

Abstract
Background Properdin (P) is a positive regulator of the alternative pathway of complement activation. Although P inhibition is expected and has been shown to ameliorate the alternative pathway of complement-mediated tissue injury in several disease models, it unexpectedly exacerbated renal injury in a murine model of C3 glomerulopathy. The role of P in atypical hemolytic uremic syndrome (aHUS) is uncertain.
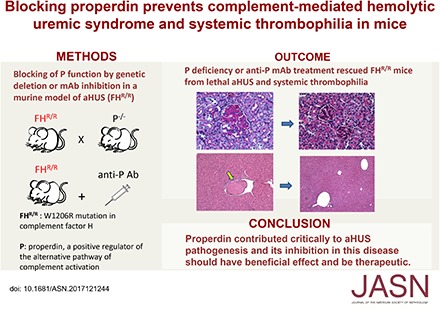
Methods We blocked P function by genetic deletion or mAb-mediated inhibition in mice carrying a factor H (FH) point mutation, W1206R (FHR/R), that causes aHUS and systemic thrombophilia with high mortality.
Results P deficiency completely rescued FHR/R mice from premature death and prevented thrombocytopenia, hemolytic anemia, and renal disease. It also eliminated macrovessel thrombi that were prevalent in FHR/R mice. All mice that received a function-blocking anti-P mAb for 8 weeks survived the experimental period and appeared grossly healthy. Platelet counts and hemoglobin levels were significantly improved in FHR/R mice after 4 weeks of anti-P mAb treatment. One half of the FHR/R mice treated with an isotype control mAb but none of the anti-P mAb-treated mice developed stroke-related neurologic disease. Anti-P mAb-treated FHR/R mice showed largely normal renal histology, and residual liver thrombi were detected in only three of 15 treated mice.
Conclusions These results contrast with the detrimental effect of P inhibition observed in a murine model of C3 glomerulopathy and suggest that P contributes critically to aHUS pathogenesis. Inhibition of P in aHUS may be of therapeutic benefit.
Keywords: complement, hemolytic uremic syndrome, Immunology and pathology
Properdin (P) is a plasma glycoprotein and the only known positive regulator of the alternative pathway (AP) of complement activation.1–5 It is composed of seven type 1 thrombospondin repeats and present in plasma at a concentration of about 5–15 μg/ml.6 P facilitates AP complement activation by binding to and stabilizing the C3 convertase C3bBb, and thereby, it significantly extends its t1/2.7,8 There is now considerable evidence to suggest that P plays a pathogenic role in AP complement–mediated autologous tissue injury. For example, P deficiency has been shown to be protective in several murine models of complement-dependent diseases, including extravascular hemolysis, abdominal aortic aneurysm, renal ischemia-reperfusion injury, inflammatory arthritis, and allergic asthma.9–12 However, it was observed in earlier studies that P deficiency or antibody neutralization unexpectedly exacerbated renal disease in murine models of C3 glomerulopathy (C3G) due to factor H (FH) knockout or C-terminal mutation.13,14 The latter finding highlighted the complex dynamics of complement activation and inhibition in the fluid phase and on target organs, and it raised intriguing questions regarding the mechanism of action of P and whether it is protective or pathogenic in atypical hemolytic uremic syndrome (aHUS), another AP complement–driven disease with renal injury as a hallmark phenotype.
Human aHUS is a form of thrombotic microangiopathy (TMA) characterized by thrombocytopenia, hemolytic anemia, and renal failure.15,16 Most aHUS cases are linked to abnormalities in the complement system, including loss of function mutations in genes encoding regulatory proteins, such as FH, or gain of functions mutations in C3 and factor B.17–21 These complement defects predispose patients to aHUS, which can be triggered by major health-related events, such as infection, pregnancy, trauma, or surgery, whereby complement activation may be induced and inadequately regulated, leading to microvascular injury.22 To better understand the aHUS pathogenesis and facilitate development of novel therapeutic options, we previously generated a high-penetrance murine model of aHUS by FH point mutation.23 Mice homozygous for the FH W1206R point mutation (FHR/R) developed characteristic aHUS symptoms, including low platelet counts, anemia, and renal disease, and they had high mortality.23 They also displayed a systemic thrombophilia phenotype with formation of macrovessel thrombi in various organs, most prominently in the liver.23 In this study, we have used the FHR/R mouse model and tested the role of P in aHUS using two separate approaches: by genetically deleting the P gene9 and by mAb neutralization of P protein.11 Our results showed that P contributed critically to the pathogenesis of aHUS in FHR/R mice and that blocking P was sufficient to rescue them from the disease. This outcome was in striking contrast with what had been observed previously in murine models of C3G, where blocking P exacerbated disease.13,14 Our data shed new light on the mechanism of action of P in different complement-mediated renal diseases and provide proof of concept in support of therapeutic targeting of P in aHUS.
Methods
Mice
The source and generation of FH W1206R mutant mice (FHR/R) and a P knockout mouse (P−/−), generated by crossing Pflox/flox and Ella-Cre mice, have been reported previously.9,23 All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Survival Curve
The survival curve was analyzed using the GraphPad Prism program (La Jolla, CA) as described previously.13,23
Measurement of BUN and Complete Blood Count
BUN was measured using serum and urea nitrogen reagents (Sigma-Aldrich, St. Louis, MO) as described previously.23 Complete blood count was measured using an automated hematology analyzer (XT-2000iV; Sysmex) at the Translational Core Laboratory of the Children’s Hospital of Philadelphia.23
Immunofluorescence Staining and Histology
Kidneys were frozen in optimal cutting temperature compound (Sakura), and 4-μm sections were cut. Sections were processed for C3, C9, and fibrin deposition as described previously.23 For C9 staining, a rabbit polyclonal anti-mouse C9 generated in house was used. Details about generation and characterization of the anti-C9 antibody will be reported elsewhere. For costaining with C3 or C9, rat anti-mouse CD31 antibody (clone MEC13.3) (0.5 mg/ml; BD Pharmingen) was used at 1:50 dilution to stain and identify endothelial cells. Rat anti-mouse CD140b (PDGF receptor-β) antibody (clone ABP5) (0.5 mg/ml; eBioscience) was used at 1:25 to stain and identify mesangial cells. For these two antibodies, Cy3-conjugated goat anti-rat IgG (0.2 mg/ml; Invitrogen) was used at 1:100 dilution as the secondary antibody. A guinea pig polyclonal anti-nephrin antibody (Progen Biotechnik, Heidelberg, Germany) was used at 1:100 dilution to stain and identify podocytes followed by the use of Alexa Flour 555 goat anti-guinea pig IgG (2 mg/ml; Invitrogen) at 1:500 dilution as the secondary antibody. Costained sections were examined on a Leica STED 3× super-resolution microscope.
To evaluate tissue pathology, isolated organs were fixed in 10% formalin-PBS–buffered saline and processed for hematoxylin and eosin and periodic acid–Schiff staining by AML Laboratories (www.amllabs.com). Kidney slides were graded by light microscopy in a blind fashion by a pathologist (M.P.) as described previously.23
Electron Microscopy
Electron microscopic analysis of kidney sections was performed at the Electron Microscopy Resource Laboratory of the University of Pennsylvania as described previously.13,23
AP and Lytic Complement Activity Assays
LPS-induced AP complement activity assay and red blood cell lysis test for lytic complement activity in mouse plasma were performed as described.23 Both assays used lupiridin (Bivalirudin; Hospira Inc., Lake Forest, IL) anticoagulated mouse plasma diluted to 10% and 50%, respectively. Nonsensitized sheep red blood cells (Complement Technology, Tyler, TX) were used in the lysis test.
Pharmacodynamics Study of mAb 14E1 in Mice
Before treatment of FHR/R mice with anti-P mAb 14E111, we determined the pharmacodynamics of mAb 14E1 in mice. C57BL/6 mice were injected with 1 mg (intraperitoneally) of mAb 14E1. Plasma samples were collected before injection (day 0) and then, at various time points after injection, and they were tested for total AP complement activity.
Treatment of FHR/R with Anti-P or Isotype Control mAb
For therapeutic studies, 4-week-old FHR/R mice of mixed sexes were treated for 8 weeks with the anti-P mAb 14E111 or an isotype murine IgG1 control mAb (MOPC 31C)24 at 1 mg per mouse administered intraperitoneally. Control mAb was administered weekly, and anti-P mAb was given either once weekly or twice weekly as specified in Results and figures.
Western Blot Analysis of Plasma Complement Proteins
Levels of complement proteins in mouse plasma were measured by Western blot analysis. Mouse plasma (0.5–2 μl) was diluted with sample buffer and boiled before loading onto 4%–12% gradient gel under reducing conditions. Samples were transferred to polyvinylidene difluoride membranes. For detection, goat anti-human factor B antibody (crossreactive with mouse factor B; 1:2000; Complement Technology, Inc.), horse radish peroxidase (HRP)-conjugated goat anti-mouse C3 antibody (1:4000; MP Biomedicals), polyclonal goat anti-human C5 sera (crossreactive with mouse C5; 1:1000; Quidel Corporation), and a rabbit anti-mouse P polyclonal antibody9 were used as primary Ab for factor B, C3, C5, and P, respectively. As secondary antibodies, HRP-conjugated goat anti-rabbit IgG (1:4000; Bio-Rad, Hercules, CA) was used for P detection, and HRP-conjugated rabbit anti-goat IgG antibody (1:4000) was used for factor B and C5 detection using the ECL chemiluminescent detection system (Amersham Pharmacia, Uppsala, Sweden). Odyssey imaging analysis system (LI-COR Bioscience, Lincoln, NE) was used to scan the membrane and quantify band intensity using ImageJ software (National Institutes of Health).
Results
P Deficiency Rescued FHR/R Mice from Lethal aHUS
We have recently described the creation of an FH mutant mouse (FHR/R) that developed high-penetrance aHUS due to a point mutation (tryptophan to arginine) at position 1206 of short consensus repeat 20 (SCR20) of FH.23 To assess the role of P in aHUS, we crossed the FHR/R mouse with a P knockout mouse (P−/−)9 and generated FHR/R P−/− mice. We then followed the double-mutant mice and compared them with FHR/R littermates for aHUS disease development. As we observed before, approximately 50% of FHR/R mice (n=74) died by 20 weeks of age, but all FHR/R P−/− mice (n=58) survived to the same age (Figure 1A). P deficiency also rescued FHR/R mice from thrombocytopenia and anemia; FHR/R P−/− mice had platelet counts and hemoglobin (Hb) levels comparable with those in wild-type (FHW/W) mice, whereas FHR/R mice exhibited severe thrombocytopenia and had significantly lower Hb levels (Figure 1, B and C). BUN increased significantly from 5 to 10 weeks of age in FHR/R mice, but an age-dependent increase in BUN was not observed in FHR/R P−/− mice (Figure 1D), suggesting that the double-mutant mice were resistant to kidney injury. These results indicated that P is critical for the development of aHUS in FHR/R mice.
Figure 1.

Properdin gene deletion prevented lethal atypical hemolytic uremic syndrome in FHR/R mice. (A) Approximately 50% of FHR/R mice (n=74) died by 20 weeks, but all FHR/R P−/− littermate mice (n=58) survived normally. (B) FHR/R mice (n=9) had thrombocytopenia, whereas FHR/R P−/−mice (n=9) had normal platelet counts similar to FHW/W (wild-type) mice (n=11). (C) FHR/R (n=9) but not FHR/R P−/−mice (n=10) had lower hemoglobin (Hb) levels than FHW/W mice (n=11). Each plot symbol in B and C represents an individual mouse (5–20 weeks old). Horizontal bars through the scatter plots in B and C are the average values in each group. (D) BUN of FHR/R mice (n=23) increased abnormally from 5 to 10 weeks of age, but BUN of FHR/R P−/−mice (n=21) remained in the normal range at both 5 and 10 weeks of age. Data shown in D are mean±SD of all mouse values. Mantel–Haenszel log rank test for A and one-way ANOVA and t test for other B–D. **P<0.01; ***P<0.001.
P Deficiency Prevented Renal TMA Pathology but Did Not Eliminate C3 or C9 Deposition in FHR/R Mice
We examined and compared renal pathology in FHW/W, FHR/R, and FHR/R P−/− mice by light, electron, and fluorescence microscopy. On periodic acid–Schiff staining of paraffin-embedded sections, the kidneys of FHR/R mice showed characteristic features of aHUS, including mesangial expansion, narrowing of capillary lumens, and microthrombi. In contrast, the kidneys of FHR/R P−/− mice were similar to those of FHW/W mice and showed essentially normal histology and glomerular integrity with no signs of TMA (Figure 2A, Table 1). Prevention of renal TMA was confirmed on electron microscopic analysis. FHR/R mouse glomeruli showed subendothelial space with fluffy granular materials, but FHR/R P−/− mice, like FHW/W mice, presented with normal glomerular basement membrane and foot processes (Figure 2B). Similarly, abundant fibrin deposition was observed only in the glomeruli of FHR/R mice but not in FHR/R P−/− or FHW/W mice (Figure 2C, Supplemental Figure 1). As described before,23 FHR/R but not FHW/W mice had granular staining of C3 in mesangium and capillary regions of glomeruli. Surprisingly, despite the absence of renal TMA pathology, significant C3 and C9 deposition remained in the glomeruli of FHR/R P−/− mice (Figure 2D, Supplemental Figures 1 and 2). Costaining experiments with nephrin, PDGF receptor-β, and CD31 used as markers for podocytes, mesangial, and vascular endothelial cells,25–29 respectively, showed that glomerular C3 or C9 deposition in FHR/R and FHR/R P−/− mice partially colocalized with podocytes and mesangial cell but not endothelial cell markers (Supplemental Figures 3–8).
Figure 2.

Properdin deficiency rescued renal thrombotic microangiopathy (TMA) pathology of FHR/R mice. (A) Periodic acid–Schiff staining of kidney sections of FHR/R mice (10 weeks of age, male) showed characteristic features of atypical hemolytic uremic syndrome, including mesangial expansion, narrowing of capillary lumen, and microthrombi, whereas FHR/R P−/− mice (10 weeks of age, male) exhibited no signs of TMA in glomeruli, similar to FHW/W mice (10 weeks of age, male). Scale bars, 25 μm. (B) On electron microscopy (EM), FHR/R mouse glomeruli (10 weeks of age, male) had typical TMA features, including subendothelial space with fluffy granular materials (yellow arrow) and no electron dense deposit, but such pathology was not present in FHW/W (10 weeks of age, male) or FHR/R P−/− mouse (10 weeks old, male) glomeruli. Scale bars, 2 μm. (C) FHR/R glomeruli showed strong fibrin staining, but fibrin was not detected in the glomeruli of FHW/W and FHR/R P−/− mice. (D) C3 staining of granular pattern in mesangium and capillaries was found in FHR/R and FHR/R P−/− mouse glomeruli but not in FHW/W mouse glomeruli. Scale bars, 25 μm in C and D.
Table 1.
Glomerular thrombotic microangiopathy scoring of FHW/W, FHR/R, and FHR/R P−/− mouse kidney sections
| Mouse Strain | Expanded Matrix and Endothelial Swelling, % | Microthrombi, % | Large Vein Thrombi, % | Glomerular Sclerosis, % | Arteriolar Hyalinosis, % |
|---|---|---|---|---|---|
| FHW/W | 0 | 0 | 0 | 0 | 0 |
| FHR/R | 54.4 | 77.3 | 54.2 | 0.9 | 79.2 |
| FHR/R P−/− | 0.3 | 0 | 0 | 0 | 0 |
At least three kidney sections from each mouse were examined for five characteristic thrombotic microangiopathy–related pathologic changes in hematoxylin and eosin and periodic acid–Schiff staining. All glomeruli on a given section were scored. Mice from each strain were mixed sexes ages 5–30 weeks old, and numbers of mice in each group are shown. FHW/W, n=9; FHR/R, n=30; FHR/R P−/−, n=20.
P Deficiency Prevented Macrovessel Thrombi and Neurologic Abnormality in FHR/R Mice
As described previously, in addition to renal TMA, FHR/R mice also displayed extrarenal phenotypes, including systemic thrombophilia with macrovessel thrombosis in multiple organs and stroke-related neurologic abnormality.23 Macrovessel thrombi in liver, lung, spleen, heart, and brain were detected in 83%, 20%, 40%, 10%, and 17% of FHR/R mice, respectively, but they were not present in any of the FHR/R P−/− mice (Figure 3, Table 2). Stroke-related neurologic abnormalities, such as hemiparalysis or circular movement,23 were observed in 17 of 30 FHR/R mice studied, but none of the 20 FHR/R P−/− mice in this cohort developed this phenotype (Table 2). These data indicated that P played a critical role in the pathogenesis of both micro- and macrovessel thrombosis in FHR/R mice.
Figure 3.

Properdin deficiency prevented macrovessel thrombosis in FHR/R mice. Hematoxylin and eosin staining of (A) liver, (B) kidney, (C) lung, and (D) spleen showed that macrovessel thrombi (yellow arrows) were present in FHR/R mice but not in FHW/W or FHR/R P−/− mice. Scale bars, 100 μm.
Table 2.
Summary of extrarenal phenotype evaluation in FHR/R and FHR/R P−/− mice
| Phenotypes | FHR/R | Incidence, % | FHR/R P−/− | Incidence, % |
|---|---|---|---|---|
| Neurologic abnormalities | 17/30 | 56.7 | 0/20 | 0 |
| Brain thrombi | 5/30 | 16.7 | 0/20 | 0 |
| Brain ischemic change | 17/30 | 56.7 | 0/20 | 0 |
| Lung thrombi | 6/30 | 20.0 | 0/20 | 0 |
| Heart thrombi | 3/30 | 10.0 | 0/20 | 0 |
| Liver thrombi | 25/30 | 83.3 | 0/20 | 0 |
| Spleen thrombi | 12/30 | 40.0 | 0/20 | 0 |
The same number of males and females ages 5–30 weeks old were examined in each strain: FHR/R, n=30; FHR/R P−/−, n=20. Serial sections from each organ were prepared, and at least three nonadjacent sections per tissue sample were examined. To avoid missing the detection of pathologic changes, several parts of each organ were harvested and examined. The total number of mice (5–30 weeks of age) examined and the number of mice showing a particular pathology are listed. Neurologic abnormality is identified using movement behavior (hemiparalysis or rapid circular movement) as a surrogate.
