

Abstract
Chronic pain is maintained in part by central sensitization, a phenomenon of synaptic plasticity and increased neuronal responsiveness in central pain pathways after painful insults. Accumulating evidence suggests that central sensitization is also driven by neuroinflammation in the peripheral and central nervous system (CNS). A characteristic feature of neuroinflammation is the activation of glial cells, such as microglia and astrocytes, in the spinal cord and brain, leading to the release of pro-inflammatory cytokines and chemokines. Recent studies suggest that central cytokines and chemokines are powerful neuromodulators and play a sufficient role in inducing hyperalgesia and allodynia after the CNS administration. Sustained increase of cytokines and chemokines in the CNS also promotes chronic widespread pain that affects multiple body sites. Thus, neuroinflammation drives widespread chronic pain via central sensitization. We also discuss sex-dependent glial/immune signaling in chronic pain and new therapeutic approaches that control neuroinflammation for the resolution of chronic pain.
Introduction
Chronic pain is a major health concern that affects one in three Americans and costs the US economy $635 billion dollars each year1,2. Acute pain is often elicited by acute inflammation and has biological significance to protect the wounded tissue. Chronic pain is maladaptive and has no beneficial biological significance and is characterized by spontaneous pain (e.g., burning) as well as evoked pain in response to noxious (hyperalgesia) or non-noxious (allodynia) stimuli. It is generally believed that neuronal plasticity in pain coding pathways and circuits results in chronic pain. Neuronal plasticity consists of peripheral sensitization in primary sensory neurons of dorsal root ganglia (DRG) and trigeminal ganglia3–5, and central sensitization of pain-processing neurons in the spinal cord and brain6–9.
The perception of pain is typically associated with inflammation, a complex biological response of the somatosensory, immune, neuronal, autonomic and vascular/circulatory system to tissue damage, pathogens, or irritants. Acute inflammation, which generally results in perception of pain, serves an important protective or survival role by removing harmful stimuli, initiating the healing process, and restoring tissue integrity (Table 1). Primary afferents that respond to tissue injury (i.e., nociceptors) include unmyelinated C-fibers and myelinated Aδ-fibers that terminate in skin, muscle, joint, and visceral organs, with their cell bodies located in DRG and trigeminal ganglia. Nociceptors are activated or sensitized by inflammatory mediators such as bradykinin, prostaglandins, nerve growth factor, as well as pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α or TNF), interleukin 1β (IL-1β) and pro-inflammatory chemokines (e.g., CCL2, CXCL5)3,10,11 that directly bind and stimulate G-protein coupled receptors (GPCRs), ionotropic receptors, and tyrosine kinase receptors. It is noteworthy that all these receptors are expressed on the terminals and/or cell bodies of nociceptors3,5. Cytokine profiles observed in human skin are also associated with inflammation and pain and thus may serve as biomarkers for chronic pain12,13. Activation of a mosaic of peripheral receptors results in hypersensitivity and hyperexcitability of nociceptor neurons (peripheral sensitization), through modulation of various ion channels, such as transient receptor potential ion channels (TRPs, such as TRPA1, TRPV1, and TRPV4), sodium channels (e.g., subtypes Nav1.7/1.8/1.9)3,4,14, and mechanosensitive piezo ion channels15,16. MicroRNA (miRNA) may serve as a novel inflammatory and pain-evoking mediator. For example, miR-let-7b induces spontaneous pain via activation of Toll-like receptor 7 (TLR7) and TRPA117. Furthermore, the activation of protein kinases such as MAP kinases, protein kinase A (PKA), and protein kinase C (PKC) in primary sensory neurons critically contributes to the induction and maintenance of peripheral sensitization. For instance, p38 MAP kinase activation in DRG neurons initiates peripheral sensitization by increasing TRPV1 activity in response to TNF18 and also by increasing NaV1.8 activity in response to IL-1β32 and, furthermore, this activation maintains peripheral sensitization and chronic pain by increasing TRPV1 expression19,20. In parallel, peripheral TNF and IL-1β have been strongly implicated in the pathogenesis of inflammatory and neuropathic pain21–25.
Table 1.
