

Abstract
Opioid peptides are key regulators in cellular and intercellular physiological responses, and could be therapeutically useful for modulating several pathological conditions. Unfortunately, the use of peptide-based agonists to target centrally located opioid receptors is limited by poor physicochemical (PC), distribution, metabolic, and pharmacokinetic (DMPK) properties that restrict penetration across the blood-brain-barrier via passive diffusion. To address these problems, the present manuscript exploits fluorinated peptidomimetics to simultaneously modify PC and DMPK properties, thus facilitating entry into the central nervous system. As an initial example, the present manuscript exploited the Tyr1-ψ[(Z)CF=CH]-Gly2 peptidomimetic to improve PC drug-like characteristics (computational), plasma and microsomal degradation, and systemic and CNS distribution of Leu-enkephalin (Tyr-Gly-Gly-Phe-Leu). Thus, the fluoroalkene replacement transformed an instable in vitro tool compound into a stable and centrally-distributed in vivo probe. In contrast, the Tyr1-ψ[CF3CH2–NH]-Gly2 peptidomimetic decreased stability by accelerating proteolysis at the Gly3–Phe4 position.
Keywords: Opioid Peptides, Leu-Enkephalin, Peptidomimetics, Fluorination, Peptide Stability, Peptide CNS-Distribution
Graphical Abstract
INTRODUCTION
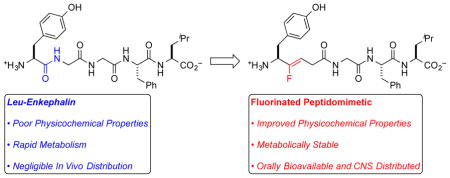
Opioid peptides are key regulators in various cellular and intercellular physiological responses, and could be therapeutically useful for modulating pain, alcohol abuse, emotional and mood disorders, and other pathological conditions.1–3 Unfortunately, the use of peptide-based agents to target centrally located opioid receptors (ORs) is limited by poor physicochemical and pharmacokinetic (PK) properties,4 including low lipophilicity (logD), high topological polar surface area (TPSA), and extensive H-bonding with H2O, which combined, lowers oral bioavailability and disfavors penetration of the blood-brain-barrier (BBB) via passive diffusion.5,6 Additionally, rapid proteolysis by endogenous peptidases provides a short half-life (t1/2) that disfavors therapeutically relevant concentrations and/or tissue distribution.4,5,7,8 Thus for medicinal application of small peptides, the ability to rationally modify both the PK and physicochemical (PC) properties, and to facilitate entry into the central nervous system (CNS) remains an important goal. To address these challenges, fluorinated peptidomimetics (FPMs) might simultaneously modulate physicochemical [logD, TPSA, and HBD/HBA (Hydrogen-Bond Donors/Hydrogen-Bond Acceptors)] and metabolic properties of neuropeptides, and in turn, improve distribution.
As a prototypical example, Leu-enkephalin (1, Tyr-Gly-Gly-Phe-Leu), agonizes the δ-opioid receptor (DOR) with 1–5-fold binding affinity over the μ receptor and >1000-fold over the κ receptor.9,10 This pharmacological profile might be useful for treating chronic pain, inflammation, and cancer pain, while avoiding respiratory depression and addiction.11 However, in vivo administration of Leu-enkephalin is limited by a poor PK profile, including rapid proteolysis of Tyr1-Gly2 by aminopeptidase N in human plasma,7,12 and of Gly3-Phe4 by angiotensin-converting enzyme at the BBB,8 both of which inhibit penetration into the CNS.5 To improve potency and selectivity, many cyclic and linear analogues of enkephalin have been developed that possess excellent affinities and selectivities for the DOR,9,13–15 and in some cases, the replacement of hydrolyzable amide bonds at various positions with peptidomimetics (e.g., trans-alkene,9 thioamide,16 ester,17 or N-methyl amide17) has modulated stability and PK properties, without major loss of agonist activity at the DOR. However, most of these peptidomimetic analogues either (a) still lack appropriate in vivo stability and CNS distribution to serve as the biological probes and/or therapeutic candidates, or (b) have not been assessed in vivo. To improve metabolic and distribution properties of Leu-enkephalin, we report the use of Tyr1-ψ[(Z)CF=CH]-Gly2 and Tyr1-ψ[CF3CH2–NH]-Gly2 analogs (2–4), of which the former displays sufficient stability to enable systemic and CNS distribution from both intravenous (IV) and oral (PO) doses.
RESULTS AND DISCUSSION
Selection of Tyr1–Gly2
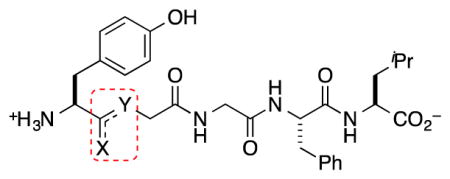
To stabilize and improve distribution of Leu-enkephalin (1), we modified Tyr1–Gly2, which undergoes rapid proteolysis by aminopeptidase N (AP-N), thus providing an insufficient half-life to sustain therapeutically useful systemic and/or CNS concentrations (Figure 1A).5–8 Additionally, this site tolerates substitution,9,18–20 which suggests that other modifications of this amide linkage might be tolerated, such as fluorinated peptidomimetics (FPMs). Specifically, FPMs, including fluoroalkenes and β,β,β-trifluoroethylamines, should improve physicochemical and biophysical properties of target molecules, and should not be substrates for the endogenous peptidases. Fluoroalkenes possess similar dipole moments, electrostatic potential,21 steric, resonance, and inductive contributions to amides.22–24 However, the sigma-electron withdrawing effect of the fluorine atom decrease the basicity of neighboring functional groups, which makes these groups less prone to H-bond with H2O and possibly the catalytic domains of metabolizing enzymes.19,25 As a result, this substitution should increase lipophilicity, and facilitate membrane permeability by passive diffusion.22,26 Trifluoroethylamines27,28 also match the polar nature of the amide, and can alter a probe by imparting greater lipophilicity, and thermal and proteolytic stability to a peptide.29 For this substructure, the strong inductive effect of the CF3 group renders neighboring amines sufficiently non-basic to remain neutral under physiological conditions, and thus, not alter the ionization state of the peptide. In addition, the strong electron-withdrawing CF3 group acidifies the N–H group, thus increasing the H-bond donating capability of the trifluoroethylamine.30 Finally, the conversion of the sp2-hybridized amide to the sp3-hybridized trifluoroethylamine can alter the conformational preference of the peptide. All combined, when judiciously applied to the appropriate location of a peptide, these FPMs should improve metabolism and distribution properties. In our previous work, Tyr1-ψ[(Z)CF=CH]-Gly2 substitution provided a 5 nM DOR full agonist (2) with similar selectivity to the endogenous peptide though 60 fold less potent than the parent peptide (1), though both (R)- and (S)-isomers of Tyr1-ψ[CF3CH2–NH]-Gly2 analogs (3–4) did not activate the DOR.31 Despite the decreased activities, the stability, metabolism, and distribution properties of all analogs were not previously assessed.
Figure 1. Hypothesis: FPMs Should Stabilize and Increase Distribution of Leu-Enkephalin.
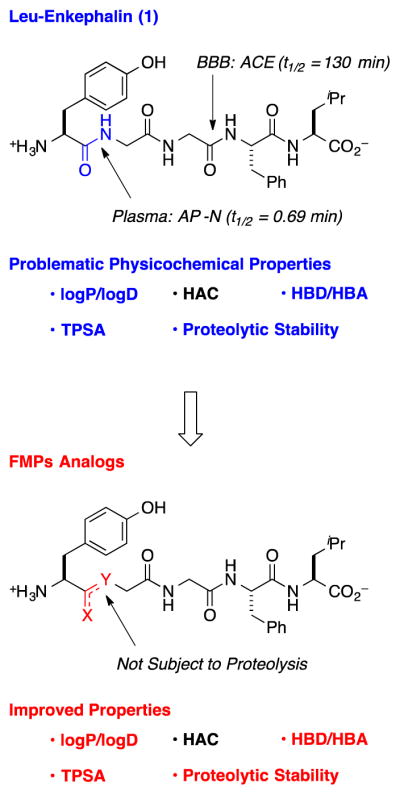
(HAC = Heavy Atom Count, HBD = Hydrogen-Bond Donors, HBA = Hydrogen-Bond Acceptors, TPSA = Topological Polar Surface Area, ACE = Angiotensin Converting Enzyme, AP-N = Aminopeptidase N)
Physicochemical Perturbations Imparted by Tyr1–Gly2 FPMs
Substitution of Tyr1–Gly2 with FPMs moves computationally predicted physicochemical properties toward CNS drug-like space (Table 1).32 Specifically, replacement of the amide linkage with FPMs increased clogD 1.1–1.6 units, and decreased topological polar surface area (TPSA) by 17–29 Å2. Replacement with Tyr1-ψ[(Z)CF=CH]-Gly2 eliminated a hydrogen-bond donor and acceptor group, while substitution with Tyr1-ψ[CF3CH2–NH]-Gly2 only reduced the hydrogen-bond acceptor count. Finally, substitution with FPMs increased overall molecular weight (MW), though considering the emerging concept of fluorine-adjusted molecular weight,33 the FPM analogs might behave as smaller molecules than the endogenous peptide (1). Further, β,β,β-trifluoroethylamine substitution at the Tyr1–Gly2 site increases the pKa of the N-terminal amine one unit, while fluoroalkene substitution does not modulate the pKa relative to 1. Combined, these PC descriptors suggest that the FPM enkephalin analogs fit within orally available drug-space for therapeutic peptides,34,35 though outside of CNS drug-like space for small molecules that enter the CNS by passive diffusion.36–38
Table 1.
Physicochemical Descriptors for FPM Analogs of Leu-Enkephalin.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Structure | clogD a | clogP a | MW [Da] | F-Adj MW | TPSA [Å2] a | HBD | HBA | pKa a |
| 1 |
|
–2.3 | –2.3 | 555 | 555 | 204 | 6 | 7 | 7.7 |
| 2 |

|
–1.2 | –1.2 | 556 | 537 | 175 | 5 | 6 | 7.7 |
| 3 |
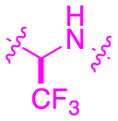
|
–0.7 | 0.7 | 609 | 552 | 187 | 6 | 7 | 8.7 |
| 4 |
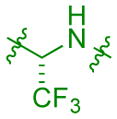
|
–0.7 | 0.7 | 609 | 552 | 187 | 6 | 7 | 8.7 |
| – | CNS-MPO36,38 (Sm. Molecules) | 2–4 | 3–5 | 360 –500 | – | 40–90 | 0–4 | – | 8–10 |
| – | PO Peptides34 80th Percentile |
– | <5.1 | <1,217 | – | <301 | <9 | <23 | – |
Calculated using MarvinSketch (https://www.chemaxon.com/products/calculators-and-predictors).
(CNS-MPO = Central Nervous System Multiparameter Optimization, PO Peptides = Calculated percentiles of main descriptors for oral peptide drugs, clogD = calculated lipophilicity in pH 7.4 buffer, clogP = calculated lipophilicity, MW = Molecular Weight, F-Adj MW = Fluorine Adjusted Molecular Weight, TPSA = Topological Polar Surface Area, HBD = Hydrogen-Bond Donors, HBA = Hydrogen-Bond Acceptors, pKa = acid dissociation constant)
FPM Analogs Demonstrate Excellent Media Stability and Solubility
All FPMs demonstrate excellent stability towards various solvents and assay media. All FPM analogs were stable over 48 h in PBS, simulated gastric fluid (pH = ~2), Captisol (200 mg/mL), 1:3 isopropanol:H2O, 1:1 acetonitrile:H2O. Thus, the investigational compounds should be stable in the acid environment of the stomach, as well as various solvents that might be useful for formulation or assay development. Further, all analogs displayed excellent solubility (>160 μM) in PBS, SGF, Captisol, IPA:H2O, MeCN:H2O, which minimizes challenges for assay-development.
Tyr1-ψ[(Z)CF=CH]-Gly2 Substitution Improves Stability
As expected, fluoroalkene analog 2 also demonstrated excellent stability in mouse, rat and human plasma (Figure 2). In mouse plasma, both Leu-enkephalin (1) and 2 showed excellent stability (>90% of peptide remaining at 4 h). However, in this experiment, the intrinsic stability of the control peptide (1) suggested that this model is not appropriate for demonstrating the improved stability of 2, which supports previous reports describing low expression of peptidases in mouse plasma.39 However, due to interspecies differences in peptidase expression, Leu-enkephalin hydrolyzes more quickly in rat and human plasma samples.7 Thus, both human and rat plasma clearly demonstrated the improved stability imparted by Tyr1-ψ[(Z)CF=CH]-Gly2 substitution. In Sprague-Dawley rat plasma, 76% of 2 remained at 4 h, while t1/2 for 1 was less than 5 min. In human plasma (female donor), 68% of 2 remained at 4 h, while t1/2 for 1 was 12 min. These rat plasma half-lives compared favorably to Leu-enkephalin (t1/2 = 4.6 min), as well as Phe4-OLeu5 (t1/2 = 3.3 min)17 and Phe4- (NMe)Leu5 (t1/2 = 10.7 min)17 analogs.
Figure 2. Tyr1-ψ[(Z)CF=CH]-Gly2 Substitution Improves Plasma Stability of Leu-Enkephalin.

(
 ) = Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin (
) = Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin (
 ) = Leu-Enkephalin
) = Leu-Enkephalin
Similarly, the Tyr1-ψ[(Z)CF=CH]-Gly2 substitution stabilized Leu-enkephalin (1) to mouse, rat and human NADPH-charged microsomes (Figure 3). Using all three microsomal samples, 2 demonstrated excellent stability with <10% of the material metabolized at 2 h. In contrast, 1 was 38–50% metabolized by all microsomal samples at 2 h. In the absence of NADPH, 2 demonstrated no microsomal degradation, while 1 slowly degraded (see Supporting Information for more details).
Figure 3. Tyr1-ψ[(Z)CF=CH]-Gly2 Substitution Improves Microsomal Stability.

(
) = Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin, (
) = Leu-Enkephalin, (▲) = Diphenhydramine (Mouse and Rat) or Verapamil (Human).
Tyr1-ψ[CF3CH2–NH]-Gly2 Substitution Decreases Stability
Considering that Tyr1–Gly2 is the primary site of proteolysis,5,6,8,40 and that the trifluoroethylamine substructure is not subject to proteolytic metabolism, we expected both (R)- and (S)- analogs of Tyr1-ψ[CF3CH2–NH]-Gly2-Leu-enkephalin (3–4) to increase stability towards proteolysis. However, 3 and 4 unexpectedly worsened stability in both plasma and microsomes (Figure 4). The main pathway of this degradation involved proteolysis of the Gly3–Phe4 linkage, as indicated by LC-MS analysis of the degradation mixture. Thus, despite stabilizing Tyr1–Gly2, this substitution accelerated metabolism at the known secondary site of metabolism,5 possibly by forcing a conformational change in the peptide that facilitates recognition by an alternate protease. As a result of the poor stability, both 3 and 4 were not promoted for further in vivo studies.
Figure 4. Tyr1-ψ[CF3CH2–NH]-Gly2 Substitution Worsens Plasma and Microsomal Stability.

(
) = Leu-Enkephalin (1), (
 ) = Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3), (
) = Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3), (
 ) = Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4), (●) = Diphenhydramine
) = Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4), (●) = Diphenhydramine
Tyr1-ψ[(Z)CF=CH]-Gly2 Improves Distribution
The excellent in vitro stability of Tyr1-ψ[(Z)CF=CH]-Gly2–Leu-enkephalin (2) also improved in vivo distribution and PK properties. Using a mouse model [BALB/c], plasma concentrations of a 50 mg/kg IV dose were detected for 2 h, while the Leu-enkephalin (1), was completely consumed within 5 min (Figure 5A). Additionally, 2 was orally bioavailable, with a 400 mg/kg PO dose providing maximum plasma concentrations of 28 ng/mL. Using the PO dose, plasma concentrations were detected up to 30 min. Despite the short half-life of 22 min, this compound provides a distinct strategy for designing orally bioavailable analogs of Leu-enkephalin. Critically, both IV and PO doses of Tyr1-ψ[(Z)CF=CH]-Gly2–Leu-enkephalin (2) crossed the BBB (Figure 5B). By IV, a 50 mg/kg dose of 2 provided measurable levels (60–140 ng/mL tissue), which eliminated to undetectable levels within 20 min. This dose of 50 mg/Kg resides within the range of previous IV doses (30–60 mg/kg) of DPDPE, [p-Cl-Phe4]DPDPE, and [p-125IPhe4]DPDPE that demonstrated in vivo antinociceptive activity (hotplate) within 5 min in Male CD1 mice.41 Additionally, the PO dose (400 mg/kg) provided detectible concentrations of 2 (5–8 ng/mL tissue) at 5 min, though the compound was no longer detected by 10 min. In both cases, the detectible concentrations of Tyr1-ψ[(Z)CF=CH]-Gly2–Leu-enkephalin show promise compared to the endogenous concentration of Leu-enkephalin (1) in the non-stressed mouse brain (4–40 ng/gm tissue, depending on region of the brain).42
Figure 5. Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin Is Orally Available and Distributes into the CNS.

(
 ) = IV 50 mg/kg Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin, (
) = IV 50 mg/kg Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin, (
 ) = PO 400 mg/kg Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin
) = PO 400 mg/kg Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-Enkephalin
For 2, maximum brain uptake was slightly lower than analogs of DPDPE and biphalin that have previously been explored in vivo (Table 2).41,43,44 Specifically, calculated brain uptake values for 2 of 0.008%/g tissue (IV) are within an order of magnitude of several related enkephalin analogs, including [3H] DPDPE41,43 (IV: 0.08%, Tail Vein Injection: 0.059–0.067%), [p-125IPhe4]DPDPE41 (Tail Vein Injection: 0.021–0.027%), and [125l-Tyr1]biphalin44 (Tail Vein Injection: 0.0045–0.053%). Though, maximum brain concentrations of 2 peaked more quickly (5 min) than [3H] DPDPE43 (60 min) and [125l-Tyr1]biphalin44 (20 min), respectively. However, due to interspecies differences (Balb/c mice vs. CD1 mice vs. Sprague–Dawley rats), and differences in assays (LCMS vs. radioactivity) and injection strategies (IV vs. tail vein injection), direct comparisons of the data are not possible.
Table 2.
Maximum Brain Concentrations of 2 Compared to Other Reported Leu-Enkephalin Analogs.
| Entry | Peptide | Time Points (min) | Brain Concentrations (% uptake/g tissue) | Comments |
|---|---|---|---|---|
| 1 | 2 | 5, 15 | IV: 0.008% (5 min) |
|
| 2 | [3H] DPDPE43 | 60, 120, 240 | IV: 0.08% at 60 min (maximum |
|
| 3 | [3H] DPDPE41 | 5, 10, 20, 40 | 0.059–0.067% |
|
| 4 | [p-125IPhe4]DPDPE41 | 5, 10, 20, 40 | 0.021–0.027% |
|
| 5 | [125l-Tyr1]biphalin44 | 5, 20, 40 | (0.0045–0.053%) |
|
Additionally, a safety/dose-escalation study suggested engagement of centrally-located DOR. Specifically, convulsions are a side-effect associated with DOR agonists, which might derive from β-arrestin2 signaling,45 and/or from rapid engagement of centrally localized DOR.45–47 At a 50 mg/kg IV dose, no side effects were observed, although higher doses induced brief seizures from which the animals recovered fully. These observations are consistent with DOR activation, though from this preliminary experiment, an appropriate safety window cannot yet be determined.
Potential Mechanisms of Membrane Transport
To demonstrate the mechanisms by which compound 2 entered systemic distribution and penetrated the BBB, standard PAMPA, Caco-2, and MDCK transwell assays were conducted. However, in these assays, minimal penetration in either the A→B or B→A directions discounted processes involving passive diffusion (see Supporting Information for more details).48,49 Further, considering that other enkephalin analogs are not substrates for P-gp,49 we tentatively believe that 2 should also not be subject to efflux.
Combined, (a) the poor penetration in transwell assays, (b) the excellent plasma and microsomal stability, (c) the rapid in vivo plasma uptake and CNS penetration (PO dose), and (d) short in vivo plasma and CNS half-live in (IV and PO doses) might best be explained by an active bidirectional transport mechanism. At present, we suspect OATP or LAT might mediate transport of 2 across the BBB. OATP facilitates entry of DPDPE and some structurally modified Leu-enkephalin analogs into the CNS,50,51 while, LAT, which normally transports large amino acids (e.g. phenylalanine, tyrosine, and leucine), can also transport enkephalin analogs (e.g. biphalin and DPDPE).52,53 This bidirectional transport mechanism might provide opportunities for lower PO and IV doses while retaining similar CNS concentrations, particularly a high concentrations of 2 that might saturate the transporter. However, future work is required to determine the mechanisms of uptake for both oral availability and CNS distribution, and to generate new analogs with improved distribution properties, which would provide concrete justification for exploring lower doses.
CONCLUSIONS
In conclusion, Tyr1-ψ[(Z)CF=CH]-Gly2 substitution of Leu-enkephalin increased plasma and microsomal stability sufficiently to provide an orally-active CNS-distributed peptide probe. Considering the improved stability and distribution, and 5 nM potency of this probe, future studies will evaluate in vitro signaling bias, in vivo anti-nociceptive and anti-alcohol abuse properties, and mechanism of transport. Combined, this data will provide a basis for designing potent, stable, long acting and CNS-distributed DOR-selective biased probes with improved safety profiles. In contrast, Tyr1-ψ[CF3CH2–NH]-Gly2 substitution decreased stability by accelerating metabolism at the Gly3–Phe4 site, which combined with poor potency, limits future investigations of this probe.
METHODS
Leu-Enkephalin Analogs
Leu-enkephalin acetate salt (1) was purchased from Sigma Aldrich. Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-enkephalin (2), Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3), and Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4) were synthesized according to previously reported procedures.31 Confirmation of the identity and purity of 2–4 was accomplished by a combination of 1H, 19F, and 13C NMR, HRMS, and UPLC.
Microsomal Stability
Microsomal stability of Leu-enkephalin (1), Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-enkephalin (2), Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3), and Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4) were monitored in vitro.54–56 Solutions of the compounds (0.50 μM) were incubated with mouse, rat or human microsomes (0.50 mg/mL, Gibco) and two cofactors UDPGA (250 μM, Sigma Aldrich) and NADPH (1 mM, Sigma Aldrich) in PBS at 37 °C. The compounds and microsomes, in the absence of co-factors, were prepared as negative controls. Samples were taken at predetermined time points, quenched with acetonitrile, and the parent compound was analyzed by LC-MS/MS. Samples were prepared in triplicate. Positive controls were set up with compounds known to be susceptible to mouse, rat, and human microsomal metabolism. These included positive controls as diphenhydramine (Sigma Aldrich) for mouse and rat microsomes, and verapamil (Sigma Aldrich) for human microsomes.
Plasma Stability
Stability of Leu-enkephalin (1), Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-enkephalin (2), Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3), and Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4) were monitored in mouse, rat, or human plasma (Innovative Research) at 37 °C. Compounds were spiked into plasma to obtain a concentration of 50 μM. At predetermined time points, samples were taken and quenched with acetonitrile, and the compounds were analyzed by LC-MS/MS. Samples were prepared in duplicate.
LCMS Analysis Method
Analysis was performed on Shimadzu 20A Series HPLC with binary pump, column oven, autosampler, controller and AB Sciex 3200 Qtrap mass spectrometer. A Zorbax SB-C18 column (2.1 mm × 50 mm, 5 μm particle size) was used for the assay. The injection volume was 5 μL. Mobile phase A consisted of 5/95/0.1 acetonitrile/water/formic acid and mobile phase B consisted of 100% methanol. The column was kept at 40 °C with a column oven and the autosampler temperature was set to 15 °C.
For Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-enkephalin (2) and Leu-enkephalin (1), the initial flow rate was 0.6 mL/min and initial mobile phase B concentration was 10%. After a 0.25 min hold, the gradient was ramped from 10% to 65% B over 4.25 min; the column was washed at 98% B and re-equilibrated to 10% B prior to the next injection. These two compounds were used as internal standards for each other. For Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-enkephalin (2) the mass transition from 557.3 → 120.2 was monitored with a collision energy of 50 eV (~Rt 3.03 min); for Leu-enkephalin (1) the mass transition 556.2 → 397.2 was monitored with a collision energy of 28 eV (~Rt 2.76 min). Additional MS/MS parameters include curtain gas (25), CAD (MED), IS (2500), TEM (600), GS1 (50), GS2 (60).
For Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3) and Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4), the initial flow rate was 0.5 mL/min and initial mobile phase B concentration was 25%. After a 0.25 min hold, the gradient was ramped from 10% to 60% B over 4.25 min; the column was washed at 98% B and re-equilibrated to 25% B prior to the next injection. These two compounds were used as internal standards for each other. For both Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3), and Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4) the mass transition from 610.3 → 120.1 was monitored with a collision energy of 62 eV. The retention time for Tyr1-ψ[(S)-CF3CH2–NH]-Gly2-Leu-enkephalin (4) was ~3.29 min and for Tyr1-ψ[(R)-CF3CH2–NH]-Gly2-Leu-enkephalin (3) was ~3.06 min). Additional MS/MS parameters were identical as for Tyr1-ψ[(Z)CF=CH]-Gly2-Leu-enkephalin (2) and Leu-enkephalin (1).
Dose Tolerance and Pharmacokinetic Studies
For the dose escalation and tolerance studies, Balb/c mice received a single IV or oral gavage dose of the compound. For the oral gavage dosing, a low dose of isoflurane was administered as a chemical restraint. The dose started at 1 mg/kg and was gradually escalated until drug related side effects were observed. The dose used in the pharmacokinetic studies was selected at a level just below where drug related side effects were observed. The mice used in the dose escalation studies were continuously monitored for 4 h and then euthanized using inhaled isoflurane to induce deep anesthesia followed by cardiac puncture. No mice exhibited severe drug related side effects.
For the pharmacokinetic studies, Balb/c mice were dosed by IV or oral gavage using a well-tolerated dose identified by the procedure above. Each mouse was used for one time point and received a single drug dose. Blood was collected at a pre-selected time point by cardiac puncture following deep anesthesia with isoflurane inhalation and exsanguination. Each time point required 3 mice and 10 time points for a total of up to 30 mice per study. Plasma was collected from each of the blood samples and assayed for the compound using a validated LCMS/MS procedure.
After death, the brains of the euthanized mice were perfused with PBS to remove as much blood as possible prior to harvesting the brains. A perfusion needle was inserted in the left ventricle of the heart and the right atrium was clipped to allow for the remaining blood from the mouse to be flushed out. Perfusion was done for approximately 20 min or until the liver became very pale and no blood was visible in the tissue around the eyes. The brain was ivory in color and had no visible signs of residual blood. The brain was harvested, and tissue was extracted. These extraction samples were assayed using a validated LCMS/MS procedure.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the US National Institute on Drug Abuse under award number DA036730. The content herein is the sole responsibility of the authors and does not necessarily represent the official views of the US National Institutes of Health. We thank Dr. Mike Rafferty for helpful discussion and reviewing a preliminary draft of this manuscript.
ABBREVIATIONS
- ACE
Angiotensin Converting Enzyme
- AP-N
Aminopeptidase N
- BBB
blood-brain-barrier
- CNS
central nervous system
- DMPK
distribution, metabolic, and pharmacokinetic
- DOR
δ-opioid receptor
- DPDPE
[D-Pen2,D-Pen5]-enkephalin
- FPMs
fluorinated peptidomimetics
- HAC
Heavy Atom Count
- HBD
Hydrogen-Bond Donors
- HBA
Hydrogen-Bond Acceptors
- IPA
isopropanol
- IV
intravenous
- LAT
large neutral amino acid transporter
- LCMS
Liquid Chromatography Mass Spectroscopy
- logD
lipophilicity at pH 7.4
- logP
lipophilicity
- NADPH
nicotinamide adenine dinucleotide phosphate
- OATP
organic-anion-transporting polypeptide
- ORs
opioid receptors
- PAMPA
parallel artificial membrane permeability assay
- PC
physicochemical
- PK
pharmacokinetic
- PO
oral
- PBS
phosphate buffered saline
- TPSA
topological polar surface area
- UPLC
Ultra-Performance Liquid Chromatography
Footnotes
Notes
The authors declare no competing financial interest.
AUTHOR CONTRIBUTIONS
R.A.A. conceived the research plan and wrote the initial draft of the manuscript. R.A.A. and K.K.S. interpreted data and revised the manuscript. L.G.R., P.C.T. and M.J.B. executed in vitro and in vivo experiments. M.P. and S.N.K. synthesized compounds 2–4.
The Supporting Information is available free of charge on the ACS Publications website at DOI: XXXXXXX. Microsomal stability without NADPH, and Caco-2, MDCK-MDR1, and PAMPA results.
References
- 1.Vallejo R, Barkin RL, Wang VC. Pharmacology of opioids in the treatment of chronic pain syndromes. Pain Physician. 2011;14:E343–E360. [PubMed] [Google Scholar]
- 2.Gentilucci L, Tolomelli A, Squassabia F. Peptides and peptidomimetics in medicine, surgery and biotechnology. Curr Med Chem. 2006;13:2449–2466. doi: 10.2174/092986706777935041. [DOI] [PubMed] [Google Scholar]
- 3.Yeomans MR, Gray RW. Opioid peptides and the control of human ingestive behaviour. Neurosci Biobehav Rev. 2002;26:713–728. doi: 10.1016/s0149-7634(02)00041-6. [DOI] [PubMed] [Google Scholar]
- 4.Witt KA, Davis TP. CNS drug delivery: Opioid peptides and the blood-brain barrier. AAPS J. 2006;8:E76–E88. doi: 10.1208/aapsj080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornford EM, Braun LD, Crane PD, Oldendorf WH. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology. 1978;103:1297–1303. doi: 10.1210/endo-103-4-1297. [DOI] [PubMed] [Google Scholar]
- 6.Pardridge WM, Mietus LJ. Enkephalin and blood-brain barrier: Studies of binding and degradation in isolated brain micro vessels. Endocrinology. 1981;109:1138–1143. doi: 10.1210/endo-109-4-1138. [DOI] [PubMed] [Google Scholar]
- 7.Weinberger SB, Martinez JLJ. Characterization of Hydrolysis of [ Leu ] enkephalin in Rat Plasma. J Pharmacol Exp Ther. 1988;247:129–135. [PubMed] [Google Scholar]
- 8.Suzanne ET, Kenneth LA. Leucine-Enkephalin Metabolism in Brain Microvessel Endothelial Cells. Peptides. 1994;15:109–116. doi: 10.1016/0196-9781(94)90178-3. [DOI] [PubMed] [Google Scholar]
- 9.Proteau-Gagné A, Bournival V, Rochon K, Dory YL, Gendron L. Exploring the backbone of enkephalins to adjust their pharmacological profile for the δ-opioid receptor. ACS Chem Neurosci. 2010;1:757–769. doi: 10.1021/cn1000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lord JAH, Waterfield AA, Hughes J, Kosterlitz HW. Endogenous opioid peptides: Multiple agonists and receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- 11.Pradhan AA, Befort K, Nozaki C, Gavériaux-Ruff C, Kieffer BL. The delta opioid receptor: An evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2011;32:581–590. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roques B, Noble F. Dual inhibitors of enkephalin-degrading enzymes (neutral endopeptidase 24.11 and aminopeptidase N) as potential new medications in the management of pain and opioid addiction. NIDA Res Monogr. 1995;147:104–145. [PubMed] [Google Scholar]
- 13.Hruby VJ, Bartosz-Bechowski H, Davis P, Slaninova J, Zalewska T, Stropova D, Porreca F, Yamamura HI. Cyclic enkephalin analogues with exceptional potency and selectivity for δ-opioid receptors. J Med Chem. 1997;40:3957–3962. doi: 10.1021/jm9704762. [DOI] [PubMed] [Google Scholar]
- 14.Deekonda S, Cole J, Sunna S, Rankin D, Largent-Milnes TM, Davis P, Bassirirad NM, Lai J, Vanderah TW, Porecca F, Hruby VJ. Enkephalin analogues with N-phenyl-N-(piperidin-2-ylmethyl)propionamide derivatives: Synthesis and biological evaluations. Bioorganic Med Chem Lett. 2016;26:222–227. doi: 10.1016/j.bmcl.2015.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosberg HI, Hurst R, Hruby VJ, Gee K, Akiyama K, Yamamura HI, Galligan JJ, Burks TF. Cyclic penicillamine containing enkephalin analogs display profound delta receptor selectivities. Life Sci. 1983;33:447–450. doi: 10.1016/0024-3205(83)90538-6. [DOI] [PubMed] [Google Scholar]
- 16.Lajoie G, Lépine F, Lemaire S, Jolicoeur F, Aubé C, Turcotte A, Belleau B. Synthesis and biological activity of monothionated analogs of leucine-enkephalin. Int J Pept Protein Res. 1984;24:316–327. doi: 10.1111/j.1399-3011.1984.tb00959.x. [DOI] [PubMed] [Google Scholar]
- 17.Rochon K, Proteau-Gagné A, Bourassa P, Nadon JF, Coîté J, Bournival V, Gobeil F, Guérin B, Dory YL, Gendron L. Preparation and evaluation at the delta opioid receptor of a series of linear Leu-enkephalin analogues obtained by systematic replacement of the amides. ACS Chem Neurosci. 2013;4:1204–1216. doi: 10.1021/cn4000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox MT, Heaton DW, Horbury J. Preparation of protected trans-olefinic dipeptide isosteres. J Chem Soc Chem Commun. 1980:799. [Google Scholar]
- 19.Hann MM, Sammes PG, Kennewell PD, Taylor JB. On the double bond isostere of the peptide bond: preparation of an enkephalin analogue. J Chem Soc Perkin Trans 1. 1982:307–314. [Google Scholar]
- 20.Garbe D, Sieber SA, Bandur NG, Koert U, Marahiel MA. Enzymatic cyclisation of peptidomimetics with incorporated (E)-alkene dipeptide isosteres. ChemBioChem. 2004;5:1000–1003. doi: 10.1002/cbic.200400042. [DOI] [PubMed] [Google Scholar]
- 21.Urban JJ, Tillman BG, Cronin WA. Fluoroolefins as peptide mimetics: A computational study of structure, charge distribution, hydration, and hydrogen bonding. J Phys Chem A. 2006;110:11120–11129. doi: 10.1021/jp062881n. [DOI] [PubMed] [Google Scholar]
- 22.Allmendinger T, Furet P, Hungerbuhler E. Fluoroolefin dipeptide isosteres - I. The synthesis of GlyΨ(CF=CH)Gly and racemic PheΨ (CF=H)Gly. Tetrahedron Lett. 1990;31:7297–7300. [Google Scholar]
- 23.Wipf P, Henninger TC, Geib SJ. Methyl- and (Trifluoromethyl)alkene Peptide Isosteres: Synthesis and Evaluation of Their Potential as β-Turn Promoters and Peptide Mimetics. J Org Chem. 1998;63:6088–6089. doi: 10.1021/jo981057v. [DOI] [PubMed] [Google Scholar]
- 24.Choudhary A, Raines RT. An Evaluation of Peptide-Bond Isosteres. ChemBioChem. 2011;12:1801–1807. doi: 10.1002/cbic.201100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bégué J-P, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. John Wiley & Sons, Inc; 2008. [Google Scholar]
- 26.Gante J. Peptidomimetics-Tailored Enzyme Inhibitors. Angew Chem Int Ed. 1994;33:1699–1720. [Google Scholar]
- 27.Volonterio A, Bellosta S, Bravin F, Bellucci MC, Bruché L, Colombo G, Malpezzi L, Mazzini S, Meille SV, Meli M, Ramírez de Arellano C, Zanda M. Synthesis, Structure and Conformation of Partially-Modified Retro- and Retro-Inversoψ[NHCH(CF3)]Gly Peptides. Chem - A Eur J. 2003;9:4510–4522. doi: 10.1002/chem.200304881. [DOI] [PubMed] [Google Scholar]
- 28.Black WC, Bayly CI, Davis DE, Desmarais S, Falgueyret JP, Léger S, Chun SL, Massé F, McKay DJ, Palmer JT, Percival MD, Robichaud J, Tsou N, Zamboni R. Trifluoroethylamines as amide isosteres in inhibitors of cathepsin K. Bioorganic Med Chem Lett. 2005;15:4741–4744. doi: 10.1016/j.bmcl.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 29.Sani M, Volonterio A, Zanda M. The trifluoroethylamine function as peptide bond replacement. ChemMedChem. 2007;2:1693–1700. doi: 10.1002/cmdc.200700156. [DOI] [PubMed] [Google Scholar]
- 30.Yamazaki T, Taguchi T, Ojima I. Fluorine in Medicinal Chemistry and Chemical Biology. John Wiley & Sons, Ltd; 2009. Unique Properties of Fluorine and their Relevance to Medicinal Chemistry and Chemical Biology; pp. 1–46. [Google Scholar]
- 31.Karad SN, Pal M, Crowley RS, Prisinzano TE, Altman RA. Synthesis and Opioid Activity of Tyr 1 - ψ [( Z )CF=CH]-Gly 2 and Tyr 1 - ψ [( S )/( R )-CF 3 CH-NH]-Gly 2 Leu-enkephalin Fluorinated Peptidomimetics. ChemMedChem. 2017;12:571–576. doi: 10.1002/cmdc.201700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Csizmadia F. JChem: Java Applets and Modules Supporting Chemical Database Handling from Web Browsers. J Chem Inf Comput Sci. 2000;40:323–324. doi: 10.1021/ci9902696. [DOI] [PubMed] [Google Scholar]
- 33.Pettersson M, Hou X, Kuhn M, Wager TT, Kauffman GW, Verhoest PR. Quantitative Assessment of the Impact of Fluorine Substitution on P-Glycoprotein (P-gp) Mediated Efflux, Permeability, Lipophilicity, and Metabolic Stability. J Med Chem. 2016;59:5284–5296. doi: 10.1021/acs.jmedchem.6b00027. [DOI] [PubMed] [Google Scholar]
- 34.Santos GB, Ganesan A, Emery FS. Oral Administration of Peptide-Based Drugs: Beyond Lipinski’s Rule. ChemMedChem. 2016;11:2245–2251. doi: 10.1002/cmdc.201600288. [DOI] [PubMed] [Google Scholar]
- 35.Doak BC, Over B, Giordanetto F, Kihlberg J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem Biol. 2014;21:1115–1142. doi: 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Wager TT, Hou X, Verhoest PR, Villalobos A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem Neurosci. 2016;7:767–775. doi: 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- 37.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wager TT, Chandrasekaran RY, Hou X, Troutman MD, Verhoest PR, Villalobos A, Will Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem Neurosci. 2010;1:420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibanoki S, Weinberger SB, Schulteis G, Ishikawa K, Martinez JL., Jr Enkephalin hydrolysis by mouse plasma in vitro. Life Sci. 1992;50:667–675. doi: 10.1016/0024-3205(92)90469-6. [DOI] [PubMed] [Google Scholar]
- 40.Bak A, Fich M, Larsen BD, Frokjaer S, Friis GJ. N-terminal 4-imidazolidinone prodrugs of Leu-enkephalin: Synthesis, chemical and enzymatic stability studies. Eur J Pharm Sci. 1999;7:317–323. doi: 10.1016/s0928-0987(98)00044-x. [DOI] [PubMed] [Google Scholar]
- 41.Weber SJ, Greene DL, Sharma SD, Yamamura HI, Kramer TH, Burks TF, Hruby VJ, Hersh LB, Davis TP. Distribution and analgesia of [3H][D-Pen2, D-Pen5]Enkephalin two halogenated analogs after intravenous administration. J Pharmacol Exp Ther. 1991;259:1109–1117. [PubMed] [Google Scholar]
- 42.Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51:211–217. doi: 10.1016/0024-3205(92)90077-3. [DOI] [PubMed] [Google Scholar]
- 43.Weber SJ, Greene DL, Hruby VJ, Yamamura HI, Porreca F, Davis TP. Whole body and brain distribution of [3H]cyclic [D-Pen2,D-Pen5] enkephalin after intraperitoneal, intravenous, oral and subcutaneous administration. J Pharmacol Exp Ther. 1992;263:1308–1316. [PubMed] [Google Scholar]
- 44.Abbruscato TJ, Thomas SA, Hruby VJ, Davis TP. Brain and spinal cord distribution of biphalin: correlation with opioid receptor density and mechanism of CNS entry. J Neurochem. 1997;69:1236–1245. doi: 10.1046/j.1471-4159.1997.69031236.x. [DOI] [PubMed] [Google Scholar]
- 45.Broom DC, Nitsche JF, Pintar JE, Rice KC, Woods JH, Traynor JR. Comparison of receptor mechanisms and efficacy requirements for delta-agonist-induced convulsive activity and antinociception in mice. J Pharmacol Exp Ther. 2002;303:723–9. doi: 10.1124/jpet.102.036525. [DOI] [PubMed] [Google Scholar]
- 46.Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Laustriat D, Cao YQ, Basbaum AI, Dierich A, Vonesh JL, Gaveriaux-Ruff C, Kieffer BL. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc Natl Acad Sci U S A. 2006;103:9691–9696. doi: 10.1073/pnas.0603359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol. 2012;167:960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2012;64:280–289. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 49.Wu S, Campbell C, Koda Y, Blanchfield JT, Toth I. Investigation of the route of absorption of lipid and sugar modified leu-enkephalin analogues and their enzymatic stability using the caco-2 cell monolayer system. Med Chem. 2006;2:203–211. doi: 10.2174/157340606776056205. [DOI] [PubMed] [Google Scholar]
- 50.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta - Biomembr. 2003;1609:1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 51.Gao B, Hagenbuch B, Kullak-Ublick Ga, Benke D, Aguzzi A, Meier PJ. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J Pharmacol Exp Ther. 2000;294:73–79. [PubMed] [Google Scholar]
- 52.Tamai I, Tsuji A. Transporter-Mediated Permeation of Drugs Across the Blood – Brain Barrier. J Pharm Sci. 2000;89:1371–1388. doi: 10.1002/1520-6017(200011)89:11<1371::aid-jps1>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 53.Egleton RD, Davis TP. Bioavailability and transport of peptides and peptide drugs into the brain. Peptides. 1997;18:1431–1439. doi: 10.1016/s0196-9781(97)00242-8. [DOI] [PubMed] [Google Scholar]
- 54.http://www.cyprotex.com/admepk/in-vitro-metabolism/microsomal-stability
- 55.https://www.thermofisher.com/us/en/home/references/protocols/drug-discovery/adme-tox-protocols/microsomes-protocol.html
- 56.Grbac RT, Stanley FA, Ambo T, Barbara JE, Haupt LJ. High Content Automated Metabolic Stability and CYP Inhibition Cocktail Screening Assays for Early Drug Development. Personal communication. 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.