

Abstract
Background Interstitial fibrosis is associated with chronic renal failure. In addition to fibroblasts, bone marrow–derived cells and tubular epithelial cells have the capacity to produce collagen. However, the amount of collagen produced by each of these cell types and the relevance of fibrosis to renal function are unclear.
Methods We generated conditional cell type–specific collagen I knockout mice and used (reversible) unilateral ureteral obstruction and adenine-induced nephropathy to study renal fibrosis and function.
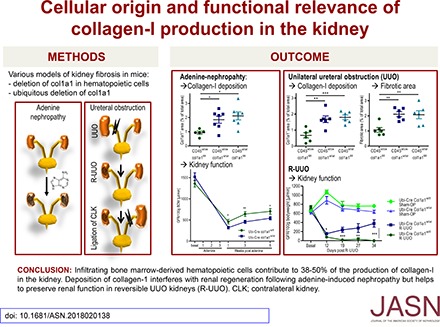
Results In these mouse models, hematopoietic, bone marrow–derived cells contributed to 38%–50% of the overall deposition of collagen I in the kidney. The influence of fibrosis on renal function was dependent on the type of damage. In unilateral ureteral obstruction, collagen production by resident fibroblasts was essential to preserve renal function, whereas in the chronic model of adenine-induced nephropathy, collagen production was detrimental to renal function.
Conclusions Our data show that hematopoietic cells are a major source of collagen and that antifibrotic therapies need to be carefully considered depending on the type of disease and the underlying cause of fibrosis.
Keywords: fibrosis, chronic kidney disease, chronic renal failure, interstitial fibrosis, immunology
Interstitial fibrosis is a common feature of various kidney diseases and associated with chronic renal failure.1,2 During recent years, it was established that collagen-producing cells can be derived from various cellular sources, including resident mesenchymal fibroblasts, pericytes, hematopoietic cells, tubular epithelial cells, and endothelial cells.3–20 This knowledge is on the basis of lineage tracing and the use of collagen I reporter mice.4,5,7,9,11,15,18 However, the number of collagen-expressing cells may not correlate with their actual production of collagen, because some of these cells may only produce very low amounts of collagen or contribute to fibrosis only indirectly (e.g., by release of profibrotic factors).21 In some studies, it is also unclear whether bone marrow–derived, collagen-producing cells are of hematopoietic origin.5 Collagen-producing hematopoietic cells are frequently called fibrocytes and thought to arise from circulating monocytes in a process called monocyte/macrophage-to-mesenchymal transition.6,8,22–27
Apart from the cellular origin of renal fibrosis, the critical question about the functional relevance of renal fibrosis is unanswered.28 A more pronounced interstitial fibrosis often correlates with a worse renal function in patients with various kidney diseases.29,30 However, it is unclear whether there is a causal relationship between fibrosis and renal function.31–33 Fibrosis may just replace damaged tissue or even help to preserve the structure of the remaining tissue.28,31,33
Thus, the functional consequences of fibrosis could differ between various types of renal disease. Unilateral ureteral obstruction (UUO) is a frequently used model of renal fibrosis. Obstruction of urine flow leads to tubular damage with rapid influx of leukocytes and results in a very reproducible course of interstitial fibrosis over a period of 5–14 days.12,34 The effect of fibrosis on renal function can be analyzed by relieving the obstruction after some days and measuring the GFR of the previously obstructed kidney.35 Adenine-induced nephropathy is a more chronic model of renal dysfunction, and it is also characterized by a considerable interstitial inflammation. Mice are fed for 3 weeks with an adenine-rich diet, leading to mild tubular damage, inflammasome-mediated activation of interstitial renal dendritic cells, considerable interstitial fibrosis, and chronic renal failure.36,37
We have generated cell type–specific and bone marrow chimeric col1a1-deficient mice to show that bone marrow–derived hematopoietic cells contribute to about 38%–50% of the deposition of collagen I in both models of kidney fibrosis. Surprisingly, the effect of fibrosis on renal function was not uniformly negative. We show that the effect of fibrosis on renal function depends on the type of renal damage. In the acute pressure–dependent model of UUO, collagen production is required to preserve renal function, whereas it is detrimental for renal function in the more chronic model of adenine-induced nephropathy. Our data identify target cells as well as pathophysiologic conditions that may be suitable to interfere with renal fibrosis.
Methods
Experimental Design
The purpose of this study was to investigate the cellular origin of renal fibrosis and the functional relevance of collagen deposition. Therefore, mice with heterozygous, inducible ubiquitous, and cell type–specific collagen knockout were used in different kidney fibrosis models (UUO, reversible unilateral ureteral obstruction [R-UUO], and adenine nephropathy). All experiments were performed with female mice at age 10–14 weeks old; they were approved by the “Regierung der Oberpfalz” and performed in accordance with the institutional guidelines. No statistical method was used to predetermine sample size in the animal studies. Sample sizes were estimated on the basis of previous experimental studies6,12 and also depended on the availability of mice from various strains. If more mice of the required genotype and age were available, the selection of mice for the experiment was at random. There were no complications, like infections, ascites, or obvious defects in wound healing, and all mice could be analyzed. No outliers were excluded in any of the experiments. Investigators were not blinded during handling of the mice, but they were fully blinded when assessing the outcome (histology, GFR measurement, real-time PCR, and flow cytometry).
Mice
All mice were bred and housed under specific pathogen-free conditions in the animal facility of the University Hospital Regensburg (Regensburg, Germany). Food and water were provided ad libitum. CH57BL/6J (CD45.1), B6.Cg-Tg(Vav1-cre)A2Kio/J38 (Vav-Cre), and B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyi/J 51 (ERT2-Cre) were obtained from The Jackson Laboratory (Bar Harbor, ME). The cd45wt/cre knock-in mice on a C57BL/6J background39 were provided by A. Medvinsky (University of Edinburgh, Edinburgh, Great Britain). C57BL/6J mice with constitutive ubiquitous expression of Cre by a cytomegalovirus promoter (Ubi-Cre) were obtained from Genoway (Lyon, France). The Pax8.rtTA;tetO.Cre mice (Pax8-Cre) on a C57BL/6 background40,41 were provided by T. Huber (University of Freiburg, Freiburg, Germany). For induction of Cre expression in ERT2-Cre mice, mice were fed with tamoxifen-supplemented food (concentration 400 mg/kg; Harlan Laboratories, Rossdorf, Germany) for 5 weeks. Mice were used after a washout of 2 weeks. To induce Cre expression in Pax8-Cre mice, doxycycline (Ratiopharm, Ulm, Germany) with a concentration of 2 mg/ml was given for 2 weeks in the drinking water. Mice were used after a washout of 4 days. All animal experiments were approved by the local authorities (Az. 2532–2-239).
Generation of Conditional col1a1 Knockout Mice
To generate a line of conditional col1a1 knockout mice, the col1a1 gene was isolated from a C57BL/6J library. LoxP sequences were inserted into the introns flanking exons 47 and 51 of col1a1. The col1a1 gene construct was introduced in C57BL/6J embryonic stem (ES) cells. ES cells were selected for homologous recombination with the help of a flippase recognition sequence flanked neomycin resistance cassette (neo) and injected into mice. The neomycin cassette and flanking flippase recognitions sequences were removed by mating chimeric mice with C57BL/6J mice expressing flip recombinase (Genoway). Progeny were mated to generate a line of homozygous mice, in which exons 47–51 were flanked by loxP sites (col1a1fl/fl) (Supplemental Figure 1). Col1a1fl/fl mice were crossed with the various Cre deleter strains described above.
Generation of Bone Marrow Chimeric Mice
To generate bone marrow chimeric mice, CD45.1 C57BL/6J wild-type mice were lethally irradiated with 9.5 Gy using a linear accelerator Lineac 2 (Siemens, Kemnath, Germany) of the University Hospital Regensburg (Regensburg, Germany) and reconstituted by intravenous transfer with 4×106 bone marrow cells from various other strains of mice (all CD45.2) as indicated. No antibiotics were used. Chimeras were used for UUO experiments 8 weeks after bone marrow reconstitution. Engraftment of donor cells was evaluated for all mice by flow cytometric analysis of splenocytes obtained at the end of the experiment (14 days after UUO). Cells were stained with a Pacific blue–labeled antibody against CD45.1 (Clone: A20; BD Biosciences, Heidelberg, Germany) and an AmCyan-labeled antibody against CD45.2 (Clone: 104; BD Biosciences) and analyzed as described below.
Murine Models of Fibrosis
To achieve UUO, the left ureter was ligated at two points under general anesthesia through a low midline abdominal incision on day 0. On day 7 or 14 after UUO, kidneys were harvested and analyzed.
R-UUO was performed according to a published protocol.35 To ensure reversibility, a displaceable clip (Fine Science Tools, Heidelberg, Germany) was used. This clip was placed on day 0, shifted in position on days 2 and 4 to avoid strictures, and removed on day 6. On day 11 after R-UUO, ureteral ligation of the contralateral kidney was performed to just measure the function of the R-UUO kidney. GFR was measured before and at several time points after R-UUO. GFR before R-UUO is calculated for one kidney only.
To achieve adenine-induced nephropathy, mice were fed with an adenine-rich diet (concentration 0.2% adenine; Altromin, Lage, Germany) for 3 weeks. Weight of the mice was measured to ensure equal food intake in all strains of mice. At several time points after termination of adenine feeding, GFR was measured. Histology of kidneys was performed at 6 weeks after termination of adenine feeding.
Measurement of GFR
The GFR of conscious mice was measured by clearance of intravenously injected FITC-labeled sinistrin (Fresenius Kabi, Graz, Austria); 1% FITC-sinistrin (3.74 × body weight of the mouse in gram × µl) was intravenously injected as a bolus, and seven small blood samples (10 μl) were obtained, covering an interval of about 3–75 minutes. Fluorescence intensity in each plasma sample was determined with a Nanodrop spectrophotometer (Nanodrop 3300; Thermo Fischer Scientific, Schwerte, Germany), and plasma concentrations of FITC-sinistrin were calculated according to a standard curve obtained for each lot of FITC-sinistrin. A two-phase exponential decay model was used to calculate the GFR.42 GFR of dead mice was set to zero.
Detection of Fibrocytes by Flow Cytometry
On day 7 or 14 after UUO, kidneys were harvested. One half of the kidney was cut into small pieces, and single-cell suspensions were obtained by digestion with 1 mg/ml collagenase (type I; Sigma-Aldrich, Deisenhofen, Germany) in HBSS for 30 minutes at 37°C and subsequent use of cell strainers with 70- and 30-μm mesh sizes. Surface molecules were stained with FITC-labeled anti-CD45 antibody (Clone: 30-F11; eBioscience, San Diego, CA) and phycoerythrin-labeled anti-CD11b antibody (Clone: M1/70; eBioscience). For intracellular staining of collagen type I, cells were fixed and permeabilized with Cytofix/Cytoperm solution (BD, Heidelberg, Germany). After two washing steps with 0.1% saponin/PBS, cells were incubated with biotinylated anti-collagen I (Clone: 7G5D2; Chondrex Redmond) or biotinylated rat IgG isotype control antibody (Clone: R35–95; BD). After another two washing steps, allophycocyanin-labeled Streptavidin (BD Biosciences) staining was performed. Counting beads were added to each sample (Invitrogen, Carlsbad, CA). Samples were measured with an FACS Canto II flow cytometer (BD). Data were analyzed with Diva Software (BD).
Isolation of Genomic DNA and Amplification by PCR
DNA was isolated from the kidney or peripheral blood cells with the DNeasy Blood and Tissue Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions and quantified by a Nanodrop spectrophotometer (Peqlab Biotechnologie GmbH, Erlangen, Germany). To determine deletion of exons 47–51 of col1a1 a PCR with the following forward and reverse collagen1a1 primers (forward primer: 5′-CCT GTC TTG TCC CCT CCT CTC TTT TAG G-3′; reverse primer: 5′-CTC AGT CCC TGT TTC TGC TGC TTG AAT-3′; Eurofins MWG Operon, Ebersberg, Germany) was performed. If exons 47–51 were deleted, the size of the PCR product decreased from 4227 to 746 bp.
RNA Isolation and Real-Time PCR
Total RNA was isolated from the kidneys with the Qiagen Midi Kit (Qiagen GmbH) according to the manufacturer’s instructions and quantified by a NanoDrop spectrophotometer. RT-PCR was performed with Oligo(dT)20 primers and M-MLV RT (Life Technologies, Carlsbad, CA). Quantitative real-time PCR was performed with the QuantiTect SYBR Green PCR Kit (Qiagen GmbH) and the ViiA7 detection system (Life Technologies). Primers for RT-PCR of col1a1 mRNA spanning exons 1 and 2 of col1a1 (forward primer: 5′-TGT TCA GCT TTG TGG ACC TC-3′; reverse primer: 5′-TCA AGC ATA CCT CGG GTT TC-3′), fibronectin mRNA (forward primer: 5′-TCC AGC CCC ACC CTA CAA GT-3′; reverse primer: 5′-CCA GAC CAA ACC ATA AGA CA-3′), and the reference gene β-actin (forward primer: 5′-ACC CGC GAG CAC AGC TTC TTT G-3′; reverse primer: 5′-ACA TGC CGG AGC CGT TGT CGA C-3′) were synthesized by Eurofins MWG Operon. Fold changes in the relative gene expression levels were determined using the 2−Δcycle threshold method.
Histologic Analysis of Kidney Sections
Kidneys were embedded in paraffin or Tissue-Tek Compound (Sakura Finetek Germany GmbH, Staufen, Germany). Paraffin sections were stained with Masson Trichrome according to the manufacturer’s protocol (Sigma, Munich, Germany). Cryosections (3 μm) were used for immunofluorescence staining. After fixation with ice-cold acetone, sections were blocked with super block blocking buffer (Thermo Fischer Scientific) and incubated with different primary antibodies: an antibody against collagen type I (ab21286; Abcam plc, Cambridge, United Kingdom), αSMA (Clone: C04018; Biocare Medical, Concord, CA), or fibronectin-1 (AV41490; Sigma). As a detection antibody, Alexa Fluor 594–labeled F(ab’)2 fragments of goat anti-rabbit IgG (Clone: A-11072; Invitrogen) were used. DNA was labeled with Hoechst 33342 (Invitrogen). Pictures of 15 visual fields of each kidney were taken with an Axio-Observer-Z1 microscope (Carl Zeiss, Oberkochen, Germany), and signals were analyzed semiautomatically with MetaMorph software (Version 4.6; Universal Imaging Corp., Downingtown, PA). All histologic sections were analyzed in a blinded manner.
Protein Preparation and Immunoblotting
Protein samples (45 μg) were separated on 8% polyacrylamide gels and transferred to nitrocellulose membranes. These were first incubated with Ponceau S (Sigma) for 5 minutes, and pictures were taken. After washing, membranes were blocked for 2 hours in 1% BSA and incubated overnight at 8°C with an antibody against collagen type I (ab21286). After washing, the membranes were incubated for 2 hours with a horseradish peroxidase–labeled goat anti-rabbit antibody (31460; Thermo Fischer Scientific). Bands were revealed by chemiluminescence and quantified with ImageJ Software in relation to the Ponceau S staining.
Statistical Analyses
Statistical analysis was performed with GraphPad Prism 5.0. Significance of the difference between two groups was assessed using unpaired two-tailed t tests for parametric distribution or two-tailed Mann–Whitney tests for nonparametric distribution. For multiple comparisons, we used one-way ANOVA with Dunnett or Bonferroni multiple comparison tests. For survival analysis, we used log rank (Mantel–Cox) tests (*P<0.05; **P<0.01; ***P<0.001).
Results
Generation and Characterization of Conditional Collagen I–Deficient Mice
To generate conditional col1a1-deficient mice, two loxP sites flanking exons 47 and 51 of the col1a1 gene were introduced by homologous recombination in C57BL/6 ES cells (Supplemental Figure 1). These 3′ exons encode for the RNA-stabilizing poly-A tail and the C-terminal propeptide that is essential for triple-helix formation of two collagen1a1 and one collagen 1a2 molecules.43 Crossbreeding of col1a1wt/fl heterozygous mice resulted in the expected ratios of col1a1wt/wt, col1a1wt/fl, and col1a1fl/fl mice.
To show that the deletion of exons 47–51 of col1a1 shuts off collagen I expression and calibrate our readout system for quantification of renal fibrosis, we crossed the col1a1fl/fl mice with constitutive ubiquitous Cre-expressing transgenic mice on C57BL/6 background (Ubi-Cre). Heterozygous Ubi-Cre×col1a1wt/fl mice were viable with no obvious phenotype under basal conditions. Crossbreeding of heterozygous Ubi-Cre×col1a1wt/fl mice containing the Cre transgene on both alleles resulted in 43% wild-type and 57% heterozygous mice (of 248 mice in total). No homozygous knockout mice (Ubi-Cre×col1a1fl/fl) were born in accordance with the previously described embryonic lethality of homozygous col1a1 deficiency.44 Heterozygous mice had no obvious phenotype; however, their birth rate was somewhat lower than the expected 66%.
We also crossed col1a1fl/fl mice with tamoxifen-inducible ubiquitous Cre-expressing transgenic mice on C57BL/6 background (insertion in the ROSA26 locus; abbreviated ERT2-Cre). Noninduced ERT2-Cre×col1a1fl/fl mice were fully viable without any obvious phenotype. At the age of 12 weeks old, the animals were fed with tamoxifen for 5 weeks, which was well tolerated and resulted in no obvious phenotype.
UUO was performed as a model of renal fibrosis. Heterozygous Ubi-Cre×col1a1wt/fl mice developed significantly less renal fibrosis than Ubi-Cre×col1a1wt/wt control mice in day 14 UUO kidneys. Deposition of collagen I quantified by immunofluorescence was reduced by about 62%, and overall fibrosis quantified by Masson Trichrome staining was reduced by about 55% (Figure 1A). There was also a reduction of about 50% in the mRNA for col1a1 in the ligated kidneys (Figure 1B). In contrast, the expression of αSMA, the deposition of fibronectin (FN1), and the amount of fibronectin mRNA were unchanged, indicating that the development of myofibroblasts and the deposition of other extracellular matrix proteins were not affected by the deletion of col1a1 (Figure 1). Nonobstructed contralateral kidneys of Ubi-Cre×col1a1wt/wt mice served as additional controls and showed basically no signs of fibrosis. Together, these data show that the deletion of collagen I was successful.
Figure 1.

Reduced fibrosis in 14-day unilateral ureteral obstruction (UUO) kidneys of mice with heterozygous ubiquitous deletion of col1a1. Nonobstructed contralateral kidneys (CLK) served as internal control. (A) Quantification of collagen I by immunofluorescence, overall fibrosis (fibrotic area) by Masson Trichrome staining, αSMA and fibronectin (FN1) by immunofluorescence, and (B) col1a1 and fibronectin mRNA expression (referred to as the housekeeping gene β-actin) by real-time RT-PCR in UUO kidneys of Ubi-Cre×col1a1wt/fl heterozygous mice (n=6) and UUO (n=9) and CLK (n=6) kidneys of Ubi-Cre×col1a1wt/wt controls. (A) Representative visual fields of renal tissue from one mouse per group are depicted. (A and B) Data are represented as mean±SEM. Unpaired two-sided t test. Scale bars, 100 μm. ***P<0.001.
UUO was also performed in tamoxifen-induced ERT2-Cre×col1a1fl/fl mice. As controls, we used tamoxifen-induced col1a1fl/fl and ERT2-Cre×col1a1wt/wt mice. Successful deletion of col1a1 in tamoxifen-induced ERT2-Cre×col1a1fl/fl mice was shown by PCR of genomic DNA from UUO kidneys (Figure 2D). Deposition of collagen I was reduced by about 70%, overall fibrosis was reduced by about 46%, and col1a1 mRNA was reduced by about 78% in day 14 UUO kidneys of the induced knockout mice (Figure 2, A and C). Expressions of αSMA and fibronectin were not altered in tamoxifen-induced ERT2-Cre×col1a1fl/fl mice (Figure 2B).
Figure 2.

Reduced fibrosis in 14-day unilateral ureteral obstruction (UUO) kidneys of mice with tamoxifen-inducible ubiquitous deletion of col1a1. For induction of Cre expression in ERT2-Cre mice, the mice were fed with tamoxifen-supplemented food for 5 weeks. After a washout of 2 weeks, UUO was performed. (A) Quantification of collagen I by immunofluorescence and overall fibrosis (fibrotic area) by Masson Trichrome staining with representative visual fields of renal tissue. (B) Analysis of αSMA and fibronectin by immunofluorescence and (C) col1a1 mRNA expression (referred to as the housekeeping gene β-actin) by real-time RT-PCR in ERT2-Cre×col1a1fl/fl (n=9), ERT2-Cre×col1a1wt/wt (n=4), and col1a1fl/fl (n=4) mice, which were all treated with tamoxifen. (D) Deletion of col1a1 in tamoxifen-induced ERT2-Cre×col1a1fl/fl mice shown by PCR of genomic DNA from UUO kidneys. (A) Representative visual fields of renal tissue from one mouse per group are depicted. (A–C) Data are represented as mean±SEM. One-way ANOVA. Scale bars, 100 μm. *P<0.05; **P<0.01; ***P<0.001.
Hematopoietic Cells Produce Substantial Amounts of Collagen I in Renal Fibrosis
We and others have shown previously that significant numbers of collagen-producing hematopoietic cells migrate into the kidney after UUO.5,6,8 However, it is currently unclear whether hematopoietic cells directly or only indirectly contribute to renal fibrosis (e.g., by the activation of resident fibroblasts) and what percentage of the deposited collagen is derived from these cells.
To assess the contribution of hematopoietic cells to renal fibrosis, col1a1fl/fl mice were crossed with cd45wt/cre C57BL/6 knock-in mice.39 CD45 is a specific marker for hematopoietic cells, and it is not expressed by other cells, such as mesenchymal fibroblasts or tubular epithelial cells.45 Cd45wt/cre×col1a1fl/fl mice were viable without any obvious phenotype under basal conditions, indicating that the homozygous deletion of collagen I in hematopoietic cells does not interfere with development of the mice; 14-day UUO was performed, and renal fibrosis was quantified in comparison with two controls: cd45wt/cre×col1a1wt/wt and cd45wt/wt×col1a1fl/fl mice. Leukocyte-specific collagen I–deficient mice exhibited about 61% less deposition of collagen I and about 49% less overall fibrosis in the UUO kidneys (Figure 3A). FACS analysis of UUO kidneys was performed to quantify the number of infiltrating fibrocytes by intracellular staining for collagen I and extracellular staining of CD45. In comparison with the control, very few CD45+ collagen I+ fibrocytes were detectable in the cd45wt/cre×col1a1fl/fl mice by flow cytometry, again showing the cell type–specific deletion of collagen I in hematopoietic cells (Figure 3B). The number of total infiltrating CD45+ cells was somewhat reduced in mice with a specific deletion of collagen type I in leukocytes. Hardly any fibrocytes or CD45+ cells were detectable in nonobstructed contralateral kidneys (Figure 3B). Expressions of fibronectin and αSMA were similar in all groups (Figure 3C).
Figure 3.

Reduced fibrosis in 14-day unilateral ureteral obstruction (UUO) kidneys of mice with leukocyte (CD45)–specific homozygous deletion of col1a1. (A) Quantification of collagen I by immunofluorescence and overall fibrosis (fibrotic area) by Masson Trichrome staining with representative visual fields of renal tissue. (B) Analysis of CD45+ collagen I+ fibrocytes related to CD45+ cells and analysis of CD45+ cells per kidney by flow cytometry with representative dot plots of UUO kidneys and nonobstructed contralateral kidneys (CLKs). (C) Quantification of αSMA and fibronectin by immunofluorescence in cd45wt/cre×col1a1fl/fl (n=6), cd45wt/cre×col1a1wt/wt (n=6), and cd45wt/wt×col1a1fl/fl (n=6) mice. (A) Representative visual fields of renal tissue from one mouse per group are depicted. (A–C) Data are represented as mean±SEM. One-way ANOVA (A–C). Scale bars, 100 μm. *P<0.05; **P<0.01; ***P<0.001.
To corroborate our findings in a second strain of mice, we crossed col1a1fl/fl mice with Vav-Cre transgenic mice on a C57BL/6 background. Vav is considered as a specific marker for hematopoietic cells, and Vav-Cre mice are frequently used to specifically delete floxed genes in the hematopoietic system.38 In 14-day UUO kidneys, Vav-Cre×col1a1fl/fl mice displayed about 41% less deposition of collagen I and about 51% less overall fibrosis compared with Vav-Cre×col1a1wt/wt and col1a1fl/fl mice (Figure 4A). Col1a1 mRNA expression was reduced by about 48% (Figure 4B). Expressions of fibronectin and αSMA were unaltered (Figure 4C).
Figure 4.

Reduced fibrosis in 14-day unilateral ureteral obstruction kidneys of mice with leukocyte (Vav)–specific homozygous deletion of col1a1. (A) Quantification of collagen I by immunofluorescence and overall fibrosis (fibrotic area) by Masson Trichrome staining with representative visual fields of renal tissue. (B) Analysis of col1a1 mRNA expression (referred to as the housekeeping gene β-actin) by real-time RT-PCR. (C) Quantification of αSMA and fibronectin by immunofluorescence in Vav-Cre×col1a1fl/fl (n=10 for histology and n=11 for RT-PCR), Vav-Cre×col1a1wt/wt (n=9), and col1a1fl/fl (n=9) mice. (A) Representative visual fields of renal tissue from one mouse per group are depicted. Data are represented as mean±SEM. (A) Mann–Whitney and (B and C) unpaired two-sided t tests. Scale bars, 100 μm. **P<0.01; ***P<0.001.
In a third approach, we generated bone marrow chimeric mice by transplanting bone marrow from tamoxifen-induced ERT2-Cre×col1a1fl/fl or tamoxifen-treated col1a1fl/fl mice into lethally irradiated wild-type C57BL/6 recipients. Reconstitution of the transplanted bone marrow and deletion of col1a1 in hematopoietic cells were shown in 14-day UUO mice by flow cytometry using CD45.1 and CD45.2 as markers to distinguish between donor and recipient hematopoietic cells (Supplemental Figure 2) and by genomic PCR of peripheral blood cells (Figure 5E). The vast majority of hematopoietic cells were of donor origin. Bone marrow chimeric collagen I–deficient mice showed an about 60% reduction of collagen I expression and an about 53% reduction of overall fibrosis in the day 14 UUO kidney (Figure 5A). Quantification of fibrocytes in the UUO kidney by flow cytometry revealed markedly reduced numbers of fibrocytes in the bone marrow chimeric collagen I–deficient mice (Figure 5B). Col1a1 mRNA expression was reduced by about 31% (Figure 5C). Expressions of αSMA and fibronectin were not different (Figure 5D). Very similar results were obtained when lethally irradiated C57BL/6 mice were reconstituted with stem cells from tamoxifen- and nontamoxifen-induced ERT2-Cre×col1a1fl/fl mice (controls) (Supplemental Figure 3).
Figure 5.

Reduced fibrosis in 14-day unilateral ureteral obstruction (UUO) kidneys of bone marrow chimeric mice with tamoxifen-induced homozygous deletion of col1a1 in hematopoietic stem cells. Lethally irradiated C56BL/6 mice were reconstituted with bone marrow from tamoxifen-induced ERT2-Cre×col1a1fl/fl (n=12) or col1a1fl/fl (n=12) mice. (A) Quantification of collagen I by immunofluorescence and overall fibrosis (fibrotic area) by Masson Trichrome staining with representative visual fields of renal tissue. (B) CD45+ collagen I+ fibrocytes analyzed by flow cytometry. (C) αSMA and fibronectin quantified by immunofluorescence and (D) col1a1 mRNA expression (referred to as the housekeeping gene β-actin) quantified by real-time RT-PCR. (E) Deletion of col1a1 shown by genomic PCR of peripheral blood cells of one representative mouse per group after 14-day UUO. (A) Representative visual fields of renal tissue from one mouse per group are depicted. Data are represented as mean±SEM. (A and C) Unpaired two-sided t test and (B) Mann–Whitney test. BM, bone marrow. Scale bars, 100 μm. ***P<0.001.
Taken together, these data provide clear evidence that bone marrow–derived hematopoietic cells directly and to a major degree contribute to the development of renal fibrosis in the UUO model. Comparing heterozygous ubiquitous deletion of collagen I with cell type–specific homozygous deletion in hematopoietic cells, we conclude that 38%–50% of the deposited collagen I in the UUO model is derived from hematopoietic cells.
Tubular Epithelial Cells Do Not Contribute to the Production of Collagen I in Renal Fibrosis
It is well established that tubular epithelial cells undergo epithelial-to-mesenchymal transformation (EMT) after renal injury (e.g., UUO) and directly or indirectly participate in the development of fibrosis.16,18 We analyzed to what extent tubular epithelial cells contribute to the direct production of collagen I. Doxycycline-inducible Pax8-Cre transgenic C57BL/6 mice (Pax8.rtTA;tetO.Cre) with inducible Cre expression in all tubular segments were crossed with col1a1fl/fl mice, allowing the specific induction of Cre and thus, the specific deletion of col1a1 in tubular epithelial cells.40,41 Pax8-Cre×col1a1fl/fl mice were treated for 2 weeks with doxycycline, and deletion of col1a1 was verified by PCR of genomic DNA (Supplemental Figure 4). As controls, we used doxycycline-treated col1a1fl/fl mice, doxycycline-treated Pax8-Cre×col1a1wt/wt, and untreated Pax8-Cre×col1a1fl/fl mice; 14-day UUO was performed in the various strains of mice. Deposition of collagen I, αSMA, fibronectin, and overall fibrosis was quantified in the UUO kidneys (Figure 6). Although there was a minor reduction in the deposition of collagen I in the doxycycline-induced Pax8-Cre×col1a1fl/fl mice, the difference to the controls was statistically NS. These data indicate that tubular epithelial cells do not produce relevant amounts of collagen I in the UUO model.
Figure 6.

Cell type–specific deletion of col1a1 in tubular epithelial cells does not reduce fibrosis in 14-day unilateral ureteral obstruction (UUO) kidneys. To induce Cre expression in Pax8-Cre mice, doxycycline was given for 2 weeks in the drinking water. UUO was performed after a washout of 4 days. Pax8-Cre×col1a1fl/fl induced with doxycycline (n=20; knockout), Pax8-Cre×col1a1fl/fl not induced with doxycycline (n=5; control), col1a1fl/fl induced with doxycycline (n=10; control), and Pax8-Cre×col1a1wt/wt induced with doxycycline (n=10; control) were analyzed for (A) deposition of collagen I, (B) fibronectin, (C) overall fibrosis (fibrotic area), and (D) αSMA. (A and C) Representative visual fields of renal tissue from one mouse per group are depicted. Data are represented as mean±SEM. One-way ANOVA test. Scale bars, 100 μm.
Expression of Collagen I Is Required to Preserve Kidney Function after Ureteral Obstruction
We used the model of R-UUO to study how changes in renal fibrosis and changes in the cell type–specific production of collagen I alter renal function and affect recovery after removal of the obstruction. Ureteral obstruction was achieved with a clip that was placed on day 0 and removed on day 6. As control, sham operations were performed on the same days. On day 11, irreversible UUO of the contralateral kidney was performed in all mice. Therefore, renal function (GFR) was measured just in the R-UUO kidney by the clearance of intravenously injected FITC-sinistrin.42 Heterozygous Ubi-Cre×col1a1wt/fl mice were compared with Ubi-Cre×col1a1wt/wt controls (Figure 7A). On day −1 before R-UUO, renal function (GFR) was identical in both strains of mice, which means that the heterozygous knockout of collagen I has no influence on normal renal function. On day 12 after R-UUO (i.e., day 6 after removal of the clip), heterozygous collagen I–deficient mice exhibited a significantly reduced GFR compared with wild-type controls. Subsequently, plasma potassium levels increased to >11 mmol/L in the heterozygous knockout mice, and all of these mice died within the next couple of days. In contrast, GFR in wild-type mice recovered from 154 μl/min at day 12 after UUO to 378 μl/min at day 34. Plasma potassium levels remained within normal range. Sham operations were equally well tolerated in heterozygous knockout mice and wild-type controls.
Figure 7.

Ubiquitous deletion of collagen I but not cell type specific deletion of collagen I in hematopoietic cells impairs renal function and survival of mice with a reversible unilateral ureteral obstruction (R-UUO). (A–C) Measurement of GFR (one kidney) per 100 g body wt and survival in various strains of mice. R-UUO was performed on day 0, obstruction was relieved on day 6, and the contralateral kidney was irreversibly obstructed on day 11. (A) GFR (one kidney) per 100 g body wt and survival in Ubi-Cre×col1a1wt/fl heterozygous mice (n=13) and Ubi-Cre×col1a1wt/wt controls (n=9). Sham operations (Sham-OP) included all procedures except R-UUO, and they were performed in Ubi-Cre×col1a1wt/fl mice (n=5) and Ubi-Cre×col1a1wt/wt controls (n=4). (B) GFR (one kidney) per 100 g body wt and survival in ERT2-Cre×col1a1fl/fl (n=5), ERT2-Cre×col1a1wt/wt (n=5), and col1a1fl/fl (n=5) mice. (C) GFR (one kidney) per 100 g body wt and survival in cd45wt/cre×col1a1fl/fl (n=8) and cd45wt/cre×col1a1wt/wt (n=8). (D) Deposition of collagen I and overall fibrosis (fibrotic area) in the R-UUO kidneys on day 35 after R-UUO with representative visual fields of renal tissue from one mouse per group. All mice were treated with tamoxifen from week −7 to −2 before R-UUO. (D) Representative visual fields of renal tissue from one mouse per group are depicted. Data are represented as mean or mean±SEM. (A and D) Unpaired two-sided t test, (B) one-way ANOVA, and log rank (Mantel–Cox) test for survival. Scale bars, 100 μm. *P<0.05; **P<0.01; ***P<0.001.
To analyze the recovery of the R-UUO kidneys and the deposition of collagen I, we performed R-UUO with Ubi-Cre×col1a1wt/fl mice and Ubi-Cre×col1a1wt/wt controls and harvested the R-UUO kidneys on day 11 (i.e., 5 days after removal of the clip). Macroscopic evaluation of R-UUO kidneys revealed that the ureteral obstruction was successfully relieved in all mice (Supplemental Figure 5A). Also, the weight of R-UUO kidneys was almost identical to that of nonobstructed healthy kidneys but much higher than the weight of 14-day UUO kidneys, again indicating that recanalization was successful in all animals (Supplemental Figure 5B).
Heterozygous Ubi-Cre×col1a1wt/fl mice displayed about 53% less deposition of collagen I and about 60% less overall fibrosis compared with Ubi-Cre×col1a1wt/wt controls. Col1a1 mRNA expression was reduced by about 40% (Supplemental Figure 6). Our data show that the reduced ability to produce collagen I during R-UUO leads to a deleterious outcome regarding renal function and survival.
We also performed R-UUO in tamoxifen-induced ERT2-Cre×col1a1fl/fl mice as well as tamoxifen-induced ERT2-Cre×col1a1wt/wt and col1a1fl/fl control mice (Figure 7B). After R-UUO and inactivation of the contralateral kidney, none of the tamoxifen-induced ERT2-Cre×col1a1fl/fl mice survived, whereas survival was between 80% and 100% in the control strains with recovery of GFR. These data show that collagen I newly deposited during UUO and not collagen I produced during development for the kidney is essential to preserve renal function in R-UUO.
As shown in Figure 3, the cell type–specific deletion of collagen I in hematopoietic cells resulted in a markedly reduced fibrosis in day 14 UUO kidneys. To assess the functional relevance of collagen I derived from hematopoietic cells, we also determined the GFR after R-UUO in cd45wt/cre×col1a1fl/fl mice. Specific deletion of collagen I in hematopoietic cells did not lead to a reduced GFR or a decreased survival (Figure 7C). On day 35 after R-UUO, we quantified renal fibrosis. Deposition of collagen I in the R-UUO kidney was significantly reduced by about 47%, and overall fibrosis was significantly reduced by about 31% in cd45wt/cre×col1a1fl/fl mice (Figure 7D). The reduction of renal fibrosis in the R-UUO kidneys of cd45wt/cre×col1a1fl/fl mice was not as pronounced as in the day 14 UUO kidneys. This could result from the fact that obstruction was relieved already after 6 days. Infiltrating hematopoietic cells may contribute to renal fibrosis primarily at later time points, because they first have to migrate into the kidney, whereas resident fibroblasts may be responsible for the immediate deposition of collagen I. We, therefore, quantified renal fibrosis on day 7 after UUO in Ubi-Cre×col1a1wt/fl mice and cd45wt/cre×col1a1fl/fl mice, each compared with the appropriate controls (Supplemental Figure 7). In day 7 UUO kidneys of Ubi-Cre×col1a1wt/fl mice, deposition of collagen I was reduced by about 61%, and overall fibrosis was reduced by about 56%, whereas the percentages were about 47% and 28%, respectively, in cd45wt/cre×col1a1fl/fl mice. Deposition of collagen I in 7-day UUO kidneys was also quantified by Western blot in various strains of mice (Supplemental Figure 8). Compared with control mice (col1a1fl/fl), the expression of collagen I protein was reduced by 58% in Ubi-Cre×col1a1wt/fl heterozygous mice, 28% in cd45wt/cre×col1a1fl/fl leukocyte-specific collagen I–deficient mice, and 94% in ERT2Cre×col1a1fl/fl induced knockout mice.
In contrast, in day 14 UUO kidneys, the reductions were comparable in the two strains of mice (62% versus 61% for collagen 1 and 55% versus 49% for overall fibrosis, respectively). These data suggest that infiltrating hematopoietic cells contribute to renal fibrosis primarily at later time points after UUO, whereas resident fibroblasts are more important for the immediate production of collagen.
Overall, the results in the R-UUO model show that the production of collagen I by resident fibroblasts but not by infiltrating hematopoietic cells is essential to maintain structural integrity and renal function in a model of acute injury with pressure-induced tubular damage.
Hematopoietic Cells Produce Substantial Amounts of Collagen I in Adenine-Induced Nephropathy—Effect of Collagen I Expression on Renal Function
Adenine-induced nephropathy leads to interstitial fibrosis and impairment of renal function in a crystal-induced, inflammasome-dependent manner.36,37 Mice were fed an adenine-rich diet for 3 weeks and analyzed after a 6-week recovery period on normal diet. As expected, heterozygous col1a1-deficient mice (Ubi-Cre×col1a1wt/fl) developed significantly less renal fibrosis than controls (Ubi-Cre×col1a1wt/wt). Deposition of collagen I was reduced by about 66% and overall fibrosis was reduced by about 45% in Ubi-Cre×col1a1wt/fl mice (Figure 8A). The expressions of αSMA and fibronectin were unchanged (Figure 8B). Cell type–specific deletion of col1a1 in hematopoietic cells was performed to study the contribution of hematopoietic cells to renal fibrosis. Cd45wt/cre×col1a1fl/fl mice displayed about 52% less collagen I deposition and about 33% less overall fibrosis compared with controls (Figure 8D), whereas αSMA and fibronectin were not different (Figure 8E). Equal body weight in the various strains of mice indicates that there are no differences in the intake of adenine-rich food that may account for differences in fibrosis (Supplemental Figure 9). These data show that hematopoietic cells also markedly contribute to renal fibrosis in adenine-induced nephropathy.
Figure 8.

Ubiquitous and cell type specific deletion of collagen I in hematopoietic cells reduces renal fibrosis and improves renal function in the model of adenine-induced nephropathy. Various strains of mice were fed for 3 weeks with an adenine-rich diet. Six weeks after termination of adenine feeding, histology was performed. (A) Quantification of collagen I and overall fibrosis (fibrotic area) with representative visual fields of renal tissue, (B) αSMA, and fibronectin in the kidneys of Ubi-Cre×col1a1wt/fl heterozygous mice (n=11) and Ubi-Cre×col1a1wt/wt controls (n=4). (C) GFR per 100 g body wt (BDW) was measured before and 1, 3, and 6 weeks after termination of adenine feeding in Ubi-Cre×col1a1wt/fl heterozygous mice (n=7) and Ubi-Cre×col1a1wt/wt controls (n=7). (D) Quantification of collagen I, overall fibrosis (fibrotic area) with representative visual fields of renal tissue, (E) αSMA, and fibronectin of cd45wt/cre×col1a1fl/fl (n=5), cd45wt/cre×col1a1wt/wt (n=7), and cd45wt/wt×col1a1fl/fl (n=7) mice. (F) GFR per 100 g BDW was measured before and 1, 3, and 6 weeks after adenine feeding in cd45wt/cre×col1a1fl/fl (n=5), cd45wt/cre×col1a1wt/wt (n=5), and cd45wt/wt×col1a1fl/fl (n=4) mice. (A and D) Representative visual fields of renal tissue from one mouse per group are depicted. Data are represented as mean±SEM. (A–C and F) Unpaired two-sided t test, and (D and E) one-way ANOVA. Scale bars, 100 μm. *P<0.05; **P<0.01; ***P<0.001.
To investigate how overall reduction of fibrosis affects renal function in the adenine model, we measured GFR at baseline and several time points after adenine feeding in heterozygous collagen I–deficient mice (Ubi-Cre×col1a1wt/fl) and controls (Ubi-Cre×col1a1wt/wt). In heterozygous collagen I–deficient mice, GFRs were 61%, 60%, and 38% better than those in the wild-type controls at 1, 3, and 6 weeks after cessation of adenine feeding, respectively (Figure 8C). These data show that fibrosis has a clear negative effect on renal function in adenine-induced nephropathy. Even a partial reduction of fibrosis may be beneficial to preserve renal function in this situation of CKDs.
In adenine-induced nephropathy, we also investigated the functional consequences of a specific deletion of collagen I in hematopoietic cells. Cd45wt/cre×col1a1fl/fl mice were compared with the appropriate controls (cd45wt/cre×col1a1wt/wt and cd45wt/wt×col1a1fl/fl). The leukocyte-specific knockout mice showed a 23% higher GFR than the controls at weeks 3 and 6 after termination of adenine feeding (Figure 8F). Less improvement of renal function in leukocyte-specific knockout mice compared with heterozygous knockout mice correlated with the lower reduction of total fibrosis (33% versus 45%). Our data show that fibrosis, independent of whether collagen I is produced by resident fibroblasts or infiltrating hematopoietic cells, impairs recovery of renal function in adenine-induced nephropathy.
Discussion
We have shown that bone marrow–derived hematopoietic cells substantially contribute to the production and deposition of collagen I in two models of renal fibrosis that are both characterized by pronounced inflammation. Hematopoietic cells do not only indirectly contribute to collagen production (e.g., by the release of profibrotic factors) and do not only appear as collagen-producing cells (e.g., by the endocytosis of extracellular collagen)21,46; hematopoietic cells directly produce and deposit collagen I. There is a longstanding controversy regarding the role of hematopoietic cells in renal fibrosis. In previous publications, the percentage of bone marrow–derived cells within the pool of collagen-expressing cells ranged from nearly 0% to 50% depending on the methods used to identify these cells.4–8,11,12,47 In addition, the frequency of collagen-expressing cells does not provide information on the amount of collagen produced by a specific cell type. Hematopoietic cells may be very weak producers of collagen. Calibration of our readouts with ubiquitous heterozygous collagen I–deficient mice enabled us to quantify the contribution of hematopoietic cells to overall production of collagen. Heterozygous ubiquitous deletion of collagen I reduced collagen I deposition by 62% in 14-day UUO kidneys and 66% in adenine-induced nephropathy. Homozygous deletion of collagen I in hematopoietic cells reduced collagen I deposition by 45%–61% in UUO kidneys and 52% in adenine-induced nephropathy. Comparing heterozygous ubiquitous depletion and homozygous leukocyte-specific deletion of collagen I, we can conclude that hematopoietic cells contribute about 38%–50% to the overall deposition of collagen I in the kidney in the two models of renal fibrosis. Time kinetic experiments of 7- and 14-day UUO kidneys also suggested that resident cells (fibroblasts/pericytes) prevail in the early production of collagen I, whereas hematopoietic cells catch up later, most likely because they need time to migrate into the kidney. Ureteral ligation and adenine-induced nephropathy both lead to a substantial leukocyte infiltration in the kidneys, which may explain why hematopoietic cells markedly contribute to the production of collagen in these models. Tubular epithelial cells have no relevant role for the direct production of collagen in UUO. This does not exclude that partial EMT of damaged tubular epithelial cells indirectly contributes to renal fibrosis.16,18,48 Partial EMT and chronic inflammation together create a profibrotic milieu that enables collagen production by resident fibroblasts/pericytes and infiltrating hematopoietic cells in the kidney. There are also multiple interactions between fibroblasts, collagen-producing hematopoietic cells (fibrocytes), and inflammatory cells that promote or inhibit fibrosis.21
Ubiquitous heterozygous deletion of col1a1 reduced deposition of collagen I in UUO kidneys by 62%–65% and overall fibrosis by 45%–55% as measured by Masson Trichrome staining. Continuous degradation of fibrotic material by proteases may explain why deposition of collagen I was reduced by >50% in heterozygous mice.49 The deletion of col1a1 was specific and had no influence on the development of αSMA+ myofibroblasts or expression of fibronectin.
We have also gained important insights into the functional relevance of renal fibrosis by directly and specifically interfering with the production of collagen I and thus avoiding the pleiotropic effects of many other approaches of interfering with fibrosis (e.g., inhibition of profibrotic cytokines, like TGF-β). So far, it was unclear whether renal fibrosis would be detrimental, irrelevant, or even beneficial for preservation and regeneration of renal function and how this would depend on the type of renal damage. We have addressed these questions by serial measurements of GFR in two different models of renal fibrosis. In the model of R-UUO with subsequent irreversible obstruction of the contralateral kidney, early production of collagen is essential to preserve renal function and enable the survival of the mice. Fibrosis seems to be necessary to withstand the increased intrarenal pressure resulting from obstruction of urine flow. In strong contrast, renal fibrosis was clearly harmful in the more chronic model of adenine-induced nephropathy. The functional relevance of collagen production was also dependent on the cellular source of the collagen. In the 6-day R-UUO model, collagen production by hematopoietic cells was not required to preserve renal function, whereas the overall production of collagen was essential. Our data indicate that the contribution of hematopoietic cells to fibrosis is lower in 7-day UUO kidneys than in 14-day UUO kidneys. Thus, at day 6 after UUO, the contribution of hematopoietic cells to overall fibrosis may be not large enough (e.g., for the support of injured epithelia in the UUO model). Alternatively, hematopoietic cells may deposit collagen at slightly different anatomic sites within the interstitium than resident fibroblasts or pericytes, which may also be relevant for the protection of tubules against pressure-induced damage. In the more chronic and pressure-independent model of adenine-induced nephropathy, deposition of collagen was detrimental, independent of the cellular source of collagen. Renal function was also improved when collagen I was specifically knocked out in hematopoietic cells.
Our findings have several implications for the therapy of renal fibrosis. We need to be aware that prevention of fibrosis may not be desirable in all types of renal diseases, because production of collagen could be protective at least under some conditions (e.g., ureteral obstruction or even tubular obstruction occurring in many types of ARF). Nevertheless, the major contribution of fibrocytes to collagen production opens up new possibilities to interfere with fibrogenesis. Targeting mesenchymal fibroblasts or their differentiation into myofibroblasts may not be sufficient to prevent fibrosis. In addition, inhibition of fibrocytes may be less risky than inhibition of fibroblasts, because knockout of collagen in fibrocytes was not harmful in the acute R-UUO model but still protective in the more chronic model of adenine-induced nephropathy. Overall, fibrocytes seem to prevail in more chronic types of fibrosis associated with prolonged inflammation.
Little progress has been made in the prevention and slow down of chronic renal failure in humans.50 Our work leads to a better understanding of the cellular origin and biologic significance of renal fibrosis and helps to identify appropriate diseases and conditions for the development of antifibrotic therapies.
Disclosures
None.
Supplementary Material
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft grants SFB/TRR57 (to P.B.), BO 3755/3-1 (to P.B.), and SFB699 (to H.C. and M.M.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018020138/-/DCSupplemental.
References
- 1.Menn-Josephy H, Lee CS, Nolin A, Christov M, Rybin DV, Weinberg JM, et al.: Renal interstitial fibrosis: An imperfect predictor of kidney disease progression in some patient cohorts. Am J Nephrol 44: 289–299, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nangaku M: Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J Am Soc Nephrol 17: 17–25, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al.: Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A 99: 12877–12882, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al.: Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al.: Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Göbel N, et al.: CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A 106: 17892–17897, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roufosse C, Bou-Gharios G, Prodromidi E, Alexakis C, Jeffery R, Khan S, et al.: Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol 17: 775–782, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Sakai N, Wada T, Yokoyama H, Lipp M, Ueha S, Matsushima K, et al.: Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci U S A 103: 14098–14103, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Deane JA, Campanale NV, Bertram JF, Ricardo SD: The contribution of bone marrow-derived cells to the development of renal interstitial fibrosis. Stem Cells 25: 697–706, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Li J, Qu X, Bertram JF: Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol 175: 1380–1388, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin SL, Kisseleva T, Brenner DA, Duffield JS: Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reich B, Schmidbauer K, Rodriguez Gomez M, Johannes Hermann F, Göbel N, Brühl H, et al.: Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int 84: 78–89, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Chen G, Lin SC, Chen J, He L, Dong F, Xu J, et al.: CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol 22: 1876–1886, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, et al.: Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 80: 1170–1181, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R: Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Arévalo M, et al.: Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 21: 989–997, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Kramann R, Fleig SV, Schneider RK, Fabian SL, DiRocco DP, Maarouf O, et al.: Pharmacological GLI2 inhibition prevents myofibroblast cell-cycle progression and reduces kidney fibrosis. J Clin Invest 125: 2935–2951, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al.: Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21: 998–1009, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, et al.: Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 121: 3981–3990, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al.: Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19: 1617–1624, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleaveland KR, Moore BB, Kim KK: Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev Respir Med 8: 163–172, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A: Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1: 71–81, 1994 [PMC free article] [PubMed] [Google Scholar]
- 23.Bucala R: Fibrocytes at 20 Years. Mol Med 21[Suppl 1]: S3–S5, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovisa S, Zeisberg M, Kalluri R: Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol Metab 27: 681–695, 2016 [DOI] [PubMed] [Google Scholar]
- 25.Pilling D, Vakil V, Cox N, Gomer RH: TNF-α-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc Natl Acad Sci U S A 112: 11929–11934, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pilling D, Zheng Z, Vakil V, Gomer RH: Fibroblasts secrete Slit2 to inhibit fibrocyte differentiation and fibrosis. Proc Natl Acad Sci U S A 111: 18291–18296, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reilkoff RA, Bucala R, Herzog EL: Fibrocytes: Emerging effector cells in chronic inflammation. Nat Rev Immunol 11: 427–435, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mack M, Yanagita M: Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int 87: 297–307, 2015 [DOI] [PubMed] [Google Scholar]
- 29.Farris AB, Adams CD, Brousaides N, Della Pelle PA, Collins AB, Moradi E, et al.: Morphometric and visual evaluation of fibrosis in renal biopsies. J Am Soc Nephrol 22: 176–186, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Servais A, Meas-Yedid V, Noël LH, Martinez F, Panterne C, Kreis H, et al.: Interstitial fibrosis evolution on early sequential screening renal allograft biopsies using quantitative image analysis. Am J Transplant 11: 1456–1463, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Kaissling B, Lehir M, Kriz W: Renal epithelial injury and fibrosis. Biochim Biophys Acta 1832: 931–939, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Sun DF, Fujigaki Y, Fujimoto T, Yonemura K, Hishida A: Possible involvement of myofibroblasts in cellular recovery of uranyl acetate-induced acute renal failure in rats. Am J Pathol 157: 1321–1335, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mack M: Inflammation and fibrosis [published online ahead of print November 28, 2017]. Matrix Biol doi: 10.1016/j.matbio.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 34.Chevalier RL, Forbes MS, Thornhill BA: Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Puri TS, Shakaib MI, Chang A, Mathew L, Olayinka O, Minto AW, et al.: Chronic kidney disease induced in mice by reversible unilateral ureteral obstruction is dependent on genetic background. Am J Physiol Renal Physiol 298: F1024–F1032, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ludwig-Portugall I, Bartok E, Dhana E, Evers BD, Primiano MJ, Hall JP, et al.: An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int 90: 525–539, 2016 [DOI] [PubMed] [Google Scholar]
- 37.Tamura M, Aizawa R, Hori M, Ozaki H: Progressive renal dysfunction and macrophage infiltration in interstitial fibrosis in an adenine-induced tubulointerstitial nephritis mouse model. Histochem Cell Biol 131: 483–490, 2009 [DOI] [PubMed] [Google Scholar]
- 38.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, et al.: Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33: 314–325, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Hills D, Taylor E, Pfeffer K, Ure J, Medvinsky A: Transgenic tools for analysis of the haematopoietic system: Knock-in CD45 reporter and deletor mice. J Immunol Methods 337: 81–87, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, et al.: Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 8: 826–837, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Traykova-Brauch M, Schönig K, Greiner O, Miloud T, Jauch A, Bode M, et al.: An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, et al.: Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Renal Physiol 292: F27–F37, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Khoshnoodi J, Cartailler JP, Alvares K, Veis A, Hudson BG: Molecular recognition in the assembly of collagens: Terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J Biol Chem 281: 38117–38121, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Schnieke A, Harbers K, Jaenisch R: Embryonic lethal mutation in mice induced by retrovirus insertion into the alpha 1(I) collagen gene. Nature 304: 315–320, 1983 [DOI] [PubMed] [Google Scholar]
- 45.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, et al.: A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 101: 6062–6067, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, et al.: Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol 193: 5229–5239, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broekema M, Harmsen MC, van Luyn MJ, Koerts JA, Petersen AH, van Kooten TG, et al.: Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol 18: 165–175, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV: Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 1p, 143, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fields GB: Interstitial collagen catabolism. J Biol Chem 288: 8785–8793, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al.: Chronic kidney disease: Global dimension and perspectives. Lancet 382: 260–272, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.