

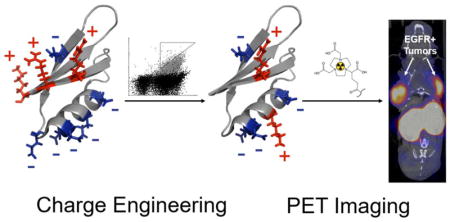
Abstract
The Gp2 domain is a protein scaffold for synthetic ligand engineering. However, the native protein function results in a heterogeneous distribution of charge on the conserved surface, which may hinder further development and utility. We aim to modulate charge, without diminishing function, which is challenging in small proteins where each mutation is a significant fraction of protein structure. We constructed rationally guided combinatorial libraries with charge-neutralizing or charge-flipping mutations and sorted them, via yeast display and flow cytometry, for stability and target binding. Deep sequencing of functional variants revealed effective mutations both in clone-dependent contexts and broadly across binders to epidermal growth factor receptor (EGFR), insulin receptor, and immunoglobulin G. Functional mutants averaged 4.3 charge neutralizing mutations per domain while maintaining net negative charge. We evolved an EGFR-targeted Gp2 mutant that reduced charge density by 33%, maintained net charge, and improved charge distribution homogeneity while elevating thermal stability (Tm = 87 ± 1 °C), improving binding specificity, and maintaining affinity (Kd = 8.8 ± 0.6 nM). This molecule was conjugated with 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid for 64Cu chelation and evaluated for physiological distribution in mice with xenografted A431 (EGFRhigh) and MDA-MB-435 (EGFRlow) tumors. Excised tissue gamma counting and positron emission tomography / computed tomography imaging revealed good EGFRhigh tumor signal (4.7 ± 0.5 %ID/g) at 2 h post-injection and molecular specificity evidenced by low uptake in EGFRlow tumors (0.6 ± 0.1 %ID/g, significantly lower than for non-charge-modified Gp2, p = 0.01). These results provide charge mutations for an improved Gp2 framework, validate an effective approach to charge engineering, and advance performance of physiological EGFR targeting for molecular imaging.
Graphical Abstract
Introduction
Small protein ligands that bind targets with high affinity and specificity are valuable resources for imaging1,2, therapeutic3–5, and diagnostic purposes6,7. Small scaffolds benefit from increased tumor extravasation8–10, tissue penetration11,12 and plasma clearance10,13 resulting in improved target-to-background contrast at early time points14–16. Protein ligands must be engineered for not only binding function but also physicochemical robustness and physiological performance. To optimize performance, several influencing factors have been studied including scaffold structure17, hydrophilicity18,19, stability20,21, and charge19,22,23. Protein charge is of considerable interest as clusters of positively charged residues can reduce solubility24 and drive non-specific binding to natively negatively charged cell membranes25–27. An increase in the fraction of charged residues on non-paratope regions can correspond to decreased target affinity28. Ligand charge can also affect transvascular flux29,30, tumor uptake31, plasma clearance32,33, and renal retention19,34. It is therefore important to be able to modulate charge without diminishing desirable ligand characteristics including high production yield and soluble recovery, strong affinity, and stability. However, these charge modifications can be particularly difficult in the context of a small ligand19,35 where such mutations would encompass a greater portion of the protein’s structure36.
A scaffold that may benefit from redistributed and reduced charge is the Gp2 domain37. Gp2 is a small, stable 45-residue scaffold with α-helical and β-strand structure whose solvent-exposed loop regions have been diversified to discover and evolve binders to numerous targets.37 Engineered Gp2 domains have been validated for molecular positron emission tomography (PET) imaging of epidermal growth factor receptor (EGFR)38 and antagonism of insulin receptor (InsR) in tamoxifen-resistant, triple-negative breast cancer cell culture39. The native Gp2 framework contains three positive and six negative residues, with each charge set regionally clustered. It is hypothesized that this charge distribution evolved in wild-type Gp2 because of its native function. In a Gp2-RNA polymerase model structure, the string of acidic residues projects into the DNA-binding channel thereby aiding inhibitory function.40 Also, the basic residues R42 and R44 were experimentally shown as necessary for Gp2 function as they aid binding to RNA polymerase.40 Yet maintenance of these charges is not necessary – and we hypothesize not optimal – for its newfound role as a scaffold for synthetic ligand engineering. These highly charged regions may drive non-specific binding41, off-target retention19,23,34,42, and decrease protein yield during ligand production due to reduced solubility43–45. Therefore, the ability to mutate such clusters without reducing Gp2 affinity or stability has considerable value.
Numerous strategies exist for charge engineering including computational design43,46,47, homology-guided consensus design48, and combinatorial library selection49. We merged versions of each of these techniques to identify charge-modifying mutations that are well tolerated in multiple Gp2 domains and applied effective mutations to an EGFR-binding Gp2 domain for PET imaging. EGFR overexpression is present in numerous cancers50, is correlated with differentiation, and is an independent prognostic indicator of poor disease-free and overall survival in colorectal cancer patients51,52. Molecular imaging of EGFR could empower patient stratification of responders versus non-responders to molecularly targeted therapy.53,54
Combinatorial libraries of Gp2 mutants targeting EGFR, InsR, and rabbit immunoglobulin G (rIgG), guided by computational stability55 as well as natural56,57 and chemical homologs58, were sorted via yeast display and flow cytometry for mutants that retained thermally stability and target affinity. Functional mutants elucidated preferred net and total charge states, determined favored amino acids at charged positions, and identified several promising charge-modified anti-EGFR Gp2 ligands. A charge-reduced clone, GαE35, exhibited reduced non-specific binding and highly functional performance in PET/CT imaging of EGFR+ tumors in murine models. The results advance Gp2 scaffold engineering, as well as charge engineering more broadly, and improve preclinical performance of an EGFR PET agent.
Results and Discussion
Mutant Library Design and Construction
To identify functional mutants of the EGFR-binding Gp2 (GαE2.2.3, also termed GαE57 since it is a Gp2 ligand targeting EGFR with five basic and seven acidic residues) with modified charge distributions, we constructed combinatorial libraries for high-throughput functional screening in which the ten charged sites present in the framework were diversified. We also aimed to identify charge mutations that are broadly acceptable within numerous engineered variants of the Gp2 scaffold (Figure 1A); thus we created additional libraries for insulin receptor and rabbit IgG binding based on previously established GαI2.2.559 and GαR3.2.337 ligands. In all three cases, charged residues in the evolved loops were conserved while ten framework residues (Figure 1B) were mutated.
Figure 1. Gp2 charge distribution.

(A) Structure of the wild-type Gp2 scaffold with acidic (blue), basic (red), and paratope (green) residues highlighted [PDB: 2WNM] (B) Sequences of wild-type, EGFR-, InsR-, and rIgG-binding clones used as parental ligands for charge modified Gp2 libraries.
Full amino acid diversity would necessitate 2010 = 1013 unique clones and surpass the transformation efficiency of yeast used for protein display and selection. Therefore, a more selective approach was undertaken to predict which newly introduced mutations would be least likely to impair protein folding and target binding. Three metrics were applied: lack of destabilization as predicted by FoldX55, frequency in natural homologs56,57, and amino acid hydrophilicity58. FoldX was used to compute the theoretical change in stability upon independent mutation of each charged site (details in Experimental Procedures), which predicted that several charged positions would be relatively intolerant to mutation without negatively affecting fold (Figure 2A). These include sites 1, 14, 24, 42, and 44 where 9–14 mutations away from wild-type amino acids increased free energy of folding in excess of 0.5 kcal mol−1. The remaining charged positions (20, 23, 27, and 30) were more amenable with only 1–6 mutations causing a similar increase in free energy.
Figure 2. Combinatorial library design.

(A) Theoretical stability upon mutation based on FoldX60 force field calculations, natural homolog frequencies from the BLAST algorithm61, and amino acid hydropathy58 (B) An example of the decision-making process behind library designs (C) EGFR-, InsR-, and rIgG-charge modified Gp2 libraries as designed.
Amino acid frequencies from Gp2 homologs (Figure 2A), identified via search of available genomes, also provided information regarding potentially acceptable mutations at charged positions. Wild-type residues were generally conserved with an average of 3.2 non-wild-type amino acids observed. This value decreased to 2.4 when excluding replacement with amino acids of identical charge. Sites 1, 14, 24, 30, 42, and 44 had high conservation of wild-type amino acids (≥ 75% consensus) whereas sites 20, 27, and 31 were more amenable to neutralization, accepting several neutral functional groups. Site 23 was highly conserved as a negatively charged residue but relatively indifferent to aspartic (56%) or glutamic (41%) acid.
