

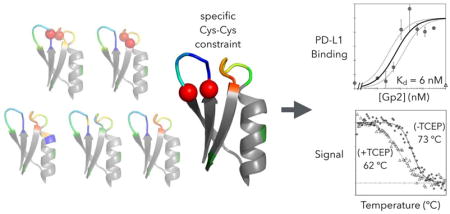
Abstract
Engineered protein ligands are used for molecular therapy, diagnostics, and industrial biotechnology. The Gp2 domain is a 45-amino acid scaffold that has been evolved for specific, high-affinity binding to multiple targets by diversification of two solvent-exposed loops. Inspired by sitewise enrichment of select amino acids, including cysteine pairs, in earlier Gp2 discovery campaigns, we hypothesized that the breadth and efficiency of de novo Gp2 discovery will be aided by sitewise amino acid constraint within combinatorial library design. We systematically constructed eight libraries and comparatively evaluated their efficacy for binder discovery via yeast display against a panel of targets. Conservation of a cysteine pair at the termini of the first diversified paratope loop increased binder discovery 16-fold (p < 0.001). Yet two other libraries with conserved cysteine pairs, within the second loop or an interloop pair, did not aid discovery thereby indicating site-specific impact. Via a yeast display protease resistance assay, Gp2 variants from the loop one cysteine pair library were 3.3 ± 2.1-fold (p = 0.005) more stable than non-constrained variants. Sitewise constraint of non-cysteine residues – guided by previously evolved binders, natural Gp2 homology, computed stability, and structural analysis – did not aid discovery. A panel of binders to programmed death ligand 1 (PD-L1), a key target in cancer immunotherapy, were discovered from the loop 1 cysteine constraint library. Affinity maturation via loop walking resulted in strong, specific cellular PD-L1 affinity (Kd = 6 – 9 nM).
Graphical Abstract
Introduction
High-affinity, specific binding ligands are valuable in molecular therapy, diagnostics, and as research reagents. Protein scaffolds enable efficient generation of binding ligands with engineered control over affinity, stability, and other biophysical properties.1 Protein scaffolds consist of a binding region, or paratope, that is engineered for each target epitope to provide selective potent interaction and supported by a larger, conserved framework. One such scaffold that has been engineered to bind multiple targets is Gp2.2 Based on the T7 RNA polymerase inhibitor, Gp2 is a 45 amino acid scaffold with a two-loop paratope and a framework consisting of three beta strands and an alpha helix (Figure 1a). Gp2 has been evolved to specifically bind multiple targets with nanomolar affinities including goat immunoglobulin G (IgG), rabbit IgG, lysozyme, epidermal growth factor receptor2, and insulin receptor3.
Figure 1. Gp2 protein scaffold structure and second generation library designs.

(a) Cartoon ribbon structure and amino acid sequence of the Gp2 scaffold (PDB ID: 2WNM). The paratope region (red) is allowed to remain wild-type length or extend by one or two amino acids. A region near paratope loop two that previously displayed higher than expected mutation rates is shown in green. (b) Combinatorial library design: codon or amino acid diversity for each site in each library. cdr+ represents base antibody CDR diversity and cdr− represents the closest match to that diversity while removing cysteine (and thus arginine, glycine, and tryptophan due to degenerate codon limitations, except when glycine is added on a separate codon (cdr− +G)). Single amino acid abbreviations are used for diversity at constrained sites, at approximately equal frequency except where denoted by underline (higher) or double underline (highest). Full details in Supplemental Tables 1 and 2.
Despite the successes, many Gp2 discovery and evolution campaigns yield a relatively small number of dominant variants. Improved combinatorial library design and evolution could generate more unique functional variants. Increased diversity of lead molecules should improve the ability to select for other desired characteristics beyond strong, specific binding including stability, solubility, or binding of a particular epitope. To develop new binding function, a sufficiently large paratope must be mutated to provide significant interaction area4,5 between the target and ligand. However, mutations are destabilizing on average6 and mutating an intramolecularly stabilizing site to a sub-optimal amino acid can eliminate function by unfolding an otherwise functional paratope. As a result, more stable starting proteins enable more efficient evolution.7 A similar, but distinct, concept is that reduced destabilization upon functional mutation enables more efficient evolution.8,9 This balancing of intermolecular binding function and intramolecular stability is especially challenging in small proteins where a large fraction of the total amino acids must be mutated to result in strong binding. Constrained diversities at each paratope site, in terms of identity and prevalence of amino acids allowed, has been shown to aid evolution in other scaffolds including fibronectin domains8,10, affibodies9, and antibodies11,12. The initial Gp2 library included amino acid bias – in the form of amino acid frequencies that mimicked the complementarity-determining region (CDR) of the third heavy chain loop of the human antibody repertoire – but lacked sitewise constraint to optimize the variance in mutational tolerance based on local environment. Previous library design in other protein scaffolds has been based on amino acids that are frequently observed at protein-protein interfaces, such as tyrosine and serine13–15, as well as the inclusion of conformationally-flexible glycine in loops. Beneficial sitewise constraint can also be identified through phylogenetic diversity16, computational stability analysis17, or deep-sequencing of high-throughput evolution for function or stability8,9.
Another approach to maintain intramolecular stability while evolving new intermolecular interactions is to stabilize the scaffold via a disulfide bond.18,19 Multiple scaffolds, including cystine knots20, antibodies21, and cyclic peptides22, have one or more disulfide bonds built in from the outset. Other scaffolds initiate as disulfide-free yet have shown an emergence of cysteine pairs during ligand discovery and evolution. Inter- and intra-helical disulfide bonds emerged in evolution of the three-helix bundle affibody.9 Interloop disulfides were found in the beta-sandwich fibronectin domain.23 Multiple earlier Gp2 engineering efforts enriched cysteine pairs within proximal sites.2,3 Thus, disulfide bonds represent an additional form of sequence constraint that warrants further investigation within Gp2 and other scaffolds. In this work, we developed constrained Gp2 libraries to investigate how focused diversity can impact ligand evolution.
We apply these Gp2 engineering strategies to discovery and evolution of binders to programmed death ligand 1 (PD-L1). Some cancers utilize the programmed death-1 (PD-1) / PD-L1 pathway for immune evasion.24 PD-L1 expression is observed in numerous cancers, both on tumor cells and tumor-infiltrating immune cells.25,26 PD-L1 expression, in many tumors but far from all, correlates with high grade, is linked to driver mutations, and is prognostic of poor survival.27 Inhibition of immune checkpoint proteins PD-1 and PD-L1 has yielded clinical efficacy in multiple cancers.27–29 Yet, autoimmune toxicity of checkpoint inhibitors is a significant concern and response rates are moderate, which motivates careful patient stratification. PD-L1 expression is predictive of response in numerous settings28,30–32 but not all. Yet PD-L1 immunohistochemistry suffers from numerous hindrances including invasiveness, confinement to the biopsy site, and inconsistency30. Conversely, positron emission tomography (PET) imaging would provide non-invasive, quantitative whole-body assessment of PD-L1 expression. PD-L1 has been imaged in preclinical models with antibodies33–39, PD-1 ectodomain40,41, affibodies42, fibronectin domains43, and nanobodies44. Gp2 domains, which have been effectively used for PET imaging of epidermal growth factor receptor45, offer an additional option for small scaffold targeting of PD-L1. Gp2 does not have appreciable structural homology to PD-1 or other natural PD-L1 binders46 but rather can be diversified and evolved to include a synthetic interface for PD-L1 interaction. Towards this goal, herein we describe discovery of PD-L1-binding Gp2 variants isolated from these second-generation libraries and their further evolution to high affinity ligands, which have potential use in biotechnology and imaging.
Results
Design of second generation library
While the first-generation Gp2 library yielded binders to each target, the discovery and evolution campaigns were typically dominated by a relatively small number of clones. We hypothesized that a different library design could provide a higher fraction of folded, functional mutants. Multiple library designs were constructed (Figure 1b, Supplemental Tables 1 and 2). The base library (named CDR+) was akin to the first-generation library with antibody CDR diversity (updated for current database values) at each site (Supplemental Figure 1).
In previous binder discovery, certain cysteine pairs occurred much more than expected based on their positional frequency (Figure 2). Such cysteine pairs can form disulfides that provide valuable stabilization. Thus, a set of libraries constrained two sites at a time to be uniquely cysteine to examine potential benefits of disulfide bonds. In particular, we tested pairs of sites that exhibited synergistic enrichment of cysteines in first-generation Gp2 evolution and are located relatively proximally in wild-type structures: C7-C12 (intraloop 1), C36-C39 (intraloop 2), and C8-C36b (interloop). These libraries have a cysteine-free analog of the CDR-inspired diversity (CDR-) at all other positions. This was achieved by elimination of guanine at the second position within each codon, which also eliminates W, R, and G. Trinucleotide synthesis,47 which could have avoided this genetic code coupling, was not used because of cost limitations for the multitude of libraries used in the study. Because of the evolutionary benefit of glycine in several sites in the first-generation evolution, glycine was uniquely allowed at sites 10, 11, 34, and 35 via an additional oligonucleotide.
Figure 2. Potential of disulfide bonded cysteines in first generation Gp2.

Observed frequencies of cysteines in evolved binders at indicated positions were multiplied to calculate predicted frequency (i.e. pairwise frequency if independent). The predicted frequency was compared to the actual pairwise frequency and the absolute difference and calculated mutual information are shown. High mutual information indicates synergistic interaction between the particular pair of cysteines. The sites with the top three values for difference and mutual information are shown in cartoon structure (PDB ID: 2WNM).
Notably, though, earlier Gp2 evolution efforts also yielded numerous functional binders that remained free of cysteines. Such variants can be desirable in some scenarios because of potentially easier protein production in the E. coli cytoplasm, functionality in downstream applications in reducing environments, and reduced potential for aggregation. Therefore, another library design eliminated cysteine and used CDR− diversity at all sites to examine if disulfide-free clones could be readily discovered. As in the paired cysteine library, G was allowed at sites 10, 11, 34, and 35. In fact, this cysteine-free library serves as a direct comparison of the evolutionary benefit of the paired cysteine libraries.
Another library design hypothesized that sitewise constraint of amino acids in the paratope region, which has been beneficial for binder discovery in other small protein scaffolds8–12, would benefit Gp2, provided we had sufficient inter- and intra-molecular interaction data to design the library. The constrained library design was guided by 3 × 104 previously evolved binding sequences against numerous epitopes on eight targets (Figure 3a), as well as sequences from homologous proteins (Figure 3b), in terms of specific amino acids and physicochemical properties at each site including size, hydrophobicity, charge, or retention of wild-type. Chemical homology was used to add in or remove amino acids that had similar properties to included or excluded amino acids, respectively. Amino acids near the cutoff of being included or excluded were decided on by computationally predicted mutational stability (Figure 3c); i.e. they were disallowed if greatly destabilizing or allowed if neutral or beneficial. Finally, sites with higher solvent accessible surface area (Figure 3d) were allowed to have a higher amount of diversity due to the potential for these exposed amino acids to form binding interactions. In depth site-by-site design decisions are described in the Experimental Procedures section. Two versions of the constrained library were built with variations in the non-constrained sites: one with full CDR+ diversity and one with cysteine-free CDR− diversity.
Figure 3. Sitewise analysis of Gp2 domain.

(a) Sitewise enrichment or depletion of each amino acid from initial naïve library to evolved binding sequences isolated from the first generation Gp2 library. 3 × 104 binding sequences against multiple epitopes on eight targets were aligned, analyzed for sitewise amino acid frequencies, and compared to the unsorted naïve library. Sites with more than a 5% change are labeled. (b) Sitewise amino acid frequencies in natural homologs to Gp2. Thirty-two Gp2 homologs were identified, aligned, and analyzed. Amino acids with above 5% prevalence are labeled. (c) Computationally estimated change in folding energy. The (de)stabilization (ΔΔGfolding in kcal/mol) upon mutation of the indicated site to the indicated amino acid was computed in FoldX48 for at least 40 random paratopes in 4 wild-type structures. Positive values represent less stable folds. (d) Sitewise solvent accessible surface area of wildtype Gp2. Values calculated from the GetArea webserver (2WNM) or the Accessible Surface Area tool from Center for Information Biology at Ochanomizu University (2WNM, 2LMC, 4LK0, 4LLG) for 4 protein data bank files were averaged. Area is normalized to surface area for each amino acid in the unfolded state and compared to the median accessibility (0.31): area(site i) – area(median). Diversified paratope is highlighted in red. The 30EWQ32 region, which is diversified in a sublibrary, is highlighted in green.
An additional library extended the second loop paratope based on previous binder sequences. A large fraction of the strongest binding clones isolated during initial sorts had mutations in the residues directly preceding loop two (Figure 1a and Supplemental Figure 2). Diversity was chosen in order to have six amino acids at each site to avoid over-diversification (Figure 1b). Amino acid diversity was selected by comparing expected (theoretical error-prone PCR mutation rate of parental codon) vs. actual (evolved) frequencies from first-generation Gp2 evolution. The extended loop library was built to have other paratope positions match the diversity of the constrained, cysteine-free library.
Increased binding specificity has been seen in other small protein scaffolds after surface charge neutralization.49 Data from charge neutralization experiments on several Gp2 clones for the purpose of affecting in vivo biodistribution indicated multiple sites where neutralization was tolerated.50 These charged framework sites were allowed to be parental or one neutral mutant in all libraries: E/A at site 20, E/Q at site 27, and R/Q at site 44 (Figure 1b).
Comparative Library Evaluation
Combinatorial libraries were built using degenerate oligonucleotides and transformed into yeast with homologous recombination into a yeast display vector.51 Based on the number of transformants, each library size contained 3–5 × 108 unique transformants. To ensure each library experienced the same selection conditions, they were combined and sorted together, resulting in a total library size of 4 × 109. Ten times the library size was used each sort so that there was >99.9% likelihood for each variant of being included at least once52. Yeast were sorted for binding to biotinylated recombinant target (PD-L1, MET, and tumor necrosis factor receptor (TNFR)) immobilized on streptavidin-coated magnetic beads. After two rounds of sorting and mutagenesis, populations of binding yeast emerged that exhibited greater than 14-fold higher yeast recovered on beads coated in target compared to beads with streptavidin or a control protein (human IgG). Enriched yeast were evaluated for higher affinity binding by labeling with 100 nM target and assessing binding by flow cytometry. Populations enriched for binding PD-L1 and MET showed significant signal over background (Supplemental Figure 3).
Naïve and binding populations were deep sequenced and unique sequences were separated by loop (339 for each loop from naïve; 53 unique loop one and 30 unique loop two from binders). Sequences with greater than 80% amino acid identity were grouped into families (339 for each loop from naïve; 38 loop one families and 18 loop two families from binders) to accentuate diversity rather than dominant clones (Supplemental Figure 4). Each family was assigned to the library that it had the highest probability of arising from based on the sequence and designed diversity. Each family was counted once after being assigned. There were 24 loop one families and 2 loop two families for MET binders, 12 loop one and 14 loop two families for PD-L1 binders, and 2 loop one and 2 loop two families for TNFR binders. This collection of Gp2 clones and families, targeting multiple epitopes on these three targets, provides a moderately diverse perspective on Gp2 evolution. Significant insights can be garnered from the data set while care must be taken when considering extrapolation to variants for additional epitopes.
Conservation of cysteine pairs
The evolutionary impact of dual cysteine constraint was evaluated by comparing libraries with broad diversity at all paratope sites but lacking C to libraries with a pair of sites conserved as C. Conservation of an intraloop C pair in loop 1 dramatically aided binder discovery as Gp2 variants were enriched from 13% of the naïve population to 47% in the functional population whereas variants from the C-free CDR− library were depleted from 23% to 5% (p < 0.001 between libraries) (Figure 4). On the contrary, conservation of an intraloop C pair in loop 2 hindered discovery as the C36-C39 library yielded reduced enrichment (+3%) relative to the C-free library (+17%; p = 0.006). An interloop C pair had undetectable impact on loop 1 enrichment but substantial hindrance to binder evolution in loop 2 (+17% for C-free vs. −21% for C8-C36b, p < 0.001). Thus, overall, constraint of a pair of cysteines can have a range of impacts on evolutionary performance — from a dramatic increase to strong decrease in binder discovery — dependent upon cysteine location.
Figure 4. Conserved cysteine pairs impact Gp2 library efficacy differently depending on location.

Cysteines were conserved at sites 7 and 12 (Intra 1), sites 36 and 39 (Intra 2) and sites 8 and 36b (Inter). Sequence frequency change from each library before (white) and after (grey) evolution were compared to the cysteine-free library (CDR−).
Cysteine-free library evaluation
While disulfide bond formation can stabilize domains and aid evolutionary efficacy, there are also potential advantages to cysteine-free domains including ease of expression, stability in reducing environments, lack of intermolecular disulfide bond formation, and availability for unique cysteine incorporation for site-specific conjugation. Notably, cysteine-free binders have been previously engineered in the Gp2 scaffold.2 Thus, we evaluated the ability to discover binders from a library lacking C. With degenerate nucleotide synthesis, this also resulted in the omission of G, R, and W. The lack of the conformationally flexible G in the paratope loops was partially addressed by inclusion of oligonucleotides encoding G at sites 10, 11, 34, and 35, which exhibited frequent G in previously evolved binders (Figure 1b). The reduced diversity library, CDR−, was compared to a full diversity counterpart, CDR+ (Figure 5). Despite the allowance of G at select sites, broad omission of C, G, R, and W was unfavorable in loop 1 as the C-free CDR− library was depleted from 23% to 5% whereas the CDR+ library including all 20 amino acids was enriched from 10% in the naïve pool to 24% in the binding population (p < 0.001). We also compared CDR− to CDR+ in sublibraries in which nine sites had diversity restricted on the basis of previous binders, homologs, theoretical stability, and accessibility (CDR−constrain vs. CDR+constrain). Similarly to the unconstrained context, the omission of C, G, R, and W hindered discovery: −14% enrichment for CDR−constrain vs. only −1% for CDR+constrain; p < 0.001. Evaluation of amino acid usage within the CDR+ variants reveals frequent G (12% in binders vs. 7% in the naïve library; albeit only with p = 0.08) whereas C, R, and W are maintained at basal levels. The converse result is observed in loop 2: Gp2 variants from the CDR− designs are nominally more frequently discovered: +17% enrichment for CDR− vs. +7% for CDR+ (p = 0.06) and +7% for CDR−constrain vs. −9% for CDR+constrain (p =0.003). In summary, omission of C, G, R, and W hinders performance in loop 1, largely because of the lack of G, but benefits performance in loop 2.
Figure 5. Allowance of C, G, R, and W aids Gp2 library efficacy in loop 1 and hinders efficacy in loop 2.

Libraries either had high diversity at each paratope site based on the antibody CDR (base) or had multiple sites constrained to lower diversity (constrained). The evaluation of a cysteine-free library (and limitations of degenerate nucleotides) resulted in libraries that contained (+) or lacked (−) the amino acids C, G, R, and W at certain sites. Change in sequence frequencies from before (white) and after (grey) evolution were compared between libraries.
Sitewise amino acid constraint
Sitewise amino acid constraint has benefited combinatorial library design for discovery of new binders in other small scaffolds.8–10 Of course, this benefit is dependent upon sitewise constraint that increases the frequency of functional amino acids and reduces the frequency of non-functional or detrimental amino acids. We biased diversity at nine sites based on previous binders, homologs, theoretical stability, and accessibility (Figure 1b). These constrained libraries were compared to their unconstrained counterparts. In the context of full diversity, constraint hindered binder discovery in loop 1 (−1% enrichment with constraint vs. +14% enrichment without, p < 0.001) and loop 2 (−9% vs. +7%, p = 0.003) (Figure 6). The impact was more modest in the absence of C, G, R, and W. One possible explanation for the lack of benefit is that first generation binders guided over-constraint or incorrect constraint. Since constraint has proven beneficial in other protein scaffold libraries, another attempt at constraining diversity in Gp2 should be performed with alternative approaches to constraint.
Figure 6. A particular sitewise amino acid constraint hinders Gp2 binder discovery.

Enrichment in sequence frequency from the combined naïve library to evolved binders is shown for libraries where nine paratope sites were constrained (red) based on first generation binder and natural homolog sequences, computational stability, and solvent accessibility, or where these sites were left as full diversity CDR (white with black lines). The CDR diversity was either cysteine-free (CDR−) or contained all amino acids (CDR+).
Extended paratope
Extending the loop two paratope to include mild diversity within 30EWQ32 (Figure 1a, green and Supplemental Figure 2) was detrimental when compared to the relevant constrained, cysteine-free CDR (−5% enrichment vs. +7%, p = 0.01) (Figure 7). The extended paratope may cause extra destabilization resulting in more unfolded proteins, or, since the library is not fully sampled, may simply have a lower fraction of beneficial target interactions.
Figure 7. Extending the loop two paratope region is detrimental to Gp2 binder discovery.
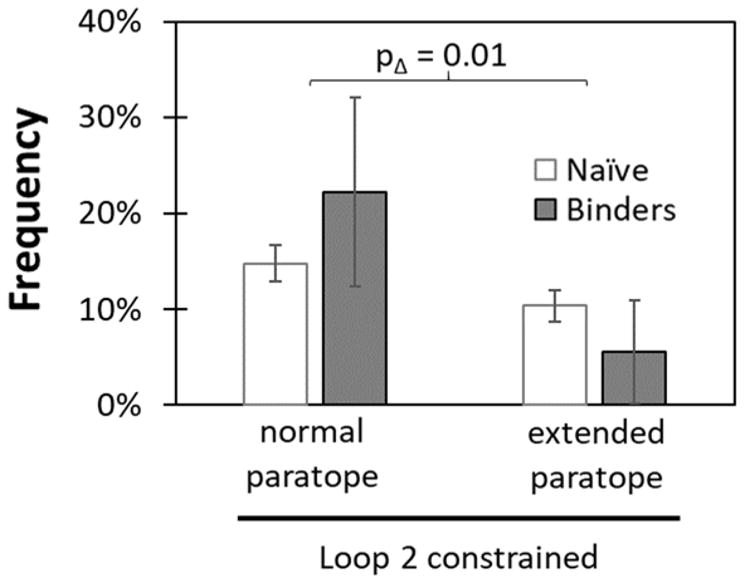
A three amino acid region (30EWQ32) near loop 2 that showed high mutation rates in the first generation library was diversified to six amino acids at each site, including wild type. The other sites in this extended paratope library were diversified identical to the cysteine-free, sitewise constrained library. The change in sequence frequency before (white) and after (grey) evolution was compared to the non-extended paratope, cysteine-free, sitewise constrained library.
Framework mutations
Although the library was designed to have equal mixtures of the wildtype and neutral mutant at the charge variant framework sites, the actual diversity varied widely (Supplemental Figure 5). Sites E20 (initially 22% wildtype) and E27 (initially 44% wildtype) preferred to revert to wildtype in evolved binders (27% enrichment and 11% enrichment of wildtype, respectively). Site R44, which started at 73% in the initial library, was relatively unchanged (−1% enrichment of wildtype). Based on the theoretical error prone PCR mutation rate, framework sites A25, L26, and L28 showed higher than expected mutation away from wildtype (−8%, −16%, and −8% relative enrichment, respectively). The large, aromatic residues F2, F13, and Y33 showed higher than expected retention as wildtype (13%, 10%, and 9% relative enrichment, respectively).
Protease Stability
Protease susceptibility of yeast surface displayed proteins has been shown to correlate with protein stability53. To determine the effect that each library design had on overall stability of the Gp2 domain, we measured the amount of full length protein remaining after exposure to proteinase K (Supplemental Figure 6). The cysteine intraloop 1 and interloop libraries were significantly more protease stable than the base CDR+ or C-free CDR− libraries (p < 0.02 in all cases). Notably, C7-C12 was the only pair (of the three tested) that was computationally predicted to form a strong disulfide54. All cysteine-free libraries and the constrained CDR+ library were nominally less stable than the first generation Gp2 library and the base CDR+ library.
Further evolution of PD-L1 ligands
High affinity PD-L1 ligands are of interest in the clinic as PET imaging agents to stratify patients and predict therapeutic response. The six most abundant variants sequenced from the PD-L1 binding population were transferred to a production vector and produced in E. coli. All six variants contained cysteines in the first loop at sites 7 and 12 (Figure 8a). When using the NEB SHuffle E. coli strain, four were able to be produced and purified from the soluble fraction. Flow cytometry verified all four PD-L1 binding Gp2 variants bound to CHO cells transfected to overexpress hPD-L1 but did not bind untransfected CHO (Figure 8b). The binding signal decreased substantially when using 150 nM Gp2 rather than 3 μM Gp2, thereby indicating a weak binding affinity. Estimating the binding affinity by fitting curves using the maximum signal from strong PD-L1 binding Gp2 leads to dissociation constants (KD) between 0.5 – 7 μM.
Figure 8. PD-L1 binding Gp2 protein characterization.

(a) The sequences of PD-L1 binding Gp2 domains recovered from library sorting in which blue lettering indicates the diversified paratope region. Clones C and G were unable to be produced in the soluble fraction. (b) The produced clones were used to label CHO-hPD-L1 cells at a medium (150 nM, gray) and high (3 μM, black concentration. Binding was detected using a fluorescently tagged anti-His6 antibody with flow cytometry. Binding is visible at 3 μM but nearly undetectable above background at 150 nM. Theoretical affinity curves were fit using data from strong PD-L1 binding Gp2 clones to determine the maximum fraction bound. Proteins were also used to label untransfected CHO-K1 cells as a negative control and showed much lower signal than on transfected cells (white).
To increase the binding strength of these lead Gp2 molecules to PD-L1 we performed extensive mutagenesis of each loop, independently within the context of the other conserved loop, and then merged the evolved loop sequences. Two sublibraries were constructed for each lead clone: one with extensively diversified loop 1 and conserved parental loop 2; and an inverse sublibrary with conserved loop 1 and extensively diversified loop 2. Amino acid diversity at each site was chosen to include homologous amino acids and to keep the library size below 107 unique members (Supplemental Figure 7 and Supplemental Table 3) so that the full library could be reliably sampled during yeast display sorting. More than 2 × 108 yeast transformants were generated, which leaves >99.9% likelihood of sampling for each variant. Sequencing indicates that the assembled library conforms with design.
Yeast were sorted by magnetic beads and fluorescence-activated cell sorting (FACS) at a PD-L1 concentration of 50 nM (Supplemental Figure 8). Deep sequencing of enhanced binders (Figure 9) reveals a range of mutational tolerances from broadly tolerant (e.g. V9 within clone E was observed as all amino acids except conformationally restricted P) to highly conserved (e.g. site T9b within the same clone E was 94% conserved). This mutational tolerance varies by site and clone. For example, in clone D, all sites in loop 2 were conserved with ≥87% frequency as the parental sequence whereas loop 1 had a single semi-conserved site and four broadly tolerant sites. The Y10F mutation was not merely tolerated but preferential relative to parental (45% to 28%, p < 0.001). In fact, each clone had at least one mutation that was observed more frequently than the parental amino acid in matured clones; clone B: M9 to R and A; clone D: aforementioned Y10F; clone E: six mutations at F10, three at T11, as well as A8S, V9S, and T9aP; and clone F: S9bT and five mutations in loop 2.
Figure 9. Deep sequencing of affinity matured PD-L1 binding Gp2 domains.

Gp2 domains that detectably bound 50 nM PD-L1 during yeast flow cytometry were grouped based on uniqueness and their frequency was quad-root damped. Observed percentages of each amino acid at each site are shown. Amino acids allowed in the initial library at that site are outlined. The parental amino acid has thick outlines.
As these mutations were all identified in the context of the other loop being conserved as parental, we then studied these beneficial and tolerated mutations in combination across loops for each clone. Collected yeast of a single parental variant had their mutated loops recombined, i.e. loop 1 of library one and loop 2 of library two were combined into a single new library. The highest affinity Gp2 mutants were isolated and Sanger sequenced after two rounds of FACS sorting (Figure 10a). Gp2 mutants were expressed in E. coli with a C-terminal His6 tag, purified by metal affinity chromatography, and titrated for binding to CHO cells transfected to express human PD-L1 as evaluated via flow cytometry. Affinity titration curves of two of the clones revealed strong, specific binding where KD = 9 nM (90% confidence interval: 4 – 24 nM, Supplemental Figure 9) for Gp2-PDL1E1 and 6 nM (3 – 13 nM) for Gp2-PDL1E4 (Figure 10b), an increase of 100x compared to the parental Gp2-PDL1E. Notably, these stronger clones do not bind untransfected CHO-K1 cells even at 1 μM.
Figure 10. Characterization of strongest binding evolved and parental PD-L1 binding Gp2.

A) Clones E1 and E4 were produced at measurable levels in the soluble fraction using E. coli. Their binding affinity (KD) was determined with n=3 trials (90% confidence interval indicated); N.E. indicates not expressed. (B) Purified Gp2-PDL1E4 was added at the indicated concentrations to CHO cells (transfected for PD-L1 expression: filled circles; untransfected: empty triangle). Binding was detected by fluorophore tagged anti-His6 antibody via flow cytometry. The best-fit estimate of the Kd (6 nM) and the 90% confidence interval (3 – 13 nM) are indicated by solid and dashed lines, respectively. (C) CHO cells transfected for PD-L1 expression were treated, at 4 °C, with 300 nM Gp2-PDL1E4 in the absence or presence of initial treatment with 67 nM atezolizumab. Binding was detected by fluorophore tagged anti-His6 antibody via flow cytometry.
Mechanistic implications of the mutational preferences (Figures 9 and 10) will require structural data and/or modeling, which is the subject of future work. Yet it is intriguing to note that available co-crystal structures of PD-L1 with cognate binders indicate overlapping (yet distinct) epitopes for five different antibodies as well as the native PD-1 domain. Pre-incubation of PD-L1 expressing cells at 4 °C with anti-PD-L1 antibody atezolizumab reduced binding of Gp2-PDL1E4 by 92% (Figure 10C), which is consistent with the evolved Gp2 domain also sharing this general epitope. Notably, the parental Gp2 variants B and D (and E) are also inhibited by atezolizumab (Supplemental Figure 10).
In addition, the impact of the putative disulfide bond between C7 and C12 was evaluated by circular dichroism under oxidized and reduced conditions. The native Gp2-PDL1E4 exhibited a midpoint of thermal denaturation (Tm) of 73 ± 2 °C whereas the presence of 200 μM tris(2-carboxyethyl)phosphine (TCEP) reduced the stability by 11 °C (p = 0.02, Supplemental Figure 11), which is consistent with the structural stability benefit of a disulfide bond. The added difficulty in producing these PD-L1 binding Gp2 domains with cysteine pairs must be balanced with potential evolutionary benefit during library design.
Conclusion
Overall, these investigations in paratope conservation advance Gp2 optimization and can be useful in library design for other protein scaffolds. Specifically, cysteine conservation at the edges of a paratope loop was beneficial to binder evolution for the epitopes targeted in this study. However, choosing the correct site (and amino acid diversity) for conservation is imperative as cysteines at other sites and conservation using different diversity schemes were detrimental to evolution. PD-L1 targeted Gp2 molecules that arose out of the loop one cysteine conservation library have been evolved to bind strongly towards cellular PD-L1 and can be utilized for molecular imaging or other applications.
Experimental Procedures
Previous Gp2-based binders
Sequences of Gp2-based binders to goat IgG, rabbit IgG, lysozyme, epidermal growth factor receptor, insulin receptor, MET (Hong Zhou and B.J.H.), interleukin family members (Milica Gakovic and K. Chris Garcia, personal communication), and oligomeric synuclein (Anthony Braun, B.J.H. and Michael Lee) were collected. To balance deep Illumina data (3.4 × 104 unique sequences) and shallow Sanger sequencing (40 unique sequences), each target campaign was equally weighted. The total collection of previous binding sequences were aligned and grouped by family (80% similarity). Amino acid frequencies at each site were calculated using quad-root dampening to decrease weight of dominant families as described previously2,55.
Gp2 homologs
Thirty-two sequences of Gp2 homologs were identified through a BLAST search of the wild type Gp2 protein sequence (in 2016). These sequences were aligned and sitewise amino acid frequencies were calculated.
Computed destabilization
To predict destabilization upon mutation, FoldX48 was used to calculate the change in folding energy for each single mutation at each site of a Gp2 with a randomized paratope. This method was repeated for 40 random paratope sequences – which was sufficient to observe < 0.1 kcal/mol absolute or < 1% relative deviation in mean with additional variants – with four unique solved structures describing T7 phage Gp2 (2WNM, 2LMC, 4LK0, 4LLG) and averaged to determine the final stability of each amino acid at each site.
Solvent accessible surface area
Solvent accessible surface area was calculated through the GetArea webserver56 (2WNM) or the Accessible Surface Area tool from Center for Information Biology at Ochanomizu University (all PDB files), using a 1.4 Å probe and averaged over four Gp2 structures (2WNM, 2LMC, 4LK0, 4LLG).
General constraint design
Site E7 had decreased levels of select amino acids in previous binders. Stability calculations suggested that diversity should not be too deleterious as 16 amino acids yield ≤ 1 kcal/mol destabilization upon mutation. Natural homologs were slightly constrained with 75% wildtype, but had a wide mixture of amino acids in the remaining 25% although this may be because wild type is important for natural function at this site 57. An average SASA of 0.35 suggests a modest likelihood for beneficial intermolecular contact upon mutation. A medium level of diversity was chosen; the design includes H, I, K, L, M, N, P, Q, R, S, T (encoded by MNK codon).
Site S8 had increased levels of the hydrophilic amino acids R, and S and decreases of hydrophobic including A, F, and L in previous binders. Stability calculations suggested that diversity may be deleterious. Natural homologs suggested small amino acids G, S, and T as well as a low level of larger amino acids are viable. An average SASA of 0.30 suggested a benefit of conservation at this site. A low level of diversity was chosen, aiming to include wild type and the upregulated hydrophilic amino acids. The design includes D, G, H, N, R, S and Y on a separate oligonucleotide (encoded by VRT + TAC codons).
Site S9 had increased levels of a variety of chemically disparate amino acids including F, G, E, L, K, W, and Y in previous binders. Stability and natural homolog analysis suggested that a wide variety of amino acids are permitted at this site. An average SASA of 0.94 suggested that high diversity may be beneficial at this site. The design chosen allows all 20 amino acids and mimicks the antibody complementarity determining region similar to generation 1 Gp2 (CDR).
The optional loop 1 insertion sites, 9a and 9b, have some amino acids that are increased in binders, especially A, H, and G at site 9b. However, due to the limited information from other sources due to these being non-natural loop length extensions, and their position in the middle of the proposed paratope, CDR was chosen for the design at both sites.
Site E10 had an increased level of hydrophilicity, especially N and D, and highly increased levels of G and P in previous binders. Stability and natural homolog data suggested that a wide variety of amino acids are permitted at this site. An average SASA of 0.78 suggested that high diversity may be beneficial at this site. Aiming for a high level of diversity with increased G, the amino acids A, D, H, N, P, S, T, Y and G on a separate oligonucleotide (encoded by NMT + GGT codons) were selected for the design at this site.
Site H11 had an overall increase in hydrophilicity and a large increase in G in previous binders. Stability and natural homologs suggested that a wide variety of amino acids are permitted at this site. An average SASA of 0.55 suggested that some diversity may be beneficial at this site. A design of CDR was chosen for this site.
Site S12 had a large increase in the frequency of hydrophobic amino acids in previous binders. Stability calculations suggested that many amino acids are permitted at this site, however natural homologs suggested more constraint with 83% as S and 6% as other small amino acids. An average SASA value of 0.47 suggested that some diversity may be beneficial. A design was chosen that increased the level of hydrophobic amino acids, while maintaining a medium level of diversity. The design includes A, G, L, M, R, S, T, V, W (encoded by DBG codon).
In loop 2, site V34 and P35 had highly increased levels of G and P, which are normally helpful in making a turn in protein secondary structure. Stability calculations and homolog analysis of V34 suggest that various amino acids are permitted. At P35, stability data suggested that any mutation would be detrimental. Yet we avoided completely locking in proline because we did not want to rely too heavily on the computational stability analysis. V34 has a medium SASA of 0.57 and P35 has a high SASA of 0.74. However, the combination of both sites showing highly increased levels of G and P lead to a constrained design with high levels of these amino acids. V34 was designed as A, E, G, P, Q, R (encoded by SVR codon). P35 was designed as A, D, H, N, P, S, T, Y and G (encoded by NMT + GGT codons).
Site A36 had an increase in total hydrophilicity in previous binders. Stability analysis suggested that a wide variety of amino acids are tolerated. Natural homologs were relatively constrained with 81% as A. An average SASA of 0.45 suggests that some diversity may be beneficial. A design was chosen to increase hydrophilicity with A, D, E, H, K, N, P, Q, S, T, Y, and a stop codon that was carried along due to the genetic code limitations (encoded by NMB codon).
Sites 36a and 36b, the non-natural loop length extension sites, had less information to use for design choice compared to the natural sites. In previous binders, 36a showed an increased hydrophilicity while 36b showed increased hydrophobicity. Due to the limited information, CDR was chosen for both sites.
Site G37 had increased hydrophobicity in previous binders, with increased levels of L, W, and Y especially, but hydrophilic D was also increased. Stability and natural homologs suggested limiting diversity at this site. However, SASA of 0.78 suggested that diversity could provide beneficial binding interactions. A design was chosen with moderate diversity and increased hydrophobicity, including D, F, I, N, V, Y, L, H (encoded by NWT codon).
Site F38 had significantly increased hydrophobicity in previous binders, with highly increased C. Stability and natural homolog analysis suggested limiting diversity at this site. An average SASA of 0.26 suggested that diversity may not be very beneficial at this site. A moderate diversity design to increase the level of hydrophobicity was chosen, including D, F, I, N, V, Y, L, H (encoded by NWT codon).
Site E39 had an increase in Y as well as other dissimilar amino acids. Stability and natural homologs both suggested that many amino acids are permitted at this site. An average SASA of 0.70 suggested that high diversity could provide beneficial binding interactions. CDR was chosen for this site.
Extended Paratope
Expected amino acid mutation rates during full gene error prone PCR were calculated based previous observed mutation rates58 with 10 μM each 8-oxo-dGTP and dPTP DNA analogues and 30 PCR rounds. The expected amino acid frequencies were normalized such that the median difference between expected enrichment and observed enrichment was zero.
Site E30 had a high percentage of G in previous binders but the ratio of actual/expected was relatively low. Conversely, V, F, A, and Q all occurred much more often than expected, and K was slightly higher than expected. The chosen design was E, Q, L, V, A, P (encoded by SHG codon) aiming to include wild type and allow for the chemically similar Q or much smaller A, V to reduce potential steric hindrance.
Site W31 had increased frequency of G, C, and L, moderately higher A, S, and slightly higher Q. The chosen design was A, G, L, S, V, W (encoded by KBG) aiming to include wild type and a variety of smaller amino acids to potentially provide additional beneficial binding interactions.
Site Q32 had lower R than expected, while L, H, K were higher than expected and E was slightly higher than expected. The design was chosen as D, E, H, L, Q, V (encoded by SWK codon) to allow for chemical homology and smaller amino acids to reduce potential steric hindrance.
See Supplemental Table 2 for oligonucleotide designs.
PD-L1 designer libraries
Cysteines at positions 7 and 12 that appeared in all parental clones were conserved as cysteine. Amino acid diversities at other sites (Supplemental Figure 7) were chosen to include parental and maximize the number of chemically similar amino acids while keeping total combinatorial diversity under 107. Four oligonucleotides that spanned the Gp2 gene were ordered (IDT) for each library.
Library construction, sorting, and sequencing
Naïve libraries were constructed and sorted as described previously2. Briefly, degenerate oligonucleotides were purchased (IDT) and assembled using overlap extension. Each library was separately transformed into yeast using homologous recombination. Yeast, ten times the library size, were sorted on magnetic beads with one wash, followed by another magnetic sort with two washes. Full length clones, isolated by flow cytometry via labeling of the C-terminal c-MYC epitope, were mutated by error-prone PCR using the dNTP analogs 8-oxo-dGTP and dPTP (Trilink Biotechnologies). First and second loops were PCR amplified separately and mixed together during homologous recombination such that loops from different variants could shuffle and recombine together. Designed framework mutations were included on oligonucleotides used in the initial construction and during amplification for error-prone PCR. Successive rounds of two magnetic bead binding sorts, one full-length flow cytometry sort, and mutagenic PCR2 was followed until selective binding, as determined by 10-fold higher recovery on target beads, was observed. This occurred after 1 full sorting round plus 1 bead sort for the MET and PD-L1 campaigns, and after 2 full rounds plus 2 beads sorts for the TNFR campaign. The initial library and isolated binders were sequenced as described previously 2. For MET and PD-L1 both bead binders (after reaching the 10:1 cutoff) and a FACS binders after an additional sort at 100 nM target were sequenced. For TNFR, only bead binders were sequenced as there was no detectable FACS binding at 100 nM target.
PD-L1 designer libraries were constructed by using an analogous method to CDR-walking 23. For each parental clone, loop one was diversified while retaining parental loop two, and vice versa. The diversity was chosen by keeping the parental amino acid and selecting chemically homologous amino acids accessible with degenerate codons. The diversified oligonucleotide for one loop and parental oligonucleotide for the other loop were constructed using overlap extension and transformed into yeast. After one standard bead sort and one flow cytometry sort at 50 nM biotin-PD-L1 the number of recovered yeast was <10,000 for each library. The functionally mutated first and second loops, from their respective libraries, were isolated by PCR and recombined into yeast for each variant. With fewer than 10,000 clones recovered, all combinations (<108) could be sampled by yeast display sorting. Yeast with combined loops were incubated with 50 nM biotin-PD-L1, washed, and then captured by beads. The enriched population was sorted by flow cytometry with 50 nM and 5 nM biotin-PD-L1 to isolate the strongest binders. A final depletion sort using three rounds of bare magnetic beads and one round of human IgG coated beads was used to remove non-PD-L1 binders.
Protein production and characterization
Protein was produced and characterized as previously described.2 Briefly, proteins were produced in T7 express, or T7 SHuffle E. coli (New England Biolabs) when proteins contained multiple cysteines, and were purified using immobilized metal affinity chromatography and, on certain clones, reverse phase high-performance liquid chromatography with a 90% buffer A (99.9% H2O, 0.1% trifluoroacetic acid (TFA))/10% buffer B (90% acetonitrile, 9.9% H2O, 0.1% TFA) to 10% A/90% B gradient over 15 minutes on a C18 column (Waters). Purity was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis using 4%–12% Bis-Tris gradient gels in MES buffer. If needed, size exclusion chromatography using an Akta Superdex 75 10/300 GL column was performed to isolate monomer from dimer or aggregates. Affinity titrations were carried out using flow cytometry to measure the level of Gp2 binding to CHO-K1 cells transfected to express human PD-L1 (CHO-hPD-L1)59. Mammalian cells were grown in F12K plus 10% fetal bovine serum with 2 mg/mL G418 or lacking G418 for CHO-K1. Purified Gp2 was added, in stoichiometric excess to PD-L1, at various concentrations, and binding was quantified via flow cytometry with fluorophore tagged anti-His6 antibody (ab1206; Abcam). The equilibrium dissociation constant was calculated as a global fit of three independent titrations using GraphPad Prism.
Specificity of Gp2 binding to human PD-L1 was assessed by pre-incubating human PD-L1 expressing CHO-K1 (CHO-hPD-L1) cells with 67 nM of anti-PD-L1 antibody, atezolizumab (Biovision), at 4 °C, followed by addition of 300 nM of Gp2-PDL1E4, or parental B, D and E clones at 4 °C. The extent of binding was calculated from the fluorescent signal of the C-terminal His6-targeting antibody by flow cytometry.
Lyophilized Gp2-PDL1E4 was resuspended in deionized water to a concentration of 80 μM. 200 μl of the resuspended sample was added into a quartz cuvette (1 mm path length). Thermal denaturation at a wavelength of 218 nm was evaluated in a Jasco J-815 spectrophotometer by measuring the ellipticity of Gp2-PDL1E4 while ramping the temperature from 25 °C to 98 °C at a rate of 2 °C min−1. The impact of the potential disulfide bond between C7 and C12 on thermal denaturation was determined by overnight incubation of 80 μM of Gp2-PDL1E4 with 200 μM of TCEP hydrochloride (Sigma-Aldrich). The midpoint of thermal denaturation was determined by minimizing the sum of squared residuals of the data assuming a two-state unfolding model.
Protease Susceptibility
1 × 106 yeast displaying Gp2 were washed with PBS (8.0 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4, pH 7.2) plus 1 g/L BSA (PBSA). Proteinase K (New England Biolabs) was serially diluted to 0.0002 units/100 μL in PBSA. The proteinase mixture was prewarmed to 37 °C for 5 min and 100 μL was added to the yeast pellet. The yeast plus proteinase mixture and a control with no proteinase were incubated at 37 °C for 10 min. Samples were washed with ice-cold PBSA two times. Yeast were labeled with an anti-HA antibody (N-terminal tag) and an anti-c-MYC antibody (C-terminal tag) and followed with fluorescent secondary antibodies as previously described2. Flow cytometry was used to analyze the percentage of HA+ and c-MYC+ cells. The proteinase concentration chosen was sufficient to retain greater than 95% HA signal after proteinase treatment.
Statistics
Sublibrary frequency means and standard deviations were determined by assuming a binomial distribution where each sequence represented a success or failure trial for a given sublibrary. For the special case of zero occurrences the rule of 360 was used to estimate a 95% confidence interval and standard error. P-values comparing change in two sublibraries from naïve to binders were calculated with a t-test assuming unequal variances.
Supplementary Material
Acknowledgments
Funding Information: This work was funded by the National Institutes of Health (R01 EB023339 to B.J.H. and R21 EB021511 to B.J.H.) and the University of Minnesota (Interdisciplinary Doctoral Fellowship to M.K.).
We thank Alex Golinski and Daniel Woldring for assistance with sequence analysis, Patrick Holec for computing stabilities, as well as Milica Gakovic and Chris Garcia (Stanford University) and Anthony Braun and Michael Lee (University of Minnesota) for sharing Gp2 sequences. CHO-hPD-L1 cells were a kind gift from Dr. Sridhar Nimmagadda (Johns Hopkins).
Abbreviations
- CDR
complementarity-determining region
- Gp2
gene 2 protein from T7 phage
- IgG
immunoglobulin G
- PD-L1
programmed death ligand 1
- PET
positron emission tomography
Footnotes
Author Contributions: M.A.K., V.S., and B.J.H. designed the experiments, M.A.K. and V.S. performed the experiments, M.A.K., V.S., and B.J.H. wrote the manuscript.
Conflict of Interest: The authors declare no conflict of interest.
Extended figures and tables of library designs, oligonucleotide sequences, and sequencing and binding data
References
- 1.Škrlec K, Štrukelj B, Berlec A. Non-immunoglobulin scaffolds: a focus on their targets. Trends Biotechnol. 2015:1–11. doi: 10.1016/j.tibtech.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Kruziki MA, Bhatnagar S, Woldring DR, Duong VT, Hackel BJ. A 45-Amino-Acid Scaffold Mined from the PDB for High-Affinity Ligand Engineering. Chem Biol. 2015;22:946–956. doi: 10.1016/j.chembiol.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan JY, Hackel BJ, Yee D. Targeting Insulin Receptor in Breast Cancer Using Small Engineered Protein Scaffolds. Mol Cancer Ther. 2017;16:1324–1334. doi: 10.1158/1535-7163.MCT-16-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chothia C, Janin J. Principles of protein-protein recognition. Nature. 1975;256:705–708. doi: 10.1038/256705a0. [DOI] [PubMed] [Google Scholar]
- 5.Kastritis PL, Rodrigues JPGLM, Folkers GE, Boelens R, Bonvin AMJJ. Proteins feel more than they see: fine-tuning of binding affinity by properties of the non-interacting surface. J Mol Biol. 2014;426:2632–52. doi: 10.1016/j.jmb.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 6.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci U S A. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woldring DR, Holec PV, Zhou H, Hackel BJ. High-Throughput Ligand Discovery Reveals a Sitewise Gradient of Diversity in Broadly Evolved Hydrophilic Fibronectin Domains. PLoS One. 2015;10:e0138956. doi: 10.1371/journal.pone.0138956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woldring DR, Holec PV, Stern LA, Du Y, Hackel BJ. A gradient of sitewise diversity promotes evolutionary fitness for binder discovery in a three-helix bundle protein scaffold. Biochemistry. 2017;56:1656–1671. doi: 10.1021/acs.biochem.6b01142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackel BJ, Ackerman ME, Howland SW, Wittrup KD. Stability and CDR Composition Biases Enrich Binder Functionality Landscapes. J Mol Biol. 2010;401:84–96. doi: 10.1016/j.jmb.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroeder HW, Kirkham PM. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol. 2003;334:733–749. doi: 10.1016/j.jmb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Hackel BJ, Wittrup KD. The full amino acid repertoire is superior to serine/tyrosine for selection of high affinity immunoglobulin G binders from the fibronectin scaffold. Protein Eng Des Sel. 2010;23:211–219. doi: 10.1093/protein/gzp083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A. 2004;101:12467–12472. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J Mol Biol. 2008;377:1518–1528. doi: 10.1016/j.jmb.2008.01.093. [DOI] [PubMed] [Google Scholar]
- 16.Steipe B, Schiller B, Pluckthun A, Steinbacher S, Steipe B, Schiller B, Plückthun A, Steinbacher S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J Mol Biol. 1994;240:188–92. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
- 17.Smith CA, Kortemme T. Predicting the Tolerated Sequences for Proteins and Protein Interfaces Using RosettaBackrub Flexible Backbone Design. PLoS One. 2011;6:e20451. doi: 10.1371/journal.pone.0020451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fass D. Disulfide Bonding in Protein Biophysics. Annu Rev Biophys. 2012;41:63–79. doi: 10.1146/annurev-biophys-050511-102321. [DOI] [PubMed] [Google Scholar]
- 19.Dombkowski AA, Sultana KZ, Craig DB. Protein disulfide engineering. FEBS Lett. 2014;588:206–212. doi: 10.1016/j.febslet.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Kintzing JR, Cochran JR. Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr Opin Chem Biol. 2016;34:143–150. doi: 10.1016/j.cbpa.2016.08.022. [DOI] [PubMed] [Google Scholar]
- 21.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 22.Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: past, present and future. Curr Opin Chem Biol. 2017;38:24–29. doi: 10.1016/j.cbpa.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Lipovsek D, Lippow SM, Hackel BJ, Gregson MW, Cheng P, Kapila A, Wittrup KD. Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: Molecular convergence with single-domain camelid and shark antibodies. J Mol Biol. 2007;368:1024–1041. doi: 10.1016/j.jmb.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 24.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Padda SK, Riess JW, Schwartz EJ, Tian L, Kohrt HE, Neal JW, West RB, Wakelee Ha. Diffuse High Intensity PD–L1 Staining in Thymic Epithelial Tumors. J Thorac Oncol. 2015;10:500–508. doi: 10.1097/JTO.0000000000000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Souza Malaspina TS, Gasparoto TH, Costa MRSN, De Melo EF, Ikoma MRV, Damante JH, Cavassani KA, Garlet GP, Da Silva JS, Campanelli AP. Enhanced programmed death 1 (PD-1) and PD-1 ligand (PD-L1) expression in patients with actinic cheilitis and oral squamous cell carcinoma. Cancer Immunol Immunother. 2011;60:965–974. doi: 10.1007/s00262-011-1007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:1–15. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn M, Felip E, Lee J, Hellmann MD, Hamid O, Goldman JW, Soria J, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 29.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P CheckMate 025 Investigators. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 31.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman Ja, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin a, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Caliò A, Cuppone F, Sperduti I, Giannarelli D, Chilosi M, Bronte V, Scarpa A, Bria E, Tortora G. Differential Activity of Nivolumab, Pembrolizumab and MPDL3280A according to the Tumor Expression of Programmed Death-Ligand-1 (PD-L1): Sensitivity Analysis of Trials in Melanoma, Lung and Genitourinary Cancers. PLoS One. 2015;10:e0130142. doi: 10.1371/journal.pone.0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heskamp S, Hobo W, Molkenboer-Kuenen JDM, Olive D, Oyen WJG, Dolstra H, Boerman OC. Noninvasive Imaging of Tumor PD-L1 Expression Using Radiolabeled Anti-PD-L1 Antibodies. Cancer Res. 2015;75:2928–36. doi: 10.1158/0008-5472.CAN-14-3477. [DOI] [PubMed] [Google Scholar]
- 34.Chatterjee S, Lesniak WG, Gabrielson M, Lisok A. A humanized antibody for imaging immune checkpoint ligand PD-L1 expression in tumors. Oncotarget. 2016;7:10215–10227. doi: 10.18632/oncotarget.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hettich M, Braun F, Bartholoma MD, Schirmbeck R, Niedermann G. High-resolution PET imaging with therapeutic antibody-based PD-1/PD-L1 checkpoint tracers. Theranostics. 2016;6:1629–1640. doi: 10.7150/thno.15253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.England CG, Ehlerding EB, Hernandez R, Rekoske BT, Graves SA, Sun H, Liu G, McNeel DG, Barnhart TE, Cai W. Preclinical Pharmacokinetics and Biodistribution Studies of 89 Zr-Labeled Pembrolizumab. J Nucl Med. 2017;58:162–168. doi: 10.2967/jnumed.116.177857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F, Tsui B, Sgouros G. Imaging, biodistribution, and dosimetry of radionuclide-labeled PD-L1 antibody in an immunocompetent mouse model of breast cancer. Cancer Res. 2016;76:472–479. doi: 10.1158/0008-5472.CAN-15-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nedrow JR, Josefsson A, Park S, Ranka S, Roy S, Sgouros G. Imaging of Programmed Cell Death Ligand 1: Impact of Protein Concentration on Distribution of Anti-PD-L1 SPECT Agents in an Immunocompetent Murine Model of Melanoma. J Nucl Med. 2017;58:1560–1566. doi: 10.2967/jnumed.117.193268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesniak WG, Chatterjee S, Gabrielson M, Lisok A, Pomper MG, Nimmagadda S. PD-L1 Detection in Tumors Using [64 Cu]Atezolizumab with PET. Bioconjug Chem. 2016;27:2103–2110. doi: 10.1021/acs.bioconjchem.6b00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG, Kimura R, Tsai JM, Manglik A, Kruse AC, Gambhir SS, Weissman IL, Ring AM. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci. 2015;112:E6506–14. doi: 10.1073/pnas.1519623112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer AT, Natarajan A, Gordon SR, Maute RL, McCracken MN, Ring AM, Weissman IL, Gambhir SS. Practical Immuno-PET Radiotracer Design Considerations for Human Immune Checkpoint Imaging. J Nucl Med. 2017;58:538–546. doi: 10.2967/jnumed.116.177659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.González Trotter DE, Meng X, McQuade P, Rubins D, Klimas M, Zeng Z, Connolly BM, Miller PJ, O’Malley SS, Lin SA, Getty KL, Fayadat-Dilman L, Liang L, Wahlberg E, Widmark O, Ekblad C, Frejd FY, Hostetler ED, Evelhoch JL. In Vivo Imaging of the Programmed Death Ligand 1 by (18)F PET. J Nucl Med. 2017;58:1852–1857. doi: 10.2967/jnumed.117.191718. [DOI] [PubMed] [Google Scholar]
- 43.Donnelly DJ, Smith RA, Morin P, Lipovsek D, Gokemeijer J, Cohen D, Lafont V, Tran T, Cole EL, Wright M, Kim J, Pena A, Kukral D, Dischino DD, Chow P, Gan J, Adelakun O, Wang XT, Cao K, Lueng D, Bonacorsi S, Hayes W. Synthesis and Biological Evaluation of a Novel 18 F-labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J Nucl Med. 2018;59:529–535. doi: 10.2967/jnumed.117.199596. [DOI] [PubMed] [Google Scholar]
- 44.Broos K, Keyaerts M, Lecocq Q, Renmans D, Nguyen T, Escors D, Liston A, Raes G, Breckpot K, Devoogdt N. Non-invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget. 2017;8:41932–41946. doi: 10.18632/oncotarget.16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kruziki MA, Case BA, Chan JY, Zudock EJ, Woldring DR, Yee D, Hackel BJ. Cu-Labeled Gp2 Domain for PET Imaging of Epidermal Growth Factor Receptor. Mol Pharm. 2016;13:3747–3755. doi: 10.1021/acs.molpharmaceut.6b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G, Holak TA. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure. 2017;25:1163–1174. doi: 10.1016/j.str.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 47.Virnekas B, Ge L, Plukthun A, Schneider KC, Wellnhofer G, Moroney SE. Trinucleotide phosphoramidites: Ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994;22:5600–5607. doi: 10.1093/nar/22.25.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Traxlmayr MW, Kiefer JD, Srinivas RR, Lobner E, Tisdale AW, Mehta NK, Yang NJ, Tidor B, Wittrup KD. Strong enrichment of aromatic residues in binding sites from a charge-neutralized hyperthermostable Sso7d scaffold library. J Biol Chem. 2016;291:22496–22508. doi: 10.1074/jbc.M116.741314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Case BA, Kruziki MA, Johnson SM, Hackel BJ. Engineered charge redistribution of Gp2 proteins through guided diversity for improved PET imaging of epidermal growth factor receptor. Bioconjug Chem. 2018;29:1646–1658. doi: 10.1021/acs.bioconjchem.8b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boder ET, Wittrup KD. Yeast Surface Display for Screening Combinatorial Polypeptide Libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 52.Reetz MT, Kahakeaw D, Lohmer R. Addressing the numbers problem in directed evolution. Chembiochem. 2008;9:1797–1804. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- 53.Rocklin GJ, Chidyausiku TM, Goreshnik I, Ford A, Houliston S, Lemak A, Carter L, Ravichandran R, Mulligan VK, Chevalier A, Arrowsmith CH, Baker D. Global analysis of protein folding using massively parallel design, synthesis, and testing. 2017;357:168–175. doi: 10.1126/science.aan0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Craig DB, Dombkowski AA. Disulfide by Design 2.0 : a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14:0–6. doi: 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woldring DR, Holec PV, Hackel BJ. ScaffoldSeq: Software for characterization of directed evolution populations. Proteins. 2016;84:869–874. doi: 10.1002/prot.25040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19:319–333. [Google Scholar]
- 57.Cámara B, Liu M, Reynolds J, Shadrin A, Liu B, Kwok K, Simpson P, Weinzierl R, Severinov K, Cota E, Matthews S, Wigneshweraraj SR. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc Natl Acad Sci U S A. 2010;107:2247–52. doi: 10.1073/pnas.0907908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol. 2008;381:1238–1252. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lesniak WG, Chatterjee S, Gabrielson M, Lisok A, Wharram B, Pomper MG, Nimmagadda S. PD-L1 Detection in Tumors Using [64Cu]Atezolizumab with PET. Bioconjug Chem. 2016;27:2103–2110. doi: 10.1021/acs.bioconjchem.6b00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanley JA, Lippman-Hand A. If nothing goes wrong, is everything alright? JAMA. 1983;249:1743–1745. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.