

Abstract
The edgewise interactions of anions with phenylalanine (Phe) aromatic rings in proteins, known as anion–quadrupole interactions, have been well studied. However, the anion–quadrupole interactions of the tyrosine (Tyr) and tryptophan (Trp) rings have been less well studied, probably because these have been considered weaker interactions than anions hydrogen bonded to Trp/Tyr sidechains. Distinguishing from such hydrogen bonding interactions, we comprehensively surveyed the edgewise interactions of certain anions (aspartate, glutamate, and phosphate) with Trp, Tyr, and Phe rings in high-resolution, non-redundant protein single chains and interfaces (protein-protein, DNA/RNA–protein, and membrane–protein). Trp/Tyr anion–quadrupole interactions are common, with Trp showing the highest propensity and average interaction energy for this type of interaction. The energy of an anion–quadrupole interaction (−15.0 −0.0 kcal/mol, based on quantum mechanical calculations) depends not only on the interaction geometry but also on the ring atom. The phosphate anions at DNA/RNA–protein interfaces interact with aromatic residues with energies comparable to that of aspartate/glutamate anion–quadrupole interactions. At DNA–protein interfaces, the frequency of aromatic ring participation in anion–quadrupole interactions is comparable to that of positive charge participation in salt bridges, suggesting an underappreciated role for anion–quadrupole interactions at DNA–protein (or membrane–protein) interfaces. Although less frequent than salt bridges in singlechain proteins, we observed highly conserved anion–quadrupole interactions in the structures of remote homologs, and evolutionary covariance-based residue contact score predictions suggest that conserved anion–quadrupole interacting pairs, like salt bridges, contribute to polypeptide folding, stability, and recognition.
TOC GRAPHICS
INTRODUCTION
Non-covalent interactions, such as Van der Waals forces, the hydrophobic effect, hydrogen bonding, and electrostatic interactions, may be weak individually, but collectively play an important role in the folding, assembly, and recognition of macromolecules1-3. Interactions involving aromatic functional groups (commonly known as π interactions), such as π–π4–6, CH–π7,13, lone pair–π14–17, cation–π18–22, and anion–π22–25 interactions, are important components of weak, non-covalent interactions and have gained considerable interest in chemistry26, for example, in the design of catalysts. In addition to synthetic chemistry, a number of recent reports suggest that weak interactions are critical in regulating protein function. For example, the CH–π switch in protein-tyrosine phosphatase 1B (PTP1B, also known as PTPN1) is critical in regulating phosphatase function27. Similarly, the repair function of the DNA glycosylase AlkD is critically dependent on CH–π interactions for the recognition of N3 methyladenosine in alkylated DNA28. Enhanced stability of engineered miniproteins29 and G-quadruplexes30 are other notable examples of CH–π interactions, not just in proteins but also in nucleic acids. While CH–π interactions are gaining prominence in the field of biological macromolecules, investigation of another potentially important non-covalent interaction in biological macromolecules, the anion–quadrupole interaction, has been rare, possibly due to the only recent recognition of its presence in proteins in the seminal study of Howell et al.25. In general, aromatic ring π-systems have a quadrupole moment as a result of the two opposing dipoles originating from each face31. The electron cloud on each face of the aromatic ring is therefore attracted to cations (by a cation–π interaction), and the edges of the ring are attracted to anions (by an anion–quadrupole interaction). In the same study25, anion–quadrupole interactions in proteins were modeled on the interactions between aspartate (Asp) or glutamate (Glu) sidechain carboxylate ions and the aromatic ring of the phenylalanine (Phe) residue in monomeric proteins. Anions could also interact with tryptophan and tyrosine ring-systems by anion–quadrupole interaction but these ring systems also have dipole moment and they make strong hydrogen bonds with anions that could influence their anion–quadrupole interactions. In general, we wanted to further understand the roles of anion–quadrupole interactions in proteins in the following categories, which were not discussed: (i) anion–quadrupole interactions involving the rings of tyrosine (Tyr) and tryptophan (Trp) residues; (ii) deconvolution of the energetic contributions of anion–quadrupole and anion-dipole interactions (iii) anion–quadrupole interactions involving the phosphate ion, an important anion in biological systems; (iv) anion–quadrupole interactions at protein–protein interfaces; (v) a comparison of the behavior of anion–quadrupole interactions with that of salt bridges in proteins and at protein–protein interfaces; and (vi) the evolutionary conservation of anion–quadrupole interactions in proteins. As salt bridges in protein structures have been well studied32–35, the behavior of anions in anion–quadrupole interactions has been compared with their behavior in salt bridges to gain a better understanding of how nature, through evolutionary conservation, utilizes anions in a range of polar interactions. Since the report on anion–quadrupole interactions in proteins referred to above, there have been other reports36–38. But these have been typically focused on a small set of a specific group/family of highly homologous proteins (or predicted interfaces) and do not cover all of the abovementioned categories. In addition, unlike the recent reports36–38, we distinguish an anion’s edgewise interactions with Trp/Tyr ring from that of an anion’s hydrogen bonded interactions with Trp/Tyr sidechains to capture the influence of different ring atoms on anion–quadrupole interactions, and the orientation preferences of aromatic rings while interacting with anions. Therefore, we systematically performed a comprehensive analysis of anion–quadrupole interactions in proteins that should also be useful in the study of other weak, non-covalent interactions in biological macromolecules. As novel weak interactions (e.g., RNA ribose–nucleobase lone pair–π stacking39, Lys-NH3+ − carbonyl linear arrangement40) in biological macromolecules continue to be reported, our analysis will be useful in the study of weak molecular interactions in general.
METHODS
Detection Procedure for Anion–quadrupole Interactions.
The procedure for detecting anion–quadrupole interactions in protein structures was developed using the PSD dataset (see Protein Datasets, SI page S3–S4). Residues with missing sidechain atoms were ignored. Sidechain–sidechain hydrogen bonded pairs of Asp/Glu and Trp/Tyr were not counted as regular anion–quadrupole interactions. We determined the distance-and-angle criterion for anion–quadrupole interactions in the following steps: (a) defined the vector nr as normal to the ring plane and passing through the ring center (Figure S1A) for each of the aromatic residues in a coordinate file; (b) defined the vector rc as the line joining the ring center and the center of carboxylate oxygen atoms of Asp/Glu sidechains (Figure S1A) for every anion-aromatic residue pair in the coordinate file; (c) determined θN the angle between nr and rc, and θP (90° - θN), the angle the anion makes with the ring plane (Figure S1A); (d) for each anion–aromatic residue pair, determined all interresidue interatomic vectors dr (the vector between the carboxylate oxygen and a ring atom); and (e) determined dS, the smallest value of dr, that is, the shortest interresidue interatomic distance (Figure S1A). The distribution of θP for dS < 5 Å (greater than the sum of van der Waal contact + 1.75 Å) showed that θρ decreases from 0° to 90°, as expected. Seventy-five percent of the population of θP values for Trp/Tyr/Phe, taken together, was < 35° (Figure S1b). The Howell study25, using quantum mechanical calculations on a benzene-formate (BF) pair, determined the interaction energies to be negative for θP < 35°. Therefore, for determining a distance criterion, we considered θP ≤ 35°, and the distribution of dS for θP ≤ 35° (Figure S1C, D) showed a bell-shaped appearance with a peak at 3.75 Å for dS ≤ 4.5 Å. For dS > 4.5 Å, the non-specific pairs kept increasing (Figure S1C, D). This behavior was similar for Trp, Tyr, and Phe residues (Figure S1C, D). Based on the distribution of dS for Trp/Tyr/Phe taken together, we set a 4.5-Å cutoff for dS for the detection of anion–quadrupole interactions. Thus, a pair of (Asp/Glu)–(Trp/Tyr/Phe) residues with dS ≤ 4.5 A and θP ≤ 35°, as well as not having a sidechain–sidechain hydrogen bond between them, was considered a regular anion–quadrupole interaction (Figure 1A, see main text). We also added two additional criteria: (i) if the dS for a carboxylate ion was > 4.5 Å and the dr distance from the anion center was ≤ 4.5 Å, there was a ~13% increase in the number of anion–quadrupole pairs; and (ii) if the θP value was > 35° and the angle with either of the oxygen atoms was ≤ 35°, there was a ~7% increase in anion–quadrupole interactions. We ignored the C-terminal carboxyl group (except for the PDZ–peptide interaction, Figure S5D).
Figure 1. Regular anion–quadrupole interactions and their energies:
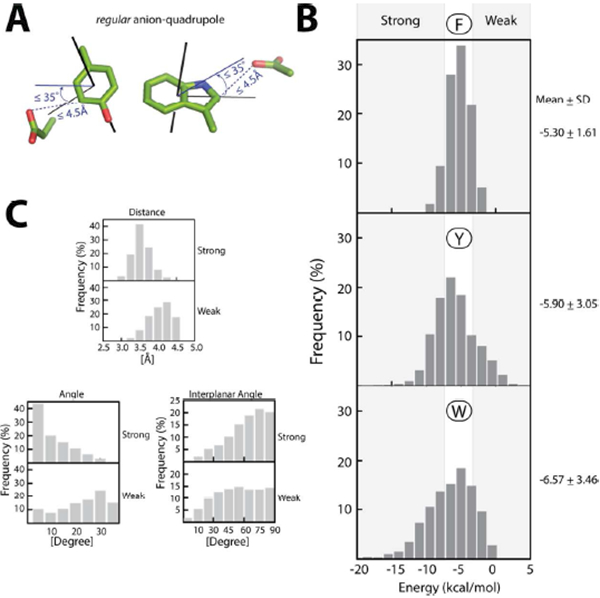
(A) The distance (≤ 4.5 Å) and angle (≤ 35°) detection criteria. (B) The distribution of the interaction energies for Phe (top), Tyr (middle), and Trp (bottom). Energy ranges for the strong and weak anion–quadrupole interactions are indicated by the gray background. (C) A comparison of the distribution of properties between strong and weak interactions: distances (top), angles (left, bottom), and interplanar angles (right, bottom).
Energy Calculations.
The energy of interactions was computed using NWChem41 6.6 quantum mechanical calculations. For computing the energy of interaction (e.g., for an anion–quadrupole pair) the coordinates of side chain atoms of the interacting pair were first extracted from a protein data bank (PDB) file. Hydrogen atoms for these side chains were added using the Open Babel version 2.3.1 chemical toolbox42. Each CB atom had three protons and setting the pH to 7.0 (in Open Babel) ensured that Asp/Glu carboxyl groups remained deprotonated. An interacting pair of the type A–B (e.g., Asp–Phe) had three input files: (i) A, (ii) B, and (iii) A–B, where the A input file has the coordinates of the atoms of A. Each of the three input files for a pair was imported to the NWChem 6.6 program for energy calculations. Single-point energies were calculated using DFT with the Grimme DFT-D3 dispersion correction43 with the AUG-CC-PVDZ basis set and B3LYP functional. The energy (in Eh units) was extracted from the corresponding NWChem log files. The energy of interaction (ΔEAB) was obtained as, ΔΕAB = E(A-B) − [E(A) + E(B)], and ΔEAB in EH units was then converted to kcal/mole. There were 13,308 anion–quadrupole and 6,256 HB-anion–quadrupole A–B pairs in total for the energy calculations. We also compared the interaction energy of aromatic ring-carboxylate anion–quadrupole to that of other types of ring-edgewise interactions (see Figure 8A). For example, we considered the edgewise interactions of aromatic rings with side chain nonpolar −CH2−/−CH3 groups (Phe-Val), polar -OH groups (Phe-Ser), polar −CONH2 groups (Phe-Asn) and the polar nucleic acid backbone anion (Phe-[−O−PO2−O−]-). There were 4399 Phe–Val, 1079 Phe–Ser, 1037 Phe–Asn, and 176 (Phe-[−O−PO2−O−]-) A–B pairs in total for the energy calculations. Energies calculated using only the dispersion corrected DFT (computed with MWChem 6.6) method were used in this study (e.g., Figures 1–4 and 7). Single-point energies were also calculated using DFT with the B3LYP functional and the 6–31G* [i.e., 6–31(d)] basis set using Gaussian 0944, but these energies were not used here. A correlation between the anion–quadrupole energies calculated by either approach is shown in Figure S12.
Figure 8. Other functional groups:
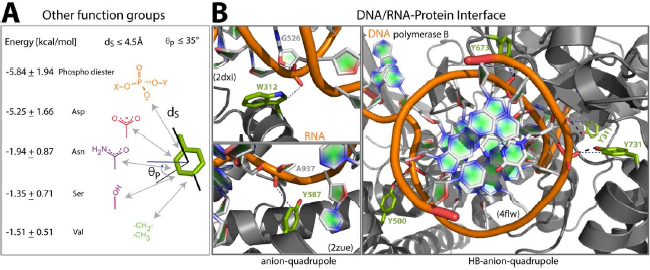
(A) The average energy of interaction (left) of other functional groups (right) with the Phe residue ring atoms. (B) Anion–quadrupole (left) and HB-anion–quadrupole (right) interactions with phosphoryl groups at DNA/RNA–protein interfaces.
Figure 4. HB-anion–quadrupole interactions:
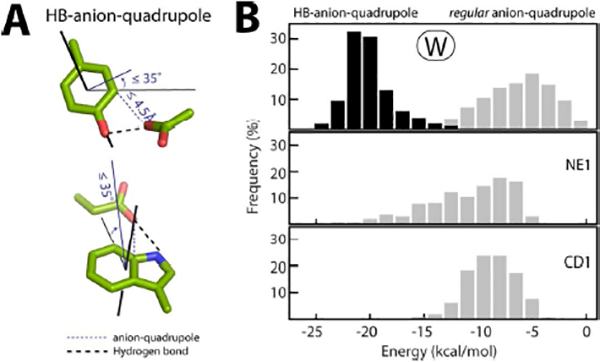
(A) HB-anion–quadrupole interactions satisfy the distance-angle detection criterion used here for anion–quadrupole interactions. (B) Comparison between the distribution of the interaction energy of anion–quadrupole and HB-anion–quadrupole interactions for Trp residues (top). The distribution of anion–quadrupole interaction energies for the Trp-NE1 (middle) and Trp-CD1 (bottom) atoms are shown below for comparison.
Figure 7. Anion–quadrupole interactions of histidine residues:
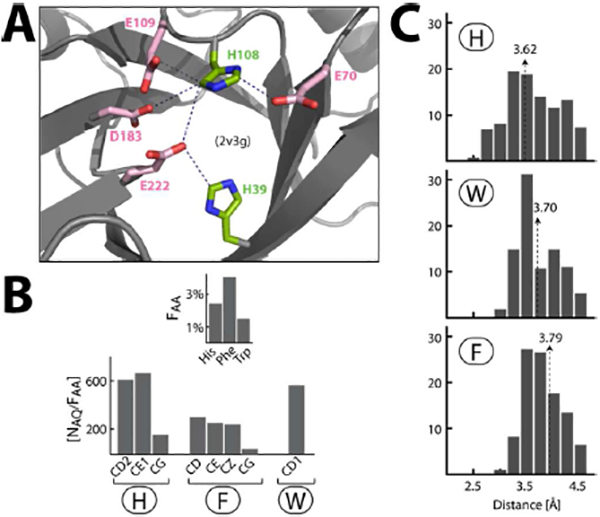
(A) Examples of histidine ring carbon atoms engaged in anion–quadrupole interactions. (B) Number of anion–quadrupole [NAQ] interaction for each ring atom is normalized by the amino acid frequency [NAQ/FAA] in the dataset. The value of [NAQ/FAA] of ring atoms is compared between histidine, phenylalanine and tryptophan. (C) The distribution of the anion–quadrupole interaction distances for histidine ring carbon atoms (top), tryptophan-CD1 atom (middle) and phenylalanine ring-carbon atoms (bottom). The arrows indicate the mean of the respective distributions. The tryptophan-CD1 serves as a reference here as the energy of anion–quadrupole interaction of tryptophan-CD1 atom was observed to be the largest among all ring carbon atoms of phenylalanine, tyrosine and tryptophan.
Hydrogen Bonds and Salt Bridges.
All hydrogen bond calculations were carried out using the HBPLUS45 program with the default settings. We considered a salt bridge pair to occur when Lys/Arg side chain atoms were hydrogen bonded to Asp/Glu side chain atoms. That is, electrostatically interacting opposite charges that were not hydrogen bonded were not considered here as salt bridges. Although this criterion underestimated the number of salt bridges, a “strict” criterion was imposed to ensure a fair comparison with anion–quadrupole interactions, which were also selected with a strict criterion. Similarly, phosphoryl group salt bridges were considered to occur when Lys/Arg side chain atoms were hydrogen bonded to a phosphoryl group.
RESULTS AND DISCUSSION
Anion–quadrupole Interactions of Tryptophan and Tyrosine.
Using highresolution (≤ 1.6 Å) crystal structures, a simple procedure was developed for detecting anion-quadrupole interactions in proteins (Figure S1 and see Methods). We considered the concept of an anion–quadrupole interacting pair to apply only when the following conditions are satisfied (Figure 1A): (a) one of the Asp/Glu carboxylate oxygen atoms is within 4.5 A of any of the ring atoms, (b) the center of carboxylate oxygen atoms subtends an angle ≤ 35° with the ring plane, and (c) a pair satisfying (a) and (b) should not also be engaged in sidechain-sidechain hydrogen bonding between them. The hydrogen bonded pairs Trp/Tyr (side chain donor) and Asp/Glu (side chain acceptor) were excluded (Figure S1A, C). Condition (c) ensured that the treatment of Trp/Tyr was similar to that of Phe, and the distance and angle distributions of Tyr and Trp were very similar to that of Phe (Figure S1B, D). Therefore, in the same manner as the Phe–Asp/Glu anion–quadrupole pairs were studied in the previously mentioned report25, Tyr and Trp were examined here for anion–quadrupole interactions. We observed 13,308 anion–quadrupole interactions in 3700 non-redundant single chains of the PISCES46 dataset. The number of anion–quadrupole interactions with Phe (~2.06 per protein) reported in the earlier study25 is higher than we observed here (~1.55 per protein), as we implemented a “strict” distance-and-angle detection criterion. Among the anion–quadrupole interactions, 43%, 36%, and 21% were respectively associated with Phe, Tyr, and Trp residues. However, the normalized occurrence frequencies (propensities, Figure S2A) were 1.38 (Trp), 0.98 (Phe), and 0.88 (Tyr), suggesting that a larger surface area and/or the indole ring dipole moment of Trp likely contributes to the interaction energy and therefore a higher propensity.
A typical upper limit for the hydrogen bond donor-acceptor distance is 3.5 A, while the distance range for the anion–quadrupole interactions is 3.25 – 4.5 A. This suggests that carboxylateions making hydrogen bonds with tryptophan (through the NE1 atom) and tyrosine (through the OH atom) will be positioned closer to the ring atoms than do the carboxylate ions engaged only in regular anion–quadrupole interactions (Figure S1E). This shorter distance and the high electronegativity of the NE1 and OH atoms would obviously make the interaction energy between an anion and an aromatic ring through hydrogen bonds much greater than that of a regular anion–quadrupole. This energy difference is consistent with the following. In the dataset, we observed 6,256 anions hydrogen bonded to tryptophan and tyrosine (i.e., with just two atoms, Trp-NE1 and Tyr-OH). By comparison, in the same dataset, we observed 7,585 anions engaged in anion–quadrupole interactions with tryptophan and tyrosine residues in which 12 ring atoms are capable of participating. If the anions hydrogen bonded to Trp-NE1 and Tyr-OH were included, the distance distribution (Figure S1E) of anion–quadrupole interactions would also have been different. Therefore, for determining the distance-angle criterion for detecting anion–quadrupole interactions, we opted for a criterion that is uniform for all the aromatic rings (Phe, Tyr and Trp). For a fair comparison of anion–quadrupole interaction energies between phenylalanine, tyrosine and tryptophan, we (unlike recent reports36–38) avoided anions hydrogen bonded to Trp-NE1 and Tyr-OH atoms. A number of structural parameters (see Supplemental Information, SI, pp. S4–S6) of the anion–quadrupole interactions of Phe residues were compared with those of Trp and Tyr residues (Figure S3). These parameters were: (a) the specific contacts, (b) the interplanar angle, (c) the presence of hydrogen bonding for anions, (d) the depth and packing, (e) the energy, and (f) the sequence separation (Figure S3A–F). Consistent with our approach of treating tryptophan and tyrosine like phenylalanine, a comparison of structural parameters showed that the behavior of tryptophan or tyrosine anion–quadrupole interactions is similar to that of phenylalanine (Figure S3), and therefore the anion–quadrupole interactions discussed here include those associated with all three aromatic residues.
Energies of Anion–quadrupole Interactions.
Interaction Energy.
We utilized dispersion-corrected density functional theory (DFT)43-47 based quantum mechanical calculations (see Methods) for computing the energy of anion–quadrupole interactions. Dispersion-corrected DFT calculations have been shown to be more accurate in capturing the behavior of the noncovalent interactions of aromatic ring systems47. The interacting functional groups were represented as a simplified organic scaffold at pH 7.0 (Figure 1A). Energy calculations carried out with the 13,308 anion–quadrupole pairs are shown in Figure 1B. The distributions of the energies suggest that the strength of the interactions, on average, is similar for all of the aromatic residues and that the average energy of Trp interactions is slightly greater (more negative) than that of the others. The observed energies are also consistent with those observed earlier with phenylalanine’s anion–quadrupole interactions25. Based on the calculated energy, we loosely defined weak (≥ −3.0 kcal/mol) and strong (≤ −8.0 kcal/mol) interactions (Figure 1B). The geometrical features of strong and weak interactions are distinct (Figure 1C). Stronger interactions tend to: (a) take place over shorter distances, (b) subtend angles closer to zero, and (c) occur preferentially in a perpendicular interplanar orientation. In addition to steric effects, the stability of conformations (e.g., trans-proline48, the polyproline II helix49, and Z-DNA15) within macromolecules is also strongly supported by stereoelectronic effects (i.e., conformational preferences that arise due to hyperconjugative delocalization), for example, nCO → π* of the succeeding >C=O in trans-proline48 and cytidine nO4’ → π* of the guanine base in CpG Z-DNA15. For anion–quadrupole interactions with a distance ≥ 3.5 Å (i.e., within orbital mixing distances), the preferences for angle (~0°, Figure 1C, bottom left) and interplanar orientation (~90°, Figure 1C, bottom right) are far stronger than in those interactions with distances > 4.0 Å (Figure 1C). Stereoelectronic effects could also contribute to the preferred geometries of anion–quadrupole interactions.
Deconvolution of Dipole-Quadrupole Energy Contributions.