Comparison of inflammation, neurogenic inflammation, and neuroinflammation, with special focus on location, features, and role in pain.
| Inflammation | Neurogenic inflammation | Neuroinflammation | |
|---|---|---|---|
| Location | Peripheral tissues | Peripheral tissues | PNS: nerves, dorsal root and trigeminal ganglia |
| Skin, muscle | Especially skin tissue | CNS: spinal cord, brain | |
| Internal organs except brain | |||
|
| |||
| Features | Disruption of vasculature and edema | Activation of C-fibers | Disruption of BBB, infiltration of immune cells |
| Infiltration of immune cells | Release of neuropeptides | Activation of peripheral & central glial cells | |
| Production of inflammatory mediators | Edema | Production of inflammatory cytokines | |
|
| |||
| Role in pain | Induction and resolution of acute pain | Induction of pain | Transition from acute to chronic pain |
| Transition from acute to chronic pain | Migraine, CRPS | Maintenance of chronic pain | |
Abbreviations: BBB, brain blood barrier; CNS, central nervous system, CRPS, complex regional pain syndrome; PNS, peripheral nervous system
Of note nociceptors and immune cells have bidirectional interactions26. Nociceptors not only listen to immune cells by responding to inflammatory mediators but also talk to immune cells and modulate the immune response to inflammation27,28. Like immune cells, nociceptors express cytokines, chemokines, and TLRs that are essential for immune modulation29–32. Release of cytokines and chemokines from nociceptors can rapidly regulate resident immune cells and attract circulating cells to the area of local inflammation that engage primary afferents and cell bodies in the nerve and DRG. For example, nociceptor-produced chemokine CCL2 regulates local macrophage activation in DRG after chemotherapy via TLR signaling, resulting in neuropathic pain32,33. Activation of nociceptors, especially C-fibers also produces neurogenic inflammation via releasing neuropeptides such as substance P and calcitonin gene related peptide (CGRP) or prostanoids27 (Table-1). Neurogenic inflammation occurs immediately following intradermal administration of capsaicin and mustard oil via respective activation of TRPA1 and TRPV1. Neurogenic inflammation results in rapid plasma extravasation and edema, even prior to the infiltration of immune cells. Neurogenic inflammation plays an important role in inflammatory diseases such as asthma and psoriasis but also contributes to pain conditions such as migraine and complex regional pain syndrome (CRPS) following bone fracture34. Nociceptors may differentially regulate inflammation in a context-dependent manner. For example, ablation of nociceptors decreases neurogenic inflammation but also enhances inflammation after bacterial infection by releasing CGRP35.
Peripheral inflammation with resulting persistent nociceptive input also leads to the increased release of neurotransmitters (glutamate, substance P, CGRP, BDNF) from the primary afferent central terminals in the spinal cord and trigeminal nucleus. Through signal transduction these neurotransmitters produce a state of neuronal hyperactivity and hyperexcitability in the spinal cord and brain known as central sensitization36,37. Activation of postsynaptic glutamate NMDA receptors and plasma cell membrane surface insertion of AMPA receptors are essential steps for the induction and maintenance of central sensitization6,38.
Neuroinflammation is associated with various insults that evoke painful sensations
A PubMed Search on September 30, 2017 using the keywords “neuroinflammation and pain” reveals a substantial increase in the number of publications in the last 10 years, jumping from 12 in 2008 to 150 in 2016. There is also a similar increase in the number of “microglia and pain” publications in last 10 years (Fig. 1). Neuroinflammation is a localized form of inflammation that occurs in the peripheral nervous system (PNS, including peripheral nerves and ganglia) and central nervous system (CNS, including the spinal cord and brain). Characteristic features of neuroinflammation are 1) vasculature changes that result in increased vascular permeability, 2) infiltration of leukocytes, 3) activation of glial cells, and 4) production of inflammatory mediators including cytokines and chemokines (Table 1). While the CNS is normally protected by the blood-brain-barrier (BBB), increased permeability of the BBB is an important feature of neuroinflammation, leading to increased leukocyte invasion to the CNS. It is becoming increasingly appreciated that neuroinflammation is a major cause of several neurological and neuropsychiatric diseases such as Alzheimer’s disease, Parkinson disease, multiple sclerosis, depression, bipolar disorder, and autism, as well as postoperative complications like neurocognitive disorders (e.g., delirium)39–41.
Figure 1.