Each charged position required individual analysis such that no metric (hydrophilicity, stability, and homolog frequency) was applied universally to identify reasonable mutants for inclusion in the library. All charged functional groups extend outside of the protein core in the wild-type structure62 making high to moderate hydrophilicity preferable. Additionally, an elevated frequency in naturally occurring homologs guided inclusion of neutral amino acids in the combinatorial library design. Finally, mutations causing a considerable increase in free energy of folding (ΔΔGfolding ≥ 1 kcal mol−1), as modeled by FoldX, were avoided unless such mutations presented high homolog frequency (> 5%) and low hydrophobicity (< −1). In some instances, the inclusion of less desirable amino acids was unavoidable due to genetic code restrictions. The following examples are used to demonstrate the overall methodology. Site 1 was allowed to mutate to glutamine for high hydrophilicity, leucine for elevated homolog frequency, and isoleucine as a byproduct of lysine and leucine codons (Figure 2B). In some cases, mutations that appeared nondetrimental in one metric would be juxtaposed by the others. For example, methionine was deemed generally acceptable at all sites of interest via stability analysis (median ΔΔGfolding,mutation = −0.4 kcal mol−1) although it is moderately hydrophobic and has a mere 4% homolog frequency at site 31 and 0% frequency elsewhere. Similarly, hydrophilic proline was predicted to be destabilizing at position 44 (ΔΔGfolding,mutation = 3.7 kcal mol−1), but presented 9% sequence identity in natural homologs. The high throughput of yeast display and flow cytometry for functional selection enabled analysis of many mutants while the combinatorics of diversifying 10 positions required exclusion of many others.
Genes containing the degenerate codons necessary to implement these charged site mutations (Figure 2C), unmutated backbone residues, and EGFR-, InsR-, or rabbit IgG-binding paratopes were constructed through overlap PCR of 8–11 oligonucleotides per library resulting in 0.75 × 106, 0.75 × 106, and 1.7 × 106 possible unique clones. Assembled genes were electroporated into EBY100 S. cerevisiae yeast along with a linearized vector to enable yeast display of Gp2 as a C-terminal fusion to Aga2p with a glycine-rich polypeptide linker.63,64 The resultant yields of 2 × 108, 1.7 × 108, and 2.1 × 108 transformants for EGFR, InsR, and rabbit IgG libraries allow for >99% sampling of theoretical sequence space.65 Illumina MiSeq deep sequencing of naïve libraries showed that 91%, 94%, 88% of EGFR, InsR, and rIgG yeast transformants presented sequences as designed. These values increase to 98–99% when single-point mutations are included, indicating high library quality.
Mutant Selection
EGFR, InsR, and rIgG libraries were sorted, via yeast display and flow cytometry, to isolate mutants that retain stability and functional binding. The yeast containing the genetic libraries were grown and induced to express the Aga2p-linker-Gp2 variants tethered to their surface via covalent linkage to the membrane-bound Aga1p mating protein.63,64 Gp2-displaying yeast libraries underwent consecutive rounds of fluorescence-activated cell sorting (FACS) in which yeast were placed at an elevated temperature to induce unfolding of unstable mutants66, incubated on ice with target protein, and evaluated for retention of activity. Parental library ligands GαE2.2.3, GαI2.2.5, and GαR3.2.3, each demonstrated target affinities in the low nanomolar range (18 ± 8, 2.4 ± 0.4, and 2.3 ± 1.4 nM) and high thermal stability (71 ± 2 °C, 78 °C, and 80 ± 2 °C) (Table S1). These values were used to guide temperature and target concentration stringency during sorts. Naïve libraries exhibited a broad distribution from mutants that lost function to mutants that tolerated thermal stress and exhibited strong target binding (Figure S1). To remove instances of highly unstable or non-binding variants, naïve libraries were initially sorted at mild conditions with thermal stress temperatures 10 °C or more below parental Tm and recombinant target at (EGFR, rIgG) or above (InsR) parental dissociation constants (Table S1 and Experimental Procedures). A subsequent set of sorts aimed to isolate populations that either demonstrate improved thermal stability while maintaining moderate affinity (via elevated thermal stress with maintained target concentration) or provide increased ligand affinity after mild thermal stress (maintained thermal stress 11–28 °C below parental Tm while reducing target concentration). To this end, the sorted populations were split and sorted separately at each condition set, then pooled. Compared to GαI and GαR libraries, anti-EGFR clones were quite robust in their response to stability and affinity selection pressures and were sorted an additional time at an elevated temperature (80 °C) and low target concentration (2 nM). Although parental GαI2.2.5 and GαR3.2.3 exhibit substantial stability (78 and 80 ± 2 °C), no appreciable target-bound populations were present above background signal in sorts exceeding 60 °C even at heightened levels of target. However, unlike GαI, GαR clones retained parental affinity at 60 °C. To deplete clones that exhibit non-specific binding, enriched populations of Gp2-displaying yeast were incubated with streptavidin- and mouse IgG (mIgG)-coated magnetic beads at saturating conditions67, and only unbound yeast were retained.
In each of the three rounds of ligand selection against recombinant EGFR, 4–6% of sorted populations were collected based on expression-normalized target binding as indicated by triangular gates (Figure 3). To prepare for use in physiological environments, we aimed to ensure that mutants bound EGFR in its native fold. Thus, the EGFR-binding population underwent two additional rounds of sorting without heating against 2 and 0.2 nM solubilized EGFR from the lysate of A431 epidermoid carcinoma cells (2 – 6% collected).
Figure 3. Enrichment of stable, EGFR-binding charge mutants.

(A) Yeast libraries displaying charge-modulated Gp2 ligands were sorted against biotinylated EGFR ectodomain (2 – 20 nM) after pre-heating the yeast for 30 minutes (60–80 °C) followed by screening against streptavidin- and mIgG-coated beads to deplete non-specific binders, and finally sorted against lysate-extracted EGFR at 2 and 0.2 nM target. EGFR-labeled and target-negative yeast are shown in red and black with collected populations in triangular gates.
Mutant Characterization
Plasmids from the starting libraries and the concluding functional populations sorted for thermal tolerance and recombinant target binding, after non-specific Gp2 ligand depletion, were sequenced via Illumina MiSeq. Additionally, 24 clones from solubilized lysate EGFR-sorted populations were sequenced using the Sanger method.68
In the EGFR case, negatively charged variants were enriched during functional selection as the mean net charge shifted from neutral (0.1 ± 1.6) in the unsorted library to −2.7 ± 1.4 in the functional pool (Figure 4A). Notably, this shift was in the direction — and past — the parental GαE57, which has a net charge of −2. This net charge shift was achieved via enrichment of wild-type acidic residues as well as neutralization of basic residues: the unsorted library neutralized more than six sites on average (GαE57 has 12 charged sites whereas the unsorted library has 5.6 ± 1.6), which only slightly increased in the functional pool (7.0 ± 1.4), indicating that the larger shift in net charge also had contributions from neutralization of basic residues (Figure 4B). Sitewise analysis reveals the specifics of these functional preferences (Figure 5), which are discussed below in the context of all domains.
Figure 4. Net and total charge distributions of clones in naïve and sorted populations.

(A, B) EGFR (naïve: 2705, sorted: 3202), (C, D) InsR (naïve: 2842, sorted: 798), and (E, F) rIgG (naïve: 596, sorted: 172) Gp2 libraries. Data presented as frequency ± standard deviation. Mean ± standard deviation of overall distribution shown in legend.
Figure 5. Sitewise amino acid functionality.

Change in frequencies between functional and naïve populations. Parental amino acids presented in black unless noted by a matching color change in legend (* indicates presence solely in the rIgG library).
The other two parental Gp2 domains exhibit divergent tolerances to charge reduction as GαI essentially maintains removed charge (6.7 ± 1.3 charged sites in functional domains relative to 6.4 ± 1.4 in the library and 12 in the parental domain) whereas GαR reverts nearly half of its removed charges (3.3 ± 1.4 naïve to 5.4 ± 1.4 in functional vs. 8 in parental). Akin to the EGFR-targeted domain, but to a lesser extent, GαR net charge evolved towards the negative parental. The lack of change in total charge of GαI is matched by an absence of movement in net charge (−3.8 ± 1.5 in naïve vs. −3.6 ± 1.5 in functional populations). Notably, unlike the other libraries, the GαI library starts as net negative because of four acidic residues and no bases in its evolved loops. One other possible explanation for the tolerance of charge removal in GαI is an increase in structural stability imparted by a predicted disulfide bond between C8 and C38 in the evolved loops.