The benzene ring possesses only a quadrupole moment, while the indole ring possesses a ring dipole moment in addition to a quadrupole moment (Figure 2B). Therefore, anion–quadrupole interactions of the tryptophan ring are expected to also be influenced by the dipole moment of the indole ring. To quantify the influence of a dipole moment, we analyzed anion–quadrupole interactions energy in the following manner. We expected that the dipole moment would influence the polarity of the ring-CH atoms, and a difference in the resulting ring-CH atom polarity also suggested that the energy of interactions would be dependent on the ring-CH atoms. We, therefore, analyzed the energy of anion–quadrupole interactions based on the ring atom that is closest to the anion. For example, the anion–quadrupole interaction with the Phe-CZ atom (Figure 2A) refers to the interactions, among all the Phe anion–quadrupole interactions, in which the CZ atom is closest to the anion. The distance dependency of the interaction energy (ion–ion, ion–dipole, dipole–dipole, ion–quadrupole) is characteristic of an interaction type, and therefore, we determined the distance dependency of the anion–quadrupole interaction energy for each of the ring atoms within the distance range 3.25 – 4.50 Å (Figure 2). For convenience, we used the distance between the ring carbon atom and the center of the carboxylate oxygen atoms. We found that the interaction energies of anions with the phenylalanine ring atoms were not identical and that the distance dependency (within this distance range) of the interaction energy for each atom type was linear and nearly parallel to that of the other atoms (Figure 2A). For benzene’s anion–quadrupole interactions, we expected all atoms to behave identically. For phenylalanine, even with a small ring dipole moment, the behavior of the interaction energies of its atoms differed from that of benzene; that is, the interaction energy of the Phe-CZ atom is different from that of the Phe-CD1/2 atom (Figure 2A). Instead of a single energy value, we considered a range of interaction energies (spanning between the Phe-CZ and Phe-CD1/CD2 parallel lines, Figure 2A) as the baseline range for anion–quadrupole interactions. We refer to this energy range as the “F-range” (Figure 2B). For convenience, we consider a deviation from the F-range to be a result of the contributions from other factors such as the ring dipole moment (Figure 2B). Consistent with the dipole moment of the indole ring, we found that the interaction energies for tryptophan ring atoms deviate from the F-range (Figure 2B). That is, anions interact with Trp-CD1 and Trp-CZ3 atoms respectively with energies much higher (more negative) and lower (more positive), respectively, than those observed in the F-range (Figure 2B, left). The value of the deviation from the F-range depends on the ring atom and provides a rough estimate of the contribution from the dipole moment for each ring atom. In other words, we propose that, in the absence of a dipole moment, the expected energies would be within the F-range, and the presence of a dipole moment would result in deviations of the energy values from the F-range.
Figure 2. Distance dependence of the anion–quadrupole interaction energy:
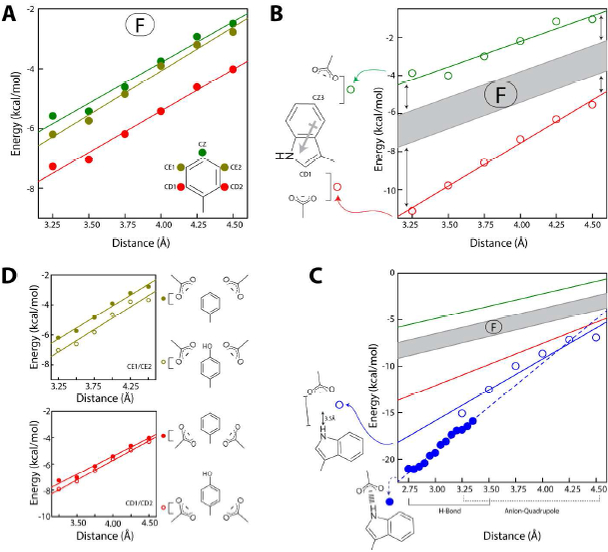
The dependencies for Phe, Trp and Tyr are shown in (A), (B) - (C), and (D), respectively, in which Phe ring atoms are represented by filled circles (green, pear and red) and Trp and Tyr are represented by empty circles. The area between the green and red lines in (A) is defined as the F-range which is shaded in gray in (B) and (C). In (C), the empty blue circles represent anion–quadrupole interactions of the Trp-NE1 atom while filled blue circles represent the energies of hydrogen bonds between the Trp-NE1 atom and anions. The green and red lines in (C) are the same as in (B). The distance range for hydrogen bonds and anion–quadrupole interactions are also indicated in (C). A comparison between the symmetry-related positions of Tyr and Phe are shown in (D).
As mentioned above, we do not consider anions hydrogen bonded to Trp-NE1 as participating in for regular anion–quadrupole interactions. However, given the polarity of the Trp-NE1 and Trp-HE1 atoms, anions within the distance range 3.25 – 4.50 Å (i.e., no hydrogen bond with Trp-NE1) can still interact with Trp-NE1 or Trp-HE1 atoms with energies greater than that of Trp-CD1 atom interactions (Figure 2C). We consider this interaction to be a part of the anion–quadrupole interactions. To distinguish this interaction from the hydrogen bonds, the interaction energy of anions hydrogen bonded to Trp-NE1 (distance range, 2.75 – 3.50 Å) is also overlaid in Figure 2C. The value and the distance dependence of the energies of hydrogen bonds and atom-specific anion–quadrupole interactions differ from one another (Figure 2C). That is, the slope and the intercept depend on the polarity and the interaction distance range. Due to the dipole moment, one end of the tryptophan ring (Trp-CZ2, Trp-CH2, Trp-CZ3) interacts with anions with energies lower than that of the phenylalanine ring, while the other end (Trp-NE1, Trp-CD1) interacts with energies greater than that of the phenylalanine ring. The anion–quadrupole interaction energy averaged over all tryptophan atoms is, however, greater (more negative) than that of phenylalanine. This is because of the higher polarity of the protons (Trp-HE1, Trp-HD1), and tryptophan’s anion–quadrupole energetics is dominated by Trp-NE1 and Trp-CD1 atoms. If the interaction energies of anions hydrogen bonded to Trp-NE1 were added, then tryptophan’s anion–quadrupole interaction energies would be even more different from that of phenylalanine. This logic is also consistent with the reason for excluding anions hydrogen bonded to the ring for a fair comparison with phenylalanine. A comparison of the energy-distance relationships for the symmetry-related positions of tyrosine and phenylalanine (Figure 2D) also shows that the presence of the −OH group in tyrosine shifts the intercept of the energy-distance plot, and the shift is more prominent for positions closer to the −OH group (Figure 2D).
Boltzmann Statistics from Protein Structures.
In evaluating the performances of alternative methods (e.g., DFT, dispersion-corrected DFT, MP2) for computing the energy of noncovalent interactions, typically an agreement between the calculated results, the experimental structure (crystalline state), and the energetic (e.g., sublimation) data is sought47. In a similar spirit, we have also attempted to correlate the computed energies of anion–quadrupole interactions to crystalline state properties. For example, we investigated whether the computed energies are consistent with the frequency with which interactions occur in protein crystal structures. In this regard, we assumed that the protein data bank (PDB) represents an equilibrium distribution of protein conformations. This assumption is based on the success of the Boltzmann hypothesis in explaining the energetics of protein structures50. All of the energy calculations described above were carried out with isolated side chain atoms (see Methods), while these interactions in real proteins actually take place among a myriad of other interactions. For example, an Asp side chain engaged in anion–quadrupole interaction may also be involved in a salt bridge interaction as well as be in contact with surface solvent water molecules by hydrogen bonds (see below). Despite these differences in context (i.e., isolated functional groups versus the entire protein), consistency between the calculated energy and observed statistics in protein structures would provide a strong rationale for basing our analyses on high-resolution protein structures. Given that the energy of anion–quadrupole interactions depends on ring atoms (as we noted above), we wanted to determine whether the occurrence frequency of anions in the vicinity of a specific ring atom is consistent with the calculated atom-specific energy. For the convenience of discussion about the occurrence frequency of anions in the vicinity of ring atoms, we utilized a measure termed FATOM_ds (see SI pp. S6–S7). The FATOM_ds value for a ring atom of a given residue type represents the percentage of time it is present at the shortest distance among all anion–quadrupole interactions of that residue type, and the sum of the FATOM_ds values of all ring atoms of a given residue type is 100% (Figure 3). It is interesting to note that the FATOM_ds values of even phenylalanine atoms (who show small energy differences, Figure 2A) differ among the phenylalanine atoms, and phenylalanine FATOM_ds values are qualitatively consistent with the atom-specific energies. For example, the FATOM_ds for Phe-CZ is 18% while that for Phe-CD1 is 22% (Figure 3A, Figure 2A). Similarly, the FATOM_ds values for tyrosine and tryptophan atoms are also consistent with the computed energy (Figure 3A, Figure 2). Due to the dipole moment of the indole ring, more than 30% of tryptophan residue interactions with anions are with its CD1 atom alone (Figure 3A). In other words, the anion–quadrupole interactions of tryptophan residues have an orientation preference. Consistent with its FATOM_ds value, the magnitude of the interaction energy of the Trp-CD1 (Figure 3B) atom was also higher than that of other Trp ring carbon atoms. In general, the computed interaction energy for ring atoms using isolated functional groups was consistent with their respective FATOM_ds values (Figure 3C). This observation is also useful in the context of molecular mechanics force fields. For example, the partial charges of Trp ring protons in the CHARMM3651 force field were better correlated with the observed ring atom participation frequency (Figure S6, and SI S6–S7) than those of the CHARMM2252 force field. Other than the HE1 atom, all of the Trp ring protons in the CHARMM22 force field are assigned a uniform partial charge equal to +0.115 (Figure S6C), while in the CHARMM36 force field, the Trp ring atom partial charges are consistent with the indole dipole moment51. As a result, a better correlation was observed with the CHARMM36 force field.
Figure 3. Ring atom participation frequency and interaction energy:
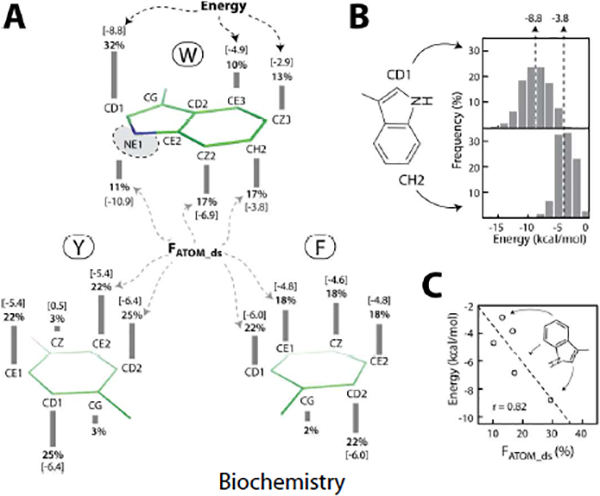
(A) The height of the bar (FATOM_ds) adjacent to the name of each atom represents the frequency (%) of its participation in anion–quadrupole interactions for that residue type, Trp (top), Tyr (bottom left), and Phe (bottom right). The number within the bracket above or below each bar represents the interaction energy (kcal/mol) when the indicated atom is closest to the interacting anion. (B) The distribution represents the energy of anion–quadrupole interactions when Trp-CD1 (upper) and Trp-CH2 (lower) atoms are closest to the interacting anion. The dotted lines with an arrow represent the mean interaction energy in kcal/mol. (C) The linear correlation (r = 0.82) between the energy and FATOM_ds for tryptophan ring carbon atoms. For a qualitative agreement between the energy and the frequency, we use FATOM_ds in place of log[FATOM_ds].
HB-anion–quadrupole Interactions.
Anion–quadrupole Interaction Types and The Energy Gap.