Anti-P mAb Treatment Rescued FHR/R Mice from Lethal aHUS Disease
To test if systemic blockade of P might have therapeutic efficacy for aHUS, we treated FHR/R mice with a function-blocking mouse anti-mouse P mAb.11 Of 15 FHR/R mice treated with anti-P antibody, ten received once weekly intraperitoneal injections and five received twice weekly intraperitoneal injections of 1 mg mAb 14E1 for 8 weeks starting at 4 weeks of age. As a control group, 15 other FHR/R mice received weekly injections of the same amount of an isotype control mAb (MOPC, clone 31C). Pharmacodynamics analysis showed that a dosage of 1 mg mAb 14E1 completely depleted plasma P and abolished LPS-dependent AP complement activity in wild-type mouse blood for at least 1 week (Supplemental Figure 9). As shown in Figure 4A, all FHR/R mice treated with the anti-P mAb survived the 8-week treatment period, whereas six of 15 FHR/R mice treated with the control mAb died during this time (60% survival rate). Complete blood count analysis showed that platelet numbers and Hb levels were significantly elevated after 4 weeks of anti-P mAb treatment (Figure 4, B and C). Platelet counts and Hb levels were maintained in the normal range in the twice weekly treated mice during 8 weeks of treatment, but they fluctuated in the once weekly treated mouse group (Figure 4, B and C).
Figure 4.

Anti-properdin (anti-P) mAb treatment rescued FHR/R mice from lethal atypical hemolytic uremic syndrome. (A) FHR/R mice treated with anti-P mAb (n=15; once weekly, ten; twice weekly, five) all survived 8 weeks of treatment, whereas control mAb-treated mice (n=15; all treated once weekly) had high mortality. (B) Platelet counts were elevated after 4 weeks of treatment in all anti-P mAb-treated mice and maintained in normal range in the twice weekly treated group until the end of the experiment. No significant change in platelet count was observed in control mAb-treated mice, and they remained thrombocytopenic throughout the experiment. (C) Hemoglobin (Hb) levels were elevated after 4 weeks of treatment in all anti-P mAb-treated mice and maintained in normal range in the twice weekly treated group. No significant change in Hb level was observed in control mAb-treated mice. Data are presented as mean±SD in B and C. Mantel–Haenszel log rank test for A and one-way ANOVA for B and C. *P<0.05 control mAb versus anti-P mAb, once weekly dosing; ***P<0.001, control mAb versus anti-P mAb, once weekly dosing; #P<0.05, control mAb versus anti-P mAb, twice weekly dosing; ##P<0.01, control mAb versus anti-P mAb, twice weekly dosing; ###P<0.001, control mAb versus anti-P mAb, twice weekly dosing.
Anti-P mAb Treatment Ameliorated Renal and Extrarenal Pathology in FHR/R Mice
Kidney sections of control mAb-treated FHR/R mice showed characteristic TMA features as previously described,23 whereas renal TMA was largely prevented in anti-P mAb-treated FHR/R mice, with residual pathology detectible only in a small percentage of the kidney glomeruli (Figure 5A, Table 3). Supporting the therapeutic efficacy of anti-P mAb, prominent fibrin deposition was detected in the glomeruli of control mAb-treated FHR/R mice, but it was completely absent in anti-P mAb-treated mouse glomeruli (Figure 5B, Supplemental Figure 1). Notably, as in FHR/R P−/− mice, significant C3 and C9 staining in mesangium and podocytes remained in anti-P mAb-treated FHR/R mice (Figure 5C, Supplemental Figures 1–8). Lastly, 73%, 33%, 7%, and 13% of the control mAb-treated FHR/R mice showed macrovessel thrombi in the liver, spleen, lung, or brain, respectively, and one half of them (seven of 15) exhibited stroke-related neurologic symptoms (Figure 5D, Supplemental Table 1). In contrast, 13 of 15 anti-P mAb-treated FHR/R mice were completely devoid of macrovessel thrombi in any of the organs examined, and only two of 15 mice had residual liver thrombi (Figure 5D, Supplemental Table 1); none of the 15 anti-P mAb-treated FHR/R mice developed stroke-related neurologic disease (Supplemental Table 1).
Figure 5.

Treatment with anti-properdin (anti-P) mAb prevented renal and extrarenal pathology in FHR/R mice. (A) Periodic acid–Schiff staining of kidney sections of control mAb-treated FHR/R mice showed thrombotic microangiopathy pathology in the glomeruli, whereas anti-P mAb-treated mice exhibited normal glomerular histology. (B) Prominent fibrin staining was present in the glomeruli of control mAb-treated mice, but fibrin staining was not detected in anti-P mAb-treated mice. (C) C3 staining of a granular pattern was detected in the glomeruli of both control and anti-P mAb-treated mice. (D) Hematoxylin and eosin staining of liver sections illustrating that large-vessel thrombi (yellow arrows) were found in the organs of control mAb-treated FHR/R mice but not in anti-P mAb-treated FHR/R mice. Scale bars, 25 μm in A–C; 100 μm in D.
Table 3.
Anti-properdin mAb treatment ameliorated renal pathology in FHR/R mice
| Treatment Group | Expanded Matrix and Endothelial Swelling, % | Microthrombi, % | Large Vein Thrombi, % | Glomerular Sclerosis, % | Arteriolar Hyalinosis, % |
|---|---|---|---|---|---|
| FHR/R + control mAb | 66.7 | 100 | 33.3 | 0.7 | 100 |
| FHR/R + anti-P mAb once weekly | 6.2 | 10 | 0 | 0 | 0 |
| FHR/R + anti-P mAb twice weekly | 5 | 0 | 0 | 0 | 0 |
At least three kidney sections from each mouse were examined for five characteristic thrombotic microangiopathy–related pathologic change in hematoxylin and eosin and periodic acid–Schiff staining. All glomeruli on a given section were scored. FHR/R + control mAb, n=15; FHR/R + anti-P mAb once weekly, n=10; FHR/R + anti-P mAb twice weekly, n=5.
AP and Terminal Complement Profile and Activity in FHR/R P−/− Mice
To correlate with the TMA-free phenotype and understand the unexpected glomerular C3 and C9 deposition in FHR/R P−/− and anti-P mAb-treated FHR/R mice, we examined plasma levels of intact C3, factor B, and C5, and we measured plasma AP and terminal complement activities in FHR/R and FHR/R P−/−mice. As expected, AP and complement lytic activity was substantially inhibited in FHR/R P−/− mice (Supplemental Figure 10), with corresponding increases in plasma C3, factor B, and C5 levels due to reduced consumption (Supplemental Figure 11). Interestingly, measurable and P-independent AP and complement lytic activity was detected in FHR/R P−/− mice (Supplemental Figure 10).
Discussion
In this study, we have shown that P played a critical role in the pathogenesis of aHUS in a murine model of the disease. The FHR/R mouse model recapitulates many key features of human aHUS, including renal TMA and systemic thrombophilia, with symptoms ranging from thrombocytopenia, hemolytic anemia, and renal failure to stroke-related ischemic brain injury and retinopathy.23 By genetic breeding with P−/− mice9 and with the use of a function-blocking anti-P mAb,11 we established here that development of these disease phenotypes was dependent on P. Our data are consistent with several recently published studies showing that P played a critical role in AP complement–mediated host tissue injury.9–12,30–32
The anti-P mAb experiment in this study used 4-week-old FHR/R mice, which validated the effectiveness of anti-P mAb treatment in preventing severe aHUS. It is likely, however, that some degree of tissue injury had already occurred in these mice at that age. Indeed, in earlier studies, we detected kidney TMA pathology and liver thrombi, even in 1-week-old FHR/R mice,23 and several mice in the anti-P mAb group of this study were already thrombocytopenic before the start of mAb treatment. The findings that renal TMA pathology and liver thrombi were largely absent at termination and that hematologic parameters were normalized in response to mAb treatment suggested that anti-P mAb could reverse as well as prevent aHUS disease. Nevertheless, it remains to be tested whether P inhibition has therapeutic effect in older FHR/R mice with more severe aHUS.
Our results here showing that blocking P prevented aHUS disease contrasted strikingly with earlier findings in a murine C3G model, whereby P inhibition exacerbated, rather than ameliorated, renal disease.13,14 In both C3G and aHUS models, disease was driven by AP complement activation as a result of FH mutation.13,14 However, the nature of FH mutation in the previous C3G models and the current aHUS model was different. In the C3G models, mutations in FH resulted in mutant mice producing no FH or a very low level of a truncated FH lacking SCR19–20.13,14 This led to uncontrolled AP complement activation in the plasma and on the cell surface with almost complete C3, factor B, and C5 consumption.13,14 This secondary complement deficiency likely has protected the mutant mice from developing lethal kidney injury.13,14 We hypothesized that, when P was inhibited, C3, factor B, and C5 consumption in extrarenal sites was reduced, and this could have made more of these proteins available in the kidney to cause P-independent AP complement activation in the absence of adequate FH protection, resulting in C3G exacerbation.13,14,33 In FHR/R mice, the W1206R point mutation did not reduce plasma FH level but impaired its interaction with red blood cells and presumably, other host cells in the vascular space, such as platelets and endothelial cells.23 This defect in FH function then led to P-dependent AP complement injury of endothelial cells and activation of platelets characteristic of aHUS. The P-dependent nature of complement activation on vascular cells explains why blocking P in FHR/R mice prevented aHUS.
Given that C3, factor B, and C5 were not exhaustively consumed and remained close to normal levels in FHR/R mice,23 one may wonder why P deficiency in these mice did not lead to lethal C3G as it did in FH SCR19–20 truncation mutant mice.13 The answer clearly is in the nature and amount present in plasma of the two FH mutant proteins in these models. As alluded to above, very little mutant FH lacking SCR19–20 was present in the C3G mouse model,13 whereas in FHR/R mice, the W1206R point mutation did not affect plasma FH level.23 It is notable that, notwithstanding the phenotype of TMA prevention, neither P deficiency nor anti-P mAb treatment eliminated glomerular C3 and C9 deposition in FHR/R mice, suggesting that the W1206R mutation in FH may nevertheless have resulted in some degree of P-independent complement activation in the glomeruli, albeit with no significant pathologic consequence. Consistent with this interpretation, we detected measurable P-independent AP and complement lytic activity in FHR/R P−/−mice (Supplemental Figure 10), the origin and mechanism of which remain to be investigated. Confocal microscopy of immunofluorescence-stained kidney sections showed that C3 and C9 deposition in FHR/R P−/−mouse glomeruli partially colocalized with mesangium and podocyte but not endothelial cell markers (Supplemental Figures 7 and 8). Surprisingly, we also did not observe colocalization of C3 or C9 with the endothelial cell marker CD31 in FHR/R mice (Supplemental Figures 7 and 8). This may be explained by CD31 downregulation on C3- or C9-positive endothelial cells in these mice, because endothelium injury is the hallmark of TMA. Collectively, this study and our previously described lethal C3G mouse model13 show the context-specific role of P in aHUS and C3G, and they highlight the need to understand the spectrum of these two diseases as well as their pathophysiologic overlap and difference to guide anti-P therapy.
Currently, aHUS is treated in the clinic with Eculizumab, a humanized anti-C5 mAb that is administered intravenously at biweekly intervals and in large doses.34 The plasma level of P, at 5–15 μg/ml or 0.1–0.3 nM, is lower than that of C5, which is present at 80 μg/ml or 0.4 nM in human blood.6,35 This may allow anti-P mAb-based drug to be given less frequently, in smaller doses, or via nonintravenous routes to both reduce cost and improve patient’s compliance. One caveat in pursuing anti-P–based therapies in aHUS is the heterogeneous nature of the underlying complement defects in patients with aHUS.15,16 The generality of the findings reported here (i.e., whether anti-P therapy is safe and effective in aHUS caused by complement dysregulation other than the case exemplified here by the FHR/R mouse model) remains to be established. Considering the clinical fact of overlapping phenotypes of TMA and C3G and in light of the existence of a P-independent mechanism of AP complement activation and glomerular injury as observed in our C3G mouse study,13,14 care must be excised, such that the benefit of aHUS amelioration by P inhibition is not offset by the development of C3G in the patients.
Disclosures
W.-C.S. is an inventor on patent applications related to anti-properdin mAbs and has received research funding from Alexion Pharmaceuticals, Inc. All other authors have no competing financial interests to declare.
Supplementary Material
Acknowledgments
We are grateful for the services provided by the Transgenic and Chimeric Mouse Facility and the Electron Microscopy Core of the Perelman School of Medicine, University of Pennsylvania for chimeric mouse production and electron microscopy, respectively, and the Clinical and Translational Research Center of the Children’s Hospital of Philadelphia for mouse complete blood count analysis.
This work is supported in part by National Institutes of Health grants AI085596, AI117410, AI44970, and EY023709 and a research grant from Alexion Pharmaceuticals, Inc.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017121244/-/DCSupplemental.
References
- 1.Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC: The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science 120: 279–285, 1954 [DOI] [PubMed] [Google Scholar]
- 2.Maves KK, Weiler JM: Properdin: Approaching four decades of research. Immunol Res 12: 233–243, 1993 [DOI] [PubMed] [Google Scholar]
- 3.Lachmann PJ: The amplification loop of the complement pathways. Adv Immunol 104: 115–149, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Lesher AM, Song WC: Review: Complement and its regulatory proteins in kidney diseases. Nephrology (Carlton) 15: 663–675, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blatt AZ, Pathan S, Ferreira VP: Properdin: A tightly regulated critical inflammatory modulator. Immunol Rev 274: 172–190, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwaeble WJ, Reid KB: Does properdin crosslink the cellular and the humoral immune response? Immunol Today 20: 17–21, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Hourcade DE: Properdin and complement activation: A fresh perspective. Curr Drug Targets 9: 158–164, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Fearon DT, Austen KF: Properdin: Binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med 142: 856–863, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura Y, Zhou L, Miwa T, Song WC: Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. J Clin Invest 120: 3545–3554, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou HF, Yan H, Stover CM, Fernandez TM, Rodriguez de Cordoba S, Song WC, et al.: Antibody directs properdin-dependent activation of the complement alternative pathway in a mouse model of abdominal aortic aneurysm. Proc Natl Acad Sci U S A 109: E415–E422, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miwa T, Sato S, Gullipalli D, Nangaku M, Song WC: Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol 190: 3552–3559, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Miwa T, Ducka-Kokalari B, Redai IG, Sato S, Gullipalli D, et al.: Properdin contributes to allergic airway inflammation through local C3a generation. J Immunol 195: 1171–1181, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Herbert AP, et al.: Combination of factor H mutation and properdin deficiency causes severe C3 glomerulonephritis. J Am Soc Nephrol 24: 53–65, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruseva MM, Vernon KA, Lesher AM, Schwaeble WJ, Ali YM, Botto M, et al.: Loss of properdin exacerbates C3 glomerulopathy resulting from factor H deficiency. J Am Soc Nephrol 24: 43–52, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noris M, Remuzzi G: Atypical hemolytic-uremic syndrome. N Engl J Med 361: 1676–1687, 2009 [DOI] [PubMed] [Google Scholar]
- 16.Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al.: Atypical aHUS: State of the art. Mol Immunol 67: 31–42, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, et al.; Itaslian Registry of Familial and Recurrent HUS/TTP : The molecular basis of familial hemolytic uremic syndrome: Mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol 12: 297–307, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, et al.: Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A 104: 240–245, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frémeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al.: Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112: 4948–4952, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, et al.: Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet 68: 485–490, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pérez-Caballero D, González-Rubio C, Gallardo ME, Vera M, López-Trascasa M, Rodríguez de Córdoba S, et al.: Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet 68: 478–484, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai HM: Thrombotic thrombocytopenic purpura and the atypical hemolytic uremic syndrome: An update. Hematol Oncol Clin North Am 27: 565–584, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Ueda Y, Mohammed I, Song D, Gullipalli D, Zhou L, Sato S, et al.: Murine systemic thrombophilia and hemolytic uremic syndrome from a factor H point mutation. Blood 129: 1184–1196, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams AL, Gullipalli D, Ueda Y, Sato S, Zhou L, Miwa T, et al.: C5 inhibition prevents renal failure in a mouse model of lethal C3 glomerulopathy. Kidney Int 91: 1386–1397, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vecchi A, Garlanda C, Lampugnani MG, Resnati M, Matteucci C, Stoppacciaro A, et al.: Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur J Cell Biol 63: 247–254, 1994 [PubMed] [Google Scholar]
- 26.Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, et al.: Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation 103: 2955–2960, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Holthöfer H, Ahola H, Solin ML, Wang S, Palmen T, Luimula P, et al.: Nephrin localizes at the podocyte filtration slit area and is characteristically spliced in the human kidney. Am J Pathol 155: 1681–1687, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holzman LB, St John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR: Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int 56: 1481–1491, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestilä M, Jalanko H, et al.: Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A 96: 7962–7967, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimitrova P, Ivanovska N, Schwaeble W, Gyurkovska V, Stover C: The role of properdin in murine zymosan-induced arthritis. Mol Immunol 47: 1458–1466, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Dimitrova P, Ivanovska N, Belenska L, Milanova V, Schwaeble W, Stover C: Abrogated RANKL expression in properdin-deficient mice is associated with better outcome from collagen-antibody-induced arthritis. Arthritis Res Ther 14: R173, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao J, Lesher AM, Miwa T, Sato S, Gullipalli D, Song WC: Tissue-specific deletion of Crry from mouse proximal tubular epithelial cells increases susceptibility to renal ischemia-reperfusion injury. Kidney Int 86: 726–737, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesher AM, Nilsson B, Song WC: Properdin in complement activation and tissue injury. Mol Immunol 56: 191–198, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al.: Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368: 2169–2181, 2013 [DOI] [PubMed] [Google Scholar]
- 35.Kohler PF, Müller-Eberhard HJ: Immunochemical quantitation of the third, fourth and fifth components of human complement: Concentrations in the serum of healthy adults. J Immunol 99: 1211–1216, 1967 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.