

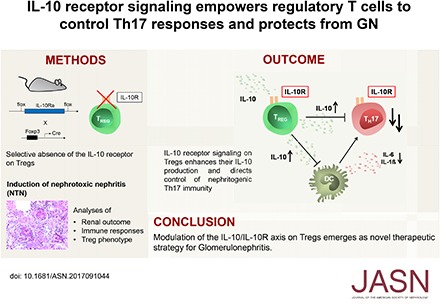
Abstract
Background Th17 cells are central pathogenic mediators of autoimmune disease, including many forms of GN. IL-10 receptor signaling (IL-10R) in regulatory T cells (Tregs) has been implicated in the downregulation of Th17 cells, but the underlying molecular mechanisms and functional relevance of this process remain unclear.
Methods We generated mice with Treg-specific IL-10Ra deficiency and subjected these mice to nephrotoxic serum–induced nephritis as a model of crescentic GN. Immune responses and Treg phenotypes were extensively analyzed.
Results Compared with controls, mice with IL-10Ra−/− Tregs showed a spontaneously overshooting Th17 immune response. This hyper-Th17 phenotype was further boosted during GN and associated with aggravated renal injury. Notably, abrogation of IL-10Ra signaling in Tregs increased dendritic cell activation and production of Th17-inducing cytokines. In contrast, Treg trafficking and expression of chemokine receptor CCR6 remained unaffected, indicating mechanisms of Th17 control, differing from those of previously identified CCR6+ Treg17 cells. Indeed, the capacity for direct in vitro suppression of Th17 responses by IL-10Ra−/− Tregs was significantly impaired. As underlying pathology, analyses conducted in vitro and in vivo using double-fluorescent reporter mice revealed strikingly decreased IL-10 production by IL-10Ra−/− Tregs. To assess, whether reduced IL-10 could explain the hyper Th17 phenotype, competitive cotransfer experiments were performed. Supporting our concept, IL-10Ra−/− T cells differentiated into Th17 cells at much higher frequencies than wild type T cells did during GN.
Conclusions IL-10R engagement optimizes Treg-mediated suppression of Th17 immunity. We hypothesize a feed-forward loop, in which IL-10Ra signaling reinforces IL-10 secretion by Tregs which potently controls Th17 development via direct and indirect mechanisms. IL-10R thus may be a promising therapeutic target for the treatment of GN.
Keywords: glomerulonephritis, cytokines, Interleukin 10, IL-10R, Treg, IL-17
The central and unique role of regulatory T cells (Tregs) for counter-regulation of autoimmunity and overshooting inflammation has been firmly established by multiple studies from the past two decades.1–4 As a consequence, the first pioneering studies have evaluated the therapeutic potential of ex vivo manipulated Tregs for human inflammatory diseases and organ transplantation.5–8 Success of this promising and trend setting therapeutic approach is presently still hampered by inefficient in vitro Treg activation and expansion as well as instability of the transferred Tregs. Research into Treg biology to better understand mechanisms and signaling cascades that enhance their activation, fitness, and stability is thus much warranted. Interestingly, it has recently become clear that Tregs are not a homogeneous population. Rather, there seem to be specialized subpopulations, which are tailor made for control of a distinct type of immune response.9–11 Koch et al.12 have identified Tregs expressing the unusual combination of the Treg master transcription factor Foxp3 together with the transcription factor T-bet. This is remarkable, because T-bet was identified before as the main transcription factor responsible for the programming of proinflammatory Th1 responses. These Th1-like, T-bet, and Foxp3 double-positive Tregs (Treg1) were shown to be optimized for downregulation of Th1 responses. Our group extended these findings and showed a crucial role for T-bet+ Treg1 cells in protection from acute crescentic GN.13 Mechanistically, we could show that Treg1 cells can colocalize to areas of Th1 inflammation by expressing the Th1 characteristic trafficking receptor CXCR3.13,14 In analogy to Treg1s, a subtype of Tregs was discovered, which is specialized for the control of Th17 immunity.15 Interestingly, these Treg17 cells also share a master transcription factor with their proinflammatory Th17 counterpart, namely Stat3.16 Activation of this transcription factor induces expression of the chemokine receptor CCR6. This, in turn, facilitates trafficking of Treg17 cells into areas of Th17 inflammation, since Th17 cells also traffic in a CCR6-dependent manner.17–19 Our group showed protective roles for Treg17 cells in both acute GN and chronic lupus nephritis.20,21 Furthermore, we showed that the concept of Th17 lineage–specific Treg17 cells is conserved in humans, which underlines their functional importance.20 Which stimuli initiate generation of CCR6+ Treg17 cells, however, remains so far elusive. In this respect, our own research has revealed that activation of RORγt, the second Th17 key transcription factor, in Tregs does not result in development of a Treg17 phenotype. Rather, these RORγt+ Tregs could be characterized as an independent Treg lineage with unique pro- and anti-inflammatory properties,22,23 including control of Th2 immunity.24,25 Another suspect for induction of a Treg17 phenotype is the multifunctional cytokine IL-10, which has been shown to be both a potent inducer of Stat326–28 and a mediator of type17 immunity.29–31 Concerning this matter, a recent study has provided compelling evidence that signaling via the IL-10 receptor (IL-10R) endows Tregs with the capacity to downregulate Th17 responses.32 Treg-specific ablation of the IL-10Ra resulted in selective hyper-Th17 immunity and spontaneous development of severe colitis. How exactly IL-10 signaling equips Tregs for control of Th17 immunity is, however, largely unclear. Potential mechanisms could include IL-10–mediated enhancement of IL-10 secretion by Tregs in the manner of an autocrine feed-forward loop. Furthermore, IL-10 could stabilize Tregs and result in higher levels of activation and general fitness.33,34 Finally, whether IL-10R signaling is responsible for expression of the CCR6 on Tregs to optimize their colocalization with Th17 cells20,21 has not been studied to date. In sum, modulation of Tregs via the IL-10/IL-10R axis might be a promising novel therapeutic strategy. However, the functional relevance of the IL-10R on Tregs for immunologically mediated diseases remains largely unclear. We thus decided to study this aspect in the Th17-dependent nephrotoxic nephritis model of acute GN.35,36
Methods
Animals
LoxP site flanked IL-10Rafl/fl mice were generated in the lab of R.A.F. (Yale University School of Medicine, New Haven, CT; details are available on request). Treg-specific deletion of the IL-10R α-chain was achieved by crossbreeding with mice expressing a yellow fluorescent protein (YFP)-Cre recombinase fusion protein under the control of the Foxp3 locus,37 which was provided by Alexander Y. Rudensky (Memorial Sloan-Kettering Cancer Center, New York, NY). Specificity of IL-10Ra knockout was tested by PCR analysis of genomic DNA prepared from FACS-sorted Tregs and T effector cells from wild-type and knockout mice (described below; primer sequences are available on request). CD4+ T cell–specific deletion of the IL-10R was induced by intercrossing IL-10Rafl/fl mice with mice expressing Cre-recombinase under the control of a transgenic CD4 enhancer-promotor-silencer sequence (B6.Cg-Tg[Cd4-cre]1Cwi/BfluJ) commercially available from The Jackson Laboratory. To analyze IL-10 production by Tregs, these mice were crossed with Foxp3 red fluorescent protein (FIR)38 and IL-10 enhanced green fluorescent protein (Tiger)39 reporter mice. Rag1−/− and CD45.1 mice were initially derived from The Jackson Laboratory. All mice are on a C57BL/6 background and were bred in our facility under specific pathogen-free conditions.
Animal Experiments and Functional Studies
Groups of naїve mice were analyzed from 9 weeks to 7 months of age as indicated. Nephrotoxic nephritis was induced in 8- to 10-week-old male Foxp3CrexIL-10Rafl/fl and Foxp3CrexIL-10Rawt/wt (referred to as Foxp3Cre) littermate controls or CD4CrexIL-10Rafl/flxFIRxTiger and CD4CrexIL-10Rawt/wtxFIRxTiger (referred to as CD4CrexFIRxTiger) littermate controls by intraperitoneal injection of nephrotoxic sheep serum,40 and organs were harvested at the indicated time points. For competitive transfer assay, spleen cells were isolated from either CD4CrexIL-10Rafl/fl (CD45.2+) or wild-type C57BL/6 (CD45.1+) donor mice. Viable cells were counted using a Biorad TC21 Counter after trypan blue staining, and 1×107 cells were intravenously injected into Rag1−/− mice in a 1:1 ratio 1 day before induction of NTN.13 Renal, splenic, and blood leukocytes were isolated 8 days after NTN induction as described below and analyzed by FACS. Urine samples were collected after housing the mice in metabolic cages. Albuminuria was determined by standard ELISA (Bethyl Laboratories). BUN and urinary creatinine were quantified using standard laboratory methods. Animal experiments were performed according to national and institutional animal care and ethical guidelines, and they were approved by local committees (approval codes 73/14, 07/15).
Morphologic Studies
Crescent formation and glomerular necrosis were determined in a minimum of 50 glomeruli per mouse in 2-μm-thick periodic acid–Schiff-stained kidney sections in a blinded manner. Semiquantitative analysis of tubulointerstitial damage was performed using ten randomly selected cortical areas (×200) as described previously.36 Paraffin-embedded sections were stained with antibodies directed against CD3 (A0452; Dako, Hamburg, Germany), F4/80 (BM8; BMA Biomedicals, Hiddenhausen, Germany), MAC2 (M3/38; Cedarlane Laboratories, Burlington, ON, Canada), Foxp3 (FJK-16s; eBiosciences, San Diego, CA), or GR-1 (NIMP-R14; Hycult Biotech, Uden, The Netherlands) and developed with a polymer-based secondary antibody-alkaline phosphatase kit (POLAP; Zytomed, Berlin, Germany). Fifty glomerular cross-sections and 30 tubulointerstitial high-power fields (magnification, ×400) per kidney section were counted in a blinded fashion.20,40 Colon tissue from naïve mice at the indicated ages was stained with hematoxylin and eosin. Each section was assigned scores for the presence and extent of tissue damage and inflammation by a semiquantitative criterion–based method.41 Scores ranged from zero to three points for tissue destruction plus from zero to three points for inflammation as follows: zero, within normal limits or absent; one, mild; two, moderate; three, severe.
Isolation of Leukocytes from Various Tissues
Spleens were harvested in HBSS and passed through 70-μm nylon meshes. After lysis of erythrocytes with ammonium-chloride, cells were washed and passed over 40-μm meshes. Cells were then washed again and counted for either culture or FACS analysis. Kidneys were minced and incubated in digestion medium (RPMI 1640 medium containing 10% FCS, 1% HEPES, 1% Penicillin/Streptomycin, 8 μg/ml Collagenase D, and 0.4 μg/ml DNase) at 37°C for 45 minutes. Tissues were then dissociated using the gentleMACS dissociator (Miltenyi Biotec) to get a single-cell suspension, and they were centrifuged at 300×g at 4°C for 8 minutes. To further purify the cells, Percoll gradient (37% Percoll; GE Healthcare, Chalfont St. Giles, Great Britain) centrifugation was performed at 500×g at room temperature for 20 minutes. Cells were washed and resuspended for staining and FACS analysis. Peripheral blood was drawn into EDTA-coated tubes, and red blood cell lysis was performed. Then, blood cells were washed and prepared for staining.20
Systemic Cellular and Humoral Immune Responses
Splenocytes (4×106 cells per 1 ml) were cultured under standard conditions in the presence of 1 mg/ml anti-CD3 (eBiosciences) or normal sheep IgG (10 μg/ml; Sigma, Taufkirchen, Germany) as indicated, and supernatants were harvested after 72 hours. Commercially available ELISAs were used for detection of IFNγ (Biolegend, San Diego, CA), IL-2 (Biolegend), IL-4 (Biolegend), IL-5 (Biolegend), IL-6 (Biolegend), IL-10 (Biolegend), TGF-β1 (R&D Systems, Minneapolis, MN), and IL-13 (eBiosciences).40 Sheep globulin-specific serum IgG titers were analyzed by ELISA (for total IgG; Biozol, Eching, Germany; for IgG1, IgG2b, IgG2c, and IgG3; Invitrogen, Frederick, MD).13,42
Flow Cytometry
Cells were surface stained for 30 minutes at 4°C with fluorochrome-labeled antibodies against CD45, CD45.1 CD45.2, CD4, CD44, CD11c, CCR7 (all BD Biosciences, Franklin Lakes, NJ), CD3, CD8, γδTCR, CD25, CD69, CD62L, CCR6, CXCR3, ICOS, PD-1, CD103, PD-L1, GITR, CTLA-4, CD39, CD73 (all Biolegend), MHCII (eBiosciences), and Granzyme B (Thermo Fischer Scientific, Waltham, MA) as previously described.20,22 For intracellular and intranuclear staining, samples were processed using a commercial intranuclear staining kit (Foxp3 Staining Kit; eBiosciences). Fluorochrome-labeled antibodies against IL-17, IFNγ, T-Bet, Gata3 (all Biolegend), Foxp3 (eBiosciences), Ki67, and RORγt (both BD Biosciences) were used as recently published.20,22 For intracellular cytokine staining, cells were activated with PMA (50 ng/ml; Sigma-Aldrich), Ionomycin (1 μg/ml; Calbiochem-Merck), and Brefeldin A (10 μg/ml; Sigma-Aldrich) for 3 hours. LIVE/DEAD staining (Invitrogen Molecular Probes, Eugene, OR) was used to exclude dead cells during flow cytometry and ensure viability of the cells after the stimulation procedure. Experiments were performed on a BD LSRII Cytometer (Becton Dickinson).
T Cell In Vitro Studies and Treg Suppression Assay
CD4+ spleen cells were enriched by using magnetic-activated cell sorting according to the manufacturer’s protocol (MACS CD4+ T Cell Kit II; Miltenyi Biotec). Tregs and T effector cells were then isolated by FACS sorting (performed on a BD ARIAIII Cytometer; Becton Dickinson). A total of 1×105 CD45+CD3+CD4+YFP− effector T cells from Foxp3Cre mice were then cultured for 72 hours in anti-CD3 mAb (5 μg/ml; BD Biosciences) precoated 96-well plates either alone or in coculture with CD45+CD4+YFP+ Tregs from Foxp3Cre or Foxp3Cre×IL-10Rafl/fl mice at the indicated ratios. Suppressive capacity and cytokine production were determined by cytokine ELISAs performed from the supernatants as recently published and FACS analysis of KI67 expression.20,22 IL-10 mRNA production of cultured cells was analyzed by real-time PCR as described below.
Dendritic Cell Preparation
Spleens were harvested from Foxp3CrexIL-10Rafl/fl and Foxp3Cre mice in the naïve state as well as 36 hours after intraperitoneal immunization with 0.5 mg sheep IgG. Each spleen was injected with 1 ml of digestion medium (RPMI 1640 medium containing 10% FCS, 1% HEPES, 1% Penicillin/Streptomycin, 8 μg/ml Collagenase D, and 0.4 μg/ml DNase), minced, and incubated in digestion medium at 37°C for 45 minutes. Subsequently, they were washed and passed over 40-μm meshes to obtain a single-cell suspension. Dead cells were removed using the Dead Cell Removal Kit according to the manufacturer’s protocol (Miltenyi Biotec). Remaining cells were stained as described above, and CD45+CD3−CD11c+MHCII+ dendritic cells (DCs) were then isolated by FACS sorting (performed on a BD ARIAIII Cytometer). mRNA expression levels of different cytokines were analyzed as described below.
Quantitative Real-Time PCR Analysis
Quantitative RT-PCR from RNA of the indicated cell populations was performed in a Stepone Plus Detector (Applied Biosystems) as described before.22 For detection of IL-10, IL-1β, TGF-β1, IL-6, IL-12a, and 18S, the SYBR green method was used (primer sequences are available on request). Samples were run in duplicates and normalized to 18S rRNA.
Statistical Analyses
Results are expressed as mean±SEM. Groups were compared by t test, and a P value <0.05 was considered statistically significant.
Results
IL-10Ra Deficiency on Tregs Causes a Spontaneous Hyper-Th17 Phenotype
To evaluate the functions of IL-10 signaling in Tregs, we generated Foxp3CrexIL-10Rafl/fl mice with Treg-selective deletion of the IL-10R α-chain. Treg specificity of the knockout was confirmed by PCR analysis of sorted splenic leukocyte populations (Supplemental Figure 1). Spleen sizes and gross leukocyte composition in spleens and blood of naïve 9- to 13-week-old Foxp3CrexIL-10Rafl/fl and Foxp3Cre control mice were similar (Supplemental Figure 2, A–C). Interestingly, however, detailed analysis of systemic immunity showed spontaneous skewing toward Th17 responses in Foxp3CrexIL-10Rafl/fl mice. Percentages of splenic Th17 cells were significantly increased as measured by CD4+ T effector cell production of IL-17 and activation of the key transcription factor RORγt (Figure 1A). No change was noted regarding splenic Th1 (Figure 1B) or Th2 (Supplemental Figure 2D) responses. Similarly, ELISA analyses of cytokine production by spleen cell cultures after T cell receptor stimulation revealed a selective increase of IL-17, whereas expression of other cytokines remained unaltered (Figure 1C). Immune responses in the naïve peripheral blood also remained unaffected (Figure 1, D and E, Supplemental Figure 2E). Furthermore, general T cell activation in blood and spleens of Foxp3CrexIL-10Rafl/fl mice was not enhanced, even with increasing age (Supplemental Figure 2, F–H). Importantly, the observed hyper-Th17 phenotype did not cause any growth retardation (Supplemental Figure 2I) or development of colitis up until 7 months of age (Supplemental Figure 2, J and K), and mice remained fit, fertile, and healthy. Finally, we found that defective IL-10R signaling in Tregs also did not result in development of spontaneous renal damage or renal inflammation as analyzed in young and aged mice (Supplemental Figure 3).
Figure 1.

IL-10 receptor a deficiency on regulatory T cells causes a spontaneous hyper-Th17 phenotype. All analyses derive from naïve 9- to 13-week-old mice (n=10 versus n=13 unless otherwise stated). (A) FACS analysis of splenic T effector cells expressing Th17 cytokine IL-17 or transcription factor RORγt as indicated. (B) FACS analysis of splenic T effector cells expressing Th1 cytokine IFNγ or transcription factor T-Bet as indicated. (C) ELISA analyses of the indicated cytokines from supernatant of anti-CD3–stimulated spleen cell cultures (n=5 mice per group). (D) FACS analysis of peripheral blood T effector cells expressing Th17 cytokine IL-17 or transcription factor RORγt as indicated. (E) FACS analysis of peripheral blood T effector cells expressing Th1 cytokine IFNγ or transcription factor T-Bet as indicated. Bar graphs show means, and error bars indicate SEM. Circles represent individual animals, and horizontal lines show mean values. *P<0.05.
Systemic and Renal Immunity Is Sequentially Skewed toward Th17 during GN
Next, we aimed to analyze immunity in Foxp3CrexIL-10Rafl/fl mice under inflammatory conditions and thus, induced the NTN model of acute GN. Our data showed sequential development of hyper-Th17 responses. At day 3 after NTN induction, a time point when nephritogenic T cell responses are just developing and structural kidney damage has not yet become evident, we found that systemic Th17 immunity in spleens of Foxp3CrexIL-10Rafl/fl mice was already significantly enhanced (Supplemental Figure 4A). Furthermore, Th17 cell frequencies in the renal lymph nodes (Supplemental Figure 4B) had also started to increase in the knockout mice. Kidneys, in contrast, did not yet contain many CD4+ T cells at this early time point, and Th17 responses were not different between the groups (Supplemental Figure 4C). During later stages of NTN at day 8, we again found overshooting Th17 immunity in FACS analyses (Figure 2A) of splenocytes and ELISA of antigen-restimulated spleen cell culture supernatant (Figure 2C). Broader analyses of systemic immunity revealed selectivity of the hyper-Th17 immunophenotype with unchanged Th1 and Th2 responses (Figure 2B, Supplemental Figure 5, A and B). Importantly, at this later time point, we also observed specific enhancement of Th17 responses in the nephritic kidneys (Figure 2, D and E), whereas again, Th1 and Th2 immunity were not affected (Figure 2F, Supplemental Figure 5C). Likewise, renal infiltration of T effector cells carrying the Th17 prototype chemokine receptor CCR6 was increased, whereas percentages of Th1-type CXCR3+ T effector cells were similar (Figure 2G). Supporting the selectivity of the hyper-Th17 phenotype, detailed analysis of humoral immunity, including anti-sheep globulin total IgG as well as the different Th1- and Th2-dependent subclasses, revealed no difference (Supplemental Figure 6).
Figure 2.

Systemic and renal immunity is skewed toward Th17 during GN. All analyses derive from NTN-treated mice at day 8 (n=16 versus n=18 unless otherwise stated). (A) FACS analysis of splenic T effector cells expressing Th17 cytokine IL-17 (n=8 versus n=11) or transcription factor RORγt. (B) FACS analysis of splenic T effector cells expressing Th1 cytokine IFNγ (n=8 versus n=11). (C) ELISA analyses of the indicated Th17 cytokines from supernatant of antigen restimulated spleen cell cultures (n=8 versus n=11). (D) FACS analysis of renal T effector cells expressing Th17 cytokine IL-17 or transcription factor RORγt. (E) Representative FACS plots of renal IL-17+ Th17 cells. (F) FACS analysis of renal T effector cells expressing Th1 cytokine IFNγ or transcription factor T-Bet. (G) FACS analyses of renal T effector cells expressing Th17-type chemokine receptor CCR6 or Th1-characteristic CXCR3. Bar graphs show means, and error bars indicate SEM. Circles represent individual animals, and horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001.
Nephritis Is Aggravated in Foxp3CrexIL-10Rafl/fl Mice
To assess the functional relevance of the observed hyper-Th17 responses, we studied renal outcome of NTN. At day 3, glomerular crescents had not yet developed, and BUN was similar between the groups (Supplemental Figure 7). Importantly, however, 8 days after NTN induction, renal tissue injury was significantly aggravated in Foxp3CrexIL-10Rafl/fl mice (Figure 3A). In addition, BUN levels were higher in the absence of IL-10Ra signaling in Tregs, indicating greater renal functional impairment (Figure 3B). Albuminuria was similar in both strains of mice (Figure 3C). In line with enhanced tissue injury, we also found increased renal infiltration of proinflammatory leukocytes. These included MAC-2+ glomerular and F4/80+ interstitial macrophages (Figure 3, D and E) as well as glomerular and interstitial CD3+ T cells (Figure 3F). Finally and in line with the elevated Th17 responses, numbers of renal neutrophils were also strikingly increased in kidneys of Foxp3CrexIL-10Rafl/fl mice (Figure 3G).
Figure 3.

Nephritis is aggravated in Foxp3CrexIL10Rafl/fl mice. All analyses derive from NTN-injected mice at day 8 (n=16 versus n=18). (A) Periodic acid–Schiff (PAS)-stained kidney sections and quantification of glomerular crescents. (B) Analyses of serum BUN levels. (C) Quantification of the urinary albumin-to-creatinine ratio. (D) Representative immunohistochemical staining and quantification of MAC-2+ glomerular macrophages. (E) Representative immunohistochemical staining and quantification of F4/80+ interstitial macrophages. (F) Representative immunohistochemical staining and quantification of CD3+ glomerular and interstitial T cells. (G) Representative immunohistochemical staining and quantification of GR-1+ glomerular and interstitial neutrophils. Circles represent individual animals, and horizontal lines show mean values. gcs, glomerular cross-section; hpf, high-power field. *P<0.05; **P<0.01; ***P<0.001.
Th17-Promoting DC Phenotype in Foxp3CrexIL10Rafl/fl Mice
Given our observation of enhanced Th17 immunity, we wanted to evaluate whether lack of IL-10Ra on Tregs might influence activation and cytokine production by DCs. Indeed, we found higher percentages of DCs in spleens from naïve Foxp3CrexIL-10Rafl/fl mice. This difference prevailed under inflammatory conditions 3.5 days after NTN induction (Figure 4, A and B). Importantly, DCs in mice lacking the IL-10Ra on Tregs also showed a more activated phenotype. MHC class 2 expression was elevated under homeostatic conditions as well as during NTN (Figure 4C). Likewise, expression of T cell coactivators CD80 and CD86 was enhanced under naïve and/or inflammatory conditions (Figure 4, D and E). To determine if higher DC activation might specifically favor development of Th17 responses, we next quantified DC cytokine production. No differences of mRNA expression were noted between the groups at baseline as evidenced by analysis of FACS-sorted DCs from spleens of naïve mice (Supplemental Figure 8). Interestingly, however, expression levels of early Th17-inducing cytokines IL-1β and IL-6 were selectively increased in DCs from Foxp3CrexIL-10Rafl/fl mice at 36 hours after in vivo antigen challenge (Figure 4F). In contrast, TGF-β1 expression was only slightly elevated, and mRNA of Th1-inducing cytokine IL-12a remained unchanged (Figure 4F). Finally, we also analyzed IL-10 transcription by stimulated DCs, which however, was not different between the groups (Figure 4F).
Figure 4.

Absence of IL-10 receptor a (IL-10Ra) on regulatory T cells results in a Th17-promoting dendritic cell (DC) phenotype. (A) FACS analysis of CD11c+MHCII+ DCs in spleens of naïve mice and at 3.5 days after induction of NTN. (B) Representative FACS plot showing DCs in spleens of naïve mice. (C) FACS analyses of MHCII mean fluorescence intensity (MFI) on splenic DCs of naïve mice and at 5.5 days after induction of NTN. FACS analyses of (D) CD80 MFIs and (E) CD86 MFIs on splenic DCs of naïve mice and at 3.5 days after induction of NTN. Group sizes in A–E were n=5 versus n=7 naïve mice and n=8 versus n=6 nephritic mice. (F) Quantitative RT-PCR analyses of indicated cytokines from splenic DCs isolated at 36 hours after antigen challenge (n=5 mice). The dotted line represents DCs from wild-type mice (n=5 mice). Bar graphs show means, and error bars indicate SEM. Circles represent individual animals, and horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001.
Treg Trafficking Is Not Impaired by Absence of IL-10R on Tregs
Another potential mechanism for overshooting Th17 responses and aggravation of GN in mice with IL-10Ra–defective Tregs might be impairment of renal Treg trafficking. However, by immunohistochemistry (Figure 5A), we found unchanged Treg numbers (Figure 5B) and Treg-to-CD3 ratios (Figure 5C) in nephritic kidneys of both strains of mice. FACS analyses even revealed higher Treg percentages among CD4+ T cells in Foxp3CrexIL-10Rafl/fl mice (Figure 5D). In congruence with preserved renal trafficking capacity, analysis of two important chemokine receptors on Tregs showed similar expression of Treg17-characteristic CCR6 as well as Treg1 hallmark chemokine receptor CXCR3 (Figure 5E). Alternatively, Treg trafficking into secondary lymphoid organs during initiation of immune responses might be affected by lack of IL-10Ra signaling. Our analyses, however, revealed equal percentages of Tregs in spleens as well as the renal lymph node at day 3 after NTN induction (Figure 5F). Furthermore, Treg expression of chemokine receptor CCR7 remained unchanged (Figure 5G), indicating preserved colocalization properties of Tregs with DCs and naïve T effector cells.
Figure 5.

Regulatory T cell (Treg) trafficking is not impaired by absence of IL-10 receptor a (IL-10Ra) on Tregs. Analyses in A–E derive from NTN-injected mice at day 8 (n=16 versus n=18). (A) Representative immunohistochemical staining and (B) quantification of Foxp3+ renal Tregs. (C) Quantification of the renal Foxp3-to-CD3 ratio as counted from immunohistochemically stained sections. (D) Representative FACS plot and quantification of renal Foxp3+ Treg infiltration. (E) FACS analyses of renal Tregs expressing the Treg17-characteristic chemokine receptor CCR6 or the Treg1 hallmark chemokine receptor CXCR3. (F) FACS analyses of Treg percentages in spleens and renal lymph nodes (LNs) at day 3 after NTN. (G) FACS analyses of CCR7 expression on Tregs in the indicated organs at day 3 after NTN; n=8 mice per group were analyzed in F and G. Circles represent individual animals, and horizontal lines show mean values. *P<0.05.
Treg Suppressive Capacity Is Reduced in Foxp3CrexIL-10Rafl/fl Mice
Given that renal Treg trafficking was unaffected, we next sought to study Treg suppressive capacity. Indeed, we found reduced in vitro suppression of IL-2 secretion in cocultures of T effector cells with IL-10Ra–deficient Tregs (Figure 6A). Likewise, suppression of in vitro T effector cell proliferation, as shown by Ki67 staining, was less effective by Tregs from Foxp3CrexIL-10Rafl/fl mice (Figure 6B). Most importantly, our in vitro analyses also showed a profound defect of IL-10Ra–deficient Tregs to downregulate production of the Th17 hallmark cytokine IL-17 (Figure 6C). Suppression of the Th1 prototype cytokine IFNγ, in contrast, remained unaffected (Figure 6D). We next aimed to assess general Treg in vivo suppressive function and analyzed splenic T cell responses at 8 days after antigen challenge with NTN. Interestingly, our analyses showed higher percentages of Tregs in spleens of mice with IL-10Ra–deficient Tregs (Figure 6E). Despite enhanced Treg percentages, we found higher Teff activation in animals harboring IL-10Ra–deficient Tregs as measured by CD69 expression (Figure 6F). Furthermore, as in our in vitro assays, suppression of Teff proliferation was less effective in Foxp3CrexIL-10Rafl/fl mice as shown by higher Ki67 levels (Figure 6G). Finally and in line with reduced in vitro suppression of IL-2 secretion, we found enhanced IL-2 production by spleen cells of Foxp3CrexIL-10Rafl/fl mice after antigen restimulation (Figure 6H).
Figure 6.

Suppressive capacity of IL-10 receptor a (IL-10Ra)–deficient regulatory T cells (Tregs) is reduced. (A) In vitro suppression assays were performed by coculturing wild-type CD4+ T effector cells (Teffs) with Tregs from Foxp3CrexIL-10Rafl/fl or Foxp3Cre control mice at the indicated ratios (n=4 per group). Cytokine levels of IL-2 were analyzed in coculture supernatants. (B) Wild-type CD4+ Teffs were cocultured with Tregs from Foxp3CrexIL-10Rafl/fl or Foxp3Cre control mice at the indicated ratios (n=4 per group), and expression of the proliferation marker Ki67 in Teff was analyzed by FACS. (C and D) Wild-type CD4+ Teffs were cocultured with Tregs from Foxp3CrexIL-10Rafl/fl or Foxp3Cre control mice at the indicated ratios (n=4 per group), and suppression of (C) IL-17 or (D) IFNγ production was analyzed in coculture supernatants (one of two representative experiments is shown). (E) FACS analysis of Treg percentages in spleens at day 8 after NTN (n=16 versus n=18). (F) FACS analyses of splenic CD69+CD62L− activated Teff at day 8 after NTN (n=8 versus n=7). (G) FACS analysis of Ki67+-proliferating splenic Teff at day 8 of NTN (n=8 versus n=7). (H) ELISA analysis of IL-2 secretion from antigen restimulated spleen cell culture supernatant (n=8 versus n=11). Bar graphs show means, and error bars indicate SEM. Circles represent individual animals, and horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001.
IL-10R Signaling Enhances IL-10 Production by Tregs
Because our results indicated reduced Treg suppressive capacity, we next aimed to determine the underlying mechanisms. Activation (Supplemental Figure 9A) and proliferation (Supplemental Figure 9B) of splenic Tregs were unchanged at day 8 after NTN. Similarly, broad analysis of a wide array of surface markers (Supplemental Figure 9C) and transcription factors (Supplemental Figure 9D) revealed no differences between the groups. Given that IL-10 has been implicated in control of Th17 immunity, we next studied this cytokine. Indeed, in vitro stimulation resulted in reduced IL-10 mRNA transcription by Tregs from Foxp3CrexIL10Rafl/fl mice (Figure 7A). Similarly, analyses of supernatant from cultured Tregs revealed significantly decreased IL-10 protein secretion (Figure 7B). Given the limitations of in vitro experiments, we next sought to determine IL-10 in vivo production. Because IL-10 staining is technically challenging and in our experience, rather unreliable, we made use of fluorescence reporters. Mice with defective IL-10Ra signaling in T cells, which at the same time, express flourochromes under the Foxp3 and IL-10 promoters (CD4CrexIL10Rafl/flxFIRxTiger), were generated. NTN was induced in these mice and their respective CD4CrexIL10Rawt/wtxFIRxTiger controls. Supporting our in vitro data, FACS analyses at day 8 revealed significantly reduced percentages of IL-10–producing Tregs in mice lacking Treg-expressed IL-10Ra (Figure 7C). Likewise, the amount of IL-10 produced by IL-10Ra–deficient Tregs was also greatly reduced as measured by IL-10 mean fluorescence intensity (Figure 7D).
Figure 7.

IL-10 receptor a (IL-10Ra) signaling enhances IL-10 production by regulatory T cells (Tregs). (A) Semiquantitative analysis of IL-10 mRNA production by the indicated FACS-sorted spleen cell populations in the naïve state (n=2 animals per group) or after in vitro stimulation (n=4 versus n=4 versus n=3 animals per group). (B) Quantification of IL-10 by ELISA from the indicated FACS-sorted cell populations after in vitro stimulation (n=4 per group). (C) Representative FACS plots and quantification of IL-10 production by Tregs from kidneys of nephritic double-reporter mice of the indicated genotypes (n=8 versus n=9). (D) Representative FACS plots and quantification of the mean fluorescence intensity (MFI) of IL-10 in Tregs of nephritic kidneys from double-reporter mice of the indicated genotypes (n=8 versus n=9). Circles represent individual animals, and horizontal lines show mean values. KO, knockout; WT, wild type*P<0.05; **P<0.01.
IL-10Ra Signaling Directly Controls Th17 Responses but Not Treg Development and Fitness
Our results so far showed reduced IL-10 production by IL-10Ra–deficient Tregs. We, therefore, wanted to explore whether a reduction of IL-10 might potentially explain the observed hyper-Th17 phenotype in Foxp3CrexIL-10Rafl/fl mice. To analyze effects of IL-10 on Th17 cell generation, we performed competitive cotransfer studies. Naïve spleen cells from wild-type donor mice carrying the congenic marker CD45.1 were mixed at a 1:1 ratio with spleen cells from CD45.2+ CD4CrexIL-10Rafl/fl mice, which lack the IL-10Ra on CD4+ T effector cells. The cell mixtures containing IL-10Ra–intact and –deficient T cells were then injected into Rag1−/− recipients. NTN was induced 1 day later, and development of Th17 and Treg responses was analyzed in blood, spleens, and kidneys at day 8 (Figure 8A). In line with our hypothesis, percentages of RORγt+ Th17 cells were dramatically higher among CD45.2 T cells lacking the IL-10Ra in all analyzed compartments (Figure 8, B and C). Likewise, expression levels of RORγt were much enhanced in the knockout T cells (Figure 8D). To extend these results, we next assessed whether lack of IL-10Ra signaling also influences Treg development and competitive fitness. Our analyses, however, showed similar percentages of Tregs from CD45.1+ wild-type and CD45.2+ CD4CrexIL-10Rafl/fl mice in blood, spleen, and kidneys (Supplemental Figure 10A). Furthermore, expression levels of Foxp3 protein by CD4+ T cells were unaffected by lack of IL-10Ra signaling (Supplemental Figure 10B). Lastly, we also found similar Treg activation in all compartments regardless of presence or absence of the IL-10Ra (Supplemental Figure 10C).
Figure 8.

IL-10 receptor a (IL-10Ra) signaling directly controls Th17 cells but not regulatory T cell development and fitness. (A) Overview of the experimental setup. Spleen cells from CD45.1+ mice with IL10Ra-sufficient CD4+ T cells and CD45.2+ mice with IL10Ra-deficient CD4+ T cells were cotransferred into n=6 Rag1−/− recipient mice, and NTN was induced. (B) Representative FACS plot of CD45.1 and CD45.2 CD4+ T cells in the kidney of Rag1−/− recipients at day 8 of NTN (left panel). Representative FACS plots indicating percentages of RORγt+ Th17 cells among both CD45 subtypes (center and right panels). (C) Quantification of RORγt+ Th17 cells among both CD45 subtypes in the indicated compartments. (D) Mean fluorescence intensity (MFI) of RORγt protein in CD4+ Teff in indicated compartments. Circles represent individual animals, and horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001.
Discussion
Our study aimed to identify and characterize mechanisms responsible for generation, activation, and functional differentiation of Tregs. A particular focus was set on advancing our knowledge of how Tregs control highly pathogenic Th17 responses. One important Treg stabilizing factor could be the multifunctional cytokine IL-10, which has been shown to be a key player of immune regulation in multiple inflammatory diseases, including GN.43,44 Supporting this notion, individuals with mutations that negatively affect the IL-10 axis develop severe spontaneous colitis early in life.45 An increasing body of evidence supports a particularly important role of IL-10 in defense against overshooting Th17 responses.29–31 However, the situation is more complex, because effects on Th144 and Th2 immunity46 and even proinflammatory functions of IL-10 have also been reported.47 It is thus not surprising that it currently remains widely unknown how IL-10 regulates Th17 immunity and in particular, how IL-10 might shape and potentiate Treg responses. In this respect, a recent study has uncovered a previously unknown role of Treg intrinsic signaling via the IL-10Rα.32 Mice deficient in IL-10Ra signaling in Tregs developed severe hyper-Th17 immunity resulting in fatal spontaneous colitis. Because the underlying mechanisms remained ill defined, we wanted to explore this topic in more detail and generated mice with Treg-selective IL-10Ra deficiency. In contrast to the aforementioned study, these mice remained healthy with no signs of colitis and showed normal development and survival in our animal house facility. This might be due to differences in the microbiota, which in our animal house, also fails to induce colitis in Treg17-deficient Foxp3CrexStat3fl/fl20,21 or IL-10 panknockout mice. Nevertheless, our mice showed spontaneously overshooting Th17 responses. In congruence, we also found selective upregulation of Th17 immunity in the absence of IL10Ra signaling in Tregs after induction of GN. Interestingly, hyper-Th17 responses developed sequentially and became primarily evident systemically in the spleen and also, the renal lymph node, which is of particular importance for immune activation in the NTN model.48 At later stages of GN coinciding with development of renal tissue damage, enhanced Th17 immunity was also detected in the inflamed kidneys. Supporting our concept of Th17 cells as pathogenic mediators in GN,20,35,36,49–51 renal inflammation, histologic damage, and functional impairment were all exacerbated in Foxp3CrexIL-10Rafl/fl mice. Subsequently, we thus aimed to explore possible underlying mechanisms. Because DCs are crucially involved in T effector cell differentiation, we analyzed potential changes of their phenotype. Interestingly, we indeed found expansion and higher activation of splenic DCs in naïve and nephritic mice with IL-10Ra–deficient Tregs. Importantly, under inflammatory conditions, DCs from knockout mice also showed selectively enhanced mRNA expression of Th17-inducing key cytokines IL-1β and IL-6. Naïve DCs, in contrast, did not show any skewing of their cytokine profile. These findings indicate that uncontrolled DC activation in nephritic mice with IL-10Ra–deficient Tregs likely supports the development of hyper-Th17 inflammation. In the naïve state, however, DCs do not seem to vitally contribute. Furthermore, one has to note that the observed changes in DC phenotype were overall relatively modest. We thus suspected that additional mechanisms are involved in generation of the overshooting Th17 responses. Because IL-10 is a known inducer of Stat3 activation, we hypothesized that lack of IL-10Ra signaling would result in loss of an Stat3-dependent Treg17 phenotype, which is hallmarked by expression of the chemokine receptor CCR6.20,21 However, we found that renal Treg trafficking was by no means impaired but rather, somewhat enhanced. In line, expressions of Treg17 characteristic CCR6 and also, Treg1 characteristic CXCR313 were maintained. Next, we investigated possible impairment of Treg trafficking into secondary lymphatic organs during initiation of immune responses. However, our analyses showed that percentages of Tregs in spleens and renal lymph nodes were not reduced. Importantly, expression of the chemokine receptor CCR7, which is known to be crucial for crosstalk of Tregs with DCs and naïve T effector cells,52,53 was also unaltered. These findings pointed toward impairment of Treg suppressive function rather than to their trafficking. We thus aimed to characterize the underlying Treg defects and define the role of direct Treg/Teff interactions for enhanced Th17 generation as opposed to indirect DC-mediated pathways. Indeed, we found significantly reduced in vitro suppression of T effector cell proliferation and cytokine secretion. Importantly, Tregs from Foxp3CrexIL-10Rafl/fl mice selectively failed to suppress Th17 immunity. Control of Th1 responses in contrast was preserved. This finding strongly suggests that impaired direct Treg interactions with T effector cells significantly contribute to the hyper-Th17 responses in Foxp3CrexIL-10Rafl/fl mice. However, given the multiple possible confounders of in vitro assays, we aimed to verify the results in vivo. Again, we observed impaired suppression of Teff activation and proliferation as well as reduced suppression of IL-2 production by spleen cells of antigen-challenged mice harboring IL-10Ra–defective Tregs. To clarify the defects underlying these observations, we analyzed a broad panel of Treg markers and transcription factors previously associated with their activation. However, our studies did not detect any significant differences. Given the suspected role of Treg-derived IL-10 in regulation of Th17 immunity, which has recently also been shown in experimental GN,31 we went on to study this aspect. Indeed, we found reduced IL-10 mRNA and protein production from knockout Tregs after in vitro stimulation. Given these results, we aimed to validate our findings in vivo. Unfortunately, FACS analysis of IL-10 production using available antibodies turned out to be unreliable and nonspecific. We, therefore, made use of complex reporter mouse systems. Mice expressing Cre recombinase under the CD4 promoter were crossed into IL-10Ra floxed mice. These mice were then mated to another strain expressing red fluorescent protein under the Foxp3 promoter and a green fluorescent protein under control of the IL-10 promoter.42 In these mice, all T cells, including Tregs, lack the IL-10Ra. In addition, Foxp3+ Tregs as well as their IL-10 production can be detected reliably by autofluorescence. Using these mice, we indeed found that lack of Treg-expressed IL-10Ra resulted in significantly lower percentages of IL-10–producing Tregs during GN. Along the same line, the amount of IL-10 produced by IL10-Ra–deficient Tregs was also significantly reduced. This interesting observation might help to explain the more activated DC phenotype in Foxp3CrexIL-10Rafl/fl mice, because IL-10 signaling has recently been reported to be required for effective DC downregulation.54 Next, the question was arising of whether reduced IL-10 production could also result in enhanced Th17 immunity by direct DC-independent effects on CD4+ T cells. We thus cotransferred wild-type spleen cells carrying the congenic marker CD45.1 together with CD45.2 spleen cells from T cell–specific IL-10Ra–deficient mice into lymphopenic Rag1−/− recipients. These mice thus contained a mixture of IL-10Ra–intact and –deficient T cells, which could be differentiated by expression of their CD45 subtype.13,22 Interestingly, after induction of GN, IL-10Ra–deficient T cells massively differentiated into Th17 cells and much outperformed Th17 cell generation among IL-10Ra–intact wild-type T cells in all compartments analyzed. Stability and competitive fitness of transferred Tregs, in contrast, were unaffected by absence of the IL-10Ra. Our data, therefore, establish that IL-10 signaling can directly control Th17 differentiation in CD4+ T cells via the IL-10Ra. This finding nicely supports previous work in which mice overexpressing a dominant negative IL-10Ra chain were analyzed.30 In summary, our data identify the IL-10Ra on Tregs as a crucial component for control of Th17 immunity during homeostasis and after induction of acute GN. Contrary to our initial hypothesis, IL-10R signaling does not induce expression of CCR6 on Tregs to facilitate their colocalization with Th17 cells in inflamed tissues. Rather, as the key mechanism, we provide evidence for a feed-forward loop, in which IL-10R signaling reinforces IL-10 production by Tregs. Enhanced availability of IL-10, in turn, results in potent and selective suppression of Th17 responses via direct and indirect mechanisms. Modulation of the IL-10/IL-10R axis on Tregs thus emerges as a novel therapeutic strategy for GN.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank M. Schaper, I. Holtze, and M. Reszka for their excellent technical help.
This work was supported by Deutsche Forschungsgemeinschaft grants SFB 1192 TPA03 (to M.A.K. and O.M.S.) and KFO 228 STE 1822/3-1 (to O.M.S.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017091044/-/DCSupplemental.
References
- 1.Sakaguchi S, Miyara M, Costantino CM, Hafler DA: FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10: 490–500, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Vignali DA, Collison LW, Workman CJ: How regulatory T cells work. Nat Rev Immunol 8: 523–532, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramsdell F, Ziegler SF: FOXP3 and scurfy: How it all began. Nat Rev Immunol 14: 343–349, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Kurts C, Panzer U, Anders HJ, Rees AJ: The immune system and kidney disease: Basic concepts and clinical implications. Nat Rev Immunol 13: 738–753, 2013 [DOI] [PubMed] [Google Scholar]
- 5.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al.: Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117: 3921–3928, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al.: Administration of CD4+CD25highCD127- regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care 35: 1817–1820, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Net JB, Bushell A, Wood KJ, Harden PN: Regulatory T cells: First steps of clinical application in solid organ transplantation. Transpl Int 29: 3–11, 2016 [DOI] [PubMed] [Google Scholar]
- 8.Hutchinson JA, Geissler EK: Now or never? The case for cell-based immunosuppression in kidney transplantation. Kidney Int 87: 1116–1124, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Feuerer M, Hill JA, Mathis D, Benoist C: Foxp3+ regulatory T cells: Differentiation, specification, subphenotypes. Nat Immunol 10: 689–695, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Campbell DJ, Koch MA: Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol 11: 119–130, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinmetz OM, Turner JE, Panzer U: Staying on top of things right from the start: The obsessive-compulsive disorder of regulatory T cells. J Am Soc Nephrol 21: 6–7, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ: The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10: 595–602, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nosko A, Kluger MA, Diefenhardt P, Melderis S, Wegscheid C, Tiegs G, et al.: T-bet enhances regulatory T cell fitness and directs control of Th1 responses in crescentic GN. J Am Soc Nephrol 28: 185–196, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paust HJ, Riedel JH, Krebs CF, Turner JE, Brix SR, Krohn S, et al.: CXCR3+ regulatory T cells control TH1 responses in crescentic GN. J Am Soc Nephrol 27: 1933–1942, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al.: CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326: 986–991, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al.: STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282: 9358–9363, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al.: Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 204: 2803–2812, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al.: Phenotypic and functional features of human Th17 cells. J Exp Med 204: 1849–1861, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner JE, Paust HJ, Steinmetz OM, Peters A, Riedel JH, Erhardt A, et al.: CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. J Am Soc Nephrol 21: 974–985, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kluger MA, Luig M, Wegscheid C, Goerke B, Paust HJ, Brix SR, et al.: Stat3 programs Th17-specific regulatory T cells to control GN. J Am Soc Nephrol 25: 1291–1302, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kluger MA, Melderis S, Nosko A, Goerke B, Luig M, Meyer MC, et al.: Treg17 cells are programmed by Stat3 to suppress Th17 responses in systemic lupus. Kidney Int 89: 158–166, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Kluger MA, Meyer MC, Nosko A, Goerke B, Luig M, Wegscheid C, et al.: RORγt(+)Foxp3(+) cells are an independent bifunctional regulatory T cell lineage and mediate crescentic GN. J Am Soc Nephrol 27: 454–465, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, et al.: Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol 9: 444–457, 2016 [DOI] [PubMed] [Google Scholar]
- 24.Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al.: Mucosal immunology. The microbiota regulates type 2 immunity through RORγt+ T cells. Science 349: 989–993, 2015 [DOI] [PubMed] [Google Scholar]
- 25.Kluger MA, Nosko A, Ramcke T, Goerke B, Meyer MC, Wegscheid C, et al.: RORγt expression in Tregs promotes systemic lupus erythematosus via IL-17 secretion, alteration of Treg phenotype and suppression of Th2 responses. Clin Exp Immunol 188: 63–78, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wehinger J, Gouilleux F, Groner B, Finke J, Mertelsmann R, Weber-Nordt RM: IL-10 induces DNA binding activity of three STAT proteins (Stat1, Stat3, and Stat5) and their distinct combinatorial assembly in the promoters of selected genes. FEBS Lett 394: 365–370, 1996 [DOI] [PubMed] [Google Scholar]
- 27.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A: Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19: 683–765, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Brockmann L, Gagliani N, Steglich B, Giannou AD, Kempski J, Pelczar P, et al.: IL-10 receptor signaling is essential for TR1 cell function in vivo. J Immunol 198: 1130–1141, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu Y, Yang J, Ouyang X, Liu W, Li H, Yang J, et al.: Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol 38: 1807–1813, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber S, Gagliani N, Esplugues E, O’Connor W Jr, Huber FJ, Chaudhry A, et al.: Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34: 554–565, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostmann A, Paust HJ, Panzer U, Wegscheid C, Kapffer S, Huber S, et al.: Regulatory T cell-derived IL-10 ameliorates crescentic GN. J Am Soc Nephrol 24: 930–942, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al.: Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34: 566–578, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al.: Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 10: 1178–1184, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al.: IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol 195: 3665–3674, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Paust HJ, Turner JE, Steinmetz OM, Peters A, Heymann F, Hölscher C, et al.: The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol 20: 969–979, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinmetz OM, Summers SA, Gan PY, Semple T, Holdsworth SR, Kitching AR: The Th17-defining transcription factor RORγt promotes glomerulonephritis. J Am Soc Nephrol 22: 472–483, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al.: Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28: 546–558, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Wan YY, Flavell RA: Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A 102: 5126–5131, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galán JE, et al.: Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25: 941–952, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Luig M, Kluger MA, Goerke B, Meyer M, Nosko A, Yan I, et al.: Inflammation-induced IL-6 functions as a natural brake on macrophages and limits GN. J Am Soc Nephrol 26: 1597–1607, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huber S, Schramm C, Lehr HA, Mann A, Schmitt S, Becker C, et al.: Cutting edge: TGF-beta signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol 173: 6526–6531, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Kluger MA, Ostmann A, Luig M, Meyer MC, Goerke B, Paust HJ, et al.: B-cell-derived IL-10 does not vitally contribute to the clinical course of glomerulonephritis. Eur J Immunol 44: 683–693, 2014 [DOI] [PubMed] [Google Scholar]
- 43.Huang XR, Kitching AR, Tipping PG, Holdsworth SR: Interleukin-10 inhibits macrophage-induced glomerular injury. J Am Soc Nephrol 11: 262–269, 2000 [DOI] [PubMed] [Google Scholar]
- 44.Kitching AR, Tipping PG, Timoshanko JR, Holdsworth SR: Endogenous interleukin-10 regulates Th1 responses that induce crescentic glomerulonephritis. Kidney Int 57: 518–525, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al.: Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361: 2033–2045, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coomes SM, Kannan Y, Pelly VS, Entwistle LJ, Guidi R, Perez-Lloret J, et al.: CD4+ Th2 cells are directly regulated by IL-10 during allergic airway inflammation. Mucosal Immunol 10: 150–161, 2017 [DOI] [PubMed] [Google Scholar]
- 47.Geginat J, Larghi P, Paroni M, Nizzoli G, Penatti A, Pagani M, et al.: The light and the dark sides of Interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev 30: 87–93, 2016 [DOI] [PubMed] [Google Scholar]
- 48.Artinger K, Kirsch AH, Aringer I, Moschovaki-Filippidou F, Eller P, Rosenkranz AR, et al.: Innate and adaptive immunity in experimental glomerulonephritis: A pathfinder tale. Pediatr Nephrol 32: 943–947, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinmetz OM, Turner JE, Paust HJ, Lindner M, Peters A, Heiss K, et al.: CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis. J Immunol 183: 4693–4704, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Summers SA, Steinmetz OM, Li M, Kausman JY, Semple T, Edgtton KL, et al.: Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol 20: 2518–2524, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gan PY, Steinmetz OM, Tan DS, O’Sullivan KM, Ooi JD, Iwakura Y, et al.: Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol 21: 925–931, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A: CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med 204: 735–745, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eller K, Weber T, Pruenster M, Wolf AM, Mayer G, Rosenkranz AR, et al.: CCR7 deficiency exacerbates injury in acute nephritis due to aberrant localization of regulatory T cells. J Am Soc Nephrol 21: 42–52, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girard-Madoux MJ, Kel JM, Reizis B, Clausen BE: IL-10 controls dendritic cell-induced T-cell reactivation in the skin to limit contact hypersensitivity. J Allergy Clin Immunol 129: 143–150.e141–110, 2012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.