Across all three targets, the six negatively charged sites exhibit enrichment of the parental acidic side chains, with an increased frequency in functional variants of 29 ± 20%, with E14 and D23 in the InsR-targeted domains being the lone exceptions. Positively charged sites showed greater variance. Positions 1 and 31 display functional preference towards neutral residues frequent in natural homologs. K1 evolved towards leucine in all campaigns, which is present in 13% of native Gp2 homologs. Position 31, which is only charged in the parental EGFR-binding domain, preferentially mutates to W, which is 56% W in native homologs. R42 exhibits nearly comparable preference of Q and R within EGFR-binding domains whereas InsR- and rIgG-binding domains strongly enrich wild-type arginine. R44 exhibits variable performance for each target. EGFR-targeted domains enrich P and tolerate Q at the expense of R; InsR-targeted domains enrich Q and tolerate R; rIgG-binding domains enrich R and tolerate Q.
The utility of the combinatorial library approach is evident from the lack of strong correlations between functional efficacy and direct design metrics. Sitewise enrichment of particular amino acids does not correlate with predicted stability by FoldX (Pearson correlation coefficient, R = 0.24) or hydrophilicity (R = 0.01). Consistent with the examples in the previous paragraph, modest correlation is evident for natural homologs (R = 0.47).
Overall, numerous members of each library were robust to thermal denaturation and sorting against concentrations of recombinant target in the low nanomolar range. Additionally, several Gp2 variants bound high picomolar concentrations of A431 lysate-derived EGFR.
Top Clones and In Vitro Characterization
In addition to revealing mutational preferences, these selections identified particular functional mutants. To determine the effects of reducing total charge, six clones, all maintaining parental or near-parental net charge, were chosen from the final EGFR lysate-sorted population to further characterize the impacts of charge modification as well as to identify compelling lead mutants for physiological study.
These variants, with charge states of +2/−2, +2/−4, +2/−5 (two clones), +3/−5, and +3/−6, were produced in T7 E. coli and evaluated in vitro for soluble production yield, target affinity, thermal stability, and secondary structure. Soluble production yield was diminished considerably for GαE24 and GαE25B with less than 0.05 mg/L recoverable for either clone. However, recovery in the remaining four mutants exceeded 1 mg/L allowing for further in vitro characterization (Table S2). As selected for, all four mutants exhibited good binding affinity (Figure S2). Compared to GαE57, charge reduced clones GαE25A and GαE36 demonstrated nominally improved target affinity against recombinant EGFR (9.4 ± 4.1 nM for parental vs. 4.9 ± 0.7 nM (GαE25A) and 2.3 ± 1.1 nM (GαE36)) while GαE22 and GαE35 maintained near parental values (6.3 ± 1.4 nM and 8.8 ± 0.6 nM). Conversely, the mutants diverged with regards to thermal stability. GαE35 improved upon parental stability (Tm = 87 ± 1 °C) whereas GαE25A, GαE22, and GαE36 saw 15–20 °C reductions in melting temperature (Figure S3). Secondary structure was reasonably maintained in all variants (Figure S4). GαE25A showed a slight shift towards increased α-helical content. Interestingly, clones that did not fully return to their original secondary structure after heating (GαE25A, GαE22, GαE36) also exhibited a demonstrable decrease in Tm when compared to fully refoldable clones (mean ± SD: 61 ± 3 °C vs 83 ± 6 °C).
Five of the six clones analyzed were either not appreciably producible or saw a significant reduction in Tm. This suggests that more stringent thermal denaturation temperatures prior to sorting against target and perhaps an increase in the number of individually assessed clones may be required to isolate more useful ligands. Nevertheless, a mutant (GαE35) was identified that reduces total charge by four and alters charge distribution (Figure S5) while maintaining net charge, elevates thermal stability, maintains binding affinity, and retains recombinant yield. In addition, a mutant (GαE22) was identified with greatly reduced total charge and maintained binding affinity albeit with notably lower thermal stability.
Physiological Distribution and Protease Degradation
To ascertain the effects that reduced and redistributed charge may impart on an EGFR-binding ligand in vivo, GαE22, GαE35, and parental GαE57 were chosen as candidates for imaging of tumor-bearing murine models. GαE35 retains parental net charge of −2, providing an opportunity to elucidate the effects of a reduction in total charge. GαE22 is the least charged anti-EGFR Gp2 isolated from the selections creating a more extreme comparison between GαE57 and a substantially charge-reduced variant. To address target specificity, A431 (2.9 ± 1.4 × 106 EGFR per cell69) and MDA-MB-435 (1.5 ± 1.1 × 104 EGFR per cell69) were incubated with 500 nM GαE22, GαE35, or GαE57 and analyzed via flow cytometry. Binding to A431 cells was substantially higher than to MDA-MB-435 cells for GαE22, GαE35, and GαE57 (p ≤ 0.001) (Figure S6). While all clones exhibited strong differentiation between the cell types, binding to EGFRlow MDA-MB-435 cells was significantly lower for GαE35 than the parental GαE57 (p < 0.001), and at a ratio consistent with reduced EGFR expression, which suggests enhanced specificity for the charge-engineered GαE35. These clones were conjugated with the metal ion chelator NODAGA through an amine-reactive crosslinking reaction with its N-hydroxysuccinimidyl ester. Labeling was non-specific due to the free amines present on lysines at position 1 of GαE57 and 27 of GαE35. However, conjugation was on average singular with 0.9 ± 0.2, 1.1 ± 0.2, and 1.1 ± 0.4 NODAGA per molecule for GαE22, GαE35, and GαE57 (Figure S7).
NODAGA-conjugated ligands were mixed with 1–2 mCi of 64Cu in 100 mM sodium acetate, pH 6.0 at 42 °C for 60 minutes. Complexed 64Cu was separated from free copper via size exclusion filtration resulting in 38–48% labeling efficiency and 87–98% radiochemical purity (Figure S8). Based on yields from previous purifications of unlabeled Gp2 ligands, approximate specific activities were calculated as 0.5–1.2 MBq nmol−1. Physiological biodistribution of each radiolabeled ligand was ascertained by injecting 1.3 ± 0.3 MBq into mice (n = 5–6) bearing EGFRhigh A431 and EGFRlow MDA-MB-435 tumors. Two hours after injection tissues were excised, weighed, and measured for radioactivity (Figure 6). 64Cu-NODAGA-GαE57 and 64Cu-NODAGA-GαE35 localized strongly to EGFR-overexpressing A431 cells, displaying 5.5 ± 0.5 %ID/g and 4.7 ± 0.5 %ID/g. Binding was target specific (p < 0.01) with 0.9 ± 0.1 %ID/g and 0.6 ± 0.1 %ID/g on EGFRlow MDA-MB-435 tumors for 64Cu-NODAGA-GαE57 and 64Cu-NODAGA-GαE35, respectively. This improved in vivo specificity of 64Cu-NODAGA-GαE35 (p = 0.01) is consistent with reduced non-specific binding in vitro (Figure S6). TumorEGFR-high:tumorEGFR-low, tumor:blood, and tumor:muscle ratios were all strong and nominally higher for the charge-reduced 64Cu-NODAGA-GαE35 (Figure 6, inset). As with many small protein probes, renal signal is elevated. 64Cu-NODAGA-GαE57 has modestly lower renal signal than 64Cu-NODAGA-GαE35, 153 ± 25 %ID/g vs. 209 ± 18 %ID/g, respectively. Liver activity is slightly elevated for both 64Cu-NODAGA-GαE57 (2.6 ± 0.1 %ID/g) and -GαE35 (3.0 ± 0.3 %ID/g), but background signal remains below 2 %ID/g for the remaining tissues sampled.
Figure 6.

Excised tissue analysis of charge modified GαE ligands. Mice xenografted with A431 (EGFRhigh) and MDA-MB-435 (EGFRlow) tumors on opposing shoulders were intravenously injected with 64Cu-NODAGA-GαE57, 64Cu-NODAGA-GαE35, or 64Cu-NODAGA-GαE22 and euthanized 2 h post-injection. Tissues or fluid of interest were extracted, weighed, and gamma decay radioactivity measured (%ID/g) (n = 5–6). *: p < 0.05. Inset represents ratios of tumor:background signal.
Conversely, 64Cu-NODAGA-GαE22 was not an effective EGFR ligand in vivo. A431 tumor signal was nearly 2-fold lower than that of GαE57 and GαE35 clones. Additionally, off-target retention was statistically higher in blood, heart, lung, liver, spleen, and bone samples (p < 0.05). Background signal was most pronounced in the liver where 64Cu-NODAGA-GαE22 displayed 8.4 ± 1.0 %ID/g, approximately 3-fold higher than 64Cu-NODAGA-GαE57 and 64Cu-NODAGA-GαE35. While this broad background could be related to slightly increased non-specific binding (Figure S6), the cooccurrence of reduced EGFRhigh tumor localization is more consistent with molecular instability. The propensity to enzymatically degrade was determined in vitro by adding 64Cu-NODAGA-GαE22, -GαE35, and –GαE57 to murine serum, human serum, or 0.25% EDTA-trypsin at 1:20 volumetric ratios and heating at 37 °C for 4 hours. Radio-thin-layer chromatography of samples taken at 1 and 4 hours post-mixing was used to determine the relative amounts of proteolytic metabolites (Figure S8). 64Cu-NODAGA-GαE22 was highly unstable with 59% and 50% cleavage in human and murine serum after 1 hour. These values increased to 80% and 68% by 4 hours. 64Cu-NODAGA-GαE35 and 64Cu-NODAGA-GαE57 were more resistant to enzymatic degradation in serum. Proteolysis of 64Cu-NODAGA-GαE35 and 64Cu-NODAGA-GαE57 in human and murine serum resulted in 10% and 17% and 10% and 26% degradation by 1 hour, respectively. All clones were degraded to near completion (≥ 92%) in EDTA-trypsin after 4 h. In retrospect, the proteolytic instability of GαE22 may have been predicted by its decreased Tm compared to GαE35 and GαE57.
Positron Emission Tomography Imaging of GαE35
In contrast to parental 64Cu-NODAGA-GαE57, 64Cu-NODAGA-GαE35 showed statistically lower MDA-MB-435 signal, similar A431 retention and off-target uptake, and nominal improvements in tumor:blood and tumor:muscle ratios. Buoyed by these results, PET/CT imaging of 64Cu-NODAGA-GαE35 was performed in murine models bearing A431 xenografts, with dual tumors to increase experimental observations of tumor uptake and retention. To further evaluate target specificity, imaging was performed with and without pre-blocking by an injection of unlabeled NODAGA-GαE35. Copper chelation of 64Cu-NODAGA-GαE35 and purification were performed as previously described resulting in 66% labeling efficiency, 97% radiochemical purity, and 1.43 ± 0.01 MBq nmol−1 specific activity. To demonstrate native EGFR specificity, four mice were intravenously injected with unlabeled NODAGA-GαE35 at 87 – 144-fold molar excess (0.7 – 0.8 mg) followed by 1.5 ± 0.4 MBq of 64Cu-NODAGA-GαE35 between 5 and 10 minutes later. Four separate mice were injected with 1.9 ± 0.7 MBq of 64Cu-NODAGA-GαE35 without blocking by unlabeled ligand. CT/PET scans were performed at 1 and 2 h post-injection (Figure 7A). Using CT scans as an anatomical guide liver, muscle, tumor, and kidney signal was quantified through PET image analysis at both time points (Figure 7B). 64Cu-NODAGA-GαE35 localized to EGFR-overexpressing A431 tumors (5.6 ± 0.6 %ID/g) and cleared background (7.2 ± 1.1 tumor:muscle ratio) within 60 minutes. Tumor signal (6.2 ± 0.6 %ID/g) and tumor:muscle ratio (6.7 ± 1.1) were essentially steady at 2 hours p.i.
Figure 7.

(A) PET/CT Imaging. Mice xenografted with two A431 (EGFRhigh) tumors on opposing shoulders were intravenously injected with either 64Cu-NODAGA-GαE35 (Non-blocked) or NODAGA-GαE35 at 102 ± 29-fold molar excess followed by 64Cu-NODAGA-GαE35 5–10 minutes later (Blocked) and imaged via PET/CT at 1 and 2 h post-injection. Coronal and transverse slices through tumors are shown for representative mice (n = 4). (B) PET imaging quantification. Signal in the liver, muscle, tumors, and kidney were quantified using Siemens Inveon research software at 1 and 2 h post-injection (**: p < 0.01).
Binding was target specific as demonstrated by a nearly 2-fold reduction in tumor signal when pre-blocked with excess NODAGA-GαE35 at both 1 (2.9 ± 0.3 %ID/g) and 2 h (2.8 ± 0.4 %ID/g) (p < 0.01). Muscle retention was low and indifferent to injection scheme at 1 h (1.3 ± 0.2 vs. 0.8 ± 0.1 %ID/g) and 2 h (0.9 ± 0.1 vs. 0.9 ± 0.1 %ID/g) for mice with or without cold NODAGA-GαE35 blocking. Hepatic signal was elevated relative to muscle in blocked (4.4 ± 0.4 %ID/g) and non-blocked (4.5 ± 0.6 %ID/g) models after 1 h, remained constant at 2 h in blocked models (4.0 ± 0.8 %ID/g), and increased modestly at 2 h in non-blocked mice (5.6 ± 0.5 %ID/g). Consistent with biodistribution analysis of other small protein ligands15,38,70–72, kidney signal is elevated due to renal processing. Renal uptake was 1.5x and 1.6x higher in non-blocked mice at 1 and 2 h. Note that due to high levels of photon emission within the kidneys, multiple coincident events may be observed within the given collection time window and therefore discarded, leading to an artificially lowered signal.
Two hours post-injection, mice were euthanized with tissues of interest excised, weighed, and measured for radioactivity (Figure 8). PET image quantifications were corroborated by tissue measurements. 64Cu-NODAGA-GαE35 exhibited strong EGFR-specific uptake (5.0 ± 0.7 %ID/g non-blocked vs. 2.5 ± 0.2 %ID/g blocked; p < 0.001), elevated tumor:blood (9.2 ± 1.9 %ID/g non-blocked vs. 2.3 ± 0.3 %ID/g blocked; p < 0.001) and tumor:muscle (25 ± 11 %ID/g non-blocked vs. 8.7 ± 2.4 %ID/g blocked; p < 0.001) ratios. 64Cu-NODAGA-GαE35 tumor signal was nominally higher when quantified via image analysis versus excised tissue scintillation (6.2 ± 0.6 %ID/g vs. 5.0 ± 0.7 %ID/g), although this difference was not statistically significant. Additionally, muscle signal was slightly increased in both blocked (0.9 ± 0.1 %ID/g vs. 0.3 ± 0.0 %ID/g) and non-blocked (0.9 ± 0.1 %ID/g vs. 0.2 ± 0.0 %ID/g) images.
Figure 8.

Excised tissue analysis of 64Cu-NODAGA-GαE35 with and without blocking by NODAGA-GαE35. Mice (n = 4) xenografted with two A431 (EGFRhigh) tumors on opposing shoulders were intravenously injected either 64Cu-NODAGA-GαE35 (Non-blocked) or NODAGA-GαE35 at 102 ± 29-fold molar excess followed by 64Cu-NODAGA-GαE35 5–10 minutes later (Blocked). Mice were euthanized 2 h post-injection. Tissues or fluid of interest were extracted, weighed, and gamma decay radioactivity measured (%ID/g) (*: p < 0.05; **: p < 0.001).
In conclusion, we created combinatorial libraries through rational design to introduce charge-neutralizing or charge-flipping mutations on the Gp2 scaffold. By briefly heating yeast-displayed Gp2 domains to denature unstable clones followed by flow cytometry to sort for the retention of binding, deep sequencing of the enriched populations revealed preferred mutations across three diverse binders (to EGFR, InsR, and rIgG). Functional populations indicated tolerance of substantial charge removal with a propensity towards re-introduction of charge and a maintenance of negative net charge. Additional screening identified a promising EGFR Gp2 ligand with improved charge distribution, reduced charge density, high stability, and strong, specific binding. Three lead EGFR binders, GαE22, GαE35, and parental GαE57, were conjugated with NODAGA, complexed with 64Cu, and analyzed in vivo using murine models bearing EGFRhigh and EGFRlow tumors. NODAGA-GαE35 demonstrated good EGFRhigh tumor localization (4.7 ± 0.5 %ID/g) and, relative to parental NODAGA-GαE57, improved specificity with reduced EGFRlow uptake (0.6 ± 0.1 vs. 0.9 ± 0.1 %ID/g, p = 0.01) and nominally improved tumor:blood (9.7 ± 1.3 vs. 6.2 ± 2.0), tumor:muscle (35 ± 5 vs. 23 ± 7), and EGFRhigh:EGFRlow (8.1 ± 1.2 vs. 6.1 ± 0.9) ratios. Further study with additional mutants will be needed to more broadly assess the impact of charge reduction and redistribution on ligand performance. Our study allows for the enhancement of the Gp2 framework through guided charge modulation, validates a method to engineer these modifications, and provides a lead clone, GαE35, to advance physiological EGFR targeting for the purpose of molecular imaging.
Experimental Procedures
FoldX and Homolog Consensus
FoldX55 was used to predict destabilizing effects of residue mutations at charged positions by estimating the change in free energy of folding for these mutations on the Gp2 scaffold. Calculations were performed at each position using 40 randomized paratopes, or until < 0.1 kcal mol−1 absolute or < 1% relative deviation in mean was achieved with each additional variant for four solved crystal structures of T7 phage Gp2 (2LMC, 2WNM, 4LLG, 4LK0) and averaged to determine the final ΔΔGfolding,mutation of each canonical amino acid at all positions.
We searched for homologs of the wild-type Gp2 protein via NCBI BLAST73 (in 2016) resulting in 32 sequences, which were aligned in ClustalW261. Sitewise amino acid frequencies were calculated.
Library Construction
Degenerate oligonucleotides containing the mixed bases necessary for desired residue mutations at charged positions and encoding for GαE2.2.3, GαI2.2.5, or GαR3.2.3 paratope regions were purchased (IDT DNA, Coralville, IA) and assembled via PCR overlap extension. Each of the three libraries was transformed into the yeast surface display strain EBY10063 using homologous recombination with a linearized pCT vector as described74. Transformation efficiency was determined by serial dilution on SD-CAA/agar plates (0.07 M sodium citrate (pH 5.3), 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 20 g/L glucose, and 15–18 g/L bacto agar in water). Sanger and Illumina sequencing of each library was performed to verify proper transformation.
Binder Selection and Illumina Sequencing
Yeast were grown in SD-CAA (0.07 M sodium citrate (pH 5.3), 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, and 20 g/L glucose in water) and induced in SG-CAA (0.1M sodium phosphate (pH 6.0), 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 19 g/L galactose, and 1 g/L glucose in water) at 30 °C, 250 rpm as described.74 Populations with sample sizes between 10x and 20x of theoretical diversity were taken from naïve libraries and sorted using flow cytometry. Yeast were washed in phosphate-buffered saline with 1 g/L bovine serum albumin (PBSA) heated to 50–80 °C for 30 minutes, cooled on ice to 4 °C, incubated with 2 – 20 nM recombinant, biotinylated target protein and anti-c-Myc antibody (Cat# 626802, BioLegend, San Diego, CA) to isolate full-length Gp2 mutants and account for differences in surface expression, labeled with streptavidin-Alexa Fluor 647 (Life Technologies, Waltham, MA) and goat anti-mouse-FITC antibody (Cat# F0257, Sigma–Aldrich, St. Louis, MO), then sorted.
Because yeast cell viability diminishes significantly above 42 °C75, the recovered populations in heat-treated sorts always exceeded 105 yeast cells. This number of cells provided sufficient plasmid reclamation for the resulting downstream plasmid recovery via Zymoprep protocol (Zymo Research, Irvine, CA), PCR amplification of Gp2 genes, and retransformation through electroporation into yeast to rebuild display libraries for continued sorting. After their final recombinant target sorts, Gp2 libraries were exposed to avidin-coated magnetic beads (Invitrogen, Waltham, MA), followed by beads with immobilized non-target mouse IgG; non-binding yeast were collected and sequenced.
The EGFR-binding population underwent two additional rounds of sorting without heating against 0.2 and 2 nM solubilized EGFR from A431 cell lysate as described previously.76 Briefly, after reaching 70–80% confluency, A431 cells were detached, washed in ice-cold PBS, incubated with NHS-PEG4-Biotin, lysed and centrifuged, and the resulting supernatant mixed with Gp2 expressing yeast recovered from bead depletion. Binding was detected via flow cytometry as for recombinant target selections.
Illumina MiSeq paired-end sequencing was used to obtain 4.7 × 105, 1.6 × 104, 5.9 × 104, and 8.9 × 105, 4.9 × 104, 5.9 × 104 reads from the naïve and sorted binding populations of EGFR, InsR, and rIgG Gp2 libraries. Forward and reverse reads were aligned using PANDAseq77 with a 90% quality cut-off resulting in 2704, 2840, 595, and 3202, 797, 171 unique sequences from naïve and enriched populations in EGFR, InsR, and rIgG libraries. Sequences were sorted, aligned, and sitewise frequencies calculated. Clones appearing more than once had frequencies dampened by where n represents the number of clonal occurrences found in the Illumina sequences.
Mammalian Cell Growth
MDA-MB-435 melanocyte (1.5 ± 1.1 × 104 EGFR per cell69) and A431 epidermoid carcinoma (2.9 ± 1.4 × 106 EGFR per cell69) cell lines were kindly provided by Dr. Tim Starr (University of Minnesota) and Dr. Daniel Vallera (University of Minnesota). Both cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1% penicillin streptomycin and 10% fetal bovine serum at 37 °C in humidified air with 5% CO2. When needed experimentally, cultures were incubated with 1–2 mL of 0.25% Trypsin-EDTA for 6–10 minutes to detach cells.
GαE Clonal Production and Characterization
Gp2-encoding regions in DNA recovered from the final lysate-extracted EGFR flow cytometry sort were amplified by polymerase chain reaction, digested with NheI-HF and BamHI-HF restriction enzymes (New England Biolabs, Ipswich, MA), and ligated into a pET-22b vector containing a C-terminal hexa-histidine (Novagen, EMD Millipore, Billerica, MA) with T4 DNA ligase (New England Biolabs). Plasmids were transformed via heat-shock into T7 Express Competent E. coli (New England Biolabs) and proper transformants selected on lysogeny broth (LB) plates containing 50 mg/L kanamycin. Surviving colonies were added to 4 mL liquid LB/kanamycin (50 mg/L) and incubated in shake flasks at 37 °C, 250 rpm for 10–16 hours. Saturated cultures were added to 100 mL of LB, incubated 37 °C, 250 rpm until reaching an optical density between 0.5 and 1.0 upon which 0.1 mL 0.5 mM isopropyl b-D-1-thiogalactopyranoside was added to induce protein expression. Induced cultures were incubated 4–24 h at 30 °C, 250 rpm, cells were pelleted and resuspended in lysis buffer (50mM sodium phosphate (pH 8.0), 0.5M sodium chloride, 5% glycerol, 5mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 25mM imidazole), frozen and thawed 4 times, centrifuged for 10 minutes at 10 °C, and the resulting cell lysate run through 0.2 mL Cobalt HisPur resin volume spin columns (Thermo Fisher Scientific, Waltham, MA) to recover Gp2 proteins. When greater levels of Gp2 production were required, as in the case of murine PET/CT imaging studies, saturated cultures were added to 1 L LB and clones were recovered using 2 mL Cobalt HisPur resin volume gravity columns. Recovered yields were assessed using absorbance at 280nm on a Synergy H1 microplate reader (BioTek, Winooski, VT).
In some cases, clones were additionally purified by reversed-phase high-performance liquid chromatography with a C18 column using a 15 min gradient of 10% to 90% elution buffer (90% acetonitrile, 9.9% water, 0.1% trifluoroacetic acid) and the remaining solution composed of running buffer (99.9% water, 0.1% trifluoroacetic acid). Recovered Gp2 was lyophilized.
Secondary structure, midpoint of thermal denaturation (Tm), and protein refolding after heating was determined using circular dichroism performed on a Jasco J-815 spectrophotometer with samples placed in a 1 mm path length quartz cuvette. Purified and lyophilized Gp2 ligands were resuspended in PBS to 0.2–1 mg/mL. Ellipticity was measured between 205 and 260 nm wavelengths at 20 °C before and after heating to 98 °C. Thermal denaturation was evaluated at 220 nm during sample heating at 2 °C/min from 20 °C to 98 °C.
At 70–80% confluence, A431 were detached as described previously, pelleted, and resuspended in 4 °C PBSA. Each Gp2 ligand was added at varying concentrations (0, 2, 20, 50, and 500 nM) to 50,000 resuspended A431 cells and incubated at 4 °C to allow for EGFR binding. Cells were again pelleted and washed with cold PBSA, then incubated with fluorescein-conjugated anti-His6 antibody (Abcam, Cambridge, MA). Cells were washed a final time and analyzed for fluorescence using flow cytometry on an Accuri C6 (BD Biosciences, Franklin Lakes, NJ). Using median fluorescence at each ligand concentration, the equilibrium dissociation constant, KD, was determined by minimizing the sum squared errors assuming a 1:1 binding interaction. Affinity titrations were performed thrice. To elucidate EGFR specificity, an analogous procedure was performed with A431 and MDA-MB-435 cell lines at 500 nM target (n = 3).
Gp2 ligands used for PET/CT imaging were resuspended in dimethyl sulfoxide, 2–3% trimethylamine v/v, and 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA)-ester (CheMatech, Dijon, France) at 5:1–10:1 molar ratios of ester:protein. Upon conjugation as verified by matrix-assisted laser desorption ionization mass spectrometry, the reaction was quenched with excess 1 M Tris buffer, pH 8.0, isolated via HPLC, freeze-dried, and stored at 4 °C until use.
Tumor Xenotransplantation
Six-week-old female (Foxn1nu/Foxn1nu) mice (Charles River Labs, Wilmington, MA) were anesthetized via inhalation of 2% isoflurane in 1–1.2 mL min−1 O2. 10 million MDA-MB-435 cells, suspended in a 50:50 v/v DMEM/Matrigel Matrix solution, were subcutaneously injected in their right shoulder. After 4.5–5 weeks, this process was repeated with 7 million A431 cells subcutaneously injected into the opposing shoulder. Xenograft tumors were grown to 5–12 mm in diameter (approximately 6 weeks after MDA-MB-435 inoculation). In pre-blocking experiments, this procedure was repeated with 7 million A431 cells subcutaneously injected into both shoulders followed by 1.5 weeks of tumor growth until tumor diameters reached 7–10 mm.
Chelation and purification
5–12 mCi 64CuCl2 (University of Wisconsin-Madison) was diluted in 100 μL 100 mM sodium acetate pH 6.0 to provide buffering, then titrated with 1M sodium hydroxide and 1M hydrochloric acid to a final pH of 6.0 as confirmed by narrow band pH paper (final volume: 200–250 μL). 1–2 mCi buffered 64CuCl2 was added to 50–120 μM NODAGA-Gp2 in 10 mM sodium acetate pH 6.0 and incubated at 42 °C for 1 h to enact complexation. 64Cu-NODAGA-Gp2 ligands were separated from free 64Cu through size exclusion chromatography with PD-10 columns pre-equilibrated with 10 mM sodium acetate pH 6.0.
RadioTLC/enzymatic degradation
64Cu-NODAGA-Gp2 purity and enzymatic stability were determined with an AR-2000 radio-thin-layer chromatography scanner (Eckert and Ziegler, Berlin, Germany) as described previously72. Briefly, 1–2 μL PD-10 elution volume 3.5–4.0 mL was spotted on chromatography paper with PBS used as the mobile phase to separate any uncomplexed copper from 64Cu-NODAGA-Gp2. Through integration analysis of the resulting position vs. scintillation count data, relative fractions of free 64Cu and 64Cu-NODAGA-Gp2 were determined.
To ascertain enzymatic stability, 1 μL from 64Cu-NODAGA-Gp2 PD-10 elution volume 3.5 – 4.0 mL was added to 20 μL normal human serum (Jackson ImmunoResearch), normal mouse serum (Jackson ImmunoResearch), or 0.25% trypsin-EDTA in Hank’s balanced salt solution and incubated at 37 °C for 4 hours. Two μL samples were taken at 1 and 4 h, spotted on chromatography paper with a PBS mobile phase, and analyzed for metabolite migration. Integration analysis was again used to determine the relative percentage of Gp2 metabolites.
Tissue quantification of 64Cu-NODAGA-GαE22, -GαE35, and -GαE57 in mice bearing A431 and MDA-MB-435 tumor xenografts
Mice were anesthetized with 2% isoflurane at 1 mL min−1 oxygen until stationary. When no longer ambulatory, 1.0–1.9 MBq 64Cu-NODAGA-GαE22, -GαE35, or -GαE57 was injected via lateral tail vein. At 2 hours p.i., mice were again anesthetized then euthanized by cervical dislocation. After confirming expiration by toe pinch, mice were dissected and blood, heart, lung, liver, pancreas, spleen, stomach, intestine, skin, brain, bone, muscle, kidney, A431, MDA-MB-435 were extracted and weighed. Resected samples were measured for radioactivity by a CRC-25W (Capintec, Florham Park, NJ) gamma counter across all energy ranges and averaged over 1–2 min. Incident counts were calibrated using serial dilutions based on the dose reported by an Atom Lab dosimeter.
PET Imaging/quantification and Tissue quantification of 64Cu-NODAGA-GαE35 in mice bearing dual A431 tumor xenografts
Mice were anesthetized as described previously. Upon loss of motor function, mice bearing dual A431 tumors were injected via lateral tail vein with 0 or 0.7 – 0.8 mg NODAGA-GαE35 followed 5–10 minutes later by 1.0–2.7 MBq 64Cu-NODAGA-GαE35. Injected radioactivity was quantified by an Atomlab 100 dosimeter (Biodex, Shirley, NY) calibrated for 64Cu measurements. Static PET scans were performed at 1 and 2 hours post-injection on an Inveon microPET/CT (Siemens, Malvern, PA) and images reconstructed with the 2D ordered-subset expectation maximization (OSEM2D) method using four iterations of Fourier rebinning. Photopeak energy cutoffs were 350–650 keV with a timing window of 3.437 nanoseconds. A 1 × 1 × 1 voxel Gaussian filter was used to smooth PET images. Concomitant CT scans were completed using 340 projections of 80 kV at 500 uA with 4 × 4 binning, 200 ms exposure over 384 s total scan time, and an effective pixel size of 85.6 μm. CT scans were reconstructed with the Feldkamp–David–Kress algorithm using slight noise reduction and Shepp-Logan filtering. PET image ROI quantification was performed on Inveon Research Workplace software (Siemens) using CT images as guides to determine 3D margins for tissues of interest including liver, thigh muscle, kidneys, and A431 tumors. The maximum average PET signal in a 3 × 3 × 3 spherical voxel (9.1 mm3) was calculated for each tissue of interest.
Two hours after 64Cu-NODAGA-GαE35 injection, mice were euthanized, tissues extracted, weighed, and radioactivity measured as described previously.
Statistics
Comparisons for statistical significance between was determined using two-tailed student’s t-tests assuming unequal variance between sample datasets. Data are presented as mean ± standard error unless otherwise noted.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R21 EB021511 and R01 EB023339 to B.J.H.) and a University of Minnesota Doctoral Dissertation Fellowship (to M.A.K.). We would also like to thank Dr. Jacob Petersburg (College of Pharmacy, University of Minnesota) for assisting in murine tail vein injections, Patrick Holec for computing FoldX stabilities, and Dr. Tim Starr (University of Minnesota) and Dr. Daniel Vallera (University of Minnesota) for providing MDA-MB-435 melanocyte and A431 epidermoid carcinoma cell lines.
Abbreviations
- CT
computed tomography
- Gp2
gene 2 protein from T7 phage
- PET
positron emission tomography
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website. Flow cytometry sorting details and parental clone biophysical properties; FACS plots; enriched anti-EGFR clone sequences and biophysical properties, thermal denaturation curves, affinity titrations, and circular dichroism spectroscopy scans; sequence and approximate structure of GαE35; EGFR binding specificity of clones GαE22, GαE35, and GαE57; MALDI spectroscopy of NODAGA-conjugated GαE22, GαE35, and GαE57; and radio-thin-layer chromatography of 64Cu-complexed NODAGA-GαE22, -GαE35, -GαE57 untreated and incubated with murine serum, human serum, or trypsin.
References
- 1.Baum RP, Prasad V, Müller D, Schuchardt C, Orlova A, Wennborg A, Tolmachev V, Feldwisch J. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules. J Nucl Med. 2010;51:892–897. doi: 10.2967/jnumed.109.073239. [DOI] [PubMed] [Google Scholar]
- 2.Stern LA, Case BA, Hackel BJ. Alternative non-antibody protein scaffolds for molecular imaging of cancer. Curr Opin Chem Eng. 2013;2:425–432. doi: 10.1016/j.coche.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K, Sandström M, Rosik D, Carlsson J, Lundqvist H, Wennborg A, Nilsson FY. Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007;67:2773–2782. doi: 10.1158/0008-5472.CAN-06-1630. [DOI] [PubMed] [Google Scholar]
- 4.Hanenberg M, McAfoose J, Kulic L, Welt T, Wirth F, Parizek P, Strobel L, Cattepoel S, Sp??ni C, Derungs R, Maier M, Pl??ckthun A, Nitsch RM. Amyloid-B peptide-specific darpins as a novel class of potential therapeutics for alzheimer disease. J Biol Chem. 2014;289:27080–27089. doi: 10.1074/jbc.M114.564013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mamluk R, Carvajal IM, Morse BA, Wong H, Abramowitz J, Aslanian S, Lim AC, Gokemeijer J, Storek MJ, Lee J, Gosselin M, Wright MC, Camphausen RT, Wang J, Chen Y, Miller K, Sanders K, Short S, Sperinde J, Prasad G, Williams S, Kerbel R, Ebos J, Mutsaers A, Mendlein JD, Harris AS, Furfine ES. Anti-tumor effect of CT-322 as an adnectin inhibitor of vascular endothelial growth factor receptor-2. MAbs. 2010;2:199–208. doi: 10.4161/mabs.2.2.11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Straw S, Ferrigno PK, Song Q, Tomlinson D, Del Galdo F. Proof of concept study to identify candidate biomarkers of fibrosis using high throughput peptide aptamer microarray and validate by enzyme linked immunosorbant assay. J Biomed Sci Eng. 2013;6:32–42. [Google Scholar]
- 7.Johnson A, Song Q, Ko Ferrigno P, Bueno PR, Davis JJ. Sensitive affimer and antibody based impedimetric label-free assays for C-reactive protein. Anal Chem. 2012;84:6553–6560. doi: 10.1021/ac300835b. [DOI] [PubMed] [Google Scholar]
- 8.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 9.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Uriell-Shoval S, Galun E, Ben-Neriah Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther. 2009;8:2861–2871. doi: 10.1158/1535-7163.MCT-09-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–1434. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29:57–61. doi: 10.1016/j.tips.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 14.Orlova A, Wållberg H, Stone-Elander S, Tolmachev V. On the selection of a tracer for PET imaging of HER2-expressing tumors: direct comparison of a 124I-labeled affibody molecule and trastuzumab in a murine xenograft model. J Nucl Med. 2009;50:417–25. doi: 10.2967/jnumed.108.057919. [DOI] [PubMed] [Google Scholar]
- 15.Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G, Dreier B, Schibli R, Binz HK, Waibel R, Plückthun A. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res. 2010;70:1595–1605. doi: 10.1158/0008-5472.CAN-09-2724. [DOI] [PubMed] [Google Scholar]
- 16.Natarajan A, Hackel BJ, Gambhir SS. A Novel Engineered Anti-CD20 Tracer Enables Early Time PET Imaging in a Humanized Transgenic Mouse Model of B-cell Non-Hodgkins Lymphoma. Clin Cancer Res. 2013;19:6820–6829. doi: 10.1158/1078-0432.CCR-13-0626. [DOI] [PubMed] [Google Scholar]
- 17.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 18.Rosik D, Thibblin A, Antoni G, Honarvar H, Strand J, Selvaraju RK, Altai M, Orlova A, Eriksson Karlström A, Tolmachev V. Incorporation of a triglutamyl spacer improves the biodistribution of synthetic affibody molecules radiofluorinated at the n-terminus via oxime formation with 18F-4-fluorobenzaldehyde. Bioconjug Chem. 2014;25:82–92. doi: 10.1021/bc400343r. [DOI] [PubMed] [Google Scholar]
- 19.Hackel BJ, Sathirachinda A, Gambhir SS. Designed hydrophilic and charge mutations of the fibronectin domain: towards tailored protein biodistribution. Protein Eng Des Sel. 2012;25:639–647. doi: 10.1093/protein/gzs036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willuda J, Honegger A, Waibel R, Schubiger PA, Stahel R, Zangemeister-Wittke U, Plückthun A. High thermal stability is essential for tumor targeting of antibody fragments. Cancer Res. 1999;59:5758–5767. [PubMed] [Google Scholar]
- 21.Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, Rollence ML, Wood JF, Schott ME, Milenic DE, Yokota T, Schlom J. An improved linker for single-chain fv with reduced aggregation and enhanced proteolytic stability. Protein Eng Des Sel. 1993;6:989–995. doi: 10.1093/protein/6.8.989. [DOI] [PubMed] [Google Scholar]
- 22.Tran T, Engfeldt T, Orlova A, Sandstroem M, Feldwisch J, Abrahmsén L, Wennborg A, Tolmachev V, Karlstroem AE. Tc-99m-maEEE-Z(HER2: 342), an affibody molecule-based tracer for the detection of HER2 expression in malignant tumors. Bioconjug Chem. 2007;18:1956–1964. doi: 10.1021/bc7002617. [DOI] [PubMed] [Google Scholar]
- 23.Tran TA, Ekblad T, Orlova A, Sandström M, Feldwisch J, Wennborg A, Abrahmsén L, Tolmachev V, Eriksson Karlström A. Effects of lysine-containing mercaptoacetyl-based chelators on the biodistribution of 99mTc-labeled anti-HER2 Affibody molecules. Bioconjug Chem. 2008;19:2568–2576. doi: 10.1021/bc800244b. [DOI] [PubMed] [Google Scholar]
- 24.Chan P, Curtis RA, Warwicker J. Soluble expression of proteins correlates with a lack of positively-charged surface. Sci Rep. 2013;3:3333. doi: 10.1038/srep03333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Mosior M, Chung LA, Wu H, McLaughlin S. Binding of peptides with basic residues to membranes containing acidic phospholipids. Biophys J. 1991;60:135–148. doi: 10.1016/S0006-3495(91)82037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murray D, Arbuzova A, Hangyás-Mihályné G, Gambhir A, Ben-Tal N, Honig B, McLaughlin S. Electrostatic properties of membranes containing acidic lipids and adsorbed basic peptides: Theory and experiment. Biophys J. 1999;77:3176–3188. doi: 10.1016/S0006-3495(99)77148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merz KM, Roux B. Biological Membranes A Molecular Perspective from Computation and Experiment 1996 [Google Scholar]
- 28.Kastritis PL, Rodrigues JPGLM, Folkers GE, Boelens R, Bonvin AMJJ. Proteins feel more than they see: fine-tuning of binding affinity by properties of the non-interacting surface. J Mol Biol. 2014;426:2632–52. doi: 10.1016/j.jmb.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 29.Dellian M, Yuan F, Trubetskoy VS, Torchilin VP, Jain RK. Vascular permeability in a human tumour xenograft: molecular charge dependence. Br J Cancer. 2000;82:1513–1518. doi: 10.1054/bjoc.1999.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stylianopoulos T, Soteriou K, Fukumura D, Jain RK. Cationic nanoparticles have superior transvascular flux into solid tumors: insights from a mathematical model. Ann Biomed Eng. 2013;41:68–77. doi: 10.1007/s10439-012-0630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krasnici S, Werner A, Eichhorn ME, Schmitt-Sody M, Pahernik SA, Sauer B, Schulze B, Teifel M, Michaelis U, Naujoks K, Dellian M. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105:561–567. doi: 10.1002/ijc.11108. [DOI] [PubMed] [Google Scholar]
- 32.Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21:2153–2163. doi: 10.1021/bc100261d. [DOI] [PubMed] [Google Scholar]
- 33.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, Hattori K. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23:385–392. doi: 10.1093/protein/gzq009. [DOI] [PubMed] [Google Scholar]
- 34.Kimura RH, Teed R, Hackel BJ, Pysz Ma, Chuang CZ, Sathirachinda A, Willmann JK, Gambhir SS. Pharmacokinetically stabilized cystine knot peptides that bind alpha-v-beta-6 integrin with single-digit nanomolar affinities for detection of pancreatic cancer. Clin Cancer Res. 2012;18:839–49. doi: 10.1158/1078-0432.CCR-11-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pace CN. Measuring and increasing protein stability. TRENDS Biotechnol. 1990;8:93–98. doi: 10.1016/0167-7799(90)90146-o. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Sawyer N, Regan L. Protein-protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013;22:510–5. doi: 10.1002/pro.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kruziki MA, Bhatnagar S, Woldring DR, Duong VT, Hackel BJ. A 45-Amino-Acid Scaffold Mined from the PDB for High-Affinity Ligand Engineering. Chem Biol. 2015;22:946–956. doi: 10.1016/j.chembiol.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kruziki MA, Case BA, Chan JY, Zudock EJ, Woldring DR, Yee D, Hackel BJ. Cu-Labeled Gp2 Domain for PET Imaging of Epidermal Growth Factor Receptor. Mol Pharm. 2016;13:3747–3755. doi: 10.1021/acs.molpharmaceut.6b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan JY, LaPara K, Hackel B, Yee D. Abstract 1881: Insulin receptor targeting in breast cancer through yeast surface display. Cancer Res. 2016;76:1881–1881. [Google Scholar]
- 40.Camara B, Liu M, Reynolds J, Shadrin A, Liu B, Kwok K, Simpson P, Weinzierl R, Severinov K, Cota E, Matthews S, Wigneshweraraj SR. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc Natl Acad Sci. 2010;107:2247–2252. doi: 10.1073/pnas.0907908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Traxlmayr MW, Kiefer JD, Srinivas RR, Lobner E, Tisdale AW, Mehta NK, Yang NJ, Tidor B, Wittrup KD. Strong enrichment of aromatic residues in binding sites from a charge-neutralized hyperthermostable Sso7d scaffold library. 2016 doi: 10.1074/jbc.M116.741314. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García Garayoa E, Schweinsberg C, Maes V, Brans L, Bläuenstein P, Tourwe DA, Schibli R, Schubiger PA. Influence of the molecular charge on the biodistribution of bombesin analogues labeled with the [99mTc(CO)3]-core. Bioconjug Chem. 2008;19:2409–2416. doi: 10.1021/bc800262m. [DOI] [PubMed] [Google Scholar]
- 43.Slovic AM, Kono H, Lear JD, Saven JG, DeGrado WF. Computational design of water-soluble analogues of the potassium channel KcsA. Proc Natl Acad Sci U S A. 2004;101:1828–1833. doi: 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jenkins TM, Hickman AB, Dyda F, Ghirlando R, Davies DR, Craigie R. Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc Natl Acad Sci U S A. 1995;92:6057–6061. doi: 10.1073/pnas.92.13.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang F, Basinski MB, Beals JM, Briggs SL, Churgay LM, Clawson DK, DiMarchi RD, Furman TC, Hale JE, Hsiung HM, Schoner BE, Smith DP, Zhang XY, Wery JP, Schevitz RW. Crystal structure of the obese protein leptin-E100. Nature. 1997;387:206–209. doi: 10.1038/387206a0. [DOI] [PubMed] [Google Scholar]
- 46.Pezzullo M, Del Vecchio P, Mandrich L, Nucci R, Rossi M, Manco G. Comprehensive analysis of surface charged residues involved in thermal stability in Alicyclobacillus acidocaldarius esterase 2. Protein Eng Des Sel. 2013;26:47–58. doi: 10.1093/protein/gzs066. [DOI] [PubMed] [Google Scholar]
- 47.Matsuura Y, Takehira M, Sawano M, Ogasahara K, Tanaka T, Yamamoto H, Kunishima N, Katoh E, Yutani K. Role of charged residues in stabilization of Pyrococcus horikoshii CutA1, which has a denaturation temperature of nearly 150 °C. FEBS J. 2012;279:78–90. doi: 10.1111/j.1742-4658.2011.08400.x. [DOI] [PubMed] [Google Scholar]
- 48.Steipe B, Schiller B, Pluckthun A, Steinbacher S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J Mol Biol. 1994;240:188–192. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
- 49.Case BA, Hackel BJ. Synthetic and natural consensus design for engineering charge within an affibody targeting epidermal growth factor receptor. Biotechnol Bioeng. 2016;113:1628–1638. doi: 10.1002/bit.25931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Huang CW, Tsai HL, Chen YT, Huang CM, Ma CJ, Lu CY, Kuo CH, Wu DC, Chai CY, Wang JY. The prognostic values of EGFR expression and KRAS mutation in patients with synchronous or metachronous metastatic colorectal cancer. BMC Cancer. 2013;13:599. doi: 10.1186/1471-2407-13-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rokita M, Stec R, Bodnar L, Charkiewicz R, Korniluk J, Smoter M, Cichowicz M, Chyczewski L, Nikliński J, Kozłowski W, Szczylik C. Overexpression of epidermal growth factor receptor as a prognostic factor in colorectal cancer on the basis of the Allred scoring system. Onco Targets Ther. 2013;6:967–76. doi: 10.2147/OTT.S42446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouché O, Reid J, Stone S, Penault-Llorca F. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27:5924–30. doi: 10.1200/JCO.2008.21.6796. [DOI] [PubMed] [Google Scholar]
- 54.Lee HJ, Seo AN, Kim EJ, Jang MH, Kim YJ, Kim JH, Kim SW, Ryu HS, Park IA, Im SA, Gong G, Jung KH, Kim HJ, Park SY. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br J Cancer. 2015;112:103–11. doi: 10.1038/bjc.2014.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steipe B. Consensus-based engineering of protein stability: from intrabodies to thermostable enzymes. Methods Enzymol. 2004;388:176–186. doi: 10.1016/S0076-6879(04)88016-9. [DOI] [PubMed] [Google Scholar]
- 57.Cochran JR, Kim YS, Lippow SM, Rao B, Wittrup KD. Improved mutants from directed evolution are biased to orthologous substitutions. Protein Eng Des Sel. 2006;19:245–253. doi: 10.1093/protein/gzl006. [DOI] [PubMed] [Google Scholar]
- 58.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 59.Chan JY, Hackel BJ, Yee D. Targeting Insulin Receptor in Breast Cancer Using Small Engineered Protein Scaffolds. Mol Cancer Ther. 2017;16:1324–1334. doi: 10.1158/1535-7163.MCT-16-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cámara B, Liu M, Reynolds J, Shadrin A, Liu B, Kwok K, Simpson P, Weinzierl R, Severinov K, Cota E, Matthews S, Wigneshweraraj SR. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc Natl Acad Sci U S A. 2010;107:2247–52. doi: 10.1073/pnas.0907908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boder ET, Wittrup KD. Yeast Surface Display for Screening Combinatorial Polypeptide Libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 64.Chen TF, de Picciotto S, Hackel BJ, Wittrup KD. Engineering fibronectin-based binding proteins by yeast surface display. Methods Enzymol. 2013;523:303–326. doi: 10.1016/B978-0-12-394292-0.00014-X. [DOI] [PubMed] [Google Scholar]
- 65.Firth AE, Patrick WM. GLUE-IT and PEDEL-AA: new programmes for analyzing protein diversity in randomized libraries. Nucleic Acids Res. 2008;36:281–285. doi: 10.1093/nar/gkn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pavoor TV, Wheasler JA, Kamat V, Shusta EV. An enhanced approach for engineering thermally stable proteins using yeast display. Protein Eng Des Sel. 2012;25:625–630. doi: 10.1093/protein/gzs041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ackerman M, Levary D, Tobon G, Hackel B, Orcutt KD, Wittrup KD. Highly avid magnetic bead capture: an efficient selection method for de novo protein engineering utilizing yeast surface display. Biotechnol Prog. 2009;25:774–783. doi: 10.1002/btpr.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stern LA, Schrack IA, Johnson SM, Deshpande A, Bennett NR, Harasymiw LA, Gardner MK, Hackel BJ. Geometry and expression enhance enrichment of functional yeast-displayed ligands via cell panning. Biotechnol Bioeng. 2016 doi: 10.1002/bit.26001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nielsen CH, Kimura RH, Withofs N, Tran PT, Miao Z, Cochran JR, Cheng Z, Felsher D, Kjaer A, Willmann JK, Gambhir SS. PET Imaging of Tumor Neovascularization in a Transgenic Mouse Model with a Novel 64Cu-DOTA-Knottin Peptide. Cancer Res. 2010;70:9022–9030. doi: 10.1158/0008-5472.CAN-10-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hackel B, Kimura R, Gambhir S. Use of 64Cu-labeled fibronectin domain with EGFR-overexpressing tumor xenograft: molecular imaging. Radiology. 2012;263:179–188. doi: 10.1148/radiol.12111504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Case BA, Kruziki MA, Stern LA, Hackel BJ. Evaluation of affibody charge modification identified by synthetic consensus design in molecular PET imaging of epidermal growth factor receptor. Mol Syst Des Eng. 2018 doi: 10.1039/C7ME00095B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Altschul S, Gish W, Miller W. Basic local alignment search tool. J Mol …. 1990 doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 74.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 75.Traxlmayr MW, Faissner M, Stadlmayr G, Hasenhindl C, Antes B, Rüker F, Obinger C. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta - Proteins Proteomics. 2012;1824:542–549. doi: 10.1016/j.bbapap.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cho YK, Shusta EV. Antibody library screens using detergent-solubilized mammalian cell lysates as antigen sources. Protein Eng Des Sel. 2010;23:567–577. doi: 10.1093/protein/gzq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.