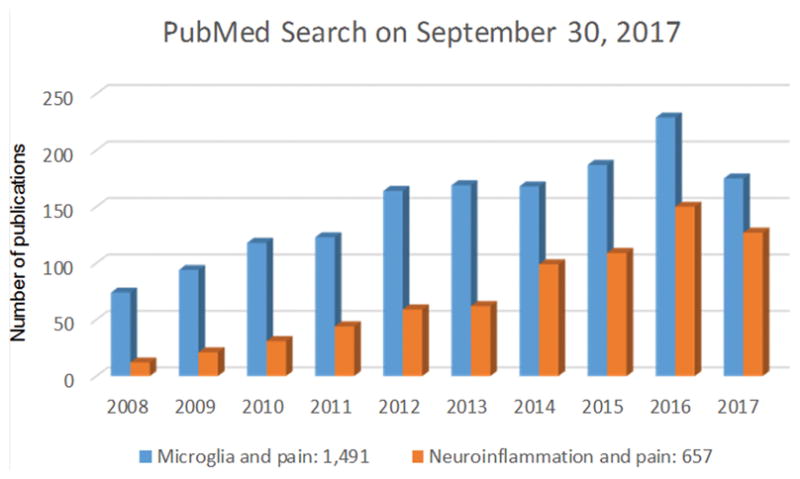
A PubMed Search on September 30, 2017 shows the number of publications on “neuroinflammation and pain” and “microglia and pain” in last 10 years. The numbers on the bottom show all time publications in PubMed.
It is noteworthy that neuroinflammation is associated with various painful insults and pathologies (Fig. 2)39, including but not limited to trauma such as traumatic brain injury (TBI), stroke, spinal cord injury (SCI), major surgeries including both cardiac and non-cardiac procedures (e.g., amputation, thoracotomy, and mastectomy), and autoimmune diseases (rheumatoid arthritis and multiple sclerosis). Other painful conditions, such as osteoarthritis, peripheral cancers (e.g., bone cancers), and viral infections (e.g., shingles/herpes zoster by varicella-zoster virus) also induce neuroinflammation in the PNS and CNS26,42. Furthermore, painful neuroinflammation (e.g., glial activation) can be induced by chemotherapy drugs such as paclitaxel, anti-HIV treatment, and chronic opioid treatment43–46. Immune therapies are increasingly appreciated for treating cancers by boosting the activities of immune cells such as T cells and other immune cells47,48. These therapies also change pain sensitivity by modulating inflammation and neuroinflammation. Deficiency of the negative immune regulator B7-H1, also called program death ligand 1 (PD-L1), enhances inflammation and neuropathic pain after chronic constriction injury of mouse sciatic nerve49. Notably, PD-1, the receptor for PD-L1, is not only present in immune cells but also expressed in nociceptor neurons of mouse and human DRG. Furthermore, PD-L1 can potently suppress nociceptor activities via regulating sodium and potassium ion channels in mouse and human nociceptors50. Many of these painful insults involve brain and spinal trauma, and nerve injury resulting from major surgeries, drug treatments, and diabetic neuropathy in neuropathic pain conditions. Neurogenic inflammation of the skin and joints is not only induced by activation of peripheral C-fibers but may also be triggered by the dorsal root reflex in the spinal cord following either orthograde or anterograde neuronal activation, which permits peripheral inflammatory responses to occur via local insults or by CNS “top-down” activation of primary afferents51,52. Furthermore, neuroinflammation in the spinal cord and brain can also be neurogenic following neuronal activation in the CNS53.
Figure 2.

Neuroinflammation is associated with various insults that evoke painful sensations. These insults include but not limited to trauma, major surgeries, drug treatments, autoimmune disease conditions, and other painful insults and tissue damage. Some of these insults such as major surgeries (breast surgery, amputation, thoracotomy), chemotherapy, and anti-viral treatment will cause nerve injury, as highlighted in red. Others will cause immune activation (highlighted in purple) and tissue injury (highlighted in light blue). Neuroinflammation results in several adverse effects, such as chronic pain and neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson disease (PD), multiple sclerosis (MS), and stroke. Neuroinflammation is also associated with chronic overlapping pain conditions. After priming of nociceptive circuit by previous injury, stress, or existing genomic, environmental, and psychological factors, acute insult may cause transition from acute pain to chronic pain.
While it is widely believed that chronic pain persists after the observable signs and symptoms of inflammation have resolved54, our understanding of neuroinflammation is changing this perspective. We now recognize that neuroinflammation is associated with and perhaps mediates the persistence and chronification of human pain conditions. Given the close proximity to pain neurocircuit, mediators of neuroinflammation are highly effective in modulating pain sensitivity. Especially, inflammation and neuroinflammation are differentially correlated with chronic pain. For example, HIV-infected patients with neuropathic pain show permanent neuroinflammation in the spinal cord55. Patients with fibromyalgia, a chronic widespread musculoskeletal pain condition56, also exhibit small fiber neuropathy together with chronic neuroninflammation57. The degree to which acute and chronic pain perception is mediated by the same cast of neuroinflammatory mediators is an interesting and open question.
It is noteworthy that a recent study by Clark and co-workers has shown a distinct role of inflammation and central neuroinflammation in acute versus chronic phase of pain behavior in a rat model of complex regional pain syndrome (CRPS)58. While peripheral IL-1β levels only increased at 4 weeks, spinal levels of IL-1β increased at both 4 weeks (acute phase) and 16 weeks (chronic phase). Importantly, systemic administration of anakinra, a peripherally restricted IL-1 receptor antagonist, only inhibited nociceptive behaviors at 4 weeks but not 16 weeks, but intrathecal injection of anakinra reduced nociceptive behaviors at both 4 and 16 weeks58. This study supports a critical role of central neuroinflammation in maintaining chronic pain in a rodent model of CRPS.
Neuroinflammation in chronic overlapping pain conditions and widespread pain
Emerging evidence suggests that neuroinflammation contributes to the pathophysiology of co-prevalent or co-existing chronic pain conditions which are referred to as chronic overlapping pain conditions (COPCs) which include but are not limited to fibromyalgia, headache, temporomandibular disorder, back pain, irritable bowel syndrome, primary headaches, pelvic pain, and vestibulodynia. COPCs are characterized by symptoms consistent with the dysregulation of sensory, inflammatory, and psychological domains13,56,59,60. There is increased evidence that patients with COPCs exhibit increased basal and stress-induced levels of catecholamines (epinephrine and norepinephrine) in circulation61–64 and reduced activity of catechol-O-methyltransferase (COMT)65,66, a ubiquitously expressed enzyme that metabolizes catecholamines67. COPC patients have functional variants in the COMT gene that result in reduced COMT activity68–72. The ‘low COMT activity’ variants are associated with increased fibromyalgia73–77 and temporomandibular disorder78 onset and increased pain in response to experimental stimuli78,79 and stressful events71,80,81. Consistent with clinical syndromes, in rodents sustained delivery of the COMT inhibitor OR486 results in pain at multiple body sites that persists for weeks and altered pain- and anxiety-related volitional behaviors68,82–84. Persistent COMT-dependent pain is initiated by peripheral β2- and β3-adrenergic receptors (β2- and β3ARs) through release of nitric oxide, TNFα, IL-1β, IL-6, and CCL2 in plasma and maintained by increased TNF in central tissues68,84–86. Higher levels of nitric oxide derivatives (e.g. nitrite and nitrate) and pro-inflammatory cytokines have been found in patients with chronic musculoskeletal pain conditions13,87–91. Of note, patients with site-specific pain conditions (e.g., localized temporomandibular disorder or localized vestibulodynia) exhibit a balance in pro- and anti-inflammatory cytokines, whereas those with COPCs fail to exhibit a compensatory increase in anti-inflammatory cytokines. Compared to patients with localized pain, those with COPCs also exhibit dysregulation in microRNAs (e.g., miR-let-7f) that augment immune response and pro-inflammatory cytokine production60. Together, these findings suggest that localized and anatomically widespread patterns of chronic pain are associated with distinct inflammatory profiles.
Widespread patterns of chronic pain exhibited by patients with COPCs may be attributed to prior injury or stressful events that produce long-lasting changes in nociceptor function, a phenomenon known as hyperalgesic priming. Pre-clinical studies have demonstrated that acute painful inflammation or nerve injury as well as non-painful stress ‘primes’ nociceptors so that they respond to a subsequent insult (e.g., PGE2 or epinephrine) for a prolonged duration due to activation of distinct kinase and gene transcription pathways4,92–95. In line with these findings, individuals with the ‘low activity’ COMT genotype report enhanced and prolonged pain after motor vehicle collisