. [Google Scholar]
- 58. Edwards H. G. M., Farwell D. W., and Webster D. (1997) FT Raman microscopy of untreated natural plant fibres. Spectrochim. Acta A 53: 2383–2392. [DOI] [PubMed] [Google Scholar]
- 59. Fischer S., Schenzel K., Fischer K., and Diepenbrock W. (2005) Applications of FT Raman spectroscopy and micro spectroscopy characterizing cellulose and cellulosic biomaterials. Macromol. Symp. 223: 41–56. [Google Scholar]
- 60. Wiley J. H., and Atalla R. H. (1987) Band assignments in the Raman spectra of celluloses. Carbohydr. Res. 160: 113–129. [Google Scholar]
- 61. Marchessault R. H. (1962) Application of infra-red spectrocopy to cellulose and wood polysaccharides. Pure Appl. Chem. 5: 107–129. [Google Scholar]
- 62. Liang C. Y., and Marchessault R. H. (1959) Infrared spectra of crystalline polysaccharides. I. Hydrogen bonds in native celluloses. J. Polym. Sci. 37: 385–395. [Google Scholar]
- 63. Liang C. Y., and Marchessault R. H. (1959) Infrared spectra of crystalline polysaccharides. II. Native celluloses in the region from 640 to 1700 cm− 1 . J. Polym. Sci. 39: 269–278. [Google Scholar]
- 64. Cael J. J., Gardner K. H., Koenig J. L., and Blackwell J. (1975) Infrared and Raman spectroscopy of carbohydrates. Paper V Normal coordinate analysis of cellulose I. J. Chem. Phys. 62: 1145–1153. [Google Scholar]
- 65. Langkilde F. W., and Svantesson A. (1995) Identification of celluloses with Fourier-Transform (FT) mid-infrared, FT-Raman and near-infrared spectrometry. J. Pharm. Biomed. Anal. 13: 409–414. [DOI] [PubMed] [Google Scholar]
- 66. Fengel D., and Ludwig M. (1991) Möglichkeiten und Grenzen der FTIR-Spektroskopie bei der Charakterisierung von Cellulose. Teil 1. Vergleich von verschiedenen Cellulosefasern und Bakterien-Cellulose Das Papier 45: 45–51. [Google Scholar]
- 67. Szymanska-Chargot M., Cybulska J., and Zdunek A. (2011) Sensing the structural differences in cellulose from apple and bacterial cell wall materials by Raman and FT-IR. Spectrosc. Sensors 11: 5543–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Horikawa Y., Itoh T., and Sugiyama J. (2006) Preferential uniplanar orientation of cellulose microfibrils reinvestigated by the FTIR technique. Cellulose 13: 309–316. [Google Scholar]
- 69. Agarwal U. P., Reiner R. S., and Ralph S. A. (2010) Cellulose I crystallinity determination using FT-Raman spectroscopy: Univariate and multivariate methods. Cellulose 17: 721–733. [Google Scholar]
- 70. Agarwal U. P., Ralph S. A., Reiner R. S., and Baez C. (2016) Probing crystallinity of never-dried wood cellulose with Raman spectroscopy. Cellulose 23: 125–144. [Google Scholar]
- 71. Agarwal U. P., Reiner R. S., and Ralph S. A. (2012) Cellulose crystallinity of biomass: Comparing the accuracy of Segal WAXS and univariate FT-Raman methods. Abstr. Pap. Am. Chem. S 243. [Google Scholar]
- 72. Kataoka Y., and Kondo T. (1999) Quantitative analysis for the cellulose Iα crystalline phase in developing wood cell walls. Int. J. Biol. Macromolecules 24: 37–41. [DOI] [PubMed] [Google Scholar]
- 73. Synytsya A., Copikova J., Matejka P., and Machovic V. (2003) Fourier transform Raman and infrared spectroscopy of pectins. Carbohydr. Polym. 54: 97–106. [Google Scholar]
- 74. Kamnev A. A., Colina M., Rodriguez J., Ptitchkina N. M., and Ignatov V. V. (1998) Comparative spectroscopic characterization of different pectins and their sources Food Hydrocolloids 12: 263–271. [Google Scholar]
- 75. Agarwal U. P., and Ralph S. A. (1997) FT-Raman spectroscopy of wood: Identifying contributions of lignin and carbohydrate polymers in the spectrum of black spruce (Picea mariana). Appl. Spectrosc. 51: 1648–1655. [Google Scholar]
- 76. Kacuráková M., Capeka P., Sasinková V., Wellner N., and Ebringerova A. (2000) FT-IR study of plant cell wall model compounds: Pectic polysaccharides and hemicelluloses. Carbohydr. Polym. 43: 195–203. [Google Scholar]
- 77. Kacurakova M., and Mathlouthi M. (1996) FTIR and laser-Raman spectra of oligosaccharides in water: Characterization of the glycosidic bond. Carbohydr. Res. 284: 145–157. [DOI] [PubMed] [Google Scholar]
- 78. Kacuráková M., Wellner N., Ebringerova A., Hromádková Z., Wilson R. H., and Belton P. S. (1999) Characterisation of xylan-type polysaccharides and associated cell wall components by FT-IR and FT-Raman spectroscopies. Food Hydrocolloids 13: 35–41. [Google Scholar]
- 79. Kacuráková M., and Wilson R. H. (2001) Developments in mid-infrared FT-IR spectroscopy of selected carbohydrates. Carbohydr. Polym. 44: 291–303. [Google Scholar]
- 80. Filippov M. P. (1992) Practical infrared spectroscopy of pectic substances. Food Hydrocolloids 6: 115–142. [Google Scholar]
- 81. Hergert H. L. (1971) Infrared spectra In Lignins. occurence, formation, structure and reactions, Sarkanen K. V. and Ludwig C. H., Eds., John Wiley & Sons, New York, Chichester, Brisbane, Toronto, Singapore, pp. 267–297. [Google Scholar]
- 82. Faix O. (1992) Fourier transform infrared spectroscopy In Methods in lignin chemistry, Lin S. Y. and Dence C. W., Eds., Springer-Verlag, New York, Berlin, Heidelberg, pp. 83–109. [Google Scholar]
- 83. Agarwal U. P., McSweeny J. D., and Ralph S. A. (2011) FT-Raman investigation of milled-wood lignins: softwood, hardwood, and chemically modified black spruce lignins. J. Wood Chem. Technol. 31: 324–344. [Google Scholar]
- 84. Barsberg S., Matousek P., and Towrie M. (2005) Structural analysis of lignin by resonance Raman spectroscopy. Macromol. Biosci. 5: 743–752. [DOI] [PubMed] [Google Scholar]
- 85. Larsen K. L., and Barsberg S. (2010) Theoretical and Raman spectroscopic studies of phenolic lignin model monomers. J. Phys. Chem. B 114: 8009–8021. [DOI] [PubMed] [Google Scholar]
- 86. Saariaho A. M., Jaaskelainen A. S., Nuopponen M., and Vuorinen T. (2003) Ultra violet resonance Raman spectroscopy in lignin analysis: Determination of characteristic vibrations of p-hydroxyphenyl, guaiacyl, and syringyl lignin structures. Appl. Spectrosc. 57: 58–66. [DOI] [PubMed] [Google Scholar]
- 87. Agarwal U. P., and Ralph S. A. (2008) Determination of ethylenic residues in wood and TMP of spruce by FT-Raman spectroscopy. Holzforschung 62: 667–675. [Google Scholar]
- 88. Agarwal U. P., and Reiner R. S. (2009) Near-IR surface-enhanced Raman spectrum of lignin. J. Raman Spectrosc. 40: 1527–1534. [Google Scholar]
- 89. Barsberg S. (2015) Identification of the best DFT functionals for a reliable prediction of lignin vibrational properties. Theory Chem. Acc. 134. [Google Scholar]
- 90. Atalla R. H., and Agarwal U. P. (1985) Raman microprobe evidence for lignin orientation in the cell walls of native woody tissue. Science 227: 636–638. [DOI] [PubMed] [Google Scholar]
- 91. Salmen L., Olsson A. M., Stevanic J. S., Simonovic J., and Radotic K. (2012) Structural organisation of the wood polymers in the wood fibre structure. Bioresources 7: 521–532. [Google Scholar]
- 92. Olsson A.-M., Bjurhager I., Gerber L., Sundberg B., and Salmen L. (2011) Ultra-structural organisation of cell wall polymers in normal and tension wood of aspen revealed by polarisation FTIR microspectroscopy. Planta 233: 1277–1286. [DOI] [PubMed] [Google Scholar]
- 93. Åkerholm M., and Salmén L. (2001) Interactions between wood polymers studied by dynamic FT-IR spectroscopy. Polymer 42: 963–969. [Google Scholar]
- 94. Simonovic J., Stevanic J., Djikanovic D., Salmen L., and Radotic K. (2011) Anisotropy of cell wall polymers in branches of hardwood and softwood: A polarized FTIR study. Cellulose 18: 1433–1440. [Google Scholar]
- 95. Stevanic J. S., and Salme L. (2009) Orientation of the wood polymers in the cell wall of spruce wood fibres. Holzforschung 63: 497–503. [Google Scholar]
- 96. Morikawa H., Hayashi R., and Senda M. (1978) Infrared Analysis of pea stem cell walls and oriented structure of matrix polysaccharides in them. Plant Cell Physiol. 19: 1151–1159. [Google Scholar]
- 97. Morikawa H., and Senda M. (1978) Infrared analysis of oat coleoptile cell walls and oriented structure of matrix polysacharides in walls. Plant Cell Physiol. 19: 327–336. [Google Scholar]
- 98. Chen L., Wilson R. H., and McCann M. C. (1997) Investigation of macromolecule orientation in dry and hydrated walls of single onion epidermal cells by FTIR microspectroscopy. J. Mol. Struct. 408–409: 257–260. [Google Scholar]
- 99. McCann1 M. C., Stacey N. J., Wilson R., and Roberts K. (1993) Orientation of macromolecules in the walls of elongating carrot cells. J. Cell Sci. 106: 1347–1356. [DOI] [PubMed] [Google Scholar]
- 100. Agarwal U. P. (2006) Raman imaging to investigate ultrastructure and composition of plant cell walls: Distribution of lignin and cellulose in black spruce wood (Picea mariana). Planta 224: 1141–1153. [DOI] [PubMed] [Google Scholar]
- 101. Schwanninger M., Rodrigues J. C., Pereira H., and Hinterstoisser B. (2004) Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib. Spectrosc. 36: 23–40. [Google Scholar]
- 102. Faix O. (1991) Classification of Lignins from different botanical origins by FTIR spectroscopy. Holzforschung 45: 21–27. [Google Scholar]
- 103. Gierlinger N., Luss S., Konig C., Konnerth J., Eder M., and Fratzl P. (2010) Cellulose microfibril orientation of Picea abies and its variability at the micron-level determined by Raman imaging. J. Exp. Botany 61: 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Burgert I., Fruhmann K., Keckes J., Fratzl P., and Stanzl-Tschegg S. (2005) Properties of chemically and mechanically isolated fibres of spruce (Picea abies [L.] Karst.). Part 2: Twisting phenomena. Holzforschung 59: 247–251. [Google Scholar]
- 105. Goswami L., Eder M., Gierlinger N., and Burgert I. (2008) Inducing large deformation in wood cell walls by enzymatic modification. J. Mater. Sci. 43: 1286–1291. [Google Scholar]
- 106. Carpita N. C., and McCann M. C. (2015) Characterizing visible and invisible cell wall mutant phenotypes. J. Exp. Botany 66: 4145–4163. [DOI] [PubMed] [Google Scholar]
- 107. Himmelsbach D. S., Khahili S., and Akin D. E. (1999) Near-infrared–Fourier-transform–Raman microspectroscopic imaging of flax stems. Vib. Spectrosc. 19: 361–367. [Google Scholar]
- 108. Himmelsbach D. S., Khalili S., and Akin D. E. (1998) FT-IR microspectroscopic imaging of flax (Linum usitatissimum L.) stems. Cell. Mol. Biol. 44: 99–108. [PubMed] [Google Scholar]
- 109. Gierlinger N., and Burgert I. (2006) Secondary cell wall polymers studied by Confocal Raman microscopy: Spatial distribution, orientation and molecular deformation New Zealand. J. Forestry Sci. 36: 60–71. [Google Scholar]
- 110. Gierlinger N., and Schwanninger M. (2006) Chemical imaging of poplar wood cell walls by confocal Raman microscopy. Plant Physiol. 140: 1246–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Agarwal U. (2006) Raman imaging to investigate ultrastructure and composition of plant cell walls: Distribution of lignin and cellulose in black spruce wood (Picea mariana). Planta 224: 1141–1153. [DOI] [PubMed] [Google Scholar]
- 112. Naumann A., and Polle A. (2006) FTIR imaging as a new tool for cell wall analysis of wood New Zealand. J. Forestry Sci. 36: 54–59. [Google Scholar]
- 113. Labbe N., Rials T. G., Kelley S. S., Cheng Z. M., Kim J. Y., and Li Y. (2005) FT-IR imaging and pyrolysis-molecular beam mass spectrometry: New tools to investigate wood tissues. Wood Sci. Technol. 39: 61–U19. [Google Scholar]
- 114. Mills E. N. C., Parker M. L., Wellner N., Toole G., Feeney K., and Shewry P. R. (2005) Chemical imaging: The distribution of ions and molecules in developing and mature wheat grain. J. Cereal Sci. 41: 193–201. [Google Scholar]
- 115. Barron C., Parker M. L., Mills E. N. C., Rouau X., and Wilson R. H. (2005) FTIR imaging of wheat endosperm cell walls in situ reveals compositional and architectural heterogeneity related to grain hardness. Planta 220: 667–677. [DOI] [PubMed] [Google Scholar]
- 116. Hanninen T., Kontturi E., and Vuorinen T. (2011) Distribution of lignin and its coniferyl alcohol and coniferyl aldehyde groups in Picea abies and Pinus sylvestris as observed by Raman imaging. Phytochemistry 72: 1889–1895. [DOI] [PubMed] [Google Scholar]
- 117. Hanninen T., Tukiainen P., Svedstrom K., Serimaa R., Saranpaa P., Kontturi E., Hughes M., and Vuorinen T. (2012) Ultrastructural evaluation of compression wood-like properties of common juniper (Juniperus communis L.). Holzforschung 66: 389–395. [Google Scholar]
- 118. Ma J., Zhang Z., Yang G., Mao J., and Xu F. (2011) Ultrastructural topochemistry of cell wall polymers in Populus nigra by transmission electron microscopy and Raman imaging. Bioresources 6: 3944–3959. [Google Scholar]
- 119. Sun L., Simmons B. A., and Singh S. (2011) Understanding tissue specific compositions of bioenergy feedstocks through hyperspectral Raman imaging. Biotechnol. Bioeng. 108: 286–295. [DOI] [PubMed] [Google Scholar]
- 120. Horvath L., Peszlen I., Gierlinger N., Peralta P., Kelley S., and Csoka L. (2012) Distribution of wood polymers within the cell wall of transgenic aspen imaged by Raman microscopy. Holzforschung 66: 717–725. [Google Scholar]
- 121. Schmidt M., Schwartzberg A. M., Carroll A., Chaibang A., Adams P. D., and Schuck P. J. (2010) Raman imaging of cell wall polymers in Arabidopsis thaliana . Biochem. Biophys. Res. Commun. 395: 521–523. [DOI] [PubMed] [Google Scholar]
- 122. Mateu B. P., Hauser M. T., Heredia A., and Gierlinger N. (2016) Waterproofing in arabidopsis: Following phenolics and lipids in situ by confocal Raman microscopy. Front Chem. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gierlinger N., Keplinger T., Harrington M., and Schwanninger M. (2013) Raman imaging of lignocellulosic feedstock In Cellulose – biomass conversion, van de Ven T., and Kadla J., Eds., InTech, Rijeka, Croatia, pp. 159–192. [Google Scholar]
- 124. Richter S., Mussig J., and Gierlinger N. (2011) Functional plant cell wall design revealed by the Raman imaging approach. Planta 233: 763–772. [DOI] [PubMed] [Google Scholar]
- 125. Blecher I. C., Seidel R., Thomann R., and Speck T. (2012) Comparison of different methods for the detection of silica inclusions in plant tissues. Int. J. Plant Sci. 173: 229–238. [Google Scholar]
- 126. Gierlinger N., Sapei L., and Paris O. (2008) Insights into the chemical composition of Equisetum hyemale by high resolution Raman imaging. Planta 227: 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sapei L., Gierlinger N., Hartmann J., Noske R., Strauch P., and Paris O. (2007) Structural and analytical studies of silica accumulations in Equisetum hyemale . Anal. Bioanal. Chem. 389: 1249–1257. [DOI] [PubMed] [Google Scholar]
- 128. Wang X., Ren H., Zhang B., Fei B., and Burgert I. (2012) Cell wall structure and formation of maturing fibres of moso bamboo (Phyllostachys pubescens) increase buckling resistance. J. R. Soc. Interf. 9: 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wang X. Q., Keplinger T., Gierlinger N., and Burgert I. (2014) Plant material features responsible for bamboo's excellent mechanical performance: A comparison of tensile properties of bamboo and spruce at the tissue, fibre and cell wall levels. Annal. Botany 114: 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Li X. L., Zhou B. X., Zhang Y., Yao Y. M., and He Y. (2016) Revealing the cell structure and formation of bamboo with confocal Raman microscopy.Spectrosc. Spectral Anal. 36: 413–418. [PubMed] [Google Scholar]
- 131. Chu L. Q., Masyuko R., Sweedler J. V., and Bohn P. W. (2010) Base-induced delignification of miscanthus x giganteus studied by three-dimensional confocal raman imaging. Bioresource Technol. 101: 4919–4925. [DOI] [PubMed] [Google Scholar]
- 132. Ma J. F., Zhou X., Ma J., Ji Z., Zhang X., and Xu F. (2014) Raman microspectroscopy imaging study on topochemical correlation between lignin and Hydroxycinnamic acids in Miscanthus sinensis . Microsc. Microanal. 20: 956–963. [DOI] [PubMed] [Google Scholar]
- 133. Chylinska M., Szymanska-Chargot M., and Zdunek A. (2014) Imaging of polysaccharides in the tomato cell wall with Raman microspectroscopy. Plant Methods 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Qin J. W., Chao K. L., and Kim M. S. (2011) Investigation of Raman chemical imaging for detection of lycopene changes in tomatoes during postharvest ripening. J. Food Eng. 107: 277–288. [Google Scholar]
- 135. Kammer M., Hedrich R., Ehrlich H., Popp J., Brunner E., and Krafft C. (2010) Spatially resolved determination of the structure and composition of diatom cell walls by Raman and FTIR imaging. Anal. Bioanal. Chem. 398: 509–517. [DOI] [PubMed] [Google Scholar]
- 136. Allouche F., Hanafi M., Jamme F., Robert P., Barron C., Guillon F., and Devaux M. F. (2012) Coupling hyperspectral image data having different spatial resolutions using multiple co-inertia analysis. Chemometr. Intell. Lab. 117: 200–212. [Google Scholar]
- 137. Zimmermann B., Bagcioglu M., Sandt C., and Kohler A. (2015) Vibrational microspectroscopy enables chemical characterization of single pollen grains as well as comparative analysis of plant species based on pollen ultrastructure. Planta 242: 1237–1250. [DOI] [PubMed] [Google Scholar]
- 138. Lasch P., and Naumann D. (2006) Spatial resolution in infrared micro spectroscopic imaging of tissues. Biochim. Et Biophys. Acta-Biomembr. 1758: 814–829. [DOI] [PubMed] [Google Scholar]
- 139. Cintron M. S., Fortier C., Hinchliffe D. J., and Rodgers J. E. (2017) Chemical imaging of secondary cell wall development in cotton fibers using a mid-infrared focal-plane array detector. Text Res. J. 87: 1040–1051. [Google Scholar]
- 140. Dadswell H. E., and Wardrop A. B. (1955) The structure and properties of tension wood. Holzforschung 9: 97–103. [Google Scholar]
- 141. Pilate G., Chabbert B., Cathala B., Yoshinaga A., Leple J. C., Laurans F., Lapierre C., and Ruel K. (2004) Lignification and tension wood. Cr. Biol. 327: 889–901. [DOI] [PubMed] [Google Scholar]
- 142. Wada M., Hidaka H., and Kim U. J. (2010) Synchrotron X-ray fiber diffraction study on the thermal expansion behavior of cellulose crystals in tension wood of Japanese poplar in the low-temperature region. Holzforschung 64: 167–171. [Google Scholar]
- 143. Donaldson L., Hague J., and Snell R. (2001) Lignin distribution in coppice poplar, linseed and wheat straw. Holzforschung 55: 379–385. [Google Scholar]
- 144. Joseleau J. P., Imai T., Kuroda K., and Ruel K. (2004) Detection in situ and characterization of lignin in the G-layer of tension wood fibres of Populus deltoides . Planta 219: 338–345. [DOI] [PubMed] [Google Scholar]
- 145. Yoshida M., Ohta H., Yamamoto H., and Okuyama T. (2002) Tensile growth stress and lignin distribution in the cell walls of yellow poplar, Liriodendron tulipifera . Linn. Trees 16: 457–464. [Google Scholar]
- 146. Lehringer C., Gierlinger N., and Koch G. (2008) Topochemical investigation on tension wood fibres of A cer spp., Fagus sylvatica L. and Quercus robur L.. Holzforschung 62: 255–263. [Google Scholar]
- 147. Czamara K., Majzner K., Pacia M. Z., Kochan K., Kaczor A., and Baranska M. (2015) Raman spectroscopy of lipids: A review. J. Raman Spectrosc. 46: 4–20. [Google Scholar]
- 148. Guo X., Wu Y. Q., and Yan N. (2017) Characterizing spatial distribution of the adsorbed water in wood cell wall of G inkgo biloba L. by mu-FTIR and confocal Raman spectroscopy. Holzforschung 71: 415–423. [Google Scholar]
- 149. Goswami L., Dunlop J. W. C., Jungnikl K., Eder M., Gierlinger N., Coutand C., Jeronimidis G., Fratzl P., and Burgert I. (2008) Stress generation in the tension wood of poplar is based on the lateral swelling power of the G-layer. Plant J. 56: 531–538. [DOI] [PubMed] [Google Scholar]
- 150. Burgert I., Eder M., Gierlinger N., and Fratzl P. (2007) Tensile and compressive stresses in tracheids are induced by swelling based on geometrical constraints of the wood cell. Planta 226: 981–987. [DOI] [PubMed] [Google Scholar]
- 151. Schreiber N., Gierlinger N., Putz N., Fratzl P., Neinhuis C., and Burgert I. (2010) G-fibres in storage roots of Trifolium pratense (Fabaceae): Tensile stress generators for contraction. Plant J. 61: 854–861. [DOI] [PubMed] [Google Scholar]
- 152. Thygesen L. G., and Gierlinger N. (2013) The molecular structure within dislocations in Cannabis sativa fibres studied by polarised Raman microspectroscopy. J. Struct. Biol. 182: 219–225. [DOI] [PubMed] [Google Scholar]
- 153. Sun L., Singh S., Joo M., Vega-Sanchez M., Ronald P., Simmons B. A., Adams P., and Auer M. (2016) Non-invasive imaging of cellulose microfibril orientation within plant cell walls by polarized Raman microspectroscopy. Biotechnol. Bioeng. 113: 82–90. [DOI] [PubMed] [Google Scholar]
- 154. Abraham Y., and Elbaum R. (2013) Quantification of microfibril angle in secondary cell walls at subcellular resolution by means of polarized light microscopy. New Phytol. 197: 1012–1019. [DOI] [PubMed] [Google Scholar]
- 155. Konnerth J., Gierlinger N., Keckes J., and Gindl W. (2009) Actual versus apparent within cell wall variability of nanoindentation results from wood cell walls related to cellulose microfibril angle. J. Mater. Sci. 44: 4399–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Witke K., Gotze J., Rossler R., Dietrich D., and Marx G. (2004) Raman and cathodoluminescence spectroscopic investigations on Permian fossil wood from Chemnitz – a contribution to the study of the permineralisation process. Spectrochim. Acta A 60: 2903–2912. [DOI] [PubMed] [Google Scholar]
- 157. Dietrich D., Hemeltjen S., Meyer N., Baucker E., Ruhle G., Wienhaus O., and Marx G. (2002) A new attempt to study biomineralised silica bodies in Dactylis glomerata L. Anal. Bioanal. Chem. 374: 749–752. [DOI] [PubMed] [Google Scholar]
- 158. Dietrich D., Hinke S., Baumann W., Fehlhaber R., Baucher E., Ruhle G., Wienhaus O., and Marx G. (2003) Silica accumulation in Triticum aestivum L. and Dactylis glomerata L. Anal. Bioanal. Chem. 376: 399–404. [DOI] [PubMed] [Google Scholar]
- 159. Merk V., Chanana M., Gaan S., and Burgert I. (2016) Mineralization of wood by calcium carbonate insertion for improved flame retardancy. Holzforschung 70: 867–876. [Google Scholar]
- 160. Merk V., Chanana M., Gierlinger N., Hirt A. M., and Burgert I. (2014) Hybrid wood materials with magnetic anisotropy dictated by the hierarchical cell structure. Acs Appl. Mater. Inter. 6: 9760–9767. [DOI] [PubMed] [Google Scholar]
- 161. Gallagher K. L., Alfonso-Garcia A., Sanchez J., Potma E. O., and Santos G. M. (2015) Plant growth conditions alter phytolith carbon. Front Plant Sci. 6: 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Chen C. H., and Lewin J. (1969) Silicon as a nutrient element for Equisetum Arvense . Can. J. Botany 47: 125–131. [Google Scholar]
- 163. Spatz H. C., Kohler L., and Speck T. (1998) Biomechanics and functional anatomy of hollow-stemmed sphenopsids. I. Equisetum giganteum (Equisetaceae). Am. J. Bot. 85: 305–314. [PubMed] [Google Scholar]
- 164. Milwich M., Speck T., Speck O., Stegmaier T., and Planck H. (2006) Biomimetics and technical textiles: Solving engineering problems with the help of nature's wisdom. Am. J. Bot. 93: 1455–1465. [DOI] [PubMed] [Google Scholar]
- 165. Perry C. C., and Fraser M. A. (1991) Silica Deposition and Ultrastructure in the Cell-Wall of Equisetum-Arvense – the importance of cell-wall structures and flow-control in biosilicification. Philos. Trans. Roy. Soc. B 334: 149–157. [Google Scholar]
- 166. Fry S. C., Nesselrode B. H. W. A., Miller J. G., and Mewburn B. R. (2008) Mixed-linkage (1 ->3,1 ->4)-beta-D-glucan is a major hemicellulose of Equisetum (horsetail) cell walls. New Phytol. 179: 104–115. [DOI] [PubMed] [Google Scholar]
- 167. Kido N., Yokoyama R., Yamamoto T., Furukawa J., Iwai H., Satoh S., and Nishitani K. (2015) The matrix polysaccharide (1;3,1;4)-beta-d-Glucan is Involved in silicon-dependent strengthening of rice cell wall. Plant Cell Physiol. 56: 268–276. [DOI] [PubMed] [Google Scholar]
- 168. Law C., and Exley C. (2011) New insight into silica deposition in horsetail (Equisetum arvense). Bmc Plant Biol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Leroux O., Leroux F., Mastroberti A. A., Santos-Silva F., Van Loo D., Bagniewska-Zadworna A., Van Hoorebeke L., Bals S., Popper Z. A., and Mariath J. E. D. (2013) Heterogeneity of silica and glycan-epitope distribution in epidermal idioblast cell walls in Adiantum raddianum laminae. Planta 237: 1453–1464. [DOI] [PubMed] [Google Scholar]
- 170. Roman M., Dobrowolski J. C., Baranska M., and Baranski R. (2011) Spectroscopic studies on bioactive polyacetylenes and other plant components in wild carrot root. J. Natural Prod. 74: 1757–1763. [DOI] [PubMed] [Google Scholar]
- 171. Baranska M., Schulz H., Baranski R., Nothnagel T., and Christensen L. P. (2005) In situ simultaneous analysis of polyacetylenes, carotenoids and polysaccharides in carrot roots. J. Agric. Food Chem. 53: 6565–6571. [DOI] [PubMed] [Google Scholar]
- 172. de Juan A., Maeder M., Hancewicz T., Duponchel L., and Tauler R. (2009) Chemometric tools for image analysis In Infrared and Raman spectroscopic imaging, Salzer R. and Siesler H. W., Eds., WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, pp. 65–108. [Google Scholar]
- 173. Geladi P., Grahn H., and Manley M. (2010) Data analysis and chemometrics for hyperspectral Imaging In Raman, infrared, and near-infrared chemical imaging, Sasic S. and Ozaki Y., Eds., John Wiley & Sons, Inc., Hoboken, New Jersey, pp. 93–109. [Google Scholar]
- 174. Shinzawa H., Awa K., Kanematsu W., and Ozaki Y. (2009) Multivariate data analysis for Raman spectroscopic imaging. J. Raman Spectrosc. 40: 1720–1725. [Google Scholar]
- 175. Næs T., Isaksson T., Fearn T., and Davies T. (2002) A user-friendly guide to multivariate calibration and classification, 1st ed., NIR Publications, Chichester. [Google Scholar]
- 176. Geladi P. (2003) Chemometrics in spectroscopy. Part 1. Classical chemometrics. Spectrochimica Acta Part B 58: 767–782. [Google Scholar]
- 177. Hastie T., Tibshirani R., and Friedman J. (2009) The elements of statistical learning, Springer, New York. [Google Scholar]
- 178. Diening T., and Ibach W. (2010) Software requirements and data analysis in confocal Raman microscopy In Confocal Raman microscopy, Diening T., Hollricher O., and Toporski J., Eds., Springer-Verlag, Berlin Heidelberg, pp. 61–89. [Google Scholar]
- 179. Nascimento J. M. P., and Dias J. M. B. (2005) Vertex component analysis: A fast algorithm to unmix hyperspectral data Ieee. T Geosci. Remote 43: 898–910. [Google Scholar]
- 180. Gierlinger N. (2014) Revealing changes in molecular composition of plant cell walls on the micron-level by Raman mapping and vertex component analysis (VCA). Front Plant Sci. 5: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Szymanska-Chargot M., Chylinska M., Pieczywek P. M., Rosch P., Schmitt M., Popp J., and Zdunek A. (2016) Raman imaging of changes in the polysaccharides distribution in the cell wall during apple fruit development and senescence. Planta 243: 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Felten J., Hall H., Jaumot J., Tauler R., de Juan A., and Gorzsas A. (2015) Vibrational spectroscopic image analysis of biological material using multivariate curve resolution-alternating least squares (MCR-ALS). Nature Protocols 10: 217–240. [DOI] [PubMed] [Google Scholar]
- 183. Liu B., Wang P., Kim J. I., Zhang D. L., Xia Y. Q., Chapple C., and Cheng J. X. (2015) Vibrational fingerprint mapping reveals spatial distribution of functional groups of lignin in plant cell wall. Anal. Chem. 87: 9436–9442. [DOI] [PubMed] [Google Scholar]
- 184. Eichhorn S. J., Sirichaisit J., and Young R. J. (2001) Deformation mechanisms in cellulose fibres, paper and wood. J. Mater. Sci. 36: 3129–3135. [Google Scholar]
- 185. Bulota M., Tanpichai S., Hughes M., and Eichhorn S. J. (2012) Micromechanics of TEMPO-Oxidized fibrillated cellulose composites. Acs Appl. Mater. Inter. 4: 331–337. [DOI] [PubMed] [Google Scholar]
- 186. Pullawan T., Wilkinson A. N., Zhang L. N. N., and Eichhorn S. J. (2014) Deformation micromechanics of all-cellulose nanocomposites: Comparing matrix and reinforcing components. Carbohydr. Polym. 100: 31–39. [DOI] [PubMed] [Google Scholar]
- 187. Quero F., Eichhorn S. J., Nogi M., Yano H., Lee K. Y., and Bismarck A. (2012) Interfaces in cross-linked and grafted bacterial cellulose/poly(lactic acid) resin composites. J. Polym. Environ. 20: 916–925. [Google Scholar]
- 188. Rusli R., and Eichhorn S. J. (2011) Interfacial energy dissipation in a cellulose nanowhisker composite. Nanotechnology 22: 32. [DOI] [PubMed] [Google Scholar]
- 189. Tanpichai S., Sampson W. W., and Eichhorn S. J. (2012) Stress-transfer in microfibrillated cellulose reinforced poly(lactic acid) composites using Raman spectroscopy. Compos. Part a-Appl. Sci. Manuf. 43: 1145–1152. [Google Scholar]
- 190. Tanpichai S., Sampson W. W., and Eichhorn S. J. (2014) Stress transfer in microfibrillated cellulose reinforced poly(vinyl alcohol) composites. Compos Part a-Appl. Sci. Manuf. 65: 186–191. [Google Scholar]
- 191. Eichhorn S. J., Young R. J., Davies R. J., and Riekel C. (2003) Characterisation of the microstructure and deformation of high modulus cellulose fibres. Polymer 44: 5901–5908. [Google Scholar]
- 192. Kong K., Wilding M. A., Ibbett R. N., and Eichhorn S. J. (2008) Molecular and crystal deformation of cellulose: Uniform strain or uniform stress? Faraday Discuss. 139: 283–298. [DOI] [PubMed] [Google Scholar]
- 193. Wanasekara N. D., Michud A., Zhu C. C., Rahatekar S., Sixta H., and Eichhorn S. J. (2016) Deformation mechanisms in ionic liquid spun cellulose fibers. Polymer 99: 222–230. [Google Scholar]
- 194. Eichhorn S. J., and Young R. J. (2003) Deformation micromechanics of natural cellulose fibre networks and composites. Compos. Sci. Technol. 63: 1225–1230. [Google Scholar]
- 195. Eichhorn S. J., and Young R. J. (2004) Composite micromechanics of hemp fibres and epoxy resin microdroplets. Compos. Sci. Technol. 64: 767–772. [Google Scholar]
- 196. Peetla P., Schenzel K. C., and Diepenbrock W. (2006) Determination of mechanical strength properties of hemp fibers using near-infrared Fourier transform Raman microspectroscopy. Appl. Spectrosc. 60: 682–691. [DOI] [PubMed] [Google Scholar]
- 197. Bakri B., and Eichhorn S. J. (2010) Elastic coils: Deformation micromechanics of coir and celery fibres. Cellulose 17: 1–11. [Google Scholar]
- 198. Gierlinger N., Schwanninger M., Reinecke A., and Burgert I. (2006) Molecular changes during tensile deformation of single wood fibers followed by Raman microscopy. Biomacromolecules 7: 2077–2081. [DOI] [PubMed] [Google Scholar]
- 199. Kong K., and Eichhorn S. J. (2005) The influence of hydrogen bonding on the deformation micromechanics of cellulose fibers. J. Macromol. Sci.-Phys. B 44: 1123–1136. [Google Scholar]
- 200. Barsberg S. (2010) Prediction of vibrational spectra of polysaccharides-simulated IR spectrum of cellulose based on density functional theory (DFT). J. Phys. Chem. B 114: 11703–11708. [DOI] [PubMed] [Google Scholar]
- 201. De Salvi D. T. B., Barud H. D., Treu O., Pawlicka A., Mattos R. I., Raphael E., and Ribeiro S. J. L. (2014) Preparation, thermal characterization, and DFT study of the bacterial cellulose. J. Therm. Anal. Calorim. 118: 205–215. [Google Scholar]
- 202. Salmen L., and Bergstrom E. (2009) Cellulose structural arrangement in relation to spectral changes in tensile loading FTIR. Cellulose 16: 975–982. [Google Scholar]
- 203. Toole G. A., Kacurakova M., Smith A. C., Waldron K. W., and Wilson R. H. (2004) FT-IR study of the Chara corallina cell wall under deformation. Carbohydrate Res. 339: 629–635. [DOI] [PubMed] [Google Scholar]
- 204. Gierlinger N., Goswami L., Schmidt M., Burgert I., Coutand C., Rogge T., and Schwanninger M. (2008) In situ FT-IR microscopic study on enzymatic treatment of poplar wood cross-sections. Biomacromolecules 9: 2194–2201. [DOI] [PubMed] [Google Scholar]
- 205. Li Z., Chu L. Q., Sweedler J. V., and Bohn P. W. (2010) Spatial correlation of confocal raman scattering and secondary ion mass spectrometric molecular images of lignocellulosic. Mater. Anal. Chem. 82: 2608–2611. [DOI] [PubMed] [Google Scholar]
- 206. Pedersen N. B., Gierlinger N., and Thygesen L. G. (2015) Bacterial and abiotic decay in waterlogged archaeological Picea abies (L.) Karst studied by confocal Raman imaging and ATR-FTIR spectroscopy. Holzforschung 69: 103–112. [Google Scholar]
- 207. Gierlinger N., Hansmann C., Roder T., Sixta H., Gindl W., and Wimmer R. (2005) Comparison of UV and confocal Raman microscopy to measure the melamine-formaldehyde resin content within cell walls of impregnated spruce wood. Holzforschung 59: 210–213. [Google Scholar]
- 208. Ermeydan M. A., Cabane E., Hass P., Koetz J., and Burgert I. (2014) Fully biodegradable modification of wood for improvement of dimensional stability and water absorption properties by poly(epsilon-caprolactone) grafting into the cell walls. Green Chem. 16: 3313–3321. [Google Scholar]
- 209. Ermeydan M. A., Cabane E., Masic A., Koetz J., and Burgert I. (2012) Flavonoid insertion into cell walls improves wood properties. Acs Appl. Mater. Inter. 4: 5782–5789. [DOI] [PubMed] [Google Scholar]
- 210. Keplinger T., Cabane E., Chanana M., Hass P., Merk V., Gierlinger N., and Burgert I. (2015) A versatile strategy for grafting polymers to wood cell walls. Acta Biomater. 11: 256–263. [DOI] [PubMed] [Google Scholar]
- 211. Prats-Mateu B., and Gierlinger N. (2017) Tip in-light on: Advantages, challenges, and applications of combining AFM and Raman microscopy on biological samples. Microsc. Res. Techniq. 80: 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Tetard L., Passian A., Farahi R. H., Thundat T., and Davison B. H. (2015) Opto-nanomechanical spectroscopic material characterization. Nat. Nanotechnol. 10: 870–877. [DOI] [PubMed] [Google Scholar]