

Abstract
Background Metabolite levels reflect physiologic homeostasis and may serve as biomarkers of disease progression. Identifying metabolites associated with APOL1 risk alleles—genetic variants associated with CKD risk commonly present in persons of African descent—may reveal novel markers of CKD progression relevant to other populations.
Methods We evaluated associations between the number of APOL1 risk alleles and 760 serum metabolites identified via untargeted profiling in participants of the African American Study of Kidney Disease and Hypertension (AASK) (n=588; Bonferroni significance threshold P<6.5×10−5) and replicated findings in 678 black participants with CKD in BioMe, an electronic medical record–linked biobank. We tested the metabolite association with CKD progression in AASK, BioMe, and the Modification of Diet in Renal Disease (MDRD) Study.
Results One metabolite, 6-bromotryptophan, was significant in AASK (P=4.7×10−5) and replicated in BioMe (P=5.7×10−3) participants, with lower levels associated with more APOL1 risk alleles. Lower levels of 6-bromotryptophan were associated with CKD progression in AASK and BioMe participants and in white participants in the MDRD Study, independent of demographics and clinical characteristics, including baseline GFR (adjusted hazard ratio per two-fold higher 6-bromotryptophan level, AASK, 0.76; 95% confidence interval [95% CI], 0.64 to 0.91; BioMe, 0.61; 95% CI, 0.43 to 0.85; MDRD, 0.52; 95% CI, 0.34 to 0.79). The interaction between the APOL1 risk alleles and 6-bromotryptophan was not significant. The identity of 6-bromotryptophan was confirmed in experiments comparing its molecular signature with that of authentic standards of other bromotryptophan isomers.
Conclusions Serum 6-bromotryptophan is a consistent and novel risk factor for CKD progression.
Keywords: AASK (African American Study of Kidney Disease and Hypertension), chronic kidney disease, end stage kidney disease, renal function decline
CKD affects nearly 15% of United States adults, and treatment options are limited.1,2 The progression of CKD can lead to ESRD, which accounted for 7.2% of United States Medicare expenditures in 2014.3 Identifying novel risk factors of CKD progression may inform our understanding of the pathophysiology of disease progression and identify potential treatment targets.
Metabolite levels reflect physiologic homeostasis and may provide early signals of disease risk.4,5 A case-control study of 200 patients with CKD demonstrated that certain metabolites were higher among patients with rapid CKD progression, but the number of metabolites investigated was small and there was no independent replication.6 Few studies have evaluated metabolites in blacks, a population with two- to four-fold higher risk of CKD progression compared with European Americans.7,8 This increased risk is partly attributable to the APOL1 risk variants, which are present almost exclusively in blacks, with the high-risk genotype having a prevalence of approximately 13%.9–11 Metabolites associated with the APOL1 risk alleles may be markers for CKD progression.
We performed untargeted metabolite profiling among participants of the African American Study of Kidney Disease and Hypertension (AASK),12 a clinical trial that enrolled black patients with CKD attributed to hypertension. We evaluated the association between the number of APOL1 risk alleles and metabolite levels, with replication of statistically significant associations in a black subcohort of BioMe, an electronic medical record–linked biobank of patients who received healthcare in the Mount Sinai Health System.13 Identified metabolites were evaluated as a risk factor for CKD progression in both AASK and the BioMe subcohort. Subsequently, we tested this association in white participants in the Modification of Diet in Renal Disease (MDRD) Study.14 For the significant metabolite, 6-bromotryptophan, we further performed a series of spiking experiments to confirm the identity of the specific isomer that was detected in these studies.
Methods
Study Design and Populations
The AASK study was a randomized controlled trial followed by a cohort phase.12,15 The randomized trial (1995 through 2001) enrolled 1094 patients, aged 18–70 years, with self-reported black race, and with CKD attributed to hypertension. The randomized controlled trial used a 3-by-2 factorial design to assign participants to one of three initial medications (ramipril, metoprolol, or amlodipine) and one of two BP control targets: intensive control (goal of mean arterial pressure ≤92 mm Hg) and standard control (goal of mean arterial pressure 102–107 mm Hg). After the trial phase, surviving patients who had not yet received a diagnosis of ESRD were invited to enroll in the cohort phase (April of 2002 through 2007), which provided BP management according to a standardized protocol on the basis of the results of the trial. We performed metabolite profiling on 963 participants using samples collected at trial enrollment. Of these, 611 participants provided written informed consent for the collection of DNA and had APOL1 risk allele genotype data. However, 23 were missing the percentage of European ancestry estimates, resulting in a sample size of 588 for the analysis of metabolite associations with APOL1 risk alleles. For analyses of CKD progression, we excluded an additional 14 patients without measures of 6-bromotryptophan and two patients without a measure of urine protein-to-creatinine ratio (PCR), resulting in an analyzed sample size of 572 (Supplemental Figure 1).
BioMe is a health care cohort established in 2007 in which patients receiving healthcare at the Mount Sinai Health System were enrolled into an electronic medical record–linked biobank.13 We selected black patients with reduced eGFR (20–75 ml/min per 1.73 m2) and APOL1 genotype data. From a population of 1525 patients meeting these criteria, we selected all with two copies of the APOL1 risk alleles (n=206) and randomly selected the remaining 474, for a final sample size of 680 for whom we conducted metabolite profiling. Of these, we excluded two patients with discordant genotype in the two missense single nucleotide polymorphisms of the G1 risk allele (rs73885319 and rs60910145), resulting in an analyzed sample size of 678. For CKD progression, the analyzed sample size was 583 after excluding 31 patients without baseline eGFR, 56 patients missing follow-up measures of eGFR for CKD progression assessment, and eight patients missing baseline systolic BP (SBP) or diastolic BP (DBP) (Supplemental Figure 2).
