

ABSTRACT
Mutations in the mitochondrial inner membrane ATPase ATAD3A result in neurological syndromes in humans. In mice, the ubiquitous disruption of Atad3 (also known as Atad3a) was embryonic lethal, but a skeletal muscle-specific conditional knockout (KO) was viable. At birth, ATAD3 muscle KO mice had normal weight, but from 2 months onwards they showed progressive motor-impaired coordination and weakness. Loss of ATAD3 caused early and severe mitochondrial structural abnormalities, mitochondrial proliferation and muscle atrophy. There was dramatic reduction in mitochondrial cristae junctions and overall cristae morphology. The lack of mitochondrial cristae was accompanied by a reduction in high molecular weight mitochondrial contact site and cristae organizing system (MICOS) complexes, and to a lesser extent in OPA1. Moreover, muscles lacking ATAD3 showed altered cholesterol metabolism, accumulation of mitochondrial DNA (mtDNA) replication intermediates, progressive mtDNA depletion and deletions. Unexpectedly, decreases in the levels of some OXPHOS components occurred after cristae destabilization, indicating that ATAD3 is not crucial for mitochondrial translation, as previously suggested. Our results show a critical early role of ATAD3 in regulating mitochondrial inner membrane structure, leading to secondary defects in mtDNA replication and complex V and cholesterol levels in postmitotic tissue.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: ATAD3, Mitochondria, Cristae, MICOS complex, Muscle, Cholesterol
Highlighted Article: Here, we show that mice without ATAD3 in muscle develop a myopathy associated with early abnormal mitochondrial cristae structure, followed by defects in lipid transport and mitochondrial DNA maintenance.
INTRODUCTION
ATPase family AAA domain-containing protein 3 (ATAD3; also known as ATAD3A) is a mitochondrial inner membrane ATPase expressed ubiquitously in plants and animals. Deletion of Atad3 and its orthologs is lethal during early embryonic development in Caenorhabditis elegans (Hoffmann et al., 2009), Drosophila melanogaster (Gilquin et al., 2010; Harel et al., 2016) and Mus musculus (Goller et al., 2013). A single copy of this gene is present in the haploid genomes of pluricellular organisms, including mouse, which has a single gene that expresses two alternatively spliced isoforms (Li et al., 2014). In contrast, in primates and humans, the gene has been duplicated twice in tandem, giving rise to ATAD3A, ATAD3B and ATAD3C. ATAD3B differs from the ancestral paralog ATAD3A by having a C-terminal extension of 62 amino acids, which is caused by a mutation in the original stop codon, whereas ATAD3C is a truncated gene, having lost the first 70 amino acids. In humans, the ATAD3A gene is expressed ubiquitously and is particularly abundant in the central nervous system. In contrast, the ATAD3B gene is not expressed in the adult brain, but seems to be preferentially expressed in embryos, adult germinal zones of the brain and human astrocytoma cell lines (Hubstenberger et al., 2008; Schaffrik et al., 2006). There are no experimental data on ATAD3C expression (Li and Rousseau, 2012).
ATAD3 is localized to the mitochondrial inner membrane and reported to interact with both the inner and outer mitochondrial membranes (Baudier, 2017; Gilquin et al., 2010). The C-terminal region of ATAD3 contains an ATPase domain in the mitochondrial matrix (Fig. S1). Towards the N-terminal region, ATAD3 has: (1) at least two transmembrane domains, (2) two coiled-coil domains, which could be important for interaction with other proteins and the oligomerization of ATAD3 monomers, and (3) a proline-rich domain of unknown function (Hubstenberger et al., 2010). ATAD3 is believed to form oligomers for two reasons: all AAA proteins investigated to date form hexameric rings (Frickey and Lupas, 2004), and overexpression of a hemagglutinin-tagged version of ATAD3, or a dominant negative mutant, resulted in the same phenotype as knockdown of the gene (Harel et al., 2016; He et al., 2012).
The family of AAA-ATPases (ATPases associated with diverse cellular activities) includes proteins with ATPase activity involved in numerous cellular processes, such as replication, transcription, translation, vesicular transport, proteolysis and others (Frickey and Lupas, 2004). ATAD3 has been implicated in several cell functions, including mitochondrial activity (Harel et al., 2016; Hoffmann et al., 2009), mitochondrial dynamics (Gilquin et al., 2010; Harel et al., 2016), lipid biosynthesis (Desai et al., 2017; Issop et al., 2015), and cell growth and cancer (Fang et al., 2010; You et al., 2013). Overexpression of the Drosophila ortholog of Atad3 (bor) resulted in increased number of elongated mitochondria, suggesting a role in mitochondrial fusion or fission (Harel et al., 2016). ATAD3 is found in mitochondrial DNA (mtDNA)-containing nucleoprotein structures know as nucleoids (Bogenhagen et al., 2008; He et al., 2007; Wang and Bogenhagen, 2006), and several studies have associated ATAD3 with the maintenance of mtDNA (Desai et al., 2017; Gerhold et al., 2015; Holt et al., 2007). Knockdown of ATAD3 resulted in disruption of the membrane-associated nucleoid fraction (Gerhold et al., 2015) and alteration of mtDNA topology (Desai et al., 2017; Holt et al., 2007). ATAD3 has also been associated with mtDNA translation, as silencing of human ATAD3A and ATAD3B genes in cultured cells abolished mitochondrial protein synthesis (He et al., 2012). Moreover, ATAD3 has been found in steroidogenic cells in the contacts between endoplasmic reticulum (ER) membranes and the outer mitochondrial membrane, termed mitochondria-associated membranes (Gilquin et al., 2010; Issop et al., 2015). Finally, ATAD3 has been suggested to regulate cholesterol metabolism in steroidogenic cells (Issop et al., 2015) and in fibroblasts from patients (Desai et al., 2017).
The mitochondrial inner membrane has two specialized domains: the cristae membranes and the inner boundary membrane, which is in close contact with the outer membrane (Vogel et al., 2006). The cristae are large invaginations of the inner membrane, where the respiratory chain complexes and ATP synthase are preferentially located (Gilkerson et al., 2003), while the inner boundary membrane is enriched in import-protein translocases (Chacinska et al., 2009; Wiedemann and Pfanner, 2017). Cristae biogenesis and maintenance strongly depend on a protein complex called the mitochondrial contact site and cristae organizing system (MICOS) (Hoppins et al., 2011) and optic atrophy protein 1 (OPA1) (Frezza et al., 2006). In humans, the MICOS is composed of six subunits: MIC10, MIC19, MIC25, MIC27, MIC28 and MIC60. The study of MICOS in Saccharomyces cerevisiae and Homo sapiens has characterized both MIC60 and MIC10 as the two functionally most important subunits of MICOS (Pfanner et al., 2014). A recent study has placed OPA1 upstream of MIC60 and in a pathway controlling cristae junction (CJ) stability (Glytsou et al., 2016). Lastly, OPA1 is essential for mtDNA maintenance as it mediates mitochondrial fusion, which results in the exchange of mitochondrial contents and maintenance of a homogeneous and balanced pool of mitochondrial proteins including the enzymes needed for mtDNA synthesis (Chen et al., 2010). In the past few years, mutations in ATAD3 genes have been associated with different neurological syndromes. Dominant point mutations (Cooper et al., 2017; Harel et al., 2016), a recessive point mutation (Harel et al., 2016) and biallelic deletions (Desai et al., 2017; Harel et al., 2016) have been described.
To address the role of ATAD3 in vivo, we investigated the consequences of its gene ablation in mouse skeletal muscle. Here, we report that knocking out the single Atad3 gene in muscle disrupts mitochondrial cristae organization, altering OPA1 steady-state levels and MICOS complex assembly, subsequently leading to cholesterol changes, mtDNA replication abnormalities and mtDNA depletion. These findings underscore the primary importance of ATAD3 in mitochondrial cristae organization in mammals.
RESULTS
Generation of Atad3 skeletal muscle knockout mice
Ubiquitous ablation of ATAD3 in mice resulted in embryonic lethality (Fig. S1; Goller et al., 2013). To understand the in vivo function of ATAD3 in mammals we generated a conditional knockout (KO) allele of the mouse Atad3 gene (Fig. S1). ATAD3 skeletal muscle-deficient mice (Atad3-Mlc1f KO) were obtained by crossing the Atad3 floxed animals (generated by us, see Materials and Methods) with Mlc1f-Cre transgenic mice, expressing Cre recombinase under the myosin light chain 1 promoter (Fig. S1; Bothe et al., 2000). The myosin light chain 1 (Mlc1f; also known as Myl1) gene is expressed strongly and selectively in differentiated skeletal muscle (Lyons et al., 1990). The presence of the various alleles and recombination was determined by PCR amplification from tail DNA and tissue samples (Fig. S1). We found the Atad3 exon 2 to be deleted selectively in various skeletal muscles, but not in liver (Fig. S1). Although a low level of the Atad3 conditional allele (Floxed-Atad3) remained detectable in the KO muscle tissue, this is compatible with close-to-full excision of the gene and contamination of muscle preparations with other cells types not expressing Mlc1f (e.g. endothelial cells, Schwann cells, adipose cells, satellite cells and fibroblasts) (Schmalbruch and Hellhammer, 1977). In mouse muscle, Atad3 is expressed as a 67 kDa isoform (Li et al., 2014), and the loss of ATAD3 was confirmed at protein level by western blot analysis in mitochondrial extracts of skeletal muscles (Fig. 1A; Fig. S1).
Fig. 1.

Creation of muscle-specific Atad3 KO mice. (A) Western blot analysis of ATAD3 protein levels in mitochondria from hind-limb skeletal muscle of 3-month-old animals. VDAC1 was used as a loading control. (B) Body weight measurement: Atad3-Mlc1f male KO mice (red) showed a significantly reduced weight compared with age-matched control (CTR) mice (blue) from 2 months of age onwards (n=12 in KO group and n=15 in control group). (C,D) Representative images of control and Atad3-Mlc1f KO male mice of 5 (C) and 18 (D) months of age, showing decreased body weight and development of kyphosis in the Atad3-Mlc1f KO male mice compared with the controls. (E) Atad3-Mlc1f KO mice showed reduced muscle strength compared with control mice at 6 months of age (n=10). (F,G) Treadmill (F, n=8) and rotarod tests (G, n=12) performed by Atad3-Mlc1f KO and control mice. Data are mean±s.e.m. P-values were calculated by Student's t-test. n.s., nonsignificant; *P<0.05, **P<0.01 and ***P<0.0001.
Atad3-Mlc1f KO mice showed progressive myopathy
Atad3-Mlc1f KO mice were born with similar body weight as their control littermates. However, at the age of 2 months in males, and 5 months in females, they showed lower body weight (Fig. 1B; Fig. S2). These differences were exacerbated throughout life, as ATAD3 muscle KO mice gained weight at a slower rate than control mice (Fig. 1B,C; Fig. S2). Atad3-Mlc1f KO mice developed a noticeable kyphosis starting at 6 months of age, a feature caused by loss of spinal muscle tonus (Fig. 1D; Fig. S2). Despite the dramatically reduced muscle tonus, mice (males and females) survived up to at least 20 months. Heterozygous knockout mice for ATAD3 were indistinguishable from controls in terms of body weight (Fig. S3).
To study the muscle phenotype in these mice, we used several behavioral assays. Maximal grip strength of the fore-limbs was measured with a grip strength analyzer at different ages, recording the best of three trials as maximum strength. Atad3-Mlc1f KO mice showed reduced maximal force at 6 months with no further worsening (Fig. 1E). Motor activity in male Atad3-Mlc1f KO mice was assessed by the treadmill test. At 2 months, Atad3-Mlc1f KO mice performance was comparable to that of their control littermates. However, from 3 months onwards Atad3-Mlc1f KO mice fell more frequently than controls, suggesting impaired locomotor activity (Fig. 1F). To study whether motor coordination was affected in Atad3-Mlc1f KO mice we performed rotarod tests on male mice. When allowed to walk on a cylinder that accelerates progressively, the KO mice could not stay on the cylinder as long as controls did (Fig. 1G).
To further characterize the muscle phenotype, we tested the mice in open field tests and noticed a progressive decline in exploratory activity (Fig. S2A,B). Atad3-Mlc1f KO mice showed decreased travelling distance, ambulatory counts and increased resting time (Fig. S2A,B). In females, we analyzed the motor phenotype only at 18 months of age and found similar impairment of motor performance and decreased strength as in males (Fig. S2C-I).
In summary, lack of ATAD3 in skeletal muscle caused a severe myopathy, as shown by reduced body weight, muscle weakness and progressive decline in locomotor activity, starting at the age of 3 months.
Loss of ATAD3 in skeletal muscle induces muscle atrophy without cytochrome oxidase-negative fibers
To determine the effect of the Atad3 deletion in skeletal muscle in mice, we dissected and weighted three hind-limb muscles of different aged control and KO males. Gross examination showed smaller muscles (Fig. 2A). The Atad3-Mlc1f KO mice showed a gradual decrease of absolute and relative muscle size with increasing age (Fig. 2A,B; Fig. S4A). Absolute muscle weight was already decreased at 3 months of age, while muscle weight/body weight ratio was detected only at 5 months. To rule out the possibility that Atad3-Mlc1f KO mice ate less than their control littermates, we measured food intake in both groups. Atad3-Mlc1f KO mice ate the same amount of food as the controls, but weighed less (Fig. S3A,B). Similar muscle wasting was observed in female KO mice (Fig. S3C). The reduction in muscle weight was associated with the lack of ATAD3 protein, as other tissues expressing ATAD3, such as heart, exhibited normal weight (Fig. S3D). Structural integrity of the skeletal muscle was assessed by detection of sarcomeric proteins. Atad3-Mlc1f KO muscles showed a significant decrease in myosin heavy chain levels at 12 months of age (Fig. S3E). Moreover, Hematoxylin and Eosin staining showed degenerating fibers in 18-month-old Atad3-Mlc1f KO mice (Fig. S3F). Muscle fibers from Atad3-Mlc1f KO mice had a significantly lower cross-sectional area compared with controls of the same age (Fig. 2C).
Fig. 2.
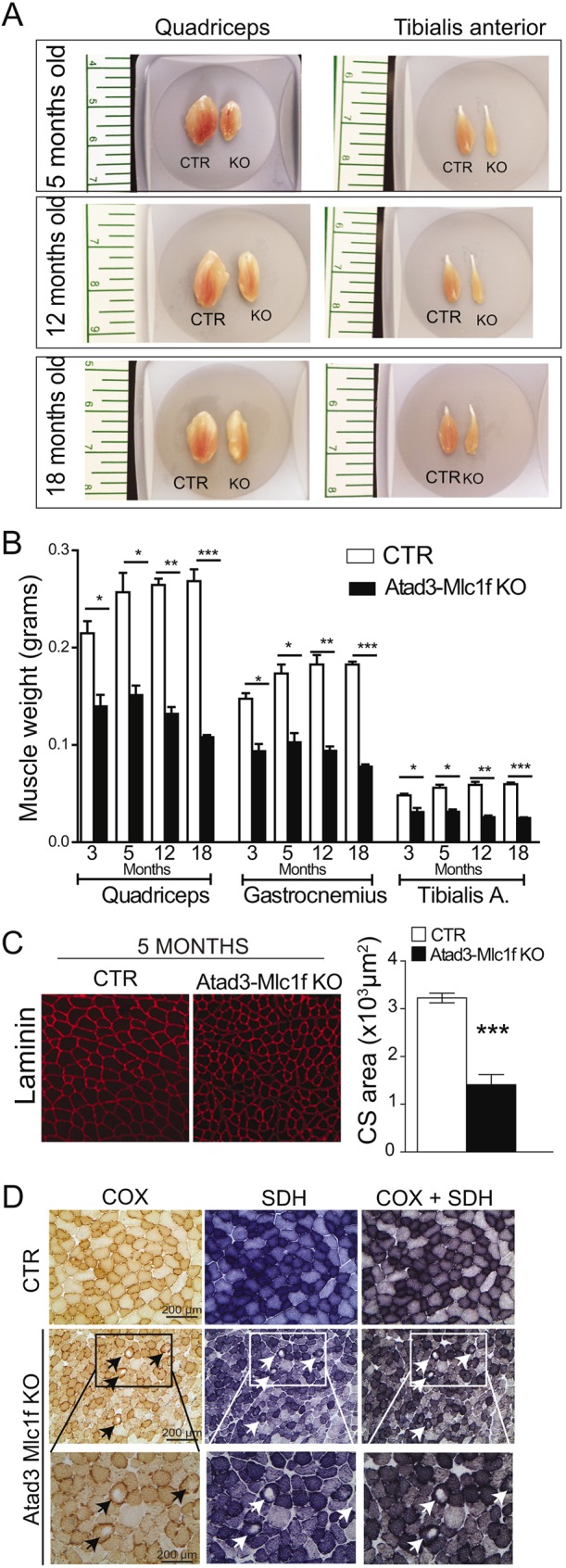
Muscle phenotype of Atad3 knockout mice. (A) Gross appearance of quadriceps and tibialis anterior of Atad3-Mlc1f KO mice. (B) Muscle weight over time of male Atad3-Mlc1f KO mice; n=6 in KO group and n=8 in control group. Data are mean±s.e.m. P-values were calculated by Student's t-test. *P<0.05, **P<0.01 and ***P<0.0001. (C) Representative laminin staining of tibialis anterior muscle from control and Atad3-Mlc1f KO mice of 5 months of age used for quantification of cross-sectional area of the fibers. (D) Representative COX, SDH and COX+SDH staining of skeletal muscle (tibialis anterior) from control and Atad3-Mlc1f KO females of 18 months of age. COX activity is shown in brown and SDH in blue. Arrows indicate central cores lacking COX and SDH activity. Scale bars: 200 µm.
Previous work by He et al. showed a dramatic decrease in mitochondrial protein synthesis upon ATAD3 knockdown in human embryonic kidney cells (He et al., 2012). Therefore, we expected the mitochondrial respiratory complexes containing subunits encoded by mtDNA, such as cytochrome c oxidase (COX), to be affected. To study the potential effect of ATAD3 knockout on OXPHOS complexes, we determined the levels of COX activity by histochemical analyses in muscle sections. Surprisingly, we found similar COX activity staining in the Atad3-Mlc1f KO mice and controls (Fig. 2D; Fig. S4B). Interestingly, we observed pale cores in the muscle fibers of the KO tissues that were not present in controls (Fig. 2D; Fig. S4B, arrows). These cores were negative for both COX and SDH (succinate dehydrogenase) activities, indicating drastically reduced mitochondrial mass in focal areas. In younger animals only one small core per fiber was detected, whereas in older animals the fibers presented larger single cores, but also more than one core (between two and four) (compare 18-month-old with 5-month-old KO mice; Fig. S4B, arrows). This result suggests that ATAD3 has a key role in the preservation of the mitochondrial network in muscle. In addition, aged Atad3-Mlc1f KO mice showed increased COX and SDH staining in the periphery of some fibers, whereas the mitochondrial staining in fibers from control mice was more homogenous, indicating subsarcolemmal mitochondrial proliferation upon ATAD3 knockout. Moreover, we observed increased empty spaces between the muscle fibers in the samples from aged KO mice (Fig. 2D; Fig. S4B), likely to be a result of muscle fiber atrophy. In agreement with the mitochondrial proliferation suggested by histochemistry, we found that the levels of mitochondrial markers (VDAC1 and SDHA) and PGC-1α (also known as Ppargc1a), a key co-transcriptional factor regulating mitochondrial proliferation, were increased in 5-month-old mice (Fig. S4C).
In summary, the lack of ATAD3 in skeletal muscle induced a progressive myopathy with an onset between 2 and 3 months of age, developing into muscle wasting, reduced fiber size and altered distribution of mitochondria in the fibers. Surprisingly, ATAD3 loss in muscle was not associated with COX-negative fibers, but with increased PGC-1α, SDHA and VDAC protein levels.
Lack of ATAD3 leads to early cristae structure disorganization, reduced cristae surface and reduced mitochondrial size
Knockdown of ATAD3 in mouse and human cultured cells was reported to induce abnormalities in mitochondrial structure (Gerhold et al., 2015; Issop et al., 2015). Therefore, we studied the mitochondrial ultrastructure in skeletal muscle from 2- and 5-month-old Atad3-Mlc1f KO mice by transmission electron microscopy (TEM). Atad3-Mlc1f KO preparations from 2-month-old animals showed that mitochondria cristae lost the contacts with the outer membrane, the CJs (Fig. 3A, arrows). We also noticed that the remaining cristae were mostly oriented parallel to the mitochondrial outer membrane, in some cases forming concentric circles (Fig. 3A; Fig. S5C). The number of cristae with CJs was reduced by 40% in the KO samples (Fig. 3B). In addition, the cristae surface per mitochondria was decreased in 2-month-old Atad3-Mlc1f KO mice (Fig. 3C). Despite the disruption, mitochondrial cristae had a lamellar morphology. However, in the preparations from 5-month-old animals, this lamellar structure of the cristae was mostly absent with predominant circular structures, indicating that mitochondria CJs were markedly lost (Fig. 3A; Fig. S5). Moreover, mitochondria presented other abnormalities including clear matrix areas, electron-dense inclusions, fragmentation and degeneration (Fig. S5). By contrast, none of these ultrastructural defects were present in control mice. At lower magnification, we observed muscle organization to be affected in preparations from 5-month-old Atad3-Mlc1f KO animals (Fig. S5E). In addition, quantification of the subsarcolemmal mitochondria on TEM pictures indicated that Atad3-Mlc1f KO muscles had reduced mitochondrial size (Fig. 3D). These findings indicate that ATAD3 is required for the integrity of mitochondrial cristae in skeletal muscle, and that cristae disruption is the first detectable phenotype in these mice.
Fig. 3.
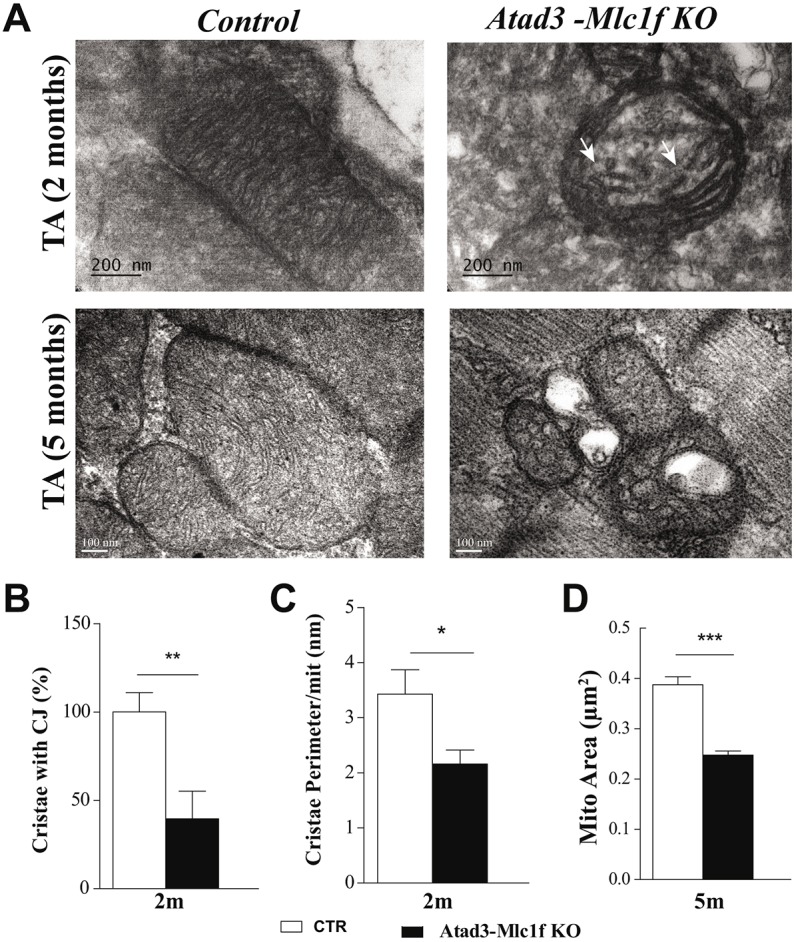
Lack of ATAD3 in muscle disrupts mitochondrial cristae structure and causes a decrease in mitochondrial size. (A) Representative electron micrographs from 2- and 5-month-old control and Atad3-Mlc1f KO tibialis anterior (TA). Mitochondria from KO tibialis anterior showed disrupted cristae (arrows). Scale bars: 200 nm (upper row), 100 nm (lower row). (B) Quantification of the cristae junctions (CJ) from >200 mitochondria from control and Atad3-Mlc1f KO mice, expressed as percentage of the control mice. (C) Quantification of the cristae surface/mitochondria from >200 mitochondria from control and Atad3-Mlc1f KO mice, expressed as cristae perimeter per mitochondria perimeter. (D) Quantification of the area of mitochondria from >400 mitochondria from control and Atad3-Mlc1f KO mice. Data are mean±s.e.m., n=3 per group. P-values were calculated by Student's t-test. *P<0.05, **P<0.01 and ***P<0.0001.
Effect of ATAD3 ablation on mitochondria metabolism and OXPHOS complexes
To further characterize the effects of ATAD3 loss on mitochondria metabolism, we measured the steady-state levels of OXPHOS subunits in the mitochondrial fractions of Atad3-Mlc1f KO and control skeletal muscle. We found that the steady-state levels of markers of complex I (NDUB8) and complex V (ATP5A) were drastically decreased in mitochondria of Atad3-Mlc1f KO mice from the age of 3 months onwards (50% lower; Fig. 4A-C). A more modest decrease in the levels of the mtDNA-encoded protein COX1 (also known as mt-Co1) was observed at 5 months, but not at earlier ages (Fig. 4A-C). Levels of complex II (SDHA) and complex III (UQCR1) markers remained unchanged at all ages tested (Fig. 4A,B). To elucidate whether the lack of ATAD3 affected the steady-state levels of OXPHOS supercomplexes, we analyzed mitochondria from skeletal muscle by blue native polyacrylamide gel electrophoresis (BN-PAGE). The main OXPHOS alterations observed in Atad3-Mlc1f KO mitochondria from muscles were reduced levels of OXPHOS supercomplexes (containing complex I and III) and reduced levels of F1F0-ATP synthase or complex V (Fig. 4D,E; Fig. S6). Complex V levels have been shown to rely on cristae morphology (Cogliati et al., 2013), supporting a role for ATAD3 in cristae structure. Atad3-Mlc1f KO mice also showed accumulation of the hydrophilic portion of the ATP-synthase, F1, which can be a consequence of decreased stability (Fig. 4D; Fig. S6). As alluded above, there were smaller decreases in complex IV at 3 months (Fig. 4D,E). Lastly, the levels of complex II, which is entirely encoded by nuclear DNA (nDNA), was found to be normal in young ATAD3 Mlc1f KO mice and increased in 18-month-old KO mice (Fig. 4D,E; Fig. S6).
Fig. 4.

Analysis of OXPHOS complexes in control and Atad3-Mlc1f KO skeletal muscle mitochondria. (A,B) Western blot analysis of mitochondrial protein levels in purified mitochondria from the muscle of 3- and 5-month-old control and Atad3-Mlc1f KO mice. (C) Quantification of the steady-state levels of respiratory subunits shown in A and B. VDAC1 was used as a loading control. (D) BN-PAGE analysis of respiratory chain supercomplexes followed by western blot analysis using antibodies against complex I (anti-NDUFA9), complex II (anti-SDHA), complex III (anti-UQCRC1), complex IV (anti-COXI), and complex V (anti-ATP5Α) in mitochondrial extract from muscles of 3-month-old control and Atad3-Mlc1f KO mice. VDAC1 was used as a loading control. (E) Quantification of relative levels of OXPHOS complexes and supercomplexes. (E) In-gel activity of complex V (ATP synthase) in muscle mitochondria from 5-month-old control and Atad3-Mlc1f KO mice. Complex V activity in gel is reduced in the mitochondrial extract from Atad3-Mlc1f KO muscle. Data are mean±s.e.m. P-values were calculated by Student's t-test. ns, nonsignificant; *P<0.05 and **P<0.01.
To determine whether complex V was affected by the loss of ATAD3, we prepared membrane extracts in conditions that preserve complex V dimers (ratio of 1:1.5, protein:digitonin), and determined the in-gel ATPase activity. Atad3-Mlc1f KO mitochondria showed reduced complex V activity in all the bands detected: multimeric, Vx; dimeric, V2; and monomeric, V1, forms of complex V (Fig. 4F). We also measured complex I and complex IV activities in mitochondrial extracts spectrophotometrically. At 18 months of age, mitochondria from Atad3-Mlc1f KO mice showed a 50% reduction in complex I activity (Fig. S6A). There were no statistically significant differences in the complex IV activity assay results between Atad3-Mlc1f KO and control mice (Fig. S7B). Lactate levels in sera of Atad3-Mlc1f KO were not increased (Fig. S7C).
Loss of ATAD3 destabilizes the large MICOSs
The MICOS and OPA1 regulate mitochondrial cristae shape (Cogliati et al., 2016; Hoppins et al., 2011). Therefore, we speculated that ATAD3 might affect the formation or stabilization of these complexes.
Although OPA1 levels were not changed in 3-month-old muscle, they were reduced at 5 months, when normalized to VDAC1 (Fig. 5A–C). To determine how this ATAD3 depletion affects larger protein complexes containing these factors, mitochondria from control and Atad3-Mlc1f KO muscle were analyzed by BN-PAGE and western blotting using antibodies against ATAD3, OPA1 and the key component of the MICOS complex, MIC60 (John et al., 2005; Odgren et al., 1996). We found that muscle ATAD3 is mainly present in a higher molecular weight complex (HMWC) of ∼900 kDa, and in small proportions in other complexes spanning 720 to 300 kDa (Fig. 5D,F). OPA1 was present mostly in a diffuse band of ∼500–600 kDa (Fig. 5E), whereas MIC60 had a fraction in high molecular weight complexes of ∼900 and 1100 kDa, and a smaller one of ∼500–600 kDa (Fig. 5G). In the absence of ATAD3, OPA1 complex levels were increased in 3-month-old muscle, to a similar extent as other mitochondrial markers such as VDAC1 (Fig. 5E). In the case of MIC60, the HMWCs of ∼1100 kDa and 900 kDa (co-migrating with a fraction of MIC60) were reduced (Fig. 5G, rectangle), and a subcomplex of ∼500–600 kDa accumulated (Fig. 5G, asterisk). At 18 months, the level of this smaller subcomplex was even higher (Fig. 5H). Two-dimensional (2D) BN-SDS corroborated that the MIC60-containing HMWCs were markedly reduced in Atad3-Mlc1f KO mitochondria (Fig. S8A). However, steady-state levels of MIC60 were not decreased in the Atad3-Mlc1f KO mitochondria (Fig. S8B). Therefore, these findings imply that ATAD3 plays a role in the formation or stabilization of the large MIC60-containing complex. Previously, MICOS components have been shown to interact with VDAC1 (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011). In 2D BN-SDS gels, we observed alterations of VDAC1 complexes in the absence of ATAD3 (Fig. S8C).
Fig. 5.

Lack of ATAD3 decreases OPA1 levels and affects MICOS complex assembly. (A,B) Western blot analysis of ATAD3 and OPA1 levels in muscle mitochondria from 3- and 5-month-old control and Atad3-Mlc1f KO mice. ‘L’ is the long form and ‘S’ the short form of OPA1. (C) Quantification of the steady-state levels shown in A and B. VDAC1 was used as a loading control. Data are mean±s.e.m. P-values were calculated by Student's t-test. **P<0.01. (D,E) BN-PAGE analysis of mitochondrial complexes followed by western blot analysis using antibody against ATAD3 and OPA1 in mitochondria from the muscles of 3-month-old control and Atad3-Mlc1f KO animals. (F,G) BN-PAGE analysis of mitochondrial protein complexes followed by western blot analysis using antibody against ATAD3 and MIC60 in mitochondria from 3-month-old control and Atad3-Mlc1f KO mice. The same samples were analyzed by SDS-PAGE to determine ATAD3 and MIC60 levels. SDHA was used as a loading control. The asterisk indicates the accumulation of a subcomplex of ∼500–600 kDa. (H) BN-PAGE analysis of MICOS complex followed by western blot analysis using antibody against MIC60 in mitochondria from muscles from 18-month-old control and Atad3-Mlc1f KO mice. VDAC1 was used as a loading control. Atad3-Mlc1f KO muscle mitochondria showed accumulation of subcomplexes containing MIC60.
This set of experiments showed that factors important for cristae structure were preferentially affected by the lack of ATAD3, including OXPHOS complex V and MICOS. OPA1 appeared to be destabilized later in the process, as cristae loss became more prominent, and mitochondria were smaller.
ATAD3-deficient muscles display altered cholesterol trafficking
Fibroblasts from patients associated with rearrangements in the ATAD3 gene cluster have impaired cholesterol metabolism (Desai et al., 2017). To evaluate the effect of ATAD3 ablation on lipid metabolism, we analyzed the lipid composition of skeletal muscle from 2- and 5-month-old animals by high-performance liquid chromatography (HPLC). In the samples extracted from 2-month-old muscle, we did not find significant differences in the lipid composition between control and Atad3 KO mice (Fig. 6A). However, at 5 months of age, the muscle of KO animals had a different distribution of the cholesterol esters (CEs) compared with control littermates (Fig. 6B). Atad3 KO muscles had decreased levels of CEs synthesized in the ER (generally containing short saturated or monounsaturated acyl chains) and increased levels of CEs obtained from the diet (generally containing longer and more unsaturated acyl chains) (Fig. 6B). On average, we detected lower levels of total esters (CEs) versus free cholesterol in the Atad3 KO muscles compared with controls (Fig. 6C, animals of 5 months of age). The ratio of CEs/free cholesterol has been shown to reflect the degree of cholesterol trafficking between plasma membrane and ER. When decreased, it indicates a lower degree of activation of acetyl-CoA acetyltransferase (ACAT), which results from decreased cholesterol transference to the ER (Härmälä et al., 1994; Slotte et al., 1990).
Fig. 6.

Loss of ATAD3 in muscle impairs cholesterol trafficking and results in mtDNA depletion, replication stalling and rearrangements. (A,B) Lipidomic analysis of the different cholesterol esters species related to total cholesterol in gastrocnemius muscle of control and Atad3-Mlc1f KO mice aged 2 (A) and 5 (B) months. Data are expressed as fold change over the controls and represented as heat maps. (C) Quantification of the total cholesterol esters/free cholesterol in muscle from 5-month-old control and Atad3-Mlc1f KO mice. Data are mean±s.e.m. n=4 in control group; n=5 in KO group. P-values were calculated by Student's t-test. (D) Analysis of mtDNA levels in skeletal muscle from Atad3-Mlc1f KO and control mice of 3, 5, 12 and 18 months age (tibialis anterior). mtDNA/nDNA levels were measured by real-time PCR from four control and five KO animals. Data are mean±s.e.m. P-values were calculated by Student's t-test. (E) Analysis of mtDNA replication intermediates (bands within the dashed blue lines) in the skeletal muscle of Atad3-Mlc1f KO and control mice. Mitochondrial nucleic acids from two Atad3-Mlc1f KO and two control quadriceps were digested with ClaI and separated by 2D-AGE; the blot was hybridized with a part of the cytochrome B sequence of the murine mitochondrial genome. (F) Frequencies of breakpoint positions in muscle from 20-month-old Atad3-Mlc1f KO and control mice obtained by next generation sequencing (NGS). Breakpoint position of 5′ bases shown in blue and 3′ bases in orange as 100 bp rolling average. The numbering corresponds to the murine mtDNA reference sequence (NC_005089). n.s., nonsignificant; * P<0.05 and **P<0.01.
Altogether, the distribution and relative levels of CEs suggest that in mutant muscle, cholesterol is internalized but does not reach the ER. ACAT activity is downregulated and, as a consequence, cholesterol would not reach the mitochondria.
Lack of ATAD3 impairs mtDNA replication and induces mtDNA depletion
To determine whether ATAD3 ablation in mice affected mtDNA content, we examined the levels of mtDNA/nDNA by quantitative PCR in skeletal muscles. The Atad3-Mlc1f KO mice showed a gradual decrease in mtDNA/nDNA levels with increasing age (Fig. 6D). A 30% reduction in mtDNA was detected at 5–6 months in the Atad3-Mlc1f KO muscle. In older animals, differences were more pronounced, with 55% and 45% reduction at 12 and 18 months of age, respectively (Fig. 6D). As shown in Fig. S4, Atad3-Mlc1f KO skeletal muscles presented increased PGC-1α and SDHA levels, probably as a result of the induction of mitochondrial biogenesis as a compensatory mechanism. Still, we detected a reduction in mtDNA levels, indicating that ATAD3 affects mtDNA maintenance. To assess the effect of ATAD3 ablation on mtDNA replication, we analyzed the intermediates of mitochondrial DNA replication using neutral 2D agarose gel electrophoresis followed by Southern blotting (2D-AGE) (Friedman and Brewer, 1995). We detected higher levels of replication intermediates in 5-month-old Atad3 KO mitochondria compared with controls (Fig. 6E). The accumulation of mtDNA replication intermediates concomitantly with copy number reduction is a hallmark of mitochondrial replication stalling (Goffart et al., 2009; Wanrooij et al., 2007).
Impaired mtDNA replication is known to induce mtDNA rearrangements (deletions and duplications) in cultured cells (Pohjoismäki et al., 2011). Therefore, we hypothesized that ablation of ATAD3 could lead to increased levels of mtDNA rearrangements. To study the effects of ATAD3 loss on mtDNA maintenance, we analyzed purified mtDNA from isolated muscle mitochondria of 20-month-old Atad3-Mlc1f KO mice and controls by next generation sequencing (NGS) (Williams et al., 2010). mtDNA from Atad3 KO muscle contained increased levels of rearranged molecules, with enriched breakpoints around the mtDNA positions 2700 bp and 15,900 bp (Fig. 6F, Fig. 7A,B). To confirm the presence of mtDNA rearrangements, PCR using primers flanking the breakpoints regions was performed (Fig. 7C). A deletion breakpoint region product was obtained from all aged Atad3-Mlc1f KO samples, but it was not present in age-matched controls or in younger KO mice (Fig. 7C). These results suggest that low levels of mtDNA rearrangements, caused by impaired replication (Tyynismaa et al., 2005), occur in skeletal muscle when ATAD3 is ablated and accumulate during aging. We were also able to detect high levels of age-related mtDNA deletions previously described from dopaminergic neurons (Neuhaus et al., 2014) in the skeletal muscle of 12- and 18-month-old Atad3-Mlc1f KO mice (Fig. 7D). Although rearranged breakpoints are commonly associated with mtDNA deletions, we cannot rule out the possibility that the mtDNA rearrangements present in Atad3-Mlc1f KO samples are caused by sequence duplication. Taken together, our results indicate that loss of ATAD3 in skeletal muscle results in mtDNA replication stalling that leads to progressive mitochondrial DNA depletion and mtDNA rearrangements in aged animals.
Fig. 7.

mtDNA deletions in Atad3-Mlc1f KO mice. (A) Representation in dots of the breakpoints positions in mtDNA muscle from 20-month-old Atad3-Mlc1f KO and control mice, obtained by NGS. Numbering in y- and x-axes corresponds to standard murine mtDNA reference sequence (NC_005089). (B) Frequency of mtDNA rearrangements between position 2700 and 1545 bp in muscle from 20-month-old control and KO mice expressed as percentage of mtDNA. (C) Schematic representation of wild-type murine mitochondrial DNA and of the deleted mtDNA based on the breakpoints detected by NGS and shown in Fig. 6F. DNA amplification of mtDNA from 6- and 20-month-old control and KO mice using primers flanking the breakpoints detected in NGS. (D) Age-related mtDNA deletion (del A) was measured in total DNA from muscle by quantitative PCR (n=5 per group). Data are mean±s.e.m. (E) FGF21 levels in serum (n=5 per group), expressed as mean±s.e.m. *P<0.05.
Finally, we determined the levels of the myokine FGF21 in sera of the mice. FGF21 was shown to be increased in sera of humans with mtDNA-related myopathies and in mouse models of mtDNA maintenance and mtDNA translation disorders (Lehtonen et al., 2016). Indeed, FGF21 levels were significantly increased in Atad3-Mlc1f KO muscle showing mtDNA depletion compared with control littermates (Fig. 7E).
DISCUSSION
In this study, we have investigated the in vivo function of the mitochondrial inner membrane ATPase, ATAD3, using a muscle-specific conditional knockout mouse model. Previous work using cultured cells had implicated ATAD3 in nucleoid structure and mtDNA metabolism, mitochondrial protein synthesis, OXPHOS function and lipid transport. However, the relevance of these changes in vivo was unknown (Desai et al., 2017; Gerhold et al., 2015; He et al., 2007). Our results showed that loss of ATAD3 in skeletal muscle results in impairment of mitochondrial ultrastructure, reduced number of CJs and decreased cristae surface. Concomitantly with the role of ATAD3 in organizing mitochondrial cristae structure, enzyme complexes known to be affected by cristae structure, such as OXPHOS complex V and MICOS, also showed abnormalities in the Atad3 KO mice. In addition, Atad3 KO resulted in mtDNA replication stalling, suggesting that the mtDNA replication process might be coupled to cristae organization. Moreover, we detected a 30% decrease in the ratio of CEs to total cholesterol in the KO muscles of 5-month-old mice, when most of the mitochondria had disrupted cristae. As cholesterol-rich membrane structures are important for tethering mtDNA nucleoids to the inner mitochondrial membranes (Gerhold et al., 2015), our data link ATAD3 to cholesterol-dependent cristae organization and reinforced the structural role of mitochondrial cristae in mtDNA maintenance. However, we cannot exclude that ATAD3 has a primary role in lipid trafficking.
Despite being required for embryonic survival, ablation of ATAD3 in skeletal muscle did not affect the survival of mice up to 20 months of age. We allowed two Atad3 KO males to age, until they succumbed at 811 and 820 days. The fact that the loss of ATAD3 in muscle does not have a major impact on lifespan suggests that skeletal muscle mitochondria are either able to compensate for the resulting functional consequences, or that in muscles, ATAD3 is not essential for function and survival. In mice, the Atad3 gene is expressed in two major isoforms, a long 67 kDa and short 57 kDa form. Both isoforms are encoded by the same gene and are expressed throughout the different stages of brain development. However, the short isoform is absent in adult muscle tissue (Li et al., 2014). Thus, one possibility is that the absence of the short 57 kDa isoform is responsible for the embryonic lethality in the total body KO models. The lack of ATAD3 in muscle causes a 50% mtDNA depletion. Commonly, 50% mtDNA depletion does not cause symptoms in patients (Gorman et al., 2016), and it was not surprising that the observed 50% reduction in mtDNA levels in muscle was not sufficient to shorten the lifespan of the ATAD3 muscle KO mice. Accordingly, other conditional mouse models that die prematurely showed much higher mtDNA depletion. TWINKLE KO mice had only 6% mtDNA in the heart (Milenkovic et al., 2013; Wang et al., 1999), and mice expressing a mitochondrial restriction nuclease became severely affected when mtDNA levels reduced by 60–70% and were sacrificed at 7 months (Srivastava and Moraes, 2005).
The degeneration of the cristae morphology began with the loss of the CJs at early ages (detected at 2 months), whereas other molecular abnormalities as well as motor symptoms manifested later. Thus, we conclude that the muscle atrophy in this model develops primarily as a mitochondrial cristae disorganization disease, which leads to the biochemical and molecular phenotypes detected. The lack of ATAD3 resulted in a decrease in the steady-state levels of OPA1, but only at 5 months, coinciding with the total lack of normal cristae. OPA1 is known to control cristae remodeling in vivo (Civiletto et al., 2015), and therefore one possibility is that ATAD3 cooperates with OPA1 in the regulation of mitochondrial cristae morphology. ATAD3 has also been reported to be located at the contact sites between inner and outer mitochondrial membrane, and to regulate the dynamic interactions between these two compartments (Baudier, 2017; Gilquin et al., 2010). Therefore, another possibility is that ATAD3 might play a direct role in the dynamics of the contact sites at the base of the cristae junctions, where MICOS is also located (Harner et al., 2011; Hoppins et al., 2011). In fact, the alteration of MICOS complex in ATAD3 KO mitochondria as early as in 3-month-old mice supports this view. However steady-state levels of MIC60 were not decreased in the ATAD3 KO mice, suggesting that the role of ATAD3 in cristae organization is independent of MIC60. Likewise, OPA1 has been shown to act upstream of MIC60 in the regulation of the cristae morphology (Glytsou et al., 2016).
Disruption of OPA1 or the MICOS complex is associated with mtDNA depletion and altered mitochondrial nucleoids size in cultured cells (Chen et al., 2010; Elachouri et al., 2011; Li et al., 2016; von der Malsburg et al., 2011). ATAD3 abundance in cultured cells affects mtDNA topology, mitochondrial nucleoid size and mtDNA levels (Desai et al., 2017; He et al., 2007, 2012; Rebelo et al., 2009). In vitro, ATAD3 binds preferentially to a DNA structure with a displacement loop (He et al., 2007). However, the fact that ATAD3 was not detected in crosslinking studies with mtDNA, suggests that ATAD3 might not interact directly with the mtDNA (He et al., 2012). In agreement, our results indicated that ATAD3 participates in the maintenance of mtDNA levels in vivo by stabilization of the mitochondrial cristae morphology.
It has recently been shown that mtDNA replication is coupled to mitochondrial division (Lewis et al., 2016), and that the ER-mitochondrial contacts are required for the replication of the mtDNA. Therefore, another possible mechanism of ATAD3 influencing mtDNA replication could be the stabilization of these platforms required for mtDNA replication. In agreement, ATAD3 has been found in steroidogenic cells in the ER-mitochondrial contacts, termed mitochondria-associated membranes (Gerhold et al., 2015; Issop et al., 2015). Given the significant role of the ER in cholesterol metabolism (Iaea and Maxfield, 2015), such contact sites were proposed to be the place for cholesterol channeling to mitochondria. The fact that muscles lacking ATAD3 protein had decreased cholesterol levels at later ages suggests that ATAD3 affects cholesterol trafficking in skeletal muscle mitochondria, which could be simply by disrupting mitochondria cristae or by a more specific role still to be discovered.
Considering the previous knowledge and the data obtained in our in vivo studies, we propose a model in which ATAD3 is located in the inner mitochondrial membrane but also interacting with the outer membrane (Fig. 8). According to this model, ATAD3 helps the MICOS complex to maintain the CJs. With the loss of ATAD3, the CJs weaken, and the MICOS complex is unable to remain in contact with other CJ factors, leading to an accumulation of the ∼600 kDa form. Also, complex V, which is enriched at the internal tips of the cristae, becomes destabilized, owing to the ongoing changes in cristae structure. Later, OPA1 becomes destabilized as well. During these changes, cholesterol transport to the mitochondria is reduced and mitochondrial replication is stalled, causing mtDNA alterations.
Fig. 8.
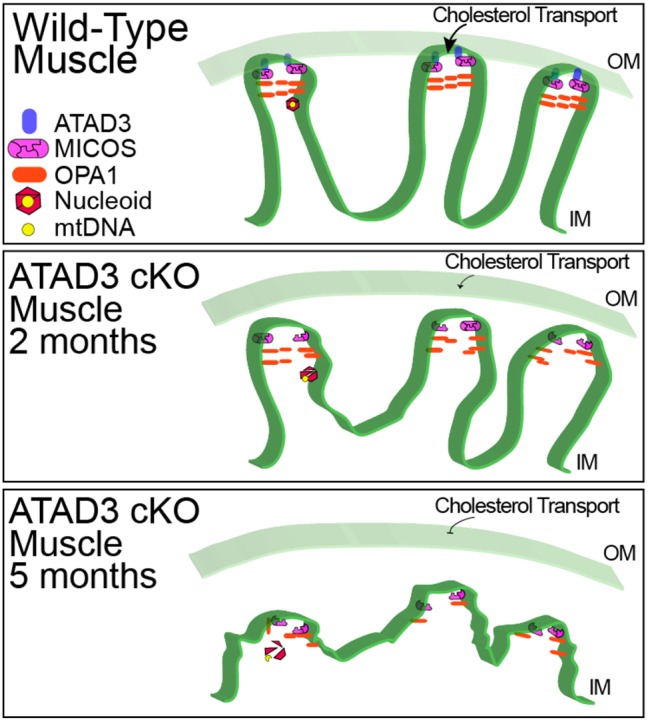
Model for mitochondrial alterations associated with ATAD3 elimination. In wild-type muscle, ATAD3 helps stabilize mitochondrial cristae, possibly as a result of its reported interactions with the outer membrane (OM). In 2-month-old KO muscle, cristae are disorganized, disturbing MICOS complexes. By 5 months, cristae are essentially absent with a block in cholesterol transport and nucleoid structure collapse. At this point, mtDNA is vulnerable to disruptions in replication and formation of deletions. IM, inner membrane.
In the ATAD3 muscle KO animals, the reduction in mtDNA was accompanied by the formation of mtDNA rearrangements, which accumulated in older animals. The retained mtDNA sequences included the D-loop region and the 12S and 16S ribosomal RNA (rRNA) genes. This deletion pattern closely resembles the rearranged molecule found in pathological states such as adult-onset progressive external ophthalmoplegia (adPEO) and MNGIE (Suomalainen et al., 1992; Wanrooij et al., 2004). However, the percentage of deletion found in our model (0.5–1%) is lower than the ones found in adPEO patients. The ATAD3 KO model had somatic point mutation loads similar to control muscle (not shown), suggesting that ATAD3 does not influence the fidelity of polymerase γ. Our results indicate that the absence of ATAD3 disturbs mtDNA replication and/or segregation of the nucleoids, leading to mtDNA rearrangements. Accordingly, lack of ATAD3 in muscle was associated with increased FGF21 levels in sera in aged animals, a biomarker associated with instability in mtDNA metabolism (Lehtonen et al., 2016).
We have made several relevant observations regarding the role of ATAD3 in OXPHOS function. ATP synthase and the supercomplexes were the first structures in which we detected alterations in the ATAD3 KO muscle. These results are compatible with the alteration of cristae observed in the KO muscle, given that supercomplex structures and complex V depend on cristae shape (Cogliati et al., 2013; Davies et al., 2011, 2012). Furthermore, we have detected an increase in the F1 subcomplex of ATP synthase in ATAD3 KO muscle. One possible explanation could be that the loss of cristae organization makes the ATP synthase complex less stable. This is supported by our observation that the same proportion of digitonin released the F1 subcomplex in the KO animals, but not in the controls. The appearance of F1 subcomplex of ATP synthase is also a characteristic feature of mtDNA maintenance defects in patients (Carrozzo et al., 2006), and was reported in numerous mouse models with impaired mtDNA expression owing to mtDNA replication defects (Milenkovic et al., 2013), impaired mtDNA transcription (Metodiev et al., 2009; Mourier et al., 2014; Park et al., 2007) or impaired mitochondrial translation (Cámara et al., 2011; Dogan et al., 2014; Iommarini et al., 2015; Szczepanowska et al., 2016).
We were also surprised by the observation that OXPHOS was not severely affected, despite the accumulation of pale cores without OXPHOS activity within muscle fibers in the KO mice. These novel observations demonstrate that ATAD3 does not have a significant role in mitochondrial translation, as previously suggested (He et al., 2012). It also shows that the primary function of ATAD3 is not crucial for OXPHOS assembly.
An interesting aspect concerning mitochondrial myopathies is the observed increased mitochondrial biogenesis in patients, which takes place to compensate for the defects (Rowland et al., 1991). Indeed, an increase in subsarcolemmal COX activity has been detected in the muscle biopsy of the patient carrying the homozygous ATAD3A missense variant (c.158C>T; p.Thr53Ile) (Harel et al., 2016). In our model, we observed increased mitochondrial markers (PGC1α, SDHA, VDAC1), and increased subsarcolemmal COX activity at 5, 12 and 18 months, indicating increased mitochondrial biogenesis. Our results suggest that the altered mitochondrial ultrastructure, together with the reduction detected in the OXPHOS supercomplexes and complex V levels in the muscle at 3 months, is sufficient to activate mitochondrial biogenesis.
In summary, in this study, we found that loss of ATAD3 in muscle primarily disrupts mitochondrial CJs and profoundly altered cristae morphology. The lack of mitochondrial ultrastructure preceded, and likely led to, downregulated cholesterol trafficking to the mitochondria, mtDNA replication stalling, and progressive mitochondrial DNA depletion and deletions. We conclude that ATAD3 influences the mtDNA replication process by regulating the dynamics of the mitochondrial cristae in cooperation, possibly, with OPA1 and the MICOS.
MATERIALS AND METHODS
Creation of Atad3 KO mice
We acquired an embryonic stem (ES) cell clone from the Knockout Mouse Project repository in which endogenous Atad3 had been modified by homologous recombination (http://www.komp.org) (Fig. S1). We injected the ES cell line containing Atad3 trap gene (clone CDS37385, derived from JM8.N4 cells, background 6N) into C57BL/6J-derived blastocysts and the embryos achieved were implanted into pseudopregnant foster mothers. To detect the chimeras, we amplify the nicotinamide nucleotide transhydrogenase (Nnt) locus, which is homozygous wild type in the 6N background (ES cells) and homozygous mutant in the 6J background (blastocysts). The primers used for amplification of Nnt gene were as follows: NntA-COMMON: 5′- GTAGGGCCAACTGTTTCTGCATGA-3′; NntD-WT: 5′- GGGCATAGGAAGCAAATACCAAGTTG -3′; and NntC-MUT: 5′- GTGGAATTCCGCTGAGAGAACTCTT -3′. From this breeding, germline transmission was obtained (48% of progeny had the transgene), and transgenic mice lines for Atad3 were established.
Heterozygous Atad3+/− mice were mated to ubiquitously expressing flipase mice (Rosa26_FLP, Jackson Laboratory, 012930) to excise the neomycin selection marker. To generate the Atad3 skeletal muscle-specific knockouts (Atad3-Mlc1f KO), homozygous Atad3(LoxP/ LoxP) were mated to double heterozygous mice Atad3(+/LoxP), Mlc1f-Cre(+/−), and the resulting progeny with the desired genotype selected for phenotypic studies. Mice were subsequently bred with C57BL6J for more than seven generations.
All experiments and animal husbandry, including care and use of experimental mice, conformed with the AAALAC regulatory standards and were performed according to a protocol approved by the University of Miami Institutional Animal Care and Use Committee. Mice were housed in a virus-antigen-free facility of the University of Miami, Division of Veterinary Resources, in a 12-h light/dark cycle at room temperature and fed ad libitum.
Behavioral tests
Treadmill test
Mice motor coordination was tested at different ages using a treadmill (Columbus Instruments Exer 3/6) set at a speed of 9 m/min. The mice were running for 3 min and the number of falls in the grid was recorded. Animals were trained in the treadmill prior to the test.
Strength test
Maximal grip strength of each mouse at different ages was measured with a grip strength analyzer (Bioseb), recording the best of three trials as maximum strength.
Rotarod test
Mice motor coordination was tested at different ages using a Rotarod (IITC 755 Life Sciences) set at a ramp speed of 6–20 rpm over 180 s. The test consisted of three trials performed for each animal at the corresponding age and the latency to fall was recorded. Mice that completed the task received a final latency time of 180 s. Animals were trained in the Rotarod two times of three trials each ∼2 weeks prior to the first test.
Activity cage test
Spontaneous ambulatory movement of mice was recorded using the Opto-M3 activity meter (Columbus Instruments) equipped with infrared beams along the cage. Movement was counted as the number of times the infrared beams were disrupted. Mice were housed individually in a new cage 30 min prior to their daily dark cycle and ambulatory counts were recorded for a period of 12 h (18:30 to 06:30).
Open field test
The open field test is a sensitive method for measuring gross and fine locomotor activity. It is performed in an open field arena (Med Associates Inc.), consisting of a chamber and a system of 16 infrared transmitters that record the position of the animal in the three-dimensional space. With this system, not only the horizontal movement can be recorded but also the rearing activity. For our study, the animals were placed in the chamber 30 min before the test and the locomotor activities were recorded for 30 min.
Histology
Skeletal muscles were frozen in isopentane in liquid nitrogen. Cryostat sections of 10 µm were stained with Hematoxylin and Eosin to study muscle structure. COX and SDH activity staining were performed as described in Peralta et al. (2016).
TEM
Tibialis anterior muscles from Atad3-Mlc1f KO mice and controls at 2 and 5 months were fixed in 2.5% glutaraldehyde overnight and treated with 1% osmium tetroxide, dehydrated in ethanol and embedded in epoxy resin. Semi-thin (1 µm) sections were stained with Richardson's stain. ImageJ software (National Institutes of Health) was used to quantify morphometric parameters of mitochondria and the mitophagic area.
Enzymatic activity assays
Mitochondria were isolated from skeletal muscle by centrifugal differentiation using mitochondria isolation buffer (10 mM HEPES, 0.5 mM EDTA, 0.5 mM EGTA, 250 mM sucrose) containing complete protease inhibitor cocktail (Roche Diagnostics). Complex I and complex IV activities were measured spectrophotometrically as described previously (Barrientos et al., 2009; Spinazzi et al., 2012). Specific activity was determined, and values are represented as nmol/min/mg of protein.
Western blotting
Mitochondria or homogenates were prepared in PBS containing protease inhibitor mixture (Roche Diagnostics) in a volume of ∼10× the weight. Sodium dodecyl sulfate (SDS) was added to the homogenate at a final concentration of 4%, and snap frozen in liquid nitrogen. Homogenates were sonicated for 3 s, centrifuged at 14,000 g at 4°C and the supernatant was collected for analysis. Approximately 20 µg protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in 4–20% acrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes. For ATAD3 detection, 40 µg protein from mitochondria was loaded and separated over an 8% acrylamide gel. Membranes were blocked with 5% nonfat milk in 0.1% Tween 20 in PBS and subsequently incubated with specific antibodies. Primary antibodies were incubated overnight at 4°C.
The following antibodies were used: anti-NDUFB8 (ab110242), anti-SDHA (ab14715), anti-UQCRC1 (ab110252), anti-COX1 (ab14705), anti-ATP5A (ab14748), anti-VDAC1 (ab14734) and anti-MIC60/mitofilin (ab110329) from Abcam; anti-TIM23 (611222) and anti-OPA1 (612606) from BD Transduction Laboratories; anti-ATAD3 (16610-1-AP) from Proteintech; anti-PGC-1α (ST1204) from Calbiochem; and anti-muscle myosin (MF-20) and anti-muscle sarcoplasmic reticulum marker (12/101) from Developmental Studies Hybridoma Bank. All antibodies were used at 1:1000 dilution (in 0.15% milk in PBST) except for anti-ATAD3, which was used at 1:1000 in 5% milk in PBST. Secondary antibodies conjugated to horseradish peroxidase (Cell Signaling Technologies) were used, and the reaction was developed by chemiluminescence using SuperSignal West reagent (Rockford).
BN-PAGE
To identify and estimate the levels of respiratory complexes, mitochondria isolated from skeletal muscle were treated with digitonin (ratio 1:8, protein:digitonin, Roche) and mitochondrial complexes separated by BN-PAGE in 3–12% acrylamide gradient gels (Invitrogen) (Diaz et al., 2009; Wittig et al., 2006). Ten micrograms of proteins were separated by PAGE, transferred to a PVDF membrane (Bio-Rad), and incubated sequentially with antibodies against several subunits of the different mitochondrial respiratory complexes. To identify the levels of ATAD3, MIC60 and OPA1 complexes mitochondria isolated from skeletal muscle were treated with digitonin (ratio 1:3, protein:digitonin, Roche) and complexes separated by BN-PAGE in 3–12% acrylamide gradient gels; 20–30 µg of mitochondrial proteins were used.
Clear native PAGE and in-gel ATP hydrolysis assay
To identify the oligomeric forms of complex V and determine the in-gel ATP hydrolysis activity of complex V, mitochondria isolated from skeletal muscle were treated with digitonin (ratio 1:1.5, protein:digitonin, Roche) and separated by clear native PAGE in 3–12% acrylamide gradient gels (Invitrogen) (Wittig and Schagger, 2005).
Quantitative PCR of genomic DNA
Genomic DNA was extracted from muscles using standard proteinase K, phenol, chloroform extraction and isopropyl alcohol precipitation. The ratio of mtDNA to nDNA was determined by quantitative real-time PCR using 10 ng of genomic DNA in a 20 µl reaction mixture using SsoFast EvaGreen Supermix (Bio-Rad) following PCR conditions stipulated by the manufacturer in a CFX96 Real Time PCR system (Bio-Rad). Primers for mtDNA were ND1-F: 5′-CAGCCTGACCCATAGCCATA-3′ and ND1-B: 5′-ATTCTCCTTCTGTCAGGTCGAA-3′ and for genomic DNA β-actin-F: 5′-GCGCAAGTACTCTGTGTGGA-3′ and β-actin-B: 5′-CATCGTACTCCTGCTTGCTG-3′. DNA amounts were quantified using the ΔΔCt method and expressed as a ratio of ND1 (also known as Avpr2)/actin. Age-related mtDNA deletions were measured in total DNA by real-time PCR, following primers described in Neuhaus et al. (2014).
2D-AGE
Mitochondrial nucleic acids were extracted from snap-frozen quadriceps femoris muscle of 5-month-old mice as previously described (Goffart et al., 2009). Five micrograms of mitochondrial nucleic acids were digested with ClaI restriction enzyme and separated over a Brewer/Fangman type 2D agarose gel as previously described (Brewer and Fangman, 1987; Friedman and Brewer, 1995). The gel was blotted under alkaline conditions onto Hybond-XL membrane, probed with a 32P-labeled probe (nucleotides 14,783–15,333 of mouse mtDNA) and exposed to film and phosphorimager.
NGS
Mitochondrial nucleic acids were extracted from muscle mitochondria isolated from mice aged 20 months as described in Williams et al. (2013). Mitochondria-enriched fractions were obtained using standard differential centrifugation of tissue homogenates (900 g, followed by 12,000 g). Total DNA was extracted using standard phenol/chloroform extraction. Double-stranded DNA was quantified by Qubit and 100 ng was used as input for PCR-free library prep using Kapa HyperPlus according to the manufacturer’s recommendations with 6-min DNA fragmentation. Libraries were sequenced on an Illumina NextSeq 500 using a mid-output flow cell with paired-end 75 bp chemistry. Base calling was run in Illumina BaseSpace and bioinformatic analysis using Qiagen CLCBio Genomics Workbench 7.5.1. mtDNA content of libraries determined as percentage of read pairs aligning to murine mtDNA reference (NC_005089) ranged from 18 to 41%, median 23%.
Analysis was performed essentially as described in Williams et al. (2013). To identify recombination breakpoints, reads were first aligned at low stringency (50% length, 95% identity) and aligned reads extracted and subsequently aligned at high stringency (97% length, 95% identity). Reads that failed to align at high stringency were used for gapped BLAST alignment against each consensus sequence with a word size of 15 bp. Data were parsed to collect reads with two aligned fragments that were not internalized nor inversions (clustering errors) extending to a minimum of read length −9 bp including overlap.
PCR of mtDNA deletion breakpoints
To amplify the sequences delimited within the breakpoints, 50 ng of total DNA were amplified using the primers: mtDNA 2567F: 5′-CGAAAAGGACAAGAGAAATAGAG-3′, and mtDNA 15,900B: 5′-ATGACACCACAGTTATGTGG-3′. For amplification of control 456 bp mtDNA fragment by PCR we used the same forward primer: mtDNA 2567F and the reversed mtDNA 3023B: .5′-TATTGGTAGGGGAACTCATAGACT-3′. Conditions of PCR were 35 cycles, each cycle consisting of 5 s of denature at 95°C, annealing at 59°C for 10 s, and 45 s of extension at 72°C.
Analysis of lipids using HPLC-mass spectrometry
Lipid extracts from gastrocnemius of 2- and 5-month-old mice were prepared using a modified Bligh and Dyer procedure as described previously (Bligh and Dyer, 1959; Chan et al., 2012), spiked with appropriate internal standards, and analyzed using a 6490 Triple Quadrupole LC/MS system (Agilent Technologies). Glycerophospholipids and sphingolipids were separated with normal-phase HPLC as described previously (Chan et al., 2012), with a few modifications. An Agilent Zorbax Rx-Sil column (inner diameter 2.1×100 mm) was used under the following conditions: mobile phase A (chloroform:methanol:1 M ammonium hydroxide, 89.9:10:0.1, v/v) and mobile phase B (chloroform:methanol:water:ammonium hydroxide, 55:39.9:5:0.1, v/v); 95% A for 2 min, linear gradient to 30% A over 18 min and held for 3 min, and linear gradient to 95% A over 2 min and held for 6 min. Sterols and glycerolipids were separated with reverse-phase HPLC using an isocratic mobile phase as described previously (Chan et al., 2012), except with an Agilent Zorbax Eclipse XDB-C18 column (4.6×100 mm). Quantification of lipid species was accomplished using multiple reaction monitoring (MRM) transitions that were developed in earlier studies (Chan et al., 2012) in conjunction with referencing of appropriate internal standards: PA 14:0/14:0, PC 14:0/14:0, PE 14:0/14:0, PI 12:0/13:0, PS 14:0/14:0, SM d18:1/12:0, D7-cholesterol, CE 17:0, MG 17:0, 4ME 16:0 diether DG, D5-TG 16:0/18:0/16:0 (Avanti Polar Lipids).
Lactate and FGF21 measurements
Blood was withdrawn from deeply anesthetized animals by cardiac puncture. Lactate levels were measured in plasma using Lactate Plus Lactate Meter (Nova Biomedical). FGF21 levels were determined using the mouse myokine panel from Millipore (MMYOMAG-74K, Luminex) and followed the Millipore protocol described in http://www.emdmillipore.com/US/en/product/MILLIPLEX-MAP-Mouse-Myokine-Bead-Panel,MM_NF-MMYOMAG-74K.
Statistical analysis
Data are presented as mean±s.em. Unpaired Student's t-test (two tailed) was used for statistical analysis and GraphPad Prism 6 software for presentation.
Supplementary Material
Acknowledgements
We are grateful to Vania Almeida at the University of Miami EM core and Aline Hida for expert technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.P., S.L.W., J.P., C.T.M.; Methodology: S.P., S. Goffart, S.L.W., F.D., S. Garcia, N.N., E.A.-G., J.P., C.T.M.; Formal analysis: S.P., S. Goffart, F.D., N.N., E.A.-G., J.P., C.T.M.; Investigation: S.P., S. Goffart, S.L.W., F.D., S. Garcia, N.N., E.A.-G., J.P.; Writing - original draft: S.P., C.T.M.; Writing - review & editing: S.P., S. Goffart, S.L.W., F.D., N.N., E.A.-G., J.P., C.T.M.; Supervision: J.P., C.T.M.; Project administration: C.T.M.; Funding acquisition: C.T.M.
Funding
This work was supported by the National Institute of Neurological Disorders and Stroke [1R01NS079965], National Institute on Aging [1R01AG036871], National Eye Institute [5R01EY010804 to C.T.M.], National Institutes of Health [P30-EY014801] and American Heart Association [16PRE30480009 to N.N.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.217075.supplemental
References
- Barrientos A., Fontanesi F. and Diaz F. (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 63, 19.3.1-19.3.14. 10.1002/0471142905.hg1903s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudier J. (2017). ATAD3 proteins: brokers of a mitochondria-endoplasmic reticulum connection in mammalian cells. Biol. Rev. Camb. Philos. Soc. 93, 827-844. 10.1111/brv.12373 [DOI] [PubMed] [Google Scholar]
- Bligh E. G. and Dyer W. J. (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911-917. 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- Bogenhagen D. F., Rousseau D. and Burke S. (2008). The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 283, 3665-3675. 10.1074/jbc.M708444200 [DOI] [PubMed] [Google Scholar]
- Bothe G. W., Haspel J. A., Smith C. L., Wiener H. H. and Burden S. J. (2000). Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26, 165-166. [DOI] [PubMed] [Google Scholar]
- Brewer B. J. and Fangman W. L. (1987). The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51, 463-471. 10.1016/0092-8674(87)90642-8 [DOI] [PubMed] [Google Scholar]
- Cámara Y., Asin-Cayuela J., Park C. B., Metodiev M. D., Shi Y., Ruzzenente B., Kukat C., Habermann B., Wibom R., Hultenby K. et al. (2011). MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 13, 527-539. 10.1016/j.cmet.2011.04.002 [DOI] [PubMed] [Google Scholar]
- Carrozzo R., Wittig I., Santorelli F. M., Bertini E., Hofmann S., Brandt U. and Schägger H. (2006). Subcomplexes of human ATP synthase mark mitochondrial biosynthesis disorders. Ann. Neurol. 59, 265-275. 10.1002/ana.20729 [DOI] [PubMed] [Google Scholar]
- Chacinska A., Koehler C. M., Milenkovic D., Lithgow T. and Pfanner N. (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628-644. 10.1016/j.cell.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R. B., Oliveira T. G., Cortes E. P., Honig L. S., Duff K. E., Small S. A., Wenk M. R., Shui G. and Di Paolo G. (2012). Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 287, 2678-2688. 10.1074/jbc.M111.274142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M. and Chan D. C. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280-289. 10.1016/j.cell.2010.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiletto G., Varanita T., Cerutti R., Gorletta T., Barbaro S., Marchet S., Lamperti C., Viscomi C., Scorrano L. and Zeviani M. (2015). Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 21, 845-854. 10.1016/j.cmet.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S., Frezza C., Soriano M. E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L. C. et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160-171. 10.1016/j.cell.2013.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S., Enriquez J. A. and Scorrano L. (2016). Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci. 41, 261-273. 10.1016/j.tibs.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Cooper H. M., Yang Y., Ylikallio E., Khairullin R., Woldegebriel R., Lin K.-L., Euro L., Palin E., Wolf A., Trokovic R. et al. (2017). ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 26, 1432-1443. 10.1093/hmg/ddx042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M., Strauss M., Daum B., Kief J. H., Osiewacz H. D., Rycovska A., Zickermann V. and Kuhlbrandt W. (2011). Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA 108, 14121-14126. 10.1073/pnas.1103621108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M., Anselmi C., Wittig I., Faraldo-Gomez J. D. and Kuhlbrandt W. (2012). Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 109, 13602-13607. 10.1073/pnas.1204593109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R., Frazier A. E., Durigon R., Patel H., Jones A. W., Dalla Rosa I., Lake N. J., Compton A. G., Mountford H. S., Tucker E. J. et al. (2017). ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 140, 1595-1610. 10.1093/brain/awx094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F., Barrientos A. and Fontanesi F. (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using blue native gel electrophoresis. Curr. Protoc. Hum. Genet. 63, 19.4.1-19.4.12. 10.1002/0471142905.hg1904s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan S. A., Pujol C., Maiti P., Kukat A., Wang S., Hermans S., Senft K., Wibom R., Rugarli E. I. and Trifunovic A. (2014). Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 19, 458-469. 10.1016/j.cmet.2014.02.004 [DOI] [PubMed] [Google Scholar]
- Elachouri G., Vidoni S., Zanna C., Pattyn A., Boukhaddaoui H., Gaget K., Yu-Wai-Man P., Gasparre G., Sarzi E., Delettre C. et al. (2011). OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 21, 12-20. 10.1101/gr.108696.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H. Y., Chang C.-L., Hsu S.-H., Huang C.-Y., Chiang S.-F., Chiou S.-H., Huang C.-H., Hsiao Y.-T., Lin T.-Y., Chiang I.-P. et al. (2010). ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci. 123, 1171-1180. 10.1242/jcs.062034 [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G. V., Rudka T., Bartoli D., Polishuck R. S., Danial N. N., De Strooper B. et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177-189. 10.1016/j.cell.2006.06.025 [DOI] [PubMed] [Google Scholar]
- Frickey T. and Lupas A. N. (2004). Phylogenetic analysis of AAA proteins. J. Struct. Biol. 146, 2-10. 10.1016/j.jsb.2003.11.020 [DOI] [PubMed] [Google Scholar]
- Friedman K. L. and Brewer B. J. (1995). Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 262, 613-627. 10.1016/0076-6879(95)62048-6 [DOI] [PubMed] [Google Scholar]
- Gerhold J. M., Cansiz-Arda S., Lõhmus M., Engberg O., Reyes A., van Rennes H., Sanz A., Holt I. J., Cooper H. M. and Spelbrink J. N. (2015). Human mitochondrial DNA-protein complexes attach to a cholesterol-rich membrane structure. Sci. Rep. 5, 15292 10.1038/srep15292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson R. W., Selker J. M. L. and Capaldi R. A. (2003). The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 546, 355-358. 10.1016/S0014-5793(03)00633-1 [DOI] [PubMed] [Google Scholar]
- Gilquin B., Taillebourg E., Cherradi N., Hubstenberger A., Gay O., Merle N., Assard N., Fauvarque M. O., Tomohiro S., Kuge O. et al. (2010). The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol. Cell. Biol. 30, 1984-1996. 10.1128/MCB.00007-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glytsou C., Calvo E., Cogliati S., Mehrotra A., Anastasia I., Rigoni G., Raimondi A., Shintani N., Loureiro M., Vazquez J. et al. (2016). Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. 17, 3024-3034. 10.1016/j.celrep.2016.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffart S., Cooper H. M., Tyynismaa H., Wanrooij S., Suomalainen A. and Spelbrink J. N. (2009). Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 18, 328-340. 10.1093/hmg/ddn359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goller T., Seibold U. K., Kremmer E., Voos W. and Kolanus W. (2013). Atad3 function is essential for early post-implantation development in the mouse. PLoS One 8, e54799 10.1371/journal.pone.0054799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman G. S., Chinnery P. F., DiMauro S., Hirano M., Koga Y., McFarland R., Suomalainen A., Thorburn D. R., Zeviani M. and Turnbull D. M. (2016). Mitochondrial diseases. Nat. Rev. Dis. Primers 2, 16080 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- Harel T., Yoon W. H., Garone C., Gu S., Coban-Akdemir Z., Eldomery M. K., Posey J. E., Jhangiani S. N., Rosenfeld J. A., Cho M. T. et al. (2016). Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am. J. Hum. Genet. 99, 831-845. 10.1016/j.ajhg.2016.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härmälä A. S., Porn M. I., Mattjus P. and Slotte J. P. (1994). Cholesterol transport from plasma membranes to intracellular membranes is inhibited by 3 beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. Biochim. Biophys. Acta 1211, 317-325. 10.1016/0005-2760(94)90156-2 [DOI] [PubMed] [Google Scholar]
- Harner M., Körner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U., Griffith J., Mann M., Reggiori F. and Neupert W. (2011). The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 30, 4356-4370. 10.1038/emboj.2011.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Mao C.-C., Reyes A., Sembongi H., Di Re M., Granycome C., Clippingdale A. B., Fearnley I. M., Harbour M., Robinson A. J. et al. (2007). The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 176, 141-146. 10.1083/jcb.200609158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Cooper H. M., Reyes A., Di Re M., Sembongi H., Litwin T. R., Gao J., Neuman K. C., Fearnley I. M., Spinazzola A. et al. (2012). Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 40, 6109-6121. 10.1093/nar/gks266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Bellance N., Rossignol R., Koopman W. J., Willems P. H., Mayatepek E., Bossinger O. and Distelmaier F. (2009). C. elegans ATAD-3 is essential for mitochondrial activity and development. PLoS One 4, e7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I. J., He J., Mao C.-C., Boyd-Kirkup J. D., Martinsson P., Sembongi H., Reyes A. and Spelbrink J. N. (2007). Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion 7, 311-321. 10.1016/j.mito.2007.06.004 [DOI] [PubMed] [Google Scholar]
- Hoppins S., Collins S. R., Cassidy-Stone A., Hummel E., Devay R. M., Lackner L. L., Westermann B., Schuldiner M., Weissman J. S. and Nunnari J. (2011). A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 195, 323-340. 10.1083/jcb.201107053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubstenberger A., Labourdette G., Baudier J. and Rousseau D. (2008). ATAD 3A and ATAD 3B are distal 1p-located genes differentially expressed in human glioma cell lines and present in vitro anti-oncogenic and chemoresistant properties. Exp. Cell Res. 314, 2870-2883. 10.1016/j.yexcr.2008.06.017 [DOI] [PubMed] [Google Scholar]
- Hubstenberger A., Merle N., Charton R., Brandolin G. and Rousseau D. (2010). Topological analysis of ATAD3A insertion in purified human mitochondria. J. Bioenerg. Biomembr. 42, 143-150. 10.1007/s10863-010-9269-8 [DOI] [PubMed] [Google Scholar]
- Iaea D. B. and Maxfield F. R. (2015). Cholesterol trafficking and distribution. Essays Biochem. 57, 43-55. 10.1042/bse0570043 [DOI] [PubMed] [Google Scholar]
- Iommarini L., Peralta S., Torraco A. and Diaz F. (2015). Mitochondrial Diseases Part II: mouse models of OXPHOS deficiencies caused by defects in regulatory factors and other components required for mitochondrial function. Mitochondrion 22, 96-118. 10.1016/j.mito.2015.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issop L., Fan J., Lee S., Rone M. B., Basu K., Mui J. and Papadopoulos V. (2015). Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology 156, 334-345. 10.1210/en.2014-1503 [DOI] [PubMed] [Google Scholar]
- John G. B., Shang Y., Li L., Renken C., Mannella C. A., Selker J. M. L., Rangell L., Bennett M. J. and Zha J. (2005). The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell 16, 1543-1554. 10.1091/mbc.e04-08-0697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen J. M., Forsstrom S., Bottani E., Viscomi C., Baris O. R., Isoniemi H., Hockerstedt K., Osterlund P., Hurme M., Jylhava J. et al. (2016). FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 87, 2290-2299. 10.1212/WNL.0000000000003374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S. C., Uchiyama L. F. and Nunnari J. (2016). ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549 10.1126/science.aaf5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. and Rousseau D. (2012). ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J. Bioenerg. Biomembr. 44, 189-197. 10.1007/s10863-012-9424-5 [DOI] [PubMed] [Google Scholar]
- Li S., Lamarche F., Charton R., Delphin C., Gires O., Hubstenberger A., Schlattner U. and Rousseau D. (2014). Expression analysis of ATAD3 isoforms in rodent and human cell lines and tissues. Gene 535, 60-69. 10.1016/j.gene.2013.10.062 [DOI] [PubMed] [Google Scholar]
- Li H., Ruan Y., Zhang K., Jian F., Hu C., Miao L., Gong L., Sun L., Zhang X., Chen S. et al. (2016). Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 23, 380-392. 10.1038/cdd.2015.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons G. E., Ontell M., Cox R., Sassoon D. and Buckingham M. (1990). The expression of myosin genes in developing skeletal muscle in the mouse embryo. J. Cell Biol. 111, 1465-1476. 10.1083/jcb.111.4.1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiev M. D., Lesko N., Park C. B., Cámara Y., Shi Y., Wibom R., Hultenby K., Gustafsson C. M. and Larsson N. G. (2009). Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 9, 386-397. 10.1016/j.cmet.2009.03.001 [DOI] [PubMed] [Google Scholar]
- Milenkovic D., Matic S., Kuhl I., Ruzzenente B., Freyer C., Jemt E., Park C. B., Falkenberg M. and Larsson N. G. (2013). TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 22, 1983-1993. 10.1093/hmg/ddt051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier A., Ruzzenente B., Brandt T., Kuhlbrandt W. and Larsson N. G. (2014). Loss of LRPPRC causes ATP synthase deficiency. Hum. Mol. Genet. 23, 2580-2592. 10.1093/hmg/ddt652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus J. F. G., Baris O. R., Hess S., Moser N., Schroder H., Chinta S. J., Andersen J. K., Kloppenburg P. and Wiesner R. J. (2014). Catecholamine metabolism drives generation of mitochondrial DNA deletions in dopaminergic neurons. Brain 137, 354-365. 10.1093/brain/awt291 [DOI] [PubMed] [Google Scholar]
- Odgren P. R., Toukatly G., Bangs P. L., Gilmore R. and Fey E. G. (1996). Molecular characterization of mitofilin (HMP), a mitochondria-associated protein with predicted coiled coil and intermembrane space targeting domains. J. Cell Sci. 109, 2253-2264. [DOI] [PubMed] [Google Scholar]
- Park C. B., Asin-Cayuela J., Camara Y., Shi Y., Pellegrini M., Gaspari M., Wibom R., Hultenby K., Erdjument-Bromage H., Tempst P. et al. (2007). MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130, 273-285. 10.1016/j.cell.2007.05.046 [DOI] [PubMed] [Google Scholar]
- Peralta S., Garcia S., Yin H. Y., Arguello T., Diaz F. and Moraes C. T. (2016). Sustained AMPK activation improves muscle function in a mitochondrial myopathy mouse model by promoting muscle fiber regeneration. Hum. Mol. Genet. 25, 3178-3191. 10.1093/hmg/ddw167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N., van der Laan M., Amati P., Capaldi R. A., Caudy A. A., Chacinska A., Darshi M., Deckers M., Hoppins S., Icho T. et al. (2014). Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 204, 1083-1086. 10.1083/jcb.201401006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjoismäki J. L., Goffart S. and Spelbrink J. N. (2011). Replication stalling by catalytically impaired Twinkle induces mitochondrial DNA rearrangements in cultured cells. Mitochondrion 11, 630-634. 10.1016/j.mito.2011.04.002 [DOI] [PubMed] [Google Scholar]
- Rebelo A. P., Williams S. L. and Moraes C. T. (2009). In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 37, 6701-6715. 10.1093/nar/gkp727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland L. P., Blake D. M., Hirano M., Di Mauro S., Schon E. A., Hays A. P. and Devivo D. C. (1991). Clinical syndromes associated with ragged red fibers. Rev. Neurol. (Paris) 147, 467-473. [PubMed] [Google Scholar]
- Schaffrik M., Mack B., Matthias C., Rauch J. and Gires O. (2006). Molecular characterization of the tumor-associated antigen AAA-TOB3. Cell. Mol. Life Sci. 63, 2162-2174. 10.1007/s00018-006-6200-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H. and Hellhammer U. (1977). The number of nuclei in adult rat muscles with special reference to satellite cells. Anat. Rec. 189, 169-175. 10.1002/ar.1091890204 [DOI] [PubMed] [Google Scholar]
- Slotte J. P., Härmälä A. S., Jansson C. and Porn M. I. (1990). Rapid turn-over of plasma membrane sphingomyelin and cholesterol in baby hamster kidney cells after exposure to sphingomyelinase. Biochim. Biophys. Acta 1030, 251-257. 10.1016/0005-2736(90)90301-4 [DOI] [PubMed] [Google Scholar]
- Spinazzi M., Casarin A., Pertegato V., Salviati L. and Angelini C. (2012). Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 7, 1235-1246. 10.1038/nprot.2012.058 [DOI] [PubMed] [Google Scholar]
- Srivastava S. and Moraes C. T. (2005). Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum. Mol. Genet. 14, 893-902. 10.1093/hmg/ddi082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A., Majander A., Haltia M., Somer H., Lönnqvist J., Savontaus M. L. and Peltonen L. (1992). Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Invest. 90, 61-66. 10.1172/JCI115856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanowska K., Maiti P., Kukat A., Hofsetz E., Nolte H., Senft K., Becker C., Ruzzenente B., Hornig-Do H. T., Wibom R. et al. (2016). CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 35, 2566-2583. 10.15252/embj.201694253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H., Mjosund K. P., Wanrooij S., Lappalainen I., Ylikallio E., Jalanko A., Spelbrink J. N., Paetau A. and Suomalainen A. (2005). Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 102, 17687-17692. 10.1073/pnas.0505551102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel F., Bornhövd C., Neupert W. and Reichert A. S. (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 175, 237-247. 10.1083/jcb.200605138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Malsburg K., Muller J. M., Bohnert M., Oeljeklaus S., Kwiatkowska P., Becker T., Loniewska-Lwowska A., Wiese S., Rao S., Milenkovic D. et al. (2011). Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 21, 694-707. 10.1016/j.devcel.2011.08.026 [DOI] [PubMed] [Google Scholar]
- Wang Y. and Bogenhagen D. F. (2006). Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 281, 25791-25802. 10.1074/jbc.M604501200 [DOI] [PubMed] [Google Scholar]
- Wang J., Wilhelmsson H., Graff C., Li H., Oldfors A., Rustin P., Bruning J. C., Kahn C. R., Clayton D. A., Barsh G. S. et al. (1999). Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 21, 133-137. 10.1038/5089 [DOI] [PubMed] [Google Scholar]
- Wanrooij S., Luoma P., van Goethem G., van Broeckhoven C., Suomalainen A. and Spelbrink J. N. (2004). Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 32, 3053-3064. 10.1093/nar/gkh634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij S., Goffart S., Pohjoismäki J. L., Yasukawa T. and Spelbrink J. N. (2007). Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 35, 3238-3251. 10.1093/nar/gkm215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N. and Pfanner N. (2017). Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 86, 685-714. 10.1146/annurev-biochem-060815-014352 [DOI] [PubMed] [Google Scholar]
- Williams S. L., Huang J., Edwards Y. J. K., Ulloa R. H., Dillon L. M., Prolla T. A., Vance J. M., Moraes C. T. and Züchner S. (2010). The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 12, 675-682. 10.1016/j.cmet.2010.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. L., Mash D. C., Züchner S. and Moraes C. T. (2013). Somatic mtDNA mutation spectra in the aging human putamen. PLoS Genet. 9, e1003990 10.1371/journal.pgen.1003990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I. and Schägger H. (2005). Advantages and limitations of clear-native PAGE. Proteomics 5, 4338-4346. 10.1002/pmic.200500081 [DOI] [PubMed] [Google Scholar]
- Wittig I., Braun H. P. and Schagger H. (2006). Blue native PAGE. Nat. Protoc. 1, 418-428. 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- You W.-C., Chiou S.-H., Huang C.-Y., Chiang S.-F., Yang C.-L., Sudhakar J. N., Lin T.-Y., Chiang I.-P., Shen C.-C., Cheng W.-Y. et al. (2013). Mitochondrial protein ATPase family, AAA domain containing 3A correlates with radioresistance in glioblastoma. Neuro Oncol. 15, 1342-1352. 10.1093/neuonc/not077 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.