

Abstract
Background:
The mechanism by which bile acids induce liver injury in cholestasis remains controversial. Although high levels of bile acids are toxic when applied to liver cells, the level of toxic bile acids in the liver of most cholestatic animals and patients is <10 μM, indicating there must be alternative mechanisms. Recent studies suggest that the inflammatory response may play an important role in bile acid-induced liver injury, as pro-inflammatory cytokine expression is stimulated by bile acids in mouse hepatocyte cultures. To elucidate the mechanisms of bile acid-induced liver injury, we assessed signs of liver damage and gene expression in Abcb4−/− mice, a well-known model for cholestasis.
Key Messages:
Elevated plasma levels of bile acids were detected as early as 10 days after birth and at all later ages in Abcb4−/− mice compared to their wild-type littermate controls. Parallel increases in expression of Tnfα, Ccl2, Cxcl1, and Cxcl2 mRNA occurred at these early time points and throughout 12 weeks in Abcb4−/− livers. Marked hepatic neutrophil infiltration was first detected in 3-week mice, whereas histological evidence of liver injury was not detected until 6-weeks of age. Subsequent in vitro studies demonstrated that normal hepatocytes but not other non-parenchymal liver cells responded to bile acids with inflammatory cytokine induction.
Conclusion:
Bile acids induce the expression of pro-inflammatory cytokines in hepatocytes in Abcb4−/− mice that initiates an inflammatory response. This inflammatory response plays an important role in the development of cholestatic liver injury in this and other cholestatic conditions. Furthermore, understanding of these inflammatory mechanisms should lead to new therapeutic approaches for cholestatic liver diseases.
Keywords: Bile acid, cholestasis, inflammation, Abcb4−/−
In the last decade, a number of studies have suggested that bile acids may injure liver cells in cholestatic liver disease, not by a direct toxic effects as a detergent, but by initiating a cytokine-mediated inflammatory response [1,2]. While there is little doubt that bile acids can injure mitochondria, produce oxidative stress and initiate Fas-dependent apoptosis, these observations have been derived primarily from in-vitro studies in isolated cell systems where the concentrations have exceeded those normally found in the serum or liver of cholestatic animal models or patients with cholestatic liver diseases [3,4,5]. As discussed by Woolbright and Jaeschke [2] and observed by Trottier et al. [6] serum concentrations of bile acids in cholestatic patients rarely exceed 150–200 μM and the highest levels are non-toxic bile acids like taurocholic and glycocholic acid, while levels of the principle toxic bile acid, glycochenodeoxycholic acid reach concentrations of no more than 30 μM. In contrast, when isolated human hepatocytes are exposed to bile acids in-vitro, little or no injury occurs until concentrations of >50 μM GCDCA are used for 24 h [7]. Similar findings are also seen after bile duct ligation in mouse models of cholestasis [7] where the most toxic bile acid, TCDCA + GCDCA only reach levels ∼25 nM during the initial 3 days of injury, whereas concentrations up to 3,000 fold are required to initiate toxicity in hepatocyte cultures from mice [7]. Thus, alternative explanations for bile acid-induced liver injury seem necessary.
A number of studies have demonstrated that bile duct ligation results in the accumulation of neutrophils within the liver parenchyma in areas of cell injury and that adhesion molecules such as ICAM-1 play an important role since ICAM-1 null mice or mice deficient in NHERF-1, an anchoring protein required for ICAM-1 expression markedly reduce liver injury and neutrophil accumulation [8,9]. Allen et al. [1] have shown that the exposure of hepatocytes in culture to levels of bile acids (both toxic and non-toxic) that are seen in-vivo, results in significant increases in mRNA expression of pro-inflammatory cytokines, particularly CXCL2 (MIP-2) and CXCL1 (KC). Importantly, toll-like receptor 4 was not involved in the initiation of this acute inflammatory response [1].
Figure 1 illustrates similar findings from our laboratory in the Abcb4−/− mouse model of cholestatic liver injury as it develops over time from birth. Note that the initial abnormalities consist of a rise in serum bile acids at 10 days of birth. This is associated with an increase in hepatic cytokine expression (mRNA), particularly Ccl2 (MCP-1) and Cxcl2 prior to any evidence of liver injury as reflected in the absence of significant elevation of aminotransferases until sometime between day 20 and 40 (fig. 2, 3). Neutrophil infiltration then follows at day 20 when liver injury appears to be initiated at that point of time (fig. 4). These findings strongly suggest that the initial injury is mediated by an increase in serum bile acids at a time that presumably reflects elevated liver bile acid levels.
Fig 1.
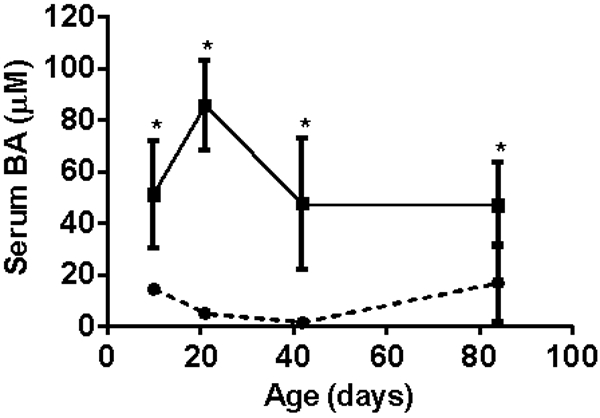
Serum bile acid levels from days of birth in Abcb4 null mice (▪) vs wild type controls (●)
Fig 2.
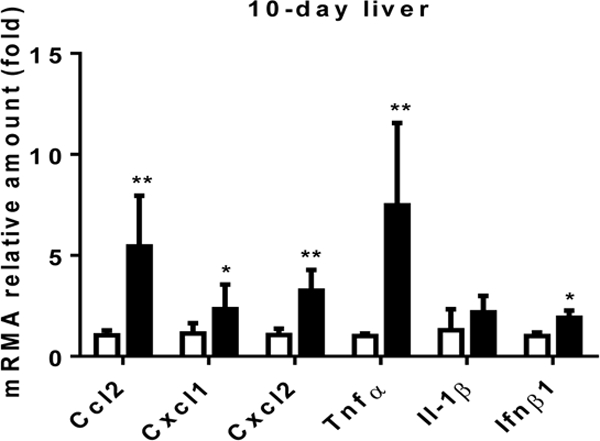
Cyctokine expression levels in liver in the ten day old Abcb4 null mice (black bars) vs wild type controls (open bars)
Fig 3.
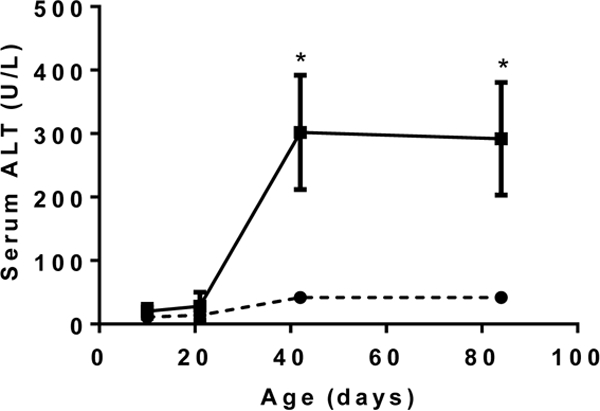
Serum Alanine aminotransferase levels as a function of age in Abcb4 null mice (▪) vs wild type controls (●)
Fig 4.
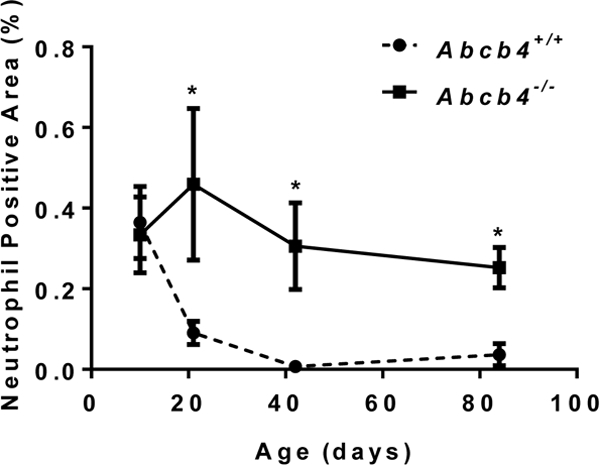
Liver neutrophil staining as a function of age in Abcb4 null mice vs wild type controls
Further detailed in vitro studies demonstrated that endogenous conjugated bile acids at pathophysiological levels cause ER stress and mitochondria damage, and stimulated cytokine expression in primary hepatocytes from mice and humans but not non-parenchymal liver cells. This cell type-specific event depends on bile acid entry to hepatocytes through its membrane transporters. Disruption of pro-inflammatory chemokines production in vivo in mice reduced liver injury after bile duct ligation [10]. This was seen in Ccl2 null mice and toll-like receptor 9 (Tlr9) null mice as well, as tlr9 can sensor damaged mitochondrial DNA and stimulated chemokine expression [11].
These series of experiments provide new insights into the mechanism by which bile acids injure the liver. First, bile acids at pathophysiologic levels seen in cholestatic liver disease accumulate in the liver where they induce ER stress and mitochondrial injury. As part of this injury response, mitochondrial DNA activates the innate immune system in part by activating TLR9, which then leads to the increase in expression of cytokines including Cxcl2, resulting in the influx of neutrophils and the onset of liver injury.
These findings suggest several novel targets for future therapeutic intervention including: (1) the inhibition of basolateral bile acid transporter, Ntcp, to prevent bile acid accumulation in the liver; (2) the application of drugs that reduce ER stress and mitochondrial injury; (3) inhibitors of cytokine production or release and finally (4) inhibitors of cytokine receptors in order to block subsequent inflammatory cell recruitment. Future studies will be needed to assess these potential targets.
Footnotes
Disclosure: All authors has nothing to disclose.
Citations:
- 1.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 2011;178:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol 2012;18:4985–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 2010;90:1165–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH, et al. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest 1999;103:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higuchi H, Bronk SF, Takikawa Y, Werneburg N, Takimoto R, El-Deiry W, Gores GJ. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. J Biol Chem 2001;276:38610–38618. [DOI] [PubMed] [Google Scholar]
- 6.Trottier j BA, Caron P, Straka RJ, Milkiewicz P and Barbier O. Profiling Circulating and Urinary Bile acids inPaatients with Biliary Obstructioin before and after Biliary Stenting. PLos one 2011;6:e22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YC, Hong JY, Rockwell CE, Copple BL, Jaeschke H, Klaassen CD. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver International 2012;32:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gujral JSLJ, Farhood A, Hinson JA, Jaeschke H. Functional. importance of ICAM-1 in the mechanism of neutrophil-induced liver injury in bile duct-ligated mice. Am J Physiol Gastrointest Liver Physiol 2004;286:G499–G507. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Mennone A, Soroka CJ, Hagey LR, Ouyang X, Weinman EJ, Boyer JL. Na(+) /H(+) exchanger regulatory factor 1 knockout mice have an attenuated hepatic inflammatory response and are protected from cholestatic liver injury. Hepatology 2015;62:1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai S-Y Ouyang X, Mennone A, Smith MR, Soroka S, Mehal WZ, Boyer JL. Ccl2-deficiency protects mice from cholestatic liver injury by blocking hepatic leukocye infiltration. Hepatology 2014;60:300A. [Google Scholar]
- 11.Cai S- Y, Ouyang X, Mennone A, Soroka S, Mehal WZ, Boyer JL. Bile acid induced cholestatic liver injury involves TLR9 initiated inflammatory signals in mouse hepatocytes. Hepatology 2015;62:601A. [Google Scholar]