

Abstract
The association between growth hormone (GH) treatment and cancer risk has not been thoroughly evaluated and there are questions about any increased risk of bone tumors. We examined cancer risk and especially bone tumor risk in a population‐based cohort study of 6874 patients treated with recombinant GH in France for isolated GH deficiency, short stature associated with low birth weight or length or idiopathic short stature. Adult mortality and morbidity data obtained from national databases and from questionnaires. Case ascertainment completeness was estimated with capture‐recapture methods. Standardized mortality and incidence ratios were calculated using national reference data. 111 875 person‐years of observation were analyzed and patients were followed for an average of 17.4 ± 5.3 years to a mean age of 28.4 ± 6.2 years. For cancer overall, mortality and incidence were not different from expected figures. Five patients developed bone tumors (chondrosarcoma, 1, Ewing sarcoma, 1, osteosarcoma, 3) of whom 3 died (Ewing sarcoma, 1, osteosarcoma, 2), whereas only 1.4 case and 0.6 deaths were expected: standardized mortality ratio, 5.0 and standardized incidence ratio from 3.5 to 3.8 accounting or not accounting for missed cases. Most patients received conventional doses of GH, although one patient with osteosarcoma had received high dose GH (60 μg/kg/d). This study confirms an increased risk of bone tumors but not overall cancer risk in subjects treated with GH in childhood for isolated GH deficiency or childhood short stature. Further work is needed to elucidate the mechanisms involved.
Keywords: Bone tumors, childhood, growth hormone treatment
1. INTRODUCTION
The risk of malignancy after growth hormone treatment during childhood remains unclear. In experimental studies, growth hormone (GH) and insulin‐like growth factors (IGFs) have mitogenic, antiapoptotic, and proliferative properties.1, 2, 3 High levels of circulating IGF‐I have been shown to be associated with increased risks of several common cancers, including breast, prostate, and colorectal cancers 3, 4, 5 and GH itself could also play a direct role in carcinogenesis6, 7; notably, patients with acromegaly have consistently been found to have increased risks of several cancers, especially colorectal cancer.8 Also, patients with GH resistance and IGF‐I deficiency due to GH resistance have a significantly decreased risk of cancer.9, 10 The risk of several cancers is linked to height with taller people having a higher risk.11, 12, 13, 14, 15, 16, 17, 18
Few studies have addressed growth hormone therapy and cancer, however, and their systematic review by Deodati et al19 suggests that cancer mortality is not increased although cancer incidence is increased; these conclusions are not definitive, given the size and the methodology of the studies. An increased risk of second neoplasm in GH‐treated patients has also been reported, in particular for bone tumors.20
The Safety and Appropriateness of Growth hormone treatments in Europe (SAGhE) project is a multinational European study that aims to evaluate long‐term mortality and cancer morbidity in subjects who were treated with recombinant GH in childhood. A preliminary report in 2012 describing the French cohort of the SAGhE study indicated that all‐type cancer‐related mortality was not higher among those treated for idiopathic short stature or isolated GH deficiency than in the general population (standardized mortality ratio [SMR] 1.02, 95% CI 0.41‐2.09). Nevertheless, the bone tumor‐related mortality was high (SMR 5.00, 95% CI 1.01‐14.63).21 More recently, the European consortium published results for cancer mortality in all eight country cohorts and results for cancer incidence for five countries (not France, Germany, and Italy).22 In GH‐treated patients without previous cancer, there was no increased risk of cancer or cancer mortality overall but there was an excess risk of cancer mortality and incidence for bone cancer, an increased mortality by prostate cancer (1 case) and an increased incidence of bladder cancer.
The French SAGhE study data is particularly high quality, being based on a national register of GH‐treated children (the France Hypophyse register), and completed by data extracted from the Center for Epidemiology on Medical Causes of Death and from the French national health insurance information system. These data allow us to examine in more detail cancer morbidity and mortality in this population.
2. PATIENTS AND METHODS
2.1. Patients
As described previously 21, 23, 24 we used the mandatory register of patients treated with GH in France [Association France‐Hypophyse] which was disbanded in 1996; we selected those patients had been treated exclusively with recombinant GH and who were born before 1 January 1990. Patients were assigned to three risk categories (low, medium, and high) for long‐term morbidity and mortality, based on the clinical condition leading to the initiation of GH treatment (Figure 1). Only low‐risk patients were included in this study, because their baseline risk of cancer is believed to be similar to or lower than that of the general population.
Figure 1.
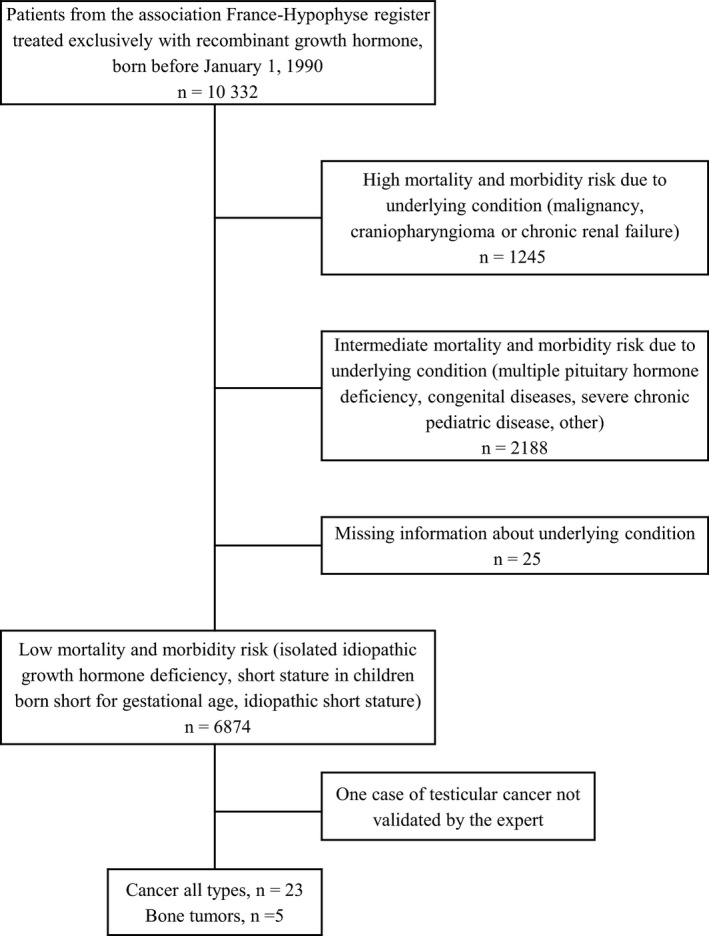
Flow chart of the SAGhE study in France and number of cancer cases identified in the low‐risk group
2.2. Data collected
2.2.1. Childhood data
Data on patient characteristics, treatment, and growth progression in the France‐Hypophyse register were routinely collected at baseline and at regular follow‐up visits until 1996, and were obtained from pediatric endocrinologists. Additional follow‐up data on GH treatment were collected from clinical centers in 2008‐2010.
2.2.2. Follow‐up data
Information on vital status was collected from the Répertoire National d'Identification des Personnes Physiques (http://www.insee.fr/fr/methodes) and the Répertoire National Inter‐régimes de l'Assurance Maladie. The cause of death indicated in death certificates was obtained from the French Center for Epidemiology on Medical Causes of Death (CépiDC, Institut National de la Santé et de la Recherche Médicale) and coded according to the 10th revision of the International Classification of Diseases (ICD‐10).
Morbidity data were collected through a health questionnaire sent to all live patients (with a response rate of 46% after several reminders: the low response rate was expected, because questionnaires were sent to the parents’ address; there had been no prior contact between the researchers and the patients; and these patients generally do not feel “sick”). Data were also extracted using patient identifiers from the French national health insurance information system (Système National d'Information Inter‐régimes de l'Assurance Maladie; SNIIRAM); this system includes the French hospital discharge database (FHDD) also called Programme de Médicalisation des Systèmes d'Information from 1 January 2008 to 31 December 2010, and long‐lasting affection statements (LLA) which identify conditions with 100% reimbursement coverage (which include cancers). We set a censor date of 31 December 2010.
2.2.3. Validation of events
Cancer events were validated using all medical and pathology reports obtained. Data about bone tumors were specifically reviewed by two oncologists specialized in these tumors (DL, JM).