Because of the shorter distance (Figure S1D), anions hydrogen bonded to tryptophan and tyrosine side chains also satisfy the distance-angle criteria (≤ 4.5 Å and ≤ 35.0°; see above) for the detection of anion–quadrupole interactions (Figure 4A). Therefore, by default, anions hydrogen bonded to tryptophan and tyrosine side chains are also considered here for anion–quadrupole interactions. We refer to these pairs as HB-anion–quadrupole interacting pairs to distinguish them from the regular anion–quadrupole pairs discussed above. There are 6,256 HB-anion–quadrupole pairs in the dataset. As expected, the energies of these interactions are much greater than those of regular anion–quadrupole interactions (Figure 4B), i.e., regular anion–quadrupole interactions are typical weak interactions. Due to dynamic motion in proteins (e.g., bond rotation), is it possible for a regular anion–quadrupole interaction to convert to an HB-anion–quadrupole interaction and vice versa? In other words, can a regular anion–quadrupole pair (distance ≤ 4.50 Å) be within hydrogen bonding distance (≤ 3.50 Å) and satisfy the hydrogen bond angle criterion during dynamic motion? Because of the significant energy gap between the two types of anion–quadrupole interactions (Figure 4B), we expected a low frequency for such conversions. To test this prediction, we created a dataset of proteins whose structures have been determined by NMR as well as X-ray crystallography and for which the NMR structure ensemble is believed to approximate the dynamics of protein motion (Figure S4). In this dataset, in only ~7% of the time are regular anion-quadrupole pairs (tryptophan, tyrosine) in the X-ray structure is observed as an HB-anion–quadrupole pair in the NMR structure of the same protein molecule. The interconversion frequency for specific ring atoms (adjacent to the ring hydrogen bond donor position) is shown in Figure S4. Other than the Trp-NE1 atom, no other ring atom has an interconversion frequency > 10% (Figure S4). The energy of the regular anion–quadrupole interaction of only the Trp-NE1 atom comes close to that of the HB-anion–quadrupole interaction (Figures 2C, 4B), and the observed higher interconversion frequency of Trp-NE1 is likely due to the small energy gap between its regular anion–quadrupole and HB-anion-quadrupole interactions. The small interconversion frequency for other ring atoms is, in general, likely due to the large energy difference between the regular anion–quadrupole and HB-anion–quadrupole interactions, and therefore we have treated these anion–quadrupole interaction types separately.
Orientation Preference of HB-anion–quadrupole Interactions.
The energy difference between the two types of interactions is also relevant for mutational analysis. For example, mutating tyrosine → phenylalanine when the tyrosine residue is engaged in an HB-anion–quadrupole interaction in the wild type protein will likely result in a regular anion–quadrupole interaction in the mutant protein. As regular anion–quadrupole interactions are weak, there can be a significant difference in stability between the wild type and the mutant in this example. As another example, an aspartate-tyrosine-mediated HB-anion–quadrupole interaction between the CDR loops of the monoclonal antibody MR78 contributes significantly to the binding affinity for its antigen53 (i.e., the Marburg virus glycoprotein), and substitution of this tyrosine residue with phenylalanine significantly compromises the binding affinity53, justifying the perception of the weak nature of anion–quadrupole interactions. Even though the energy of the anion–quadrupole component of the HB-anion–quadrupole interaction is low, an anion–quadrupole interaction can still make a significant contribution to the behavior of an HB-anion–quadrupole interaction. Here we show that an anion–quadrupole interaction contributes to the orientation preferences of HB-anion–quadrupole interactions (Figures 5). For example, tyrosine residues have an orientation preference for Asp/Gly carboxylate ions when hydrogen bonded to the Tyr-OH group (Figure 5). The orientation preference can be assessed by a measure referred to here as the pseudo-dihedral angle (Figures 5, S7A). The distribution of pseudo-dihedral angles for Tyr-OH was quite different from that of the Thr/Ser-OH group, suggesting that the carboxylate oxygen atoms prefer proximity to tyrosine ring edges (Figures 5, S7B). This is likely due to anion–quadrupole interactions, as we observed that the distances of anions to the tyrosine ring edges were shorter than the respective carbon-anion distances of Thr/Ser residues (Figure S7B).
Figure 5. Tyr HB-anion–quadrupole orientation:
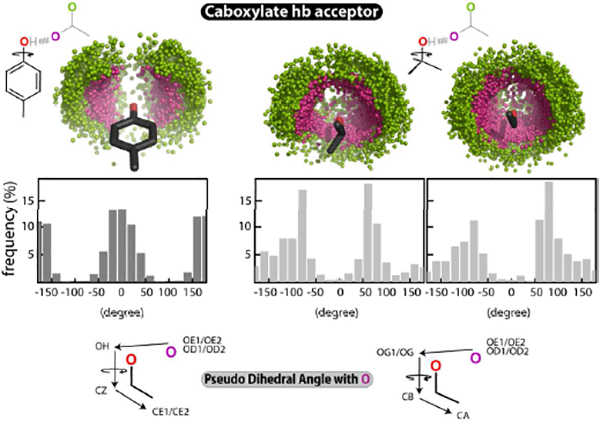
The spatial distribution of the carboxylate oxygen atoms (magenta and green, top) when hydrogen bonded with the donor -OH of Tyr (left), Thr (middle), and Ser (right). The acceptor carboxylate O-atom is in magenta, while the other carboxylate O-atom is in green. The distribution of the pseudo-dihedral angle (θD) between the 4 atoms O-O-CZ-CE1/CE2 (Tyr, left) and O-O-CB-CA (Thr/Ser, center right) is shown in the middle. A drawing representing the θD is shown at the bottom, with Tyr (left) and Thr/Ser (right).
Investigation of the above orientation preference was prompted by the following observation. Asp/Glu sidechain carboxylates were the most frequent hydrogen bond acceptors for each type of side chain hydrogen bond donor in proteins (Figure S7C). The Asp/Glu-acceptor frequency for Tyr was much higher than that of the uncharged side chain donors (Figure S7C), that is to say, the frequency was midway between that of the positively charged and uncharged side chain donors. Although the partial charges of the Tyr–OH group (Figure S7C) may contribute to this, the observed orientation preferences (Figure 5) suggest that anion–quadrupole interactions also contribute to the observed higher Asp/Glu-acceptor frequency for the Tyr–OH group. The observed higher frequency of the Tyr hydrogen bonds with Asp/Glu also suggested that the strength of these hydrogen bonds (HB-anion-quadrupole) is likely to be greater than typical hydrogen bonds, and the following examples support this notion: (i) in the anti-Marburg virus glycoprotein monoclonal antibody (MR78)53, a kink in the HCDR3 loop, critical for antigen recognition, is stabilized by an Asp–Tyr HB-anion–quadrupole interaction in place of a canonical Asp–Arg/Lys salt bridge53 (Figure S5F), (ii) the nanobody Nb60, by virtue of an interfacial Glu–Tyr HB-anion–quadrupole interaction, locks the ligand-(carazolol)-bound conformation of the G protein coupled receptor β2AR54 (Figure S5G). In general, we expect that the strength of noncovalent interactions is likely to follow the order: salt bridge > HB-anion–quadrupole > typical hydrogen bond > anion–quadrupole interactions.
Hydrogen Bonds of Anion and Interaction Clusters.
Interaction Clusters.
Anions participating in HB-anion–quadrupole interactions are always hydrogen bonded to the side chain atoms of the partner aromatic residue (Tyr–OH and Trp-NE1); i.e., the anions of 100% of HB-anion–quadrupole pairs are hydrogen bonded. By comparison, anions of ~70% and ~50% of regular anion–quadrupole and salt bridge pairs, respectively, were also hydrogen bonded to other hydrogen bond donors. In addition to the hydrogen bonds with Tyr–OH and Trp-NE1 atoms, 32% of anions of HB-anion–quadrupole pairs also engage in additional hydrogen bonds. These observations suggest that all of these interactions tend to be part of interaction networks (Figure 6A, left). In these networks, the anion and the aromatic ring of one anion–quadrupole interaction can be part of another anion–quadrupole interaction. For example, ~7% of aromatic residues of anion-quadrupole interactions interact with more than one anion (Figure 6A, right), and ~7% of anions of anion–quadrupole interactions similarly interact with more than one aromatic residue. Even though the energy difference between the regular anion–quadrupole and HB-anion–quadrupole interactions is significant, in certain homologous families we find that a regular anion–quadrupole in one member of the family can be replaced by an HB-anion–quadrupole in another member (Figure 6B). Given that these interactions are part of a cluster of interactions, such amenability to substitution seems reasonable, because the loss/gain of energy due to replacement of one type of interaction is likely compensated by a corresponding replacement in another interaction within the interaction network.
Figure 6. Interaction network and hydrogen bonding of anion–quadrupole interactions:
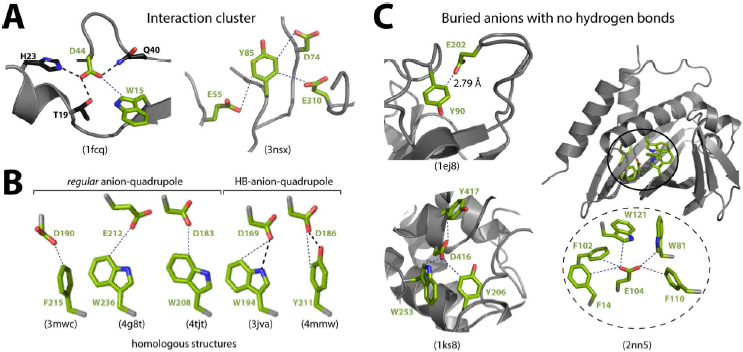
(A) The anion of an anion–quadrupole pair involved in three hydrogen bonds (left) and the aromatic ring of an anion–quadrupole pair interacting with three anions (right). (B) Regular anion–quadrupole and HB-anion–quadrupole interactions observed between conserved positions in homologous protein structures of Pfam MR_MLE_C domain. (C) Buried anions having no hydrogen bonds are surrounded by aromatic rings engaged in anion–quadrupole interactions. The pdb IDs are in braces.
Buried Anions.
Polar groups, buried within the protein interior, must be hydrogen bonded to satisfy the hydrogen bonds with solvent water molecules that were broken upon folding55. As anions of ~30% regular anion–quadrupole interactions are not hydrogen bonded, we determined the solvent accessible surface area (ASA) of these anions to look for buried anions without a hydrogen bond. We found an ASA < 2.0 Å2 for each of its side chain carboxyl-oxygen atoms of 158 anions, and we considered these anions to be buried with an unsatisfied hydrogen bond. The remaining anions of the 30% pairs are considered solvent accessible and expected to be hydrogen bonded to solvent water molecules. How does one account for the lost or missing hydrogen bonds for these 158 buried anions? For the convenience of discussion on this we refer to these 158 anions as BAUH_AQ (i.e., buried anions with unsatisfied hydrogen bonds participating in anion–quadrupole interactions). We examined the distance distribution of BAUH_AQ pairs and found that 50% of them are ≤ 3.5 Å away from the ring, and therefore there is a possibility of forming CH—O hydrogen bonds (see Figure 6C, top left). By comparison, the distances of between 36% of the regular anion–quadrupole interacting pairs in the dataset are ≤ 3.5 Å. Relaxing the hydrogen bond detection criterion could lower the number of BAUH_AQ pairs. However, the fact that the observed proportion (50%) of distances of BAUH_AQ pairs is ≤ 3.5 Å instead of the expected proportion (36%) suggests that regular anion–quadrupole interactions could be a solution for satisfying the lost hydrogen bonds of buried anions. To see whether this could be true, we found that there are 252 BAUH anions in this dataset and 158 of them are BAUH_AQ (~63%). We also checked whether the anions of BAUH_AQ pairs participate in more than one regular anion–quadrupole interaction, such that the sum of two or more weak interactions could make up for the lost hydrogen bond. We found that ~22% of the BAUH_AQ anions are involved in two or more interactions of the anion–quadrupole. This proportion is much higher than the expected ~7% in the dataset (see above). Thus, anion–quadrupole interaction clusters are expected to be more common for BAUH_AQ anions (Figure 6C), and we have observed as many as five aromatic rings surrounding a single BAUH_AQ anion engaged in as many anion–quadrupole interactions (Figure 6C). These observations suggest that such strategies could be employed to stabilize buried anions in the design of “non-ideal” proteins, as discussed recently in the context of designing binding antibodies56, in which buried charges and unpaired polar group are common57.
Comparison with Salt Bridge.
Anions predominantly interact with cations, typically forming salt bridges. The number of salt bridges in the dataset was 28,589. By comparison, anions interacting with aromatic residues in regular anion–quadrupole and HB-anion–quadrupole interactions in the dataset was 19,561 (13,308 + 6,256). On average, we observed ~5 anion–quadrupole and ~8 salt bridge interacting pairs per protein, and 17% of aromatic residues (Phe, Tyr, Trp) were engaged in regular anion–quadrupole interactions, and 14% of Tyr/Trp residues were engaged in HB-anion-quadrupole interactions. The fraction of aromatic residues engaged in anion–quadrupole and HB-anion–quadrupole interactions taken together (17% + 14%) was comparable to the 32% of Lys/Arg residues engaged in salt bridges. Like salt bridges, anion–quadrupole interactions may also play an important role in macromolecular folding, assembly, and recognition, which is supported by the set of examples in Figure S5. These include apoptosis-inducing factor (AIF)58, redesigned Tiam1 PDZ59 (Figure S5A), the extracellular receptor of the kinase WalK (erWalk) of the Staphylococcus aureus two-component signal-transduction system60, and others. In the NADH-driven allosteric regulation of AIF, the release of the C-loop region from the rest of the polypeptide chain is a critical step58 (Figure S5B). AIF residues, crucial for the C-loop release58, interact by the anion–quadrupole interaction (Figure S5B). It is also interesting to see that two residues (Y165, D119) of the three key signal-transducing residues of erWalK are engaged in anion–quadrupole interactions (Figure S5C) and that substitutions of these residues impair WalKR function60.
Anion–quadrupole Interactions of Histidine Residues.
For the aromaticity of the imidazole ring, histidine residues are also expected to participate in anion–quadrupole interactions (like the cation–π interactions of histidine residues). However, the protonation status of the imidazole ring can significantly influence the energy of cation-π interactions of a histidine residue61. Similarly, we expect that protonation status of a histidine ring will likely influence the energy its anion–quadrupole interactions due to electrostatics. Therefore, here we had focused on analyzing the anion–quadrupole interactions of histidine residues that were made only with the ring carbon atoms (Figure 7A). That is, we ignored the interactions of anions with the histidine-ring nitrogen atoms for: (a) the uncertainty of their protonation status, (b) hydrogen bonding as discussed above. Using the same distance-angle detection criterion, we observed 3445 histidine-mediated (i.e., with CG, CD2 and CE1 ring-atoms) anion–quadrupole interactions in the same dataset. The distance and angle distributions of the histidine-anion–quadrupole interactions were respectively very similar to those of phenylalanine, tyrosine and tryptophan suggesting that anions interact with histidine ring edges in a manner very similar to those of the three aromatic rings. In this dataset, the frequency of histidine, phenylalanine and tryptophan residues respectively were 2.4%, 4.1% and 1.5%. Normalizing the occurrence frequency of anion–quadrupole interactions of the ring carbon atoms with the respective amino acid frequency (Figure 7B) suggested that histidine-ring carbon (CD2 and CE1) atoms participate in anion–quadrupole interactions more frequently than the others. Thus, we expect the energies of histidine-anion–quadrupole interactions to be higher (more negative) than those of the other aromatic amino acids. This is also consistent with the shorter distances of histidine-anion–quadrupole interactions in comparison with others (Figure 7C). For the following sections, however, we discuss anion-quadrupole interactions of only the three aromatic amino acids (phenylalanine, tyrosine and tryptophan).
Other Functional Groups and Interfaces.
Other Functional Groups.
Like carboxylate ions, the phosphoryl group, –(PO4−)–, may also participate in anion–quadrupole interactions. We also estimated the energy of the phosphoryl group interacting with the Phe ring edges (Figure 8A). For comparison, the energy of interaction of other amino acid sidechain functional groups (–OH, –CONH2, and –CH2–/CH3–) with Phe edges was also calculated (Figure 8A). Like the carboxylate anion–quadrupole interaction, we imposed the same distance-and-angle geometric criteria when selecting these functional groups from protein structures. The phosphoryl diester, CH3–O– (PO2−)–O–sCH3, functional groups included here for the calculations are those present at DNA/RNA–protein interfaces, and they were selected by imposing the geometric criterion on the OP1 and OP2 atoms (see Methods). The average energy of interaction of the phosphoryl group was comparable with that of the carboxylate ion (Figure 8A). As expected, the energy of interaction for other functional groups was much smaller (Figure 8A), justifying the conclusion that, irrespective of the nature of the anion, the energetic preference of ring edges is to interact with anions. Therefore, using the same detection criterion, we checked for the presence of anion–quadrupole and HB-anion–quadrupole interactions of nucleic acid backbone phosphate ions with protein aromatic residues (Phe, Tyr, and Trp) (Figure 8B) in a high-resolution (≤ 2.20 Å), non-redundant dataset of 252 DNA–protein and 126 RNA–protein interfaces. For the DNA-protein dataset, there were 347 anion–quadrupole and 179 HB-anion–quadrupole pairs present at the interface, and for comparison there were 904 pairs of salt bridges with the backbone phosphate (Table S1). Even though the number of anion–quadrupole and HB-anion–quadrupole interactions for the DNA–protein interfaces was fewer than the number of salt bridges, it is interesting to note that the fraction of interfacial aromatic residues engaging in anion–quadrupole interactions (30.0%, Table S1) at DNA–protein interfaces was considerably higher than that seen in single-chain proteins (17.0%, see above). The same was true for HB-anion–quadrupole interactions (Table S1). The fraction of interfacial Arg/Lys residues at DNA-protein interfaces engaging in salt bridges (32.6%, Table S1) was comparable to that seen in single-chain proteins (31.9%). Even though the percentage of Arg/Lys residues more than doubled (10.6% → 25.2%) going from the single-chain set to the DNA–protein interfaces, the fraction of Arg/Lys residues in physical contact with negative changes did not seem to alter. The percentage of aromatic residues (9.2%) in the single-chain dataset was comparable to that in the DNA–protein interfaces (10.5%), and yet the fraction of aromatic residues in physical proximity to negative charges (phosphoryl groups) was nearly double (16.9% → 30.0%), suggesting a role for anion–quadrupole interactions in DNA recognition. The numbers for RNA–protein interfaces, 130 (anion–quadrupole) and 19 (salt bridge) interactions (Table S1), and, similarly, the enhanced fraction of aromatic residues engaged in anion–quadrupole interactions both support this conclusion (Table S1).
The role of aromatic residues in membrane protein structure at the lipid-water interface is well known62. The abovementioned interactions of phosphate ions with aromatic residues may therefore also be important for the structure of membrane proteins at the lipid–water interface. Four recent 3.2-Å structures of SERCA1 (a Ca2+-ATPase pump), which were elucidated using X-ray solvent contrast modulation, captured the structure of the lipid-bilayer-like membrane around a protein for the first time63 (Figure 9). In these four structures, representing the four conformational states of SERCA1, we observed 26 anion–quadrupole and 6 HB-anion–quadrupole interactions of the SERCA1 aromatic residues with the O1P and O2P atoms of the phospholipid head groups (Figure 9). This study highlighted the roles of the Arg/Lys residues when forming salt bridges with the phospholipid head groups. By comparison, in these four structures we observed 17 salt bridges between SERCA1 and the O1P/O2P atoms of the phospholipids. Thus, our observation of anion–quadrupole interactions at the bilayer–protein interface may also suggest an underappreciated role for anion–quadrupole interactions in membrane proteins. With the recent report on the functional contact between the broadly neutralizing anti-HIV-1 antibody (4E10) and the membrane-proximal external region (MPER) of the HIV gp41 protein, which extends into the membrane64, it is tempting to speculate that anion-quadrupole interactions between the Trp residues of the “long” antibody CDRH3 loop and the membrane phospholipid head groups contribute to this recognition (Figure S8A). Like other membrane anion transporters (e.g., the fluoride channel Phe box65), we also observed anion–quadrupole interactions between the multidrug resistance protein MRP166 and its physiological substrate, leukotriene C4 (a negatively charged small molecule, Figure S8B). Similarly, other organic small molecules may also utilize the anion–quadrupole mechanism to interact with proteins, for example, the cereblon:GSPTI assembly67 and kinase suppressor of RAS (KSR2)68 in complex with their respective small-molecule modulators (Figure S8C).
Figure 9. Anion–quadrupole interactions at the lipid-water interface of membrane proteins:

Anion–quadrupole interactions between the phospholipid phosphoryl anion and the aromatic residues (green) at the lipid-water interface of SERC1A. Only one of the four structures of SERCIA is shown. The membrane bilayer is represented as a shaded region.
Interfaces.
We also checked a set of non-redundant, high-resolution protein interfaces (Table S1) for the presence of anion–quadrupole interactions. In all of the different interfaces, anion–quadrupole interactions were invariably present (Table S1, Figure 10), even though the fraction of interfacial aromatic residues engaging in interchain anion–quadrupole and HB-anion–quadrupole interactions was lower than that of intrachain interactions. The examples in Figure 10, however, suggest that key interfacial contacts in many complexes do involve anion–quadrupole interactions. Some of these examples are: (i) the E. coli periplasmic molecular chaperone SurA PPIase P1 domain, which binds the consensus Ar-x-Ar-x motif69 (Ar, aromatic residue) using anion–quadrupole interactions (Figure 10A); (ii) bapineuzumab70, a humanized antibody (3D6 Fab) that targets amyloid plaques and binds amyloid peptide engaged in anion–quadrupole interactions (Figure 10B); and (iii) the interface between the S and L chain of Desulfovibrio desulfuricans iron hydrogenase, which exhibits an unusually high number of anion–quadrupole interactions (Figure 10C). Redox-active proteins are known to utilize aromatic residues to transport oxidizing equivalents (holes) to the protein surface71 to mitigate protein interior damage, and the D. desulfuricans hydrogenase likely utilizes the interfacial aromatic residues, oriented by anion–quadrupole interactions, to protect from such damage. It is also interesting to observe that at the interfaces of antibody-protein complexes (Table S1), the number of anion–quadrupole pairs was comparable to the number of salt bridges. This observation on anion–quadrupole interactions at the antibody interfaces is also consistent with the recent antibody-antigen interface amino acid compositional analysis72 (e.g., enriched Trp, Asp, Arg, etc.). In general, other than peptide-binding interfaces, the number of interchain salt bridges was ~2-fold higher than the number of anion–quadrupole and HB-anion–quadrupole interactions combined. However, like interfacial aromatic residues, the fraction of interfacial Arg/Lys residues participating in interchain salt bridges was also lower than that of intrachain salt bridges (Table S1), suggesting that interactions of aromatic and Arg/Lys residues with Asp/Glu residues are likely to be more prominent in the context of polypeptide chain folding, and therefore we considered probing the behavior of their conservation in the context of intramolecular interactions (see below).
Figure 10. Anion–quadrupole interactions at protein-protein interfaces:
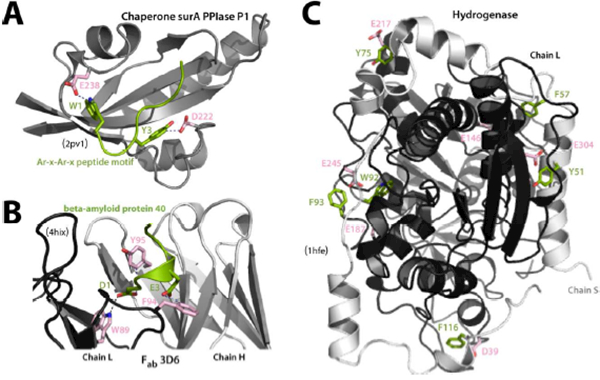
(A) Peptide recognition by the SurA PPIase domain and (B) antibody 3D6 involving anion–quadrupole interactions. (C) The hydrogenase small chain (chain S, light gray) interacts with the long chain (chain L, dark gray) by a number of anion–quadrupole interactions.
Interaction Conservation.
Conservation of Weak Interactions.
How conserved is a weak interaction? How does the conservation of a weak interaction compare with that of a salt bridge, an interaction considered be to be energetically stronger than an anion–quadrupole interaction. A non-redundant set of 4835 high-resolution protein structures belonging to 776 pfam domains was chosen to address this in our conservation analysis (see SI pp. S8–S10). A position in a structure in this set was first mapped to a column of the corresponding pfam domain alignment (Figure S9). For an anion–quadrupole interaction observed in a structure, the occurrence frequency of the residue types (e.g., Trp/Tyr/Phe) in the corresponding alignment columns was determined. We represented the occurrence frequency as a probability (0.0 ≤ p ≤ 1.0), and the distributions of the occurrence frequencies of Asp/Glu in anion–quadrupole, HB-anion–quadrupole, and salt bridge interactions were not only similar but also tended to show low levels of conservation (p ≤ 0.3) in general (Figure 11A). The trend was similar for the aromatic residues of anion–quadrupole and HB-anion–quadrupole interactions and the positively charged residues of salt bridges (Figure S9C), suggesting that an interaction observed in one structure can be replaced by a different type of interaction or be absent in another structure. Even though less conserved in general, we wanted to probe the correlation between the interacting positions. We utilized a simple measure termed the conditional probability (detailed in SI pp. S9–S10 and Figure S10) for probing the correlation between interacting positions. We have also used conditional probability in describing the coevolving nature of the binding-site residues of the PHD-finger module73. The probability of aromatic residues (W75 in the structure, Figure 11B) in column number 73 of the pfam CMD domain alignment increases (0.24 → 0.67) when the alignment was perturbed by column number 91 (D93 in the structure), in which the W75 and D93 residues interacted by the HB-anion-quadrupole interaction (Figure 11B). In the pfam CMD example, the conditional probability (p = 0.67) was greater than the probability (p = 0.24), and we considered an enhancement of the occurrence frequency as an indication of positional correlation; that is, if a residue is retained, the interacting partner is likely to be retained as well. For anion–quadrupole, HB-anion–quadrupole, and salt bridge residue pairs, the conditional probability on average was always greater than the probability (Figure 11C), suggesting that there is a preference for retention of the interacting partner. The average conditional probability generally followed the order salt bridge > HB-anion–quadrupole > anion-quadrupole interaction (Figure 11C), and the observed energetics (Figure 4B) likely contributed to the observed order, in which energetically stronger interactions were more likely to retain the interacting partner during evolution.
Figure 11. Conservation of anion–quadrupole, HB-anion–quadrupole, and salt bridge interactions:
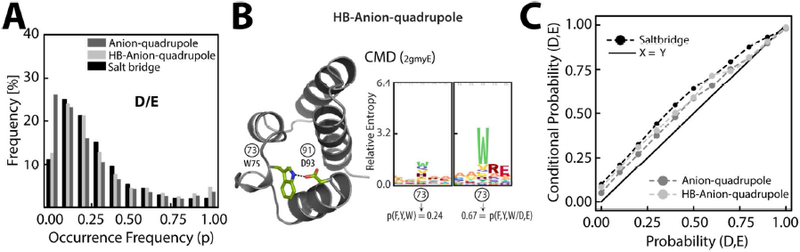
(A) The distribution (bars) of the occurrence frequency (0.0 ≤ p ≤ 1.0) of residues (D/E) engaged in anion-quadrupole (dark gray), HB-anion–quadrupole (light gray), and salt bridge (black) interactions. (B) Sequence logos (right) representing the correlation between the positions of the CMD pfam domain (left) that are engaged in HB-anion–quadrupole interactions. The right panel of the sequence logos represents the increase in the occurrence frequency of aromatic residues [p(F,Y,W)] upon sequence perturbation (see Figure S9). The left panel of the logos represents unperturbed alignments. Circled numbers (73 and 91) represent alignment positions. (C) The correlation between the probability and the conditional probability of anion–quadrupole (dark gray), HB-anion–quadrupole (light gray) and salt bridge (black) interactions.
Conservation Among Remote Homologs.
From the set of 4835 protein structures, we created a subset of structures, termed pfam_nr40, for each of the pfam domains (see SI pp. S3). The pfam_nr40 subset for each domain contained at least two structures, and the sequences of all the structures within pfam_nr40 shared less than 40% sequence identity, and we considered pfam_nr40 to consist of structures of remote homologs. We observed many instances in which an interacting pair was retained at the respective positions of the pfam_nr40 structures, for example, anion–quadrupole interactions of the pfam Peripla_BP_4 (Figure 12A) and MR_MLE_C (Figure S11C) families. Despite structural deviations among the pfam_nr40 structures, conserved interacting residues (p ≤ 0.5) participated in anion–quadrupole interactions in each of the structures of Peripla_BP_4 (Figure 12A). In the pfam_nr40 analysis, anion–quadrupole interactions were first detected in each structure and then mapped to the pfam alignment columns (see above, and Figure S9A) to check whether an alignment column was common among the pfam_nr40 structures. This means that the distance-and-angle detection criteria first need to be satisfied in the corresponding positions of the pfam_nr40 structures for a common column to be observed for a pfam domain family (Figure S9). This strict criterion restricted accounting for positions, not merely for the amino acid’s presence but for its presence with a well-defined geometry, even among remote homologs. For the common alignment columns of pfam_nr40 structures (Figure S9), we also obtained the occurrence frequency (p). A distribution of these frequency values for the interacting residues (anion–quadrupole, HB-anion–quadrupole, and salt bridge interactions combined) in this case (i.e., conserved among pfam_nr40 structures) showed a much higher level of conservation, p ≤ 0.35 (Figure 12B).
Figure 12. Conservation among remote homologs:
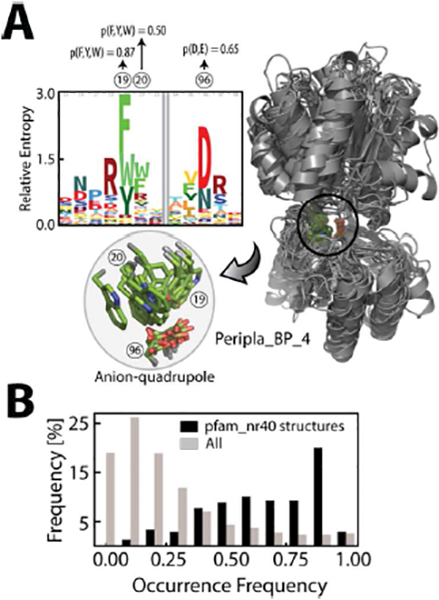
(A) Conserved anion–quadrupole-interacting residues (inset) in the pfam_nr40 structures of the Peripla_BP_4 domain. The occurrence frequency of these residues is displayed above the sequence logo. (B) The combined distribution (black bar) of the occurrence frequency of anion–quadrupole, HB-anion–quadrupole (D/E and F/Y/W), and salt bridge (D/E and R/K) residue positions when they are observed in the corresponding positions in at least two of the pfam_nr40 structures. The gray bars represent the combined background distributions of Figure 11A.
Protein Folding.
Based on the observation of a highly conserved pair of interacting residues (anion–quadrupole, HB-anion–quadrupole, and salt bridge) observed among diverse sequences that retain the same fold (Figures 12A, S11C), it is tempting to believe that interacting residues or residue positions conserved among remote homologs contribute towards folding and fold determination. Probing this question would, however, require detailed experimentation and has already been the subject of folding studies in the past decade74-76. The availability of in silico folding algorithms77–78 based on evolutionary covariance information encouraged us to check the predicted residue–residue contact scores for the interacting residues, particularly for interactions that were conserved in two structures of the remote homologs (Figure 13C). We utilized the GREMLIN79, 80 interresidue contact scores (see SI pp. S10-S11) available for the pfam domain sequences to map the scores onto the structures of pfam_nr40 (Figure S11A). The distribution of the interresidue contact scores for interacting residues (anion–quadrupole, HB-anion–quadrupole, and salt bridge interactions) that were conserved among remote homologs was shifted towards higher scores when compared with the background distribution of the contact scores (Figure 13C). This suggests that, at least in the in silico folding of a protein sequence, the distance restraints obtained from the conserved anion–quadrupole, HB-anion–quadrupole, and salt-bridge pairs can make predominant contributions, even though these interactions are on the surface. These observations are physically meaningful in the context of the pfam_nr40 structures (Figure 13B). For example, in the pfam_nr40 structures of the bromodomain and MR_MLE_N domains, the anion–quadrupole and HB-anion–quadrupole interacting pairs with large interresidue contact scores were in the regions having the least structural deviation among pfam_nr40 structures (Figure 13B). In general, anion–quadrupole, HB-anion–quadrupole, and salt bridge pairs that were conserved among remote homologs appeared within regions showing small structural deviations (Figures 13B, S11B). This observation was quantified by the distribution of the root-mean-square deviation (RMSD) of Cα atoms of conserved anion–quadrupole, HB-anion–quadrupole, and salt bridge pairs, which was shifted to lower RMSD values when compared with that of equivalent Cα atoms of remote homologs (Figure 13A). These observations suggest that anion–quadrupole and HB-anion–quadrupole interactions likely play important roles in folding and stability.
Figure 13. Structural deviations and residue-residue contact score:
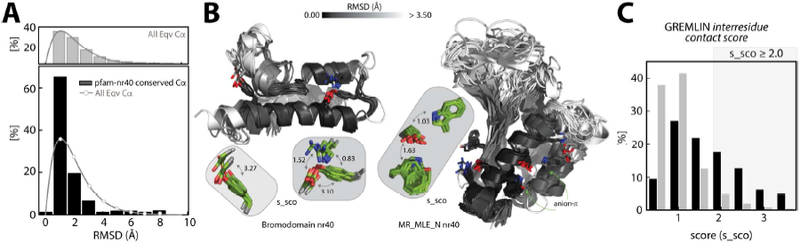
(A) Root-mean-square deviation (RMSD) distribution (top, gray bars) of all equivalent Cα atoms in the structural alignment of pfam_nr40 structures. The RMSD distribution of Cα atoms (bottom, black bars) of conserved interacting positions (anion-quadrupole, HB-anion–quadrupole, and salt bridge) in the pfam_nr40 structures. The background distribution (gray line) of all equivalent Cα atoms is overlaid. (B) Structural deviation by RMSD (white, ≥ 3.5 Å and black, 0.0 A) in the pfam_nr40 structures of the bromodomain (left) and MR_MLE_N (right) domain. Anion–quadrupole-interacting positions in these structures are also shown in the inset in green with their GREMLIN s_sco scores indicated with curved, double-headed arrows. (C) The distribution of the s_sco scores (black bars) for positions conserved in at least two of the pfam_nr40 structures. Gray bars represent the background distribution of the GREMLIN s_sco scores in the dataset.
Concluding Remarks.
Like the Phe-Asp/Glu pairs, anion–quadrupole interactions with His, Tyr and Trp residues are also very common, and such interactions often occur within networks of other interactions. The energies of anion–quadrupole interactions depend on the geometry between the functional groups and the ring atom type (e.g., on the presence or absence of a dipole moment). Highly conserved anion–quadrupole interactions can be observed, even among remote homologs, suggesting that such interactions could contribute to folding and stability of proteins. The recent success in designing miniature monomeric proteins based on conserved and stabilizing structural motifs (e.g., cation–π in TrpPlexus81, CH–π in PPα-Tyr29) suggests that the conserved anion–quadrupole structural motifs observed here could also be exploited for similar design studies. In general, standard protein design algorithms typically lack energy functions for interactions such as cation–π or water-mediated stabilization. To function as well as natural proteins, in many cases designed proteins require multiple rounds of laboratory evolution in which such interactions can be introduced (e.g., in a recently redesigned ubiquitin variant82). Several examples of anion–quadrupole interactions in natural proteins cited in this study encourage the incorporation of energy functions for this kind of interaction in design algorithms. As anion–quadrupole interactions are a weak in nature, they likely contribute cooperatively with other interaction. For example, the highly conserved anion–quadrupole pairs (among remote homologs) appear in a network of other interactions (Figure 13B) and parsing the energy contributions of an anion–quadrupole interaction in a network of other interactions may not be straightforward.
However, the consistency between computed energy (calculated using isolated functional groups) and ring atom participation frequency and geometry (both obtained from folded proteins) suggests that statistics on the structures of weak interactions reasonably approximate interaction energies obtained from quantum mechanical calculations. The statistics on the orientation preferences of Tyr/Trp residues with anions observed here could be useful for describing such interaction potentials83-86 in protein design, refinement, and structure prediction applications. Since aromatic scaffolds are common in small organic molecules such as the Trp residue, substituted/unsubstituted organic aromatic rings with a net dipole moment can also exploit edgewise interactions with anions on the surface of a protein molecule. It is also encouraging to observe the correlation between molecular mechanics force field parameters and the structural statistics of weak interactions (e.g., Trp partial charges in the CHARMM36 force field51). Our observation thus provides a rationale for probing the performance of CHARMM3687 (for proteins) compared with other force fields in the study of anion–quadrupole-like interactions by molecular dynamics simulations. In general, reports about a novel structure or mode of interaction (e.g., Fcab88) often involve analysis of residue interaction networks (e.g., using the RING 2.0 server89). Such analysis does not typically include anion–quadrupole interactions. However, since we observed that anion–quadrupole interactions can be important components of interaction networks, inclusion of such interactions will enrich the analysis of residue interaction networks. For example, buried anions lacking hydrogen bonds were observed to be frequently involved in anion–quadrupole interaction networks, and one or more edgewise interactions with aromatic residues could stabilize buried anions lacking a hydrogen-bonded partner. Finally, anion–quadrupole interactions are observed not just in proteins but also in DNA/RNA–protein and lipid–water–membrane-protein interfaces. Although less frequent than salt bridges, our conservation analysis, especially among remote homologs, suggests that in many instances anion–quadrupole-interacting residues remain highly conserved and likely contribute to the folding of the polypeptide chain. Many critical contacts at interfaces mediated by anion–quadrupole interactions have also been observed here, and, in general, the numerous examples we came across provide a good source of information with which one can experimentally further probe the role of anion–quadrupole interactions in molecular recognition, folding, and assembly.
Supplementary Material
ACKNOWLEDGEMENTS
This study has received funding support from the National Science Foundation/EPSCoR Award No. AII-1355423 and the state of South Dakota, through BioSNTR (Biochemical Spatiotemporal NeTwork Resource). Additional support was provided by the National Institutes of Health (NIH) grant 1R15GM116040–01A1 to SC and the Department of Chemistry & Biochemistry startup funds to SC. Authors thank the SDSU academic computing facility for the access to the Linux cluster for all the energy calculations. Authors also thank the anonymous referees for their valuable suggestions.
The authors declare no competing financial interest.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of authors and do not necessarily reflect the views of the National Science Foundation and National Institutes of Health.
Supporting Information (SI)
The supporting information (SI) attached with the text contains: (i) Extended experimental section, (ii) Table S1, and (iii) Supplemental Figures S1 – S12.
REFERENCES
- (1).Kim KS, Tarakeshwar P, and Lee JY (2000) Molecular Clusters of pi-Systems: Theoretical Studies of Structures, Spectra, and Origin of Interaction Energies, Chem. Rev 100, 4145–4186. [DOI] [PubMed] [Google Scholar]
- (2).Jain A, Ramanathan V, and Sankararamakrishnan R (2009) Lone pair … pi interactions between water oxygens and aromatic residues: quantum chemical studies based on high-resolution protein structures and model compounds, Protein Sci. 18, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dougherty DA (1996) Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp, Science 271, 163–168. [DOI] [PubMed] [Google Scholar]
- (4).Sinnokrot MO, and Sherrill CD (2006) High-accuracy quantum mechanical studies of pi-pi interactions in benzene dimers, J. Phys. Chem. A 110, 10656–10668. [DOI] [PubMed] [Google Scholar]
- (5).McGaughey GB, Gagne M, and Rappe AK (1998) pi-Stacking interactions. Alive and well in proteins, J. Biol. Chem 273, 15458–15463. [DOI] [PubMed] [Google Scholar]
- (6).Martinez CR, and Iverson BL (2012) Rethinking the term “pi-stacking”, Chemical Sci. 3, 2191–2201. [Google Scholar]
- (7).Bartlett GJ, Choudhary A, Raines RT, and Woolfson DN (2010) n→ π * interactions in proteins, Nat. Chem. Biol 6, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nishio M, Umezawa Y, Fantini J, Weiss MS, and Chakrabarti P (2014) CH-pi hydrogen bonds in biological macromolecules, Phys. Chem. Chem. Phys 16, 12648–12683. [DOI] [PubMed] [Google Scholar]
- (9).Ozawa T, and Okazaki K (2008) CH/pi hydrogen bonds determine the selectivity of the Src homology 2 domain to tyrosine phosphotyrosyl peptides: an ab initio fragment molecular orbital study,J. Comp. Chem 29, 2656–2666. [DOI] [PubMed] [Google Scholar]
- (10).Tsuzuki S, Honda K, Uchimaru T, Mikami M, and Tanabe K (2000) The magnitude of the CH/π interaction between benzene and some model hydrocarbons, J. Am. Chem. Soc 122, 3746–3753. [Google Scholar]
- (11).Steiner T, and Koellner G (2001) Hydrogen bonds with pi-acceptors in proteins: frequencies and role in stabilizing local 3D structures, J. Mol. Biol 305, 535–557. [DOI] [PubMed] [Google Scholar]
- (12).Brandl M, Weiss MS, Jabs A, Suhnel J, and Hilgenfeld R (2001) C-H...pi-interactions in proteins, J. Mol. Biol 307, 357–377. [DOI] [PubMed] [Google Scholar]
- (13).Mitchell JB, Nandi CL, McDonald IK, Thornton JM, and Price SL (1994) Amino/aromatic interactions in proteins: is the evidence stacked against hydrogen bonding?, J. Mol. Biol 239, 315–331. [DOI] [PubMed] [Google Scholar]
- (14).Singh SK, and Das A (2015) The n ⤍ pi* interaction: a rapidly emerging non-covalent interaction, Phys. Chem. Chem. Phys 17, 9596.-. [DOI] [PubMed] [Google Scholar]
- (15).Egli M, and Gessner RV (1995) Stereoelectronic effects of deoxyribose 04’ on DNA conformation, Proc. Natl. Acad. Sci. U. S. A 92, 180.-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Egli M, and Sarkhel S (2007) Lone pair-aromatic interactions: To stabilize or not to stabilize, Acc. Chem. Res 40, 197–205. [DOI] [PubMed] [Google Scholar]
- (17).Mooibroek TJ, Gamez P, and Reedijk J (2008) Lone pair-π interactions: a new supramolecular bond?, CrystEngComm 10, 1501–1515. [Google Scholar]
- (18).Gallivan JP, and Dougherty DA (1999) Cation-pi interactions in structural biology, Proc. Natil. Acad. Sci. U. S. A 96, 9459.-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Crowley PB, and Golovin A (2005) Cation-pi interactions in protein-protein interfaces, Proteins 59, 231.-. [DOI] [PubMed] [Google Scholar]
- (20).Wintjens R, Lievin J, Rooman M, and Buisine E (2000) Contribution of cation-pi interactions to the stability of protein-DNA complexes, J. Mol. Biol 302, 395–410. [DOI] [PubMed] [Google Scholar]
- (21).Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, and Dougherty DA (1998) From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor, Proc. Natl. Acad. Sci. U. S. A 95, 12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xiu X, Puskar NL, Shanata JA, Lester HA, and Dougherty DA (2009) Nicotine binding to brain receptors requires a strong cation-pi interaction, Nature 458, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Estarellas C, Frontera A, Quinonero D, and Deya PM (2011) Relevant anion-pi interactions in biological systems: the case of urate oxidase, Angew. Chem. Int. Ed. Engl 50, 415–418. [DOI] [PubMed] [Google Scholar]
- (24).Zhao Y, Domoto Y, Orentas E, Beuchat C, Emery D, Mareda J, Sakai N, and Matile S (2013) Catalysis with anion-π interactions, Angewandte Chemie 125, 10124–10127. [DOI] [PubMed] [Google Scholar]
- (25).Philip V, Harris J, Adams R, Nguyen D, Spiers J, Baudry J, Howell EE, and Hinde RJ (2011) A survey of aspartate-phenylalanine and glutamate-phenylalanine interactions in the protein data bank: searching for anion-pi pairs, Biochemistry 50, 2939–2950. [DOI] [PubMed] [Google Scholar]
- (26).Neel AJ, Hilton MJ, Sigman MS, and Toste FD (2017) Exploiting non-covalent pi interactions for catalyst design, Nature 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Choy MS, Li Y, Machado LE, Kunze MB, Connors CR, Wei X, Lindorff-Larsen K, Page R, and Peti W (2017) Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery, Mol. cell 65, 644–658 e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mullins EA, Shi R, Parsons ZD, Yuen PK, David SS, Igarashi Y, and Eichman BF (2015) The DNA glycosylase AlkD uses a non-base-flipping mechanism to excise bulky lesions, Nature 527, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Baker EG, Williams C, Hudson KL, Bartlett GJ, Heal JW, Porter Goff KL, Sessions RB, Crump MP, and Woolfson DN (2017) Engineering protein stability with atomic precision in a monomeric miniprotein, Nat. Chem. Biol 13, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tateishi-Karimata H, Ohyama T, Muraoka T, Podbevsek P, Wawro AM, Tanaka S, Nakano SI, Kinbara K, Plavec J, and Sugimoto N (2017) Newly characterized interaction stabilizes DNA structure: oligoethylene glycols stabilize G-quadruplexes CH-pi interactions, Nucleic Acids Res. 45, 7021–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gallivan JP, and Dougherty DA (2000) A Computational Study of Cation-π Interactions vs Salt Bridges in Aqueous Media: Implications for Protein Engineering, J. Am. Chem. Soc 122, 870–874. [Google Scholar]
- (32).Waldburger CD, Schildbach JF, and Sauer RT (1995) Are buried salt bridges important for protein stability and conformational specificity?, Nat. Struct. Mol. Biol 2, 122–128. [DOI] [PubMed] [Google Scholar]
- (33).Sindelar CV, Hendsch ZS, and Tidor B (1998) Effects of salt bridges on protein structure and design, Protein Sci. 7, 1898–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kumar S, and Nussinov R (1999) Salt bridge stability in monomeric proteins, J. Mol. Biol 293, 1241–1255. [DOI] [PubMed] [Google Scholar]
- (35).Tomlinson JH, Ullah S, Hansen PE, and Williamson MP (2009) Characterization of salt bridges to lysines in the protein G B1 domain, J. Am. Chem. Soc 131, 4674–4684. [DOI] [PubMed] [Google Scholar]
- (36).Estarellas C, Frontera A, Quiñonero D, and Deyà PM (2011) Anion⍰ π Interactions in Flavoproteins, Chemistry-An Asian Journal 6, 2316.-. [DOI] [PubMed] [Google Scholar]
- (37).Breberina LM, Milčić MK, Nikolić MR, and Stojanović SĐ (2015) Contribution of anion-π interactions to the stability of Sm/LSm proteins, JBIC J. Biol. Inorg. Chem 20, 475–485. [DOI] [PubMed] [Google Scholar]
- (38).Zlatović MV, Borozan SZ, Nikolić MR, and Stojanović SĐ (2015) Anion-π interactions in protein-porphyrin complexes, RSC Advances 5, 38361–38372. [Google Scholar]
- (39).Chawla M, Chermak E, Zhang Q, Bujnicki JM, Oliva R, and Cavallo L (2017) Occurrence and stability of lone pair-pi stacking interactions between ribose and nucleobases in functional RNAs, Nucleic Acids Res. 45, 11019–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rogacheva ON, Izmailov SA, Slipchenko LV, and Skrynnikov NR (2017) A new structural arrangement in proteins involving lysine NH3(+) group and carbonyl, Scientific Rep. 7, 16402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Valiev M, Bylaska EJ, Govind N, Kowalski K, Straatsma TP, Van Dam HJ, Wang D, Nieplocha J, Apra E, and Windus TL (2010) NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations, Comp. Phys. Comm 181, 1477–1489. [Google Scholar]
- (42).O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR (2011) Open Babel: An open chemical toolbox, J Cheminform 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Grimme S, Antony J, Ehrlich S, and Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys 132, 154104. [DOI] [PubMed] [Google Scholar]
- (44).Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Mennucci B, and Petersson G (2008) Gaussian 09, Revision C. 01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed;(b) Y. Zhao and DG Truhlar, Theor. Chem. Acc 120, 215. [Google Scholar]
- (45).McDonald IK, and Thornton JM (1994) Satisfying hydrogen bonding potential in proteins, J. Mol. Biol 238, 777–793. [DOI] [PubMed] [Google Scholar]
- (46).Wang G, and Dunbrack RL Jr. (2003) PISCES: a protein sequence culling server, Bioinformatics 19, 1589.-. [DOI] [PubMed] [Google Scholar]
- (47).Ehrlich S, Moellmann J, and Grimme S (2013) Dispersion-corrected density functional theory for aromatic interactions in complex systems, Acc. Chem. Res 46, 916–926. [DOI] [PubMed] [Google Scholar]
- (48).Hinderaker MP, and Raines RT (2003) An electronic effect on protein structure, Protein Sci. 12, 1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Horng JC, and Raines RT (2006) Stereoelectronic effects on polyproline conformation, Protein Sci. 15, 74.-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shortle D (2003) Propensities, probabilities, and the Boltzmann hypothesis, Protein Sci. 12, 1298–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Macias AT, and Mackerell AD Jr. (2005) CH/pi interactions involving aromatic amino acids: refinement of the CHARMM tryptophan force field, J. Comp. Chem 26, 1452–1463. [DOI] [PubMed] [Google Scholar]
- (52).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, and Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (53).Sangha AK, Dong J, Williamson L, Hashiguchi T, Saphire EO, Crowe JE Jr., and Meiler J (2017) Role of Non-local Interactions between CDR Loops in Binding Affinity of MR78 Antibody to Marburg Virus Glycoprotein, Structure 25, 1820–1828 e1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, Kruse AC, Pardon E, Steyaert J, Weis WI, Prosser RS, Kobilka BK, Costa T, and Lefkowitz RJ (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation, Nature 535, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Panasik N Jr., Fleming PJ, and Rose GD (2005) Hydrogen-bonded turns in proteins: the case for a recount, Protein Sci. 14, 2910–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Baran D, Pszolla MG, Lapidoth GD, Norn C, Dym O, Unger T, Albeck S, Tyka MD, and Fleishman SJ (2017) Principles for computational design of binding antibodies, Proc. Natl. Acad. Sci. U. S. A 114, 10900–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ferreiro DU, Komives EA, and Wolynes PG (2014) Frustration in biomolecules, Q. Rev. Biophys 47, 285.-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Brosey CA, Ho C, Long WZ, Singh S, Burnett K, Hura GL, Nix JC, Bowman GR, Ellenberger T, and Tainer JA (2016) Defining NADH-Driven Allostery Regulating Apoptosis-Inducing Factor, Structure 24, 2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Liu X, Speckhard DC, Shepherd TR, Sun YJ, Hengel SR, Yu L, Fowler CA, Gakhar L, and Fuentes EJ (2016) Distinct Roles for Conformational Dynamics in Protein-Ligand Interactions, Structure 24, 2053–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ji Q, Chen PJ, Qin G, Deng X, Hao Z, Wawrzak Z, Yeo WS, Quang JW, Cho H, Luo GZ, Weng X, You Q, Luan CH, Yang X, Bae T, Yu K, Jiang H, and He C (2016) Structure and mechanism of the essential two-component signal-transduction system WalKR in Staphylococcus aureus, Nature Commun. 7, 11000. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (61).Tencer AH, Gatchalian J, Klein BJ, Khan A, Zhang Y, Strahl BD, van Wely KHM, and Kutateladze TG (2017) A Unique pH-Dependent Recognition of Methylated Histone H3K4 by PPS and DIDO, Structure 25, 1530–1539 e1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Killian JA, and von Heijne G (2000) How proteins adapt to a membrane-water interface, Trends Biochem. Sci 25, 429.-. [DOI] [PubMed] [Google Scholar]
- (63).Norimatsu Y, Hasegawa K, Shimizu N, and Toyoshima C (2017) Protein-phospholipid interplay revealed with crystals of a calcium pump, Nature 545, 193–198. [DOI] [PubMed] [Google Scholar]
- (64).Rujas E, Insausti S, Garcia-Porras M, Sanchez-Eugenia R, Tsumoto K, Nieva JL, and Caaveiro JM (2017) Functional Contacts between MPER and the Anti-HIV-1 Broadly Neutralizing Antibody 4E10 Extend into the Core of the Membrane, J. Mol. Biol 429, 1213–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Stockbridge RB, Kolmakova-Partensky L, Shane T, Koide A, Koide S, Miller C, and Newstead S (2015) Crystal structures of a double-barrelled fluoride ion channel, Nature 525, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Johnson ZL, and Chen J (2017) Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1, Cell 168, 1075–1085 e1079. [DOI] [PubMed] [Google Scholar]
- (67).Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, Fang W, Wang NY, Nguyen D, Houston J, Carmel G, Tran T, Riley M, Nosaka L, Lander GC, Gaidarova S, Xu S, Ruchelman AL, Handa H, Carmichael J, Daniel TO, Cathers BE, Lopez-Girona A, and Chamberlain PP (2016) A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase, Nature 535, 252–257. [DOI] [PubMed] [Google Scholar]
- (68).Dhawan NS, Scopton AP, and Dar AC (2016) Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling, Nature 537, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Xu X, Wang S, Hu YX, and McKay DB (2007) The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues, J. Mol. Biol 373, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Miles LA, Crespi GA, Doughty L, and Parker MW (2013) Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation, Scientific Rep. 3, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gray HB, and Winkler JR (2015) Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage, Proc. Natl. Acad. Sci. U. S. A 112, 10920–10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Nguyen MN, Pradhan MR, Verma C, and Zhong P (2017) The interfacial character of antibody paratopes: analysis of antibody-antigen structures, Bioinformatics 33, 2971–2976. [DOI] [PubMed] [Google Scholar]
- (73).Boamah D, Lin T, Poppinga FA, Basu S, Rahman S, Essel F, and Chakravarty S (2018) Characteristics of a PHD Finger Subtype, Biochemistry 57, 525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Zarrine-Afsar A, Larson SM, and Davidson AR (2005) The family feud: do proteins with similar structures fold via the same pathway?, Curr. Opin. Struct. Biol 15, 42–49. [DOI] [PubMed] [Google Scholar]
- (75).Plaxco KW, Larson S, Ruczinski I, Riddle DS, Thayer EC, Buchwitz B, Davidson AR, and Baker D (2000) Evolutionary conservation in protein folding kinetics, J. Mol. Biol 298, 303–312. [DOI] [PubMed] [Google Scholar]
- (76).Gunasekaran K, Eyles SJ, Hagler AT, and Gierasch LM (2001) Keeping it in the family: folding studies of related proteins, Curr. Opin. Struct. Biol 11, 83–93. [DOI] [PubMed] [Google Scholar]
- (77).Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, and Sander C (2011) Protein 3D structure computed from evolutionary sequence variation, PloS One 6, e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Anishchenko I, Ovchinnikov S, Kamisetty H, and Baker D (2017) Origins of coevolution between residues distant in protein 3D structures, Proc. Natl. Acad. Sci. U. S. A 114, 9122–9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Kamisetty H, Ovchinnikov S, and Baker D (2013) Assessing the utility of coevolutionbased residue-residue contact predictions in a sequence- and structure-rich era, Proc. Natil. Acad. Sci. U. S. A 110, 15674–15679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ovchinnikov S, Kamisetty H, and Baker D (2014) Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information, eLife 3, e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Craven TW, Cho MK, Traaseth NJ, Bonneau R, and Kirshenbaum K (2016) A Miniature Protein Stabilized by a Cation-pi Interaction Network, J. Am. Chem. Soc 138, 1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Biel JT, Thompson MC, Cunningham CN, Corn JE, and Fraser JS (2017) Flexibility and Design: Conformational Heterogeneity along the Evolutionary Trajectory of a Redesigned Ubiquitin, Structure 25, 739–749 e733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Kortemme T, Morozov AV, and Baker D (2003) An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes, J. Mol. Biol 326, 1239–1259. [DOI] [PubMed] [Google Scholar]
- (84).Misura KM, Morozov AV, and Baker D (2004) Analysis of anisotropic side-chain packing in proteins and application to high-resolution structure prediction, J. Mol. Biol 342, 651–664. [DOI] [PubMed] [Google Scholar]
- (85).Zhang J, and Zhang Y (2010) A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction, PloS One 5, e15386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Zhou H, and Skolnick J (2011) GOAP: a generalized orientation-dependent, all-atom statistical potential for protein structure prediction, Biophys. J 101, 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Huang J, and MacKerell AD Jr. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data, J. Comp. Chem 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Lobner E, Humm AS, Goritzer K, Mlynek G, Puchinger MG, Hasenhindl C, Ruker F, Traxlmayr MW, Djinovic-Carugo K, and Obinger C (2017) Fcab-HER2 Interaction: a Menage a Trois. Lessons from X-Ray and Solution Studies, Structure 25, 878–889 e875. [DOI] [PubMed] [Google Scholar]
- (89).Piovesan D, Minervini G, and Tosatto SC (2016) The RING 2.0 web server for high quality residue interaction networks, Nucleic Acids Res. 44, W367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.