The MDRD study was a multicenter clinical trial to assess the effects of dietary protein restriction and BP control in patients with progressive kidney disease.14 The MDRD study was used as a third, confirmatory cohort to assess the association of the replicated metabolite with CKD progression in patients with CKD of other ethnicities. This analysis was performed in a sample of 390 white patients with GFR≥20 ml/min per 1.73 m2 at the 12-month visit when 6-bromotryptophan was measured (Supplemental Figure 3).
Metabolite Profiling
Metabolite profiling was performed by Metabolon, Inc. (Morrisville, NC) using serum samples collected at the baseline visit of AASK, at enrollment of BioMe, and at the 12-month visit of MDRD using untargeted mass spectrometry (MS).16 Metabolite profiling was performed using the following four liquid chromatography/MS analyses: two separate reverse phase ultraperformance liquid chromatography (UPLC) tandem mass spectrometry (RP/UPLC-MS/MSn) methods using positive ion mode electrospray ionization (ESI), an additional RP/UPLC-MS/MSn method using negative ion mode ESI, and a hydrophilic interaction UPLC tandem mass spectrometry method with negative ion mode ESI. Metabolites were identified by matching detected features to an in-house spectral and chromatographic library of authentic reference standards containing the retention time/index, retention time index, mass-to-charge ratio (m/z), and chromatographic data (including MS/MS spectral data). The levels of each metabolite were quantified using area-under-the-curve of the MS peaks after interday normalization.
Genotyping of APOL1 Risk Variants
The APOL1 risk alleles consist of the G1 allele, which comprises two missense variants: rs73885319-G and rs60910145-G, which are in almost perfect positive linkage disequilibrium on one chromosome and the G2 allele (two amino acid deletion, rs71785313, which is in complete negative disequilibrium with the G alleles in G1). The genotyping was performed using ABI TaqMan assays in AASK and a clinical genetic test in BioMe.17 The number of risk alleles was defined as the sum of the number of G1 alleles (G allele at rs738885319) and the number of G2 deletion alleles. Because G1 and G2 are in negative disequilibrium, the numbers of risk alleles have values of 0, 1, and 2. The APOL1 high-risk status was defined as having two copies of the G1 and/or G2 risk alleles and low-risk as having 0 or 1 risk allele.
Definitions of CKD Progression
In all cohorts, baseline was considered the visit of the blood draw for the profiling of serum metabolites. In AASK, CKD progression was defined as a doubling of serum creatinine or the incidence of ESRD.18 Patients were followed until June of 2007.19 In BioMe and the MDRD Study, CKD progression was defined as a 57% decline in eGFR (BioMe) or measured GFR (MDRD), which is equivalent to the doubling of serum creatinine,20 or incidence of ESRD. Patients in BioMe were followed until September of 2016; patients in MDRD, until June of 1993. Patients without CKD progression were censored at the date of death or the last measure of kidney function.
Measurement of Covariates
In AASK and the MDRD Study, all baseline variables were ascertained using standardized protocols.14,21 GFR was measured using urinary clearance of 125I-iothalamate. Urine PCR was on the basis of 24-hour urine collection. In BioMe, baseline variables were ascertained from electronic medical records. eGFR was calculated on the basis of serum creatinine using the CKD Epidemiology Collaboration creatinine equation.22 Urinary albumin-to-creatinine ratio (ACR) was on the basis of spot urine collection. Baseline eGFR, ACR, SBP, DBP, diabetes, and hypertension diagnosis were defined as the value reported closest to the enrollment date and within 1 year before and up to 1 day after enrollment. The proportions of these values recorded within 90 days of enrollment were 89% for eGFR, 99% for SBP, and 99% for DBP.
Statistical Analyses
Metabolite Associations with APOL1 Risk Alleles
To reduce skewness on both sides of the distribution, we applied inverse normal transformation, a rank-based transformation, to the measures of the metabolites.23,24 Linear regression was then used to evaluate the association between the number of APOL1 risk alleles and metabolites. In keeping with previous studies of the metabolite profiling of genetic variants, we modeled the APOL1 risk allele using an additive genetic model,25–28 regressing metabolites (dependent variable) on the number of APOL1 risk alleles (independent variable), age, and sex, as well as percentage of European ancestry, study medication assignment, and BP goal in AASK. The significance thresholds were set using the Bonferroni method: 6.6×10−5 (=0.05/760) metabolites for the discovery (AASK) and 0.05 for the one metabolite tested in BioMe.
Correlates of 6-Bromotryptophan Levels
To elucidate the correlates of 6-bromotryptophan (the replicated metabolite), we calculated the Spearman correlation between 6-bromotryptophan and the other 759 metabolites in AASK. To detect potential clinical correlates of 6-bromotryptophan levels, we evaluated univariate associations between tertiles of 6-bromotryptophan levels and baseline variables using t tests, Kruskal–Wallis, and chi-squared tests as appropriate in AASK, BioMe, and MDRD.
Association between 6-Bromotryptophan Levels and CKD Progression
Cox regression was used to evaluate the association between serum 6-bromotryptophan levels and CKD progression. For ease of interpretation of the hazard ratio (HR) and deriving a common unit for 6-bromotryptophan levels among the three cohorts, we modeled 6-bromotryptophan levels using log base 2 transformation, winsorizing values outside of 3 SDs, such that the unit of the HR was per two-fold higher levels of 6-bromotryptophan. We investigated the assumption of a log-linear relationship between 6-bromotryptophan levels and CKD progression using cubic splines with knots at tertiles of 6-bromotryptophan levels. The spline term was NS (P>0.05) and thus we retained our original transformation. The proportional hazards assumption was confirmed using an interaction term between 6-bromotryptophan and follow-up time (P>0.05 for all models tested).
In AASK, covariates used to adjust the analysis of CKD progression included age, sex, and randomization arm (model 1). In BioMe, the covariates of model 1 included age, sex, and baseline diabetes and hypertension diagnosis (variables which were uniform in AASK, given the enrollment criteria). In model 2, we added baseline measured GFR, coronary heart disease (CHD), and DBP in AASK and baseline eGFR, CHD, and DBP in BioMe. In model 3, we added APOL1 high-risk status in both AASK and BioMe. We performed meta-analysis combining the results of models 1, 2, and 3 in AASK and BioMe using the fixed effect inverse variance-weighted method. In AASK, we constructed model 4 by adding natural log–transformed baseline PCR.
In MDRD, the model covariates were similar to those used in BioMe with the addition of study-specific variables. Model 1 adjusted for age, sex, diabetes, hypertension, cause of CKD, estimated protein intake, and study randomization (diet and BP goal assignment). Model 2 added baseline measured GFR, coronary artery disease, and DBP; and Model 3 added natural log–transformed baseline PCR.
Assessment of Potential Mediating and Modifying Relation between the APOL1 High-Risk Status and 6-Bromotryptophan for CKD Progression
To explore whether 6-bromotryptophan might mediate the association between the APOL1 high-risk status and CKD progression, we assessed the association between the APOL1 high-risk status and CKD progression with and without controlling for 6-bromotryptophan levels in AASK and BioMe. To evaluate whether the APOL1 high-risk status might modify the association between 6-bromotryptophan and CKD progression, we stratified by APOL1 high-risk status and tested for the interaction between the APOL1 high-risk status and 6-bromotryptophan in AASK and BioMe. The analysis of CKD progression was conducted using Stata 14.0 (StataCorp LLC, College Station, TX), and other analyses were conducted using R 3.2.5.
Validation of the Identity of 6-Bromotryptophan
Like all known metabolites quantified by Metabolon, 6-bromotryptophan was first identified on the basis of a three-criteria match to an authentic standard, namely accurate mass ±10 ppm, fragmentation spectrum, and retention time. After identifying bromotryptophan as the top metabolite, we further verified that the bromine was located at the position 6 in the indole ring of tryptophan through a series of spiking experiments in which a plasma sample with endogenous bromotryptophan was spiked with authentic reference standards of the various available bromotryptophan isomers. These isomers included 4-, 5-, 6-, and 7-bromotryptophan (Sigma Aldrich for 5-bromotryptophan and American Customs Chemical Corporation for the others). In these experiments, 100 µl of an experimental plasma sample containing an endogenous peak of bromotryptophan was extracted and analyzed in triplicate using the RP/Neg (RP/UPLC-MS/MSn method using negative ion mode ESI) method. This same plasma sample, postextraction, was spiked individually with 5 µg/ml neat solutions of each of the above bromotryptophan isomers and run in duplicate. The experimental signature of the endogenous peak in the unspiked plasma was compared with the signature of the peak when spiked with the various authentic standards with respect to retention time, m/z, and signal intensity.
Results
Baseline Characteristics of the AASK and BioMe Cohorts
The mean participant age was 54 years in AASK and 60 years in BioMe (Table 1). The proportion of men was 59.7% in AASK and 69.6% in BioMe. The mean measured GFR was 47 ml/min per 1.73 m2 in AASK, and the mean eGFR was 59 ml/min per 1.73 m2 in BioMe. All AASK participants had hypertension but no diabetes at enrollment; in BioMe, 75.5% of participants had hypertension, and 41% had diabetes. There were 139 and 206 participants with the APOL1 high-risk genotype in AASK and BioMe, respectively.
Table 1.
Baseline characteristics of participants in the AASK and BioMe studies
| Variable | AASK | BioMe |
|---|---|---|
| N | 588 | 678 |
| Mean age, year, (SD) | 54.2 (10.5) | 60.3 (13.1) |
| Men, n (%) | 351 (59.7) | 472 (69.6) |
| Mean body mass index, kg/m2, (SD)a | 31.1 (6.7) | 31.2 (8.2) |
| GFRa,b mL/min per 1.73m2 | 47.4 (13.4) | 58.8 (13.9) |
| Urine PCR, mg/g, median (first, third quartile)c | 72 (27, 308) | – |
| Urine ACR, mg/g, median (first, third quartile)a | – | 13 (4.0, 84) |
| CHD, n (%)a | 294 (50.0) | 47 (6.9) |
| Mean SBP (SD), mm Hga | 150.1 (24.6) | 134.7 (23.0) |
| Mean DBP (SD), mm Hga | 95.6 (15) | 76.8 (12.5) |
| Hypertension, n (%)a | 588 (100.0) | 512 (75.5) |
| Diabetes, n (%)a | 0 (0) | 278 (41.0) |
| % European ancestrya | 0.2 (0.1) | 0.2 (0.2) |
| APOL1 risk allele, n (%) | ||
| 0 copies | 194 (33.0) | 246 (36.3) |
| 1 copy | 255 (43.4) | 226 (33.3) |
| 2 copies | 139 (23.6) | 206 (30.4) |
| G1 (rs73885319) | ||
| AA | 302 (51.4) | 370 (54.6) |
| AG | 224 (38.1) | 221 (32.6) |
| GG | 59 (10.0) | 87 (12.8) |
| G2 (rs7178513) | ||
| Wild type | 419 (71.3) | 473 (69.8) |
| One deletion | 147 (25.0) | 167 (24.6) |
| Two deletion | 21 (3.6) | 38 (5.6) |
BioMe sample size: body mass index (484), eGFR (647), urinary albumin-to-creatine ratio (177), SBP (665), DBP (671), CHD (677), hypertension (677), and diabetes (677); % European ancestry (478).
AASK used measured GFR and BioMe used eGFR calculated from serum creatinine using the CKD Epidemiology Collaboration equation.
AASK sample size: proteinuria (586).
Quantification and Quality Control of Metabolites
A total of 1228 metabolites were identified (833 with known structural identity and 395 unknown) via untargeted profiling. We focused on the 760 known metabolites that were missing in <20% of samples. For overall quality control, we assessed 20 blind duplicate pairs, where the median Spearman correlation (ρ) was 0.94 (25th and 75th percentile: 0.87, 0.98). The metabolite 6-bromotryptophan had a Spearman ρ of 0.90 and a coefficient of variation of 0.08 in the 20 blind duplicates in AASK, and Spearman ρ of 0.97 and a coefficient of variation of 0.06 in the 20 blind duplicates in BioMe.
Associations between APOL1 Risk Alleles and Untargeted Metabolites
Relating the levels of 760 metabolites in AASK to the number of APOL1 risk alleles identified a single association that met Bonferroni-corrected statistical significance (P<6.5×10−5) after adjusting for age, sex, study assignment, and percentage of European ancestry (n=588). Greater numbers of APOL1 risk alleles were associated with lower levels of 6-bromotryptophan (β per copy of risk allele=−0.23 SD; 95% confidence interval [95% CI], −0.33 to −0.12; P=4.7×10−5). No additional metabolites were statistically significant (Figure 1, Supplemental Tables 3 and 4). The association between the number of APOL1 risk alleles and 6-bromotryptophan was replicated in BioMe adjusting for age and sex (n=678; β per copy of risk allele=−0.13 SD; 95% CI, −0.22 to −0.04; P=5.7×10−3).
Figure 1.

Lower 6-bromotryptophan was significantly associated with the higher number of APOL1 risk alleles. Y axis represents the -log10(P value) in the association between the number of APOL1 risk alleles and inverse normal transformed metabolite levels in AASK.
Cross-Sectional Correlates of 6-Bromotryptophan
When stratified by tertile of 6-bromotryptophan level, lower 6-bromotryptophan levels were associated with lower measured GFR and higher proteinuria levels in AASK (Table 2). In BioMe, lower 6-bromotryptophan levels were associated with lower eGFR, lower DBP, and higher proportion of diabetes.
Table 2.
Study population characteristics, by tertile of 6-bromotryptophan
| Variable | AASK | BioMe | ||||
|---|---|---|---|---|---|---|
| Tertile 1 | Tertile 2 | Tertile 3 | Tertile 1 | Tertile 2 | Tertile 3 | |
| Mean age, year, (SD) | 54.4 (10.2) | 54.3 (10.2) | 54.0 (11.2) | 60.24 (12.3) | 60.08 (13.0) | 60.67 (13.9) |
| Men, n (%) | 107 (54.6) | 116 (59.2) | 128 (65.3) | 148 (65.5) | 160 (70.8) | 164 (72.6) |
| Mean body mass index, kg/m2 (SD)a | 30.8 (7.6) | 31.2 (6.1) | 31.3 (6.5) | 30.4 (8.4) | 32.24 (8.5) | 31.88 (7.2) |
| GFRa,b mL/min per 1.73m2 | 43.0 (13.6) | 47.6 (13.8) | 51.5 (11.5) | 56.03 (16.2) | 59.57 (12.1) | 60.72 (12.6) |
| Urine PCR, mg/g, median (25th, 75th percentile)c | 121 (37, 512) | 65 (28, 311) | 54 (22, 196) | – | – | – |
| Urine ACR, mg/g, median (25th, 75th percentile)a | – | – | – | 20 (3, 147) | 12 (5, 41) | 8.5 (3, 31) |
| CHD, n (%)a | 103 (52.6) | 89 (45.4) | 102 (52) | 14 (6.2) | 14 (6.2) | 19 (8.4) |
| Mean SBP (SD), mm Hga | 148.6 (22.5) | 150.3 (24.1) | 151.4 (27.0) | 131.5 (20.5) | 137.6 (25.0) | 135.0 (22.9) |
| Mean DBP (SD), mm Hga | 94.7 (14.1) | 94.8 (15.3) | 97.3 (15.4) | 74.66 (11.2) | 77.55 (12.6) | 78.29 (13.3) |
| Hypertension, n (%)a | 196 (100) | 196 (100) | 196 (100) | 159 (70.4) | 176 (78.2) | 177 (78.3) |
| Diabetes, n (%)a | 0 (0) | 0 (0) | 0 (0) | 118 (52.2) | 80 (35.6) | 80 (35.4) |
| % European ancestrya | 0.16 (0.12) | 0.17 (0.14) | 0.17 (0.14) | 0.18 (0.15) | 0.18 (0.16) | 0.19 (0.18) |
| APOL1 risk allele, n (%) | ||||||
| 0 copies | 53 (27) | 65 (33.2) | 76 (38.8) | 73 (32.3) | 85 (37.6) | 88 (38.9) |
| 1 copy | 78 (39.8) | 90 (45.9) | 87 (44.4) | 73 (32.3) | 74 (32.7) | 79 (35.0) |
| 2 copies | 65 (33.2) | 41 (20.9) | 33 (16.8) | 80 (35.4) | 67 (29.6) | 59 (26.1) |
| G1 (rs73885319) | ||||||
| AA | 87 (44.4) | 100 (51.3) | 115 (59.3) | 116 (51.3) | 124 (54.9) | 130 (57.5) |
| AG | 82 (41.8) | 76 (39.0) | 66 (34.0) | 72 (31.9) | 83 (36.7) | 66 (29.2) |
| GG | 27 (13.8) | 19 (9.7) | 13 (6.7) | 38 (16.8) | 19 (8.4) | 30 (13.3) |
| G2 (rs7178513) | ||||||
| Wild type | 11 (5.6) | 4 (2.0) | 6 (3.1) | 158 (69.9) | 151 (66.8) | 164 (72.6) |
| One deletion | 50 (25.6) | 49 (25.0) | 48 (24.5) | 51 (22.6) | 63 (27.9) | 53 (23.5) |
| Two deletion | 134 (68.7) | 143 (73.0) | 142 (72.4) | 17 (7.5) | 12 (5.3) | 9 (4) |
BioMe sample size: body mass index (545), eGFR (658), urinary albumin-to-creatine ratio (177), SBP (673), DBP (677), CHD (677), hypertension (677), diabetes (677); % European ancestry (478).
AASK used measured GFR, BioMe used eGFR.
AASK sample size for proteinuria: 586.
Among the other 759 metabolites identified in the serum samples, 6-bromotryptophan was most strongly correlated with tryptophan (Spearman ρ: 0.54) (Supplemental Table 5).
Associations of 6-Bromotryptophan with CKD Progression in AASK and BioMe
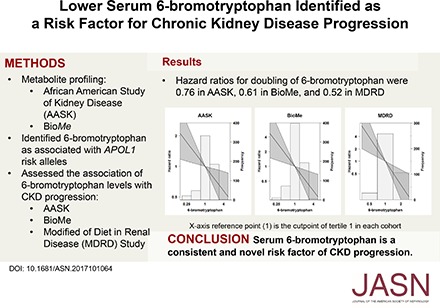
In AASK, over a median follow-up time of 9.0 years, there were 236 CKD progression events among 572 participants with available data. Lower 6-bromotryptophan levels were significantly associated with CKD progression adjusting for baseline age, sex, and randomization arm (model 1, HR per two-fold higher 6-bromotryptophan levels, 0.64; 95% CI, 0.55 to 0.75; Table 3). With the additional adjustment of baseline GFR, DBP, and CHD status in model 2 and APOL1 high-risk status in model 3, this association remained significant with some attenuation (model 2 HR, 0.73; 95% CI, 0.61 to 0.86; model 3 HR, 0.76; 95% CI, 0.64 to 0.91; Figure 2).
Table 3.
Risk of CKD progression per two-fold higher serum 6-bromotryptophan levels in AASK (n=586) and BioMe (n=583)
| Variable | HR (95% CI) | P Value |
|---|---|---|
| AASK | ||
| Model 1 | 0.64 (0.55 to 0.75) | <0.001 |
| Model 2 | 0.73 (0.61 to 0.86) | <0.001 |
| Model 3 | 0.76 (0.64 to 0.91) | 0.002 |
| Model 4 | 0.79 (0.66 to 0.95) | 0.01 |
| BioMe | ||
| Model 1 | 0.57 (0.42 to 0.79) | 0.001 |
| Model 2 | 0.60 (0.43 to 0.85) | 0.003 |
| Model 3 | 0.61 (0.43 to 0.85) | 0.004 |
| Combined analysis | ||
| Model 1 | 0.63 (0.55 to 0.72) | <0.001 |
| Model 2 | 0.70 (0.60 to 0.82) | <0.001 |
| Model 3 | 0.73 (0.63 to 0.85) | <0.001 |
Covariates: Model 1. AASK: age, sex, study drug, and BP assignment; BioMe: age, sex, baseline diabetes, and hypertension. Model 2. Model 1+baseline GFR in AASK and eGFR in BioMe, CHD, DBP. Model 3: Model 2 + APOL1 high-risk status. Model 4. AASK: Model 3+log(PCR).
Figure 2.

Lower 6-bromotryptophan was significantly associated with higher risk of CKD progression. Hazard ratio was estimated per two-fold change in 6-bromotryptophan levels. The reference point (1) is the cut point for tertile 1 of 6-bromotryptophan levels within each cohort. The HRs were estimated from model 3. Covariates in AASK: age, sex, study drug and BP assignment, baseline GFR, CHD, DBP, and APOL1 high-risk status. Covariates in BioMe: age, sex, baseline diabetes and hypertension status, eGFR, CHD, DBP, and APOL1 high-risk status.
In BioMe, over a median follow-up time of 4.6 years, there were 100 CKD progression events among 583 participants with available data. Similar to the results in AASK, lower 6-bromotryptophan levels were significantly associated with CKD progression adjusting for baseline age, sex, and baseline diabetes and hypertension status (model 1 HR, 0.57; 95% CI, 0.42 to 0.79). With the additional adjustment of baseline eGFR, DBP, and CHD status in model 2 and APOL1 high-risk status in model 3, this association remained significant (model 2 HR, 0.60; 95% CI, 0.43 to 0.85; model 3 HR, 0.61; 95% CI, 0.43 to 0.85). In an analysis combining the results of model 3 from the AASK and BioMe, two-fold higher 6-bromotryptophan levels were associated with 27% reduction in the risk of CKD progression (HR, 0.73; 95% CI, 0.62 to 0.85). In AASK, adding proteinuria as a covariate did not substantially alter the association between lower levels of 6-bromotryptophan and CKD progression (model 4 HR, 0.79; 95% CI, 0.66 to 0.95).
The association between APOL1 high-risk status and CKD progression was slightly attenuated after controlling for 6-bromotryptophan levels in AASK and BioMe (model 2, in AASK, not controlling for 6-bromotryptophan: HR, 1.80; 95% CI, 1.35 to 2.38; controlling for bromotryptophan: HR, 1.68; 95% CI, 1.26 to 2.24; in BioMe, not controlling for 6-bromotryptophan: HR, 1.14; 95% CI, 0.74 to 1.76; controlling for 6-bromotryptophan: HR, 1.05; 95% CI, 0.68 to 1.62; Supplemental Table 6). There was no significant interaction between 6-bromotryptophan and APOL1 high-risk status in either AASK or BioMe (Supplemental Table 7).
Association between 6-Bromotryptophan and CKD Progression in the MDRD Study
The mean participant age was 52 years in MDRD, 64.6% were men, mean measured GFR was 35 ml/min per 1.73 m2, 83% had hypertension, and 4% had diabetes (Supplemental Table 8). Correlates of lower 6-bromotryptophan were lower GFR and higher proteinuria. Lower levels of 6-bromotryptophan were associated with CKD progression after adjusting for age, sex, study randomization arms, cause of CKD, estimated protein intake, history of diabetes and hypertension, DBP, coronary artery disease, GFR, and proteinuria (model 3 HR, 0.52; 95% CI, 0.34 to 0.79) (Supplemental Table 9).
Validation of the Identity of 6-Bromotryptophan
The experimental isotope signature matched the theoretic isotope pattern of bromotryptophan with respect to m/z and relative abundance (Supplemental Figure 4) with high confidence, particularly given the very characteristic isotope signature associated with a bromine atom. The experimentally detected fragmentation spectrum matched that of the 6-bromotryptophan authentic reference standard very well (Supplemental Figure 5). We confirmed that the 6-isoform coeluted with the endogenous peak (Supplemental Figure 6) and had a distinct retention time from the other isomers (Supplemental Figure 7). Finally, bromotryptophan was detected with the most sensitivity and precision using the RP/Neg method; however, it was also detected, albeit with less sensitivity, using the two separate RP/UPLC-MS/MSn methods using positive ion mode ESI. The experimental correlations were high between these different liquid chromatography/MS methods (Supplemental Figure 8).
Discussion
In this study using untargeted metabolite profiling and a discovery and replication approach in two deeply phenotyped cohorts of blacks with CKD, we identified a metabolite, 6-bromotryptophan, of which lower levels were associated with greater number of APOL1 risk alleles as well as CKD progression. However, we observed no mediating nor modifying effect of 6-bromotryptophan on the association between APOL1 high-risk status and CKD progression. In addition, the association between 6-bromotryptophan and CKD progression was evident in the white participants of the MDRD Study. Thus, although our initial intent was to identify metabolites that may inform the effects of the APOL1 risk allele and CKD progression, our results may suggest that serum 6-bromotryptophan is a general marker of CKD risk rather than a pathway-specific mediator of APOL1-related disease.
Bromotryptophan is the bromination product of tryptophan, but little has been reported on its presence or role in humans. There are many haloperoxidases that catalyze chlorination and bromination reactions of amino acids in biologic systems, and animal models have suggested that hypohalous acids might be overproduced in disease states such as diabetes, resulting in destabilization of extracellular matrix proteins in kidney tissue.29,30 Several bromotryptophan isoforms are known to be inhibitors of hemoglobin S polymerization, and the most potent, 5-bromotryptophan, has been studied as an antigelation agent for sickle cell anemia in mice, with effectiveness in reducing the aggregation of deoxyhemoglobin S but an additional effect of decreased locomotor activity at high intraperitoneal doses.31,32 Another recent study using the untargeted metabolomics approach of Metabolon linked lower bromotryptophan levels to higher gut permeability in children.33
Serum 6-bromotryptophan may be a marker of levels of circulating bromide (Br−), which is the ionic form of bromine. Bromine is a member of the halogen family (along with fluorine, chlorine, and iodine) and is thought to be an essential element, although its function in humans has not been established.34 The typical daily dietary intake range of Br− is estimated to be 2–8 mg, and it is excreted in the urine.35 The normal range of Br− level in whole blood is estimated to be 2.5–11.7 mg/L,36 and higher levels are considered to be toxic and due to nondietary sources, such as from medications and exposure to fumigant.37–39 Lower levels of Br− have been noted in patients receiving dialysis and hypothesized to play a role in dialysis-associated sleep disturbances.40,41 In Drosophila, Br− is an essential cofactor in the catalytic activity of peroxidasin for the formation of sulfilimine, a crosslink protein integral to the assembly of collagen IV scaffolds.42 Br− deficiency in Drosophila results in altered basement membrane morphologies and lethality. Type IV collagen is one of main components of the glomerular and tubular basement membrane,43 and defects in type IV collagen assembly can induce fibrosis, a common pathway in CKD progression.44 If 6-bromotryptophan is indeed a marker for Br− levels, and the role of Br− in the formation of sulfilimine crosslink in collagen IV assembly exists in the human glomerular and tubular basement membrane, this might suggest a need for studying adequate circulating Br− levels in the general population and in patients with CKD, as has previously been suggested.40
Strengths of this study include the unbiased approach to metabolite identification and the use of two deeply phenotyped but geographically and temporally distinct cohorts of blacks with CKD for discovery and replication as well as a third white cohort. The consistent associations in these three cohorts support the robustness of the results. Metabolite measures had high reliability as shown by the correlation between blind duplicates. In addition, the identity of 6-bromotryptophan was further verified using spiking experiments with authentic reference standards. However, our study also has limitations. Untargeted metabolite profiling does not provide absolute quantification of metabolite levels, limiting comparisons across populations. Another limitation is that in BioMe, albuminuria was not included as a covariate in the analysis of CKD progression due to the lack of albuminuria measures in a large proportion of participants. However, in AASK and MDRD, the association between 6-bromotryptophan and CKD progression remained similar after controlling for proteinuria.
In summary, lower levels of serum 6-bromotryptophan were associated with CKD progression independent of the APOL1 risk alleles and baseline kidney measures. These findings may point to actionable targets for preventing CKD progression.
Disclosures
Assay costs were discounted as part of a collaboration agreement between Metabolon and J.C., L.A.I., and A.S.L. to develop metabolomics estimates of GFR and for which they have a provisional patent filed on August 15, 2014, entitled “Precise estimation of GFR from multiple biomarkers” (no. PCT/US2015/044567). The technology is not licensed in whole or in part to any company. A.M.E. is a full-time employee of Metabolon, Inc.
Supplementary Material
Acknowledgments
This work is supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R01 DK108803. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (NIH), under contract HHSN26120080001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. C.M.R. is supported by a mentored research scientist development award from the NIDDK (K01 DK107782).
An abstract reporting the results of this study was presented at the American Society of Nephrology Kidney Week, November 4, 2017, in New Orleans, LA.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Study design: A.T., J.C., and M.E.G. Data collection: G.N., E.B., M.E.G., and L.J.A. Metabolite profiling and validation study: A.M.E. Data analysis: A.T. and A.M.E. Draft manuscript: A.T., A.M.E., and M.E.G. Critical review of the manuscript: all authors. All authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017101064/-/DCSupplemental.
References
- 1.United States Renal Data System 2017 USRDS annual data report: Epidemiology of kidney disease in the United States, volume 1: Chronic kidney disease, Chapter 1: CKD in the general population. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2017 [Google Scholar]
- 2.Murphy D, McCulloch CE, Lin F, Banerjee T, Bragg-Gresham JL, Eberhardt MS, et al.; Centers for Disease Control and Prevention Chronic Kidney Disease Surveillance Team : Trends in prevalence of chronic kidney disease in the United States. Ann Intern Med 165: 473–481, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.United States Renal Data System 2017 USRDS annual data report: Epidemiology of kidney disease in the United States, volume 2: End-stage renal disease, Chapter 11: Medicare expenditures for persons with ESRD. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2017 [Google Scholar]
- 4.Kalim S, Rhee EP: An overview of renal metabolomics. Kidney Int 91: 61–69, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hocher B, Adamski J: Metabolomics for clinical use and research in chronic kidney disease. Nat Rev Nephrol 13: 269–284, 2017 [DOI] [PubMed] [Google Scholar]
- 6.Rhee EP, Clish CB, Wenger J, Roy J, Elmariah S, Pierce KA, et al.: Metabolomics of chronic kidney disease progression: A case-control analysis in the chronic renal insufficiency cohort study. Am J Nephrol 43: 366–374, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbour SJ, Schachter M, Er L, Djurdjev O, Levin A: A systematic review of ethnic differences in the rate of renal progression in CKD patients. Nephrol Dial Transplant 25: 2422–2430, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Derose SF, Rutkowski MP, Levin NW, Liu IL, Shi JM, Jacobsen SJ, et al.: Incidence of end-stage renal disease and death among insured African Americans with chronic kidney disease. Kidney Int 76: 629–637, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR: Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol 22: 2098–2105, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Seaghdha CM, Parekh RS, Hwang SJ, Li M, Köttgen A, Coresh J, et al.: The MYH9/APOL1 region and chronic kidney disease in European-Americans. Hum Mol Genet 20: 2450–2456, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, et al.: APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 24: 1484–1491, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Appel LJ, Middleton J, Miller ER 3rd, Lipkowitz M, Norris K, Agodoa LY, et al.: The rationale and design of the AASK cohort study. J Am Soc Nephrol 14[Suppl 2]: S166–S172, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Tayo BO, Teil M, Tong L, Qin H, Khitrov G, Zhang W, et al.: Genetic background of patients from a university medical center in Manhattan: Implications for personalized medicine. PLoS One 6: e19166, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modification of Diet in Renal Disease Study Group: Design, methods, and results from the feasibility study. Am J Kidney Dis 20: 18–33, 1992 [DOI] [PubMed] [Google Scholar]
- 15.Gassman JJ, Greene T, Wright JT Jr, Agodoa L, Bakris G, Beck GJ, et al.: Design and statistical aspects of the African American Study of Kidney Disease and Hypertension (AASK). J Am Soc Nephrol 14[Suppl 2]: S154–S165, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E: Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem 81 (16): 6656–6667, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Fedick A, Wasserman S, Zhao G, Edelmann L, Bottinger EP, et al.: Analytical validation of a personalized medicine APOL1 genotyping assay for nondiabetic chronic kidney disease risk assessment. J Mol Diagn 18: 260–266, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parsa A, Kao WH, Xie D, Astor BC, Li M, Hsu CY, et al.; AASK Study Investigators; CRIC Study Investigators : APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 369: 2183–2196, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Appel LJ, Wright JT Jr, Greene T, Agodoa LY, Astor BC, Bakris GL, et al.; AASK Collaborative Research Group : Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med 363: 918–929, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levey AS, Inker LA, Matsushita K, Greene T, Willis K, Lewis E, et al.: GFR decline as an end point for clinical trials in CKD: A scientific workshop sponsored by the National Kidney Foundation and the US Food and Drug Administration. Am J Kidney Dis 64: 821–835, 2014 [DOI] [PubMed] [Google Scholar]
- 21.Wright JT Jr, Bakris G, Greene T, Agodoa LY, Appel LJ, Charleston J, et al.; African American Study of Kidney Disease and Hypertension Study Group : Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: Results from the AASK trial. JAMA 288: 2421–2431, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al.; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) : A new equation to estimate glomerular filtration rate. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beasley TM, Erickson S, Allison DB: Rank-based inverse normal transformations are increasingly used, but are they merited? Behav Genet 39: 580–595, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aulchenko Y: GenABEL package, Comprehensive R Archive Network, 2015. Available at: https://cran.r-project.org/. Accessed: October 3, 2016 [Google Scholar]
- 25.Rhee EP, Ho JE, Chen MH, Shen D, Cheng S, Larson MG, et al.: A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab 18: 130–143, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin SY, Fauman EB, Petersen AK, Krumsiek J, Santos R, Huang J, et al.; Multiple Tissue Human Expression Resource (MuTHER) Consortium : An atlas of genetic influences on human blood metabolites. Nat Genet 46: 543–550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rueedi R, Ledda M, Nicholls AW, Salek RM, Marques-Vidal P, Morya E, et al.: Genome-wide association study of metabolic traits reveals novel gene-metabolite-disease links. PLoS Genet 10: e1004132, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kettunen J, Demirkan A, Würtz P, Draisma HH, Haller T, Rawal R, et al.: Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat Commun 7: 11122, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown KL, Darris C, Rose KL, Sanchez OA, Madu H, Avance J, et al.: Hypohalous acids contribute to renal extracellular matrix damage in experimental diabetes. Diabetes 64: 2242–2253, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pattison DI, Davies MJ: Kinetic analysis of the reactions of hypobromous acid with protein components: Implications for cellular damage and use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry 43: 4799–4809, 2004 [DOI] [PubMed] [Google Scholar]
- 31.De Croos PZ, Sangdee P, Stockwell BL, Kar L, Thompson EB, Johnson ME, et al.: Hemoglobin S antigelation agents based on 5-bromotryptophan with potential for sickle cell anemia. J Med Chem 33: 3138–3142, 1990 [DOI] [PubMed] [Google Scholar]
- 32.Poillon WN: Noncovalent inhibitors of sickle hemoglobin gelation: Effects of aryl-substituted alanines. Biochemistry 21: 1400–1406, 1982 [DOI] [PubMed] [Google Scholar]
- 33.Semba RD, Trehan I, Li X, Moaddel R, Ordiz MI, Maleta KM, et al.: Environmental enteric dysfunction is associated with carnitine deficiency and altered fatty acid oxidation. EBioMedicine 17: 57–66, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuenca RE, Pories WJ, Bray J: Bromine levels in human serum, urine, hair. Short communication. Biol Trace Elem Res 16: 151–154, 1988 [DOI] [PubMed] [Google Scholar]
- 35.Nielsen FH: How should dietary guidance be given for mineral elements with beneficial actions or suspected of being essential? J Nutr 126[Suppl]: 2377S–2385S, 1996 [DOI] [PubMed] [Google Scholar]
- 36.Olszowy HA, Rossiter J, Hegarty J, Geoghegan P: Background levels of bromide in human blood. J Anal Toxicol 22: 225–230, 1998 [DOI] [PubMed] [Google Scholar]
- 37.Müller M, Reinhold P, Lange M, Zeise M, Jürgens U, Hallier E: Photometric determination of human serum bromide levels--a convenient biomonitoring parameter for methyl bromide exposure. Toxicol Lett 107: 155–159, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Hsieh PF, Tsan YT, Hung DZ, Hsu CL, Lee YC, Chang MH: Bromism caused by mix-formulated analgesic injectables. Hum Exp Toxicol 26: 971–973, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Hung YM: Bromide intoxication by the combination of bromide-containing over-the-counter drug and dextromethorphan hydrobromide. Hum Exp Toxicol 22: 459–461, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Canavese C, De Costanzi E, Stratta P, Sabbioni E: A role for bromine deficiency in sleep disturbances of long-term dialysis patients. Am J Kidney Dis 48: 1018–1019, author reply 1019, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Wallaeys B, Cornelis R, Mees L, Lameire N: Trace elements in serum, packed cells, and dialysate of CAPD patients. Kidney Int 30: 599–604, 1986 [DOI] [PubMed] [Google Scholar]
- 42.McCall AS, Cummings CF, Bhave G, Vanacore R, Page-McCaw A, Hudson BG: Bromine is an essential trace element for assembly of collagen IV scaffolds in tissue development and architecture. Cell 157: 1380–1392, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeisberg M, Bonner G, Maeshima Y, Colorado P, Müller GA, Strutz F, et al.: Renal fibrosis: Collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am J Pathol 159: 1313–1321, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y: Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7: 684–696, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.