2.3. Statistical analyses
The overall risk of cancer was assessed by calculating standardized incidence ratios (SIR), with adjustment for age and sex, using reference rates for all cancer combined from population‐based registries of cancer in France, centralized by the FRANCIM (FRANce‐Cancer‐Incidence et Mortalité) network, between 1985 and 2010.25 The risk of bone tumor was assessed using data provided by FRANCIM combined six general registries of bone tumors in France. The number of person‐years at risk was calculated for GH‐treated subjects, for each 5‐years age class, separately for men and women, from the date of first administration of GH to the date of cancer, death, loss to follow‐up, or 31 December 2010. The expected number of events was then calculated for GH‐treated subjects by multiplying the age‐ and sex‐specific incidence rates by the number of person‐years at risk. SIRs were estimated by dividing the number of observed events by the number of expected events. Significance tests and 95% confidence intervals (CI) for the SIR were calculated with Byar's approximation to the exact Poisson test and the exact Poisson limits. Standardized mortality ratios (SMR) and 95% CIs were calculated as reported previously.21
The CépiDC source for fatal events is exhaustive but the three sources (SNIIRAM — LLA, SNIIRAM — FHDD — and questionnaires) used to identify nonfatal events are not and therefore some events could have been missed by all sources. We used capture‐recapture methods 26, 27 to estimate the number of nonfatal events missed by all three sources. These methods are commonly used in epidemiology to estimate the completeness of ascertainment of disease registries and to estimate the number of cases that could have been missed by all sources of ascertainment. Thus, they allow improved accuracy of estimation of disease or event rates. Log‐linear modeling was used to adjust for source dependence by including the corresponding interaction term into the model,23 and the significance of the interaction was assessed using likelihood ratio statistics. A confidence interval (CI) for the estimated number of cases missed was computed by the profile likelihood method. The Akaike Information Criterion (AIC) was used for selection of the model. Two analyses were carried out: one with observed events, giving the crude SIR, the other including estimated events using the capture‐recapture method, giving the corrected SIR. Because of the small number of events for bone tumors, the ratio of estimated/observed nonfatal events for the whole cancer group was used to estimate the number of nonfatal events of bone tumors.
This study was approved by the Comité Consultatif sur le Traitement de l'Information en Matière de Recherche dans le Domaine de la Santé and the Commission Nationale de l'Informatique et des Libertés (the national data protection agency). The use of the Registre National Inter‐Régimes de l'Assurance Maladie was approved by a specific statute.
3. RESULTS
Most of the low‐risk mortality and morbidity group (n = 6874) were patients treated for the indication of isolated GH deficiency, based on GH stimulation tests (peak below 10 ng/mL) (n = 4600, 67%) (Table 1). In this group, there were over 111 000 person‐years at risk and the mean follow‐up between the beginning of GH treatment and the date of last follow‐up, event, or death was 17.4 years. The mean (±SD) treatment duration was 3.9 (±2.6) years, with mean doses slightly below the current recommendations for isolated GH deficiency.
Table 1.
Main characteristics of patients and GH treatment for studied sample. N = 6874
| Number of male patients (%) | 4510 (66) |
| Indication for GH treatment. number (%) | |
| Isolated GH deficiency | |
| Maximum peak GH <3 μg/L | 295 (4) |
| Maximum peak GH ≥3 μg/L and <7 μg/L | 1557 (23) |
| Maximum peak GH ≥7 μg/L and <10 μg/L | 2748 (40) |
| Missing value for maximum peak GH | 516 (8) |
| Maximum peak GH ≥10 μg/L | |
| Neurosecretory dysfunction | 547 (8) |
| Idiopathic short stature | 868 (13) |
| Small for gestational age | 343 (5) |
| Year of treatment start. number (%) | |
| 1985‐1987 | 506 (7.4) |
| 1988‐1990 | 2470 (36) |
| 1991‐1993 | 2362 (34) |
| 1994‐1996 | 1536 (22.6) |
| Birth length (SDS for gestational age) | −1.2 ± 1.2 (n = 4875) |
| Birth weight (SDS for gestational age) | −0.6 ± 1.2 (n = 5130) |
| Children born small for gestational age (birth weight or length ≤−2 SDS for gestational age) | |
| Yes | 1298 (19) |
| No | 3864 (56) |
| Missing data | 1712 (25) |
| Chronological age at start of treatment (y) | 11.0 ± 3.4 (n = 6874) |
| Height at start of treatment (SDS) | −2.7 ± 0.8 (n = 6285) |
| Weight at start of treatment (SDS) | −1.6 ± 0.9 (n = 6242) |
| Mean dose (μg/kg/d) | 24.5 ± 12.3 (n = 6212) |
| Treatment duration (y) | 3.9 ± 2.6 (n = 6380) |
| Chronological age at end of treatment (y) | 15.1 ± 2.7 (n = 6380) |
| Person‐years of observation (n) | 111 875 |
| Chronological age at the time of census or event or death (y) | 28.4 ± 6.2 |
| Duration of follow‐up from start of GH to time of census or event or deaths (y) | 17.4 ± 5.3 (n = 6616) |
Mean ± SD or n (%) are shown. GH, growth hormone; SDS, SD score.
Twenty‐four cancer events were identified in this group through the different sources, including one case which was not validated (Table 2). The most common cancers were bone tumors (n = 5), lymphoma (n = 4), and acute leukemia (n = 3). Overall, the patients were treated with conventional doses of GH (mean 23.4 μg/kg/d) with only one patient receiving high GH doses (60 μg/kg/d). None of the patients presented particularly severe short stature at the beginning of treatment or exhibited substantial height gain during treatment. Eight patients, including two with osteosarcoma and one with Ewing sarcoma, died at a mean age of 17.3 years.
Table 2.
Clinical characteristics and GH treatments in the 23 patients who developed cancer after GH treatment
| # | Type of cancer | Died (Y/N) | Age at event or death (y)a | Sex | Diagnosis leading to GH treatment | Age at start of GH treatment (y) | Height (SD) before treatment | GH treatment duration (y) | Height (SD) at the end of treatment | Adult height (SD) | Mean GH dose (μg/kg/d) | Source of information for cancer |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Acute leukemia | Y | 12.9 | F | GHDI | 11.1 | ‐ | 1.4 | ‐ | ‐ | ‐ | CépiDC |
| 2 | Acute leukemia | Y | 14.8 | M | GHDI | 12.7 | −2.1 | 0.8 | −1.8 | ‐ | 23.5 | CépiDC |
| 3 | Acute leukemia | Y | 16.5 | F | GHDI | 9.2 | ‐ | 1.9 | ‐ | ‐ | ‐ | CépiDC |
| 4 | Lymphoma | N | 29.0 | F | GHDI | 12.0 | −2.5 | 3.3 | −2.1 | −1.5 | 19.2 | Questionnaire |
| 5 | Lymphoma | N | 11.5 | M | GHDI | 7.0 | −1.9 | 3.7 | −1.0 | −2.1 | 17.1 | Questionnaire |
| 6 | Lymphoma | N | 24.4 | M | GHDI | 9.9 | −2.2 | 5.6 | −0.6 | ‐ | 23.4 | LLA. Questionnaire |
| 7 | Lymphoma | N | 22.4 | M | GHDI | 8.6 | −2.0 | 4.0 | −1.9 | ‐ | 21.1 | LLA. FHDD |
| 9 | Ewing sarcoma | Y | 17.3 | M | GHDI | 10.3 | −3.5 | 5.1 | −2.4 | ‐ | 24.4 | CépiDC |
| 8 | Osteosarcoma | Y | 14.4 | M | SGA | 8.5 | −3.6 | 3.0 | −2.4 | ‐ | 60.0 | CépiDC |
| 10 | Osteosarcoma | Y | 20.2 | F | GHDI | 4.5 | −2.7 | 8.2 | ‐ | ‐ | 26.6 | CépiDC |
| 11 | Osteosarcoma | N | 7.4 | M | GHDI | 6.1 | −3.3 | 1.1 | −2.9 | −3.6 | 24.1 | LLA. Questionnaire |
| 12 | Chondrosarcoma | N | 21.8 | M | GHDI | 15.8 | −1.3 | 1.2 | −0.7 | −0.7 | 26.1 | Questionnaire |
| 13 | Malignant brain tumor | N | 18.2 | M | GHDI | 2.4 | ‐ | 9.0 | ‐ | ‐ | ‐ | LLA |
| 14 | Melanoma | Y | 28.1 | M | GHDI | 11.1 | −2.6 | 6.1 | −1.8 | −2.0 | 17.3 | CépiDC |
| 15 | Melanoma | N | 31.5 | F | GHDI | 15.1 | ‐ | 1.9 | ‐ | −1.5 | ‐ | Questionnaire |
| 16 | Nasopharyngeal carcinoma | Y | 19.3 | M | GHDI | 11.2 | ‐ | 1.4 | ‐ | ‐ | ‐ | CépiDC |
| 17 | Malignant tumor of the mouth | N | 26.4 | M | GHDI | 8.5 | −2.0 | 7.1 | −0.6 | −0.9 | 21.5 | LLA. FHDD |
| 18 | Malignant tumor of the kidney | N | 29.5 | F | GHDI | 9.6 | ‐ | 2.4 | ‐ | −2.7 | ‐ | LLA. FHDD. Questionnaire |
| 19 | Malignant tumor of the testis | N | 31.6 | M | GHDI | 12.2 | ‐ | 2.0 | −1.8 | −0.9 | 15.8 | LLA. FHDD. Questionnaire |
| 20 | Malignant tumor of the testis | N | 15.0 | M | GHDI | 12.7 | −1.9 | 3.5 | −1.3 | 27.9 | Questionnaire | |
| 21 | Sweat gland carcinoma | N | 32.6 | F | GHDI | 13.9 | −2.7 | 2.5 | −2.2 | −2.3 | 28.0 | Questionnaire |
| 22 | Pulmonary carcinoma | N | 26.9 | M | GHDI | 8.0 | −1.7 | 3.7 | −1.2 | −1.7 | 17.2 | FHDD. Questionnaire |
| 23 | Malignant pancreatic tumor | N | 19.4 | M | GHDI | 8.2 | −2.2 | 6.6 | −0.5 | −1.1 | 16.7 | LLA. FHDD. Questionnaire |
LLA, long‐lasting affection; FHDD, French hospital discharge database; F, female; M, male; GHDI, idiopathic growth hormone deficiency; SGA, Born small for gestational age.
Age at event was used for live patients, and age at death was used for those who died.
Bone tumor characteristics are shown in Table 3. There were three osteosarcomas occurring 1.1, 2.9, and 15 years after the start of GH treatment; the diagnosis of cancer led to the interruption of GH treatment in the first two cases. Two of the patients with osteosarcoma died, 6 months‐3 years, after diagnosis. There was one fatal case of Ewing sarcoma, with bone and bone marrow metastases, 5.1 years after GH treatment initiation and 1.5 years after GH treatment interruption. There was one case of chondrosarcoma, 6 years after GH treatment initiation and 4.8 years after GH treatment interruption, and this patient is alive 8 years after surgery. One of the patients with osteosarcoma had received high GH doses (60 μg/kg/d).
Table 3.
Clinical characteristics, treatment and evolution related to GH and bone tumors in the 5 patients with bone tumors
| # | Type of cancer | Localization | Age at diagnosis of cancer (y) | Delay between start of GH and cancer diagnosis (y) | Clinical manifestations of cancer | Metastasis | Metastasis localization | Initial cancer treatment | Complete remission | Relapse | Treatment of cancer relapse | Died | Medical and pathology reports available |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | Ewing sarcoma | Sacrum | 15.9 | 6.6 | Dysuria, constipation, pain | Y | Bone and bone marrow | Chemotherapy, surgery and radiotherapy | Y | Y | Oral chemotherapy (VP16) | Y | Y |
| 9 | Osteosarcoma | Left lower tibia | 11.5 | 2.9 | Pain | N | Chemotherapy, surgery and radiotherapy | Y | Y | CHD | Y | Y | |
| 10 | Osteosarcoma | Iliac wing | 19.5 | 15 | Pain | N | Chemotherapy and radiotherapy (no surgery because of deep vein thrombosis) | N | Y | Y | |||
| 11 | Osteosarcoma | Lower end of the right femur | 7.4 | 1.1 | U | U | U | U | N | N | |||
| 12 | Chondrosarcoma | Inter‐tibio‐fibular | 21.8 | 6 | U | N | Surgery | Y | N | N | Y |
Y, Yes; N, No; U, Unknown.
SIRs and SMRs for all cancers and bone tumors are presented in Table 4. The risk of cancer overall was not different from expected values in patients treated with GH (SIR 0.7, 95% CI 0.5‐1.1). The risk of bone tumor was significantly higher than expected in this population (SIR 3.5, 95% CI 1.1‐8.1). After capture‐recapture analysis (Table 4 and S1), the estimated number of missed cases of cancer overall was 3.5 (95% CI 0.8‐15.3) and the corrected SIR were 0.8 (95% CI 0.5‐1.2) for all cancers and 3.8 (95% CI 1.3‐8.6) for bone tumors. The SMR was 1.0 (95% CI 0.4‐2.1) for cancer overall and 5.0 (95% CI 1.0‐14.6) for bone tumors.
Table 4.
Crude SMR and SIR and corrected SIR for all‐type cancer and bone tumors in young adults treated with growth hormone in childhood
| Crude | Corrected | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Observed deaths (n) | Expected deaths (n) | Crude SMR (95% CI) | Observed events (n) | Expected events (n) | Crude SIR (CI 95%) | Estimated events (n) | Expected events (n) | Corrected SIR (CI 95%) | |
| All cancers | 7 | 6.9 | 1.0 (0.4‐2.1) | 23 | 32.3 | 0.7 (0.5‐1.1) | 26.5 | 32.3 | 0.8 (0.5‐1.2) |
| Bone tumors | 3 | 0.6 | 5.0 (1.0‐14.6) | 5 | 1.4 | 3.5 (1.1‐8.1) | 5.5 | 1.4 | 3.8 (1.3‐8.6) |
Bold values indicate statistical significance.
4. DISCUSSION
We report an analysis using the largest national population‐based register of patients treated with GH during childhood. Our study shows a 3.5‐3.8‐fold higher incidence of and a 5‐fold higher mortality from bone tumors in patients treated with GH for idiopathic isolated GH deficiency, idiopathic short stature, or short stature in children born short for gestational age, than in the general population. This raises issues about the mechanism, possible confounders, and causal effect of GH treatment. In contrast, the incidence and mortality of cancer overall were not different from those in the general population.
Growth hormone and IGF‐I act on bone and cartilage, specifically on chondrocytes and osteoblasts 28, 29, 30 and the GH receptor is strongly expressed on these cell types. Osteosarcomas occur preferentially in rapidly growing bones, during the rapid bone growth of puberty and in tall adolescents (patients are 0.3 SD taller than the general population).31, 32, 33 Observations of childhood cancer survivors indicate that GH treatment is associated with a large increase in the risk of osteosarcoma20: 15% of second neoplasms in this population are osteosarcomas.34, 35 The European SAGhE study has analyzed cancer morbidity using data from five countries (excluding France, due to methodological issues) 22 and also found an excess risk of bone tumors (SIR of 4.1) albeit without a clear dose‐effect relationship. Thus, evidence is growing that GH treatment may have a biological effect on bone tumor initiation, promotion, and/or progression.
The risk of cancer overall in our population of GH‐treated subjects with an SIR of 0.7‐0.8 and an SMR of 1.0 tended to be lower than that in the general population. This is consistent with the results of the meta‐analysis by Deodati et al19 — who provided summary tables of studies of cancer incidence and mortality following GH administration, with or without a cancer history — and those of the European SAGhE study.22 It is also consistent with the evidence that cancer incidence is lower in short stature than general populations, with hazard ratios applied to our cohort giving an estimated SIR of 0.8‐0.9.11, 12, 13, 14, 15, 16, 17, 18 Our observations for bone tumors are therefore even more striking, as the SIR would be expected to be lower than normal in a population of short individuals.
Our study has several potential limitations. First, noncompleteness of the sources of ascertainment of cancer was a limitation for the analysis of the incidence of cancer. We addressed this limitation by using a capture‐recapture method which gave reassuring results on the validity of our crude analysis. Capture‐recapture methods have been successfully applied in a wide range of epidemiologic fields to estimate the completeness of ascertainment of registries. Such estimation of the potential number of cases (sometimes high) missed by all sources of ascertainment improves the accuracy of assessment of disease rates.27 The second limitation is that the numbers of events and especially of fatal events (both in the study cohort and in the reference registries) are small such that the CIs, in particular CIs of SMRs, were wide. The number of events was too small to allow testing for a relationship between the dose of GH treatment and the incidence of bone tumors. Another limitation was that we were unable to include a comparable reference group of short‐stature individuals not treated with GH. Also there is the issue of whether patients with a genetic predisposition to bone tumors were inadvertently included in our low‐risk group of patients; this issue is important because retrospective genetic analysis of families was not possible in our study for legal and ethical reasons. Several genetic syndromes, some of them associated with short stature (Bloom, Rothmund‐Thompson, Werner, hereditary retinoblastoma) are risk factors for osteosarcomas 32 but would not commonly be misdiagnosed for idiopathic isolated GH deficiency. Li‐Fraumeni syndrome, by contrast, could go clinically unnoticed if questions about familial history of tumor in childhood or young adults are not asked before beginning GH treatment but is generally not associated with short stature. Mild Fanconi anemia, which could be misdiagnosed for some time in short‐stature children, does not seem to be associated with an increased of bone tumors, contrary to other forms of malignancy (leukemias, myelodysplastic syndromes, solid tumors of head, neck, skin, gastrointestinal tract, and genitourinary tract).36 These rare genetic syndromes, none of which are strongly associated with both the risk of short stature and the risk of bone tumor, are therefore unlikely to confound our results. Finally, the reference data for bone tumors we used was based on six regional registries and not national data.
In conclusion, our analyses support the view that subjects treated with GH in childhood for isolated GH deficiency or childhood short stature are at increased risk of bone tumors but not of cancer overall. A causal relationship between GH treatment and bone tumors is biologically plausible but the mechanisms remain to be elucidated and the dose‐effect relationship, if any, is yet to be described. We believe that our results should be taken into consideration in the evaluation of the risk‐benefit balance of childhood GH treatments.
CONFLICT OF INTEREST
A. Poidvin received a master grant from the French Endocrine Society (Société Française d'Endocrinologie) funded by Lilly. J.C. Carel has the following conflicts of interest to declare, all outside the scope of this work: investigator in clinical trials using GH sponsored by Pfizer and Lilly and in postmarketing studies using several brands of GH, and support for travel to international meetings from several GH manufacturers. Emmanuel Ecosse, Dominique Levy, Jean Michon, and Joël Coste report no disclosures relevant to the manuscript.
Supporting information
ACKNOWLEDGMENTS
The authors are indebted to Fabienne Landier for data collection and organization of the cohort study, Alain Weill and Philippe Ricordeau for help with the data from the French national health insurance information system (Système National d'Information Inter‐régimes de l'Assurance Maladie; SNIIRAM), and Grégoire Rey for help with the data from the French Center for Epidemiology on Medical Causes of Death (CépiDC, Institut National de la Santé et de la Recherche Médicale).
Poidvin A, Carel J‐C, Ecosse E, Levy D, Michon J, Coste J. Increased risk of bone tumors after growth hormone treatment in childhood: A population‐based cohort study in France. Cancer Med. 2018;7:3465–3473. 10.1002/cam4.1602
REFERENCES
- 1. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20‐47. [DOI] [PubMed] [Google Scholar]
- 2. Seccareccia E, Brodt P. The role of the insulin‐like growth factor‐I receptor in malignancy: An update. Growth Horm IGF Res. 2012;22:193‐199. [DOI] [PubMed] [Google Scholar]
- 3. Clayton PE, Banerjee I, Murray PG, Renehan AG. Growth hormone, the insulin‐like growth factor axis, insulin and cancer risk. Nat Rev Endocrinol. 2011;7:11‐24. [DOI] [PubMed] [Google Scholar]
- 4. Swerdlow AJ. Does growth hormone therapy increase the risk of cancer? Nat Clin Pract Endocrinol Metab. 2006;2:530‐531. [DOI] [PubMed] [Google Scholar]
- 5. Pollak MN, Schernhammer ES, Hankinson SE. Insulin‐like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505‐518. [DOI] [PubMed] [Google Scholar]
- 6. Perry JK, Liu D‐X, Wu Z‐S, Zhu T, Lobie PE. Growth hormone and cancer: an update on progress. Curr Opin Endocrinol Diabetes Obes. 2013;20:307‐313. [DOI] [PubMed] [Google Scholar]
- 7. Lea RW, Dawson T, Martinez‐Moreno CG, El‐Abry N, Harvey S. Growth hormone and cancer: GH production and action in glioma? Gen Comp Endocrinol. 2015;220:119‐123. [DOI] [PubMed] [Google Scholar]
- 8. Dal J, Leisner MZ, Hermansen K, et al. Cancer Incidence in Patients with Acromegaly: a cohort study and meta‐analysis of the literature. J Clin Endocrinol Metab. 2018; (in press). https://academic.oup.com/jcem/advance-article/doi/10.1210/jc.2017-02457/4951496. [DOI] [PubMed] [Google Scholar]
- 9. Guevara‐Aguirre J, Balasubramanian P, Guevara‐Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro‐aging signaling, cancer and diabetes in humans. Sci Transl Med. 2011;3:70ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shevah O, Laron Z. Patients with congenital deficiency of IGF‐I seem protected from the development of malignancies: a preliminary report. Growth Horm IGF Res. 2007;17:54‐57. [DOI] [PubMed] [Google Scholar]
- 11. Green J, Cairns BJ, Casabonne D, Wright FL, Reeves G, Beral V. Height and cancer incidence in the Million Women Study: prospective cohort, and meta‐analysis of prospective studies of height and total cancer risk. Lancet Oncol. 2011;12:785‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sung J, Song Y‐M, Lawlor DA, Smith GD, Ebrahim S. Height and site‐specific cancer risk: a cohort study of a korean adult population. Am J Epidemiol. 2009;170:53‐64. [DOI] [PubMed] [Google Scholar]
- 13. Gunnell D, May M, Ben‐Shlomo Y, Yarnell J, Smith GD. Height, leg length, and cancer: the Caerphilly Study. Nutr Cancer. 2003;47:34‐39. [DOI] [PubMed] [Google Scholar]
- 14. Batty GD, Barzi F, Woodward M, et al. Adult height and cancer mortality in Asia: the Asia Pacific Cohort Studies Collaboration. Ann Oncol. 2010;21:646‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Batty GD, Shipley MJ, Langenberg C, Marmot MG, Smith GD. Adult height in relation to mortality from 14 cancer sites in men in London (UK): evidence from the original Whitehall study. Ann Oncol. 2006;17:157‐166. [DOI] [PubMed] [Google Scholar]
- 16. Emerging Risk Factors Collaboration . Adult height and the risk of cause‐specific death and vascular morbidity in 1 million people: individual participant meta‐analysis. Int J Epidemiol. 2012;41:1419‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kabat GC, Heo M, Kamensky V, Miller AB, Rohan TE. Adult height in relation to risk of cancer in a cohort of Canadian women. Int J Cancer. 2013;132:1125‐1132. [DOI] [PubMed] [Google Scholar]
- 18. Walter RB, Brasky TM, Buckley SA, Potter JD, White E. Height as an explanatory factor for sex differences in human cancer. J Natl Cancer Inst. 2013;105:860‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deodati A, Ferroli BB, Cianfarani S. Association between growth hormone therapy and mortality, cancer and cardiovascular risk: systematic review and meta‐analysis. Growth Horm IGF Res. 2014;24:105‐111. [DOI] [PubMed] [Google Scholar]
- 20. Sklar CA, Mertens AC, Mitby P, et al. Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: a report from the Childhood Cancer Survivor Study. J Clin Endocrinol Metab. 2002;87:3136‐3141. [DOI] [PubMed] [Google Scholar]
- 21. Carel J‐C, Ecosse E, Landier F, et al. Long‐term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. J Clin Endocrinol Metab. 2012;97:416‐425. [DOI] [PubMed] [Google Scholar]
- 22. Swerdlow AJ, Cooke R, Beckers D, et al. Cancer risks in patients treated with growth hormone in childhood: the SAGhE European cohort study. J Clin Endocrinol Metab. 2017;102:1661‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poidvin A, Touze E, Ecosse E, et al. Growth hormone treatment for childhood short stature and risk of stroke in early adulthood. Neurology. 2014;83:780‐786. [DOI] [PubMed] [Google Scholar]
- 24. Swerdlow AJ, Cooke R, Albertsson‐Wikland K, et al. Description of the SAGhE cohort: a large European study of mortality and cancer incidence risks after childhood treatment with recombinant growth hormone. Horm Res Pædiatrics. 2015;84:172‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. InVS > Cancers > Surveillance épidémiologique des cancers en France > Principaux acteurs et partenaires institutionnels. http://www.invs.sante.fr/surveillance/cancers/acteurs.htm#3 Accessed February 7, 2018.
- 26. Hook EB, Regal RR. Recommendations for presentation and evaluation of capture‐recapture estimates in epidemiology. J Clin Epidemiol. 1999;52:917‐926; discussion 929‐933. [DOI] [PubMed] [Google Scholar]
- 27. Hook EB, Regal RR. Capture‐recapture methods in epidemiology: methods and limitations. Epidemiol Rev. 1995;17:243‐264. [DOI] [PubMed] [Google Scholar]
- 28. Slootweg MC, van Buul‐Offers SC, Herrmann‐Erlee MP, van der Meer JM, Duursma SA. Growth hormone is mitogenic for fetal mouse osteoblasts but not for undifferentiated bone cells. J Endocrinol. 1988;116:R11‐R13. [DOI] [PubMed] [Google Scholar]
- 29. Lindahl A, Nilsson A, Isaksson OG. Effects of growth hormone and insulin‐like growth factor‐I on colony formation of rabbit epiphyseal chondrocytes at different stages of maturation. J Endocrinol. 1987;115:263‐271. [DOI] [PubMed] [Google Scholar]
- 30. Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin‐like growth factors, and the skeleton. Endocr Rev. 2008;29:535‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gelberg KH, Fitzgerald EF, Hwang S, Dubrow R. Growth and development and other risk factors for osteosarcoma in children and young adults. Int J Epidemiol. 1997;26:272‐278. [DOI] [PubMed] [Google Scholar]
- 32. Broadhead ML, Clark JCM, Myers DE, Dass CR, Choong PFM. The molecular pathogenesis of osteosarcoma: a review. Sarcoma. 2011;2011:95924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Longhi A, Pasini A, Cicognani A, et al. Height as a risk factor for osteosarcoma. J Pediatr Hematol Oncol. 2005;27:314‐318. [DOI] [PubMed] [Google Scholar]
- 34. Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. Long‐term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab. 2010;95:167‐177. [DOI] [PubMed] [Google Scholar]
- 35. Ergun‐Longmire B, Mertens AC, Mitby P, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab. 2006;91:3494‐3498. [DOI] [PubMed] [Google Scholar]
- 36. Mehta PA, Tolar J. Fanconi anemia In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993. ‐2018. https://www.ncbi.nlm.nih.gov/books/NBK1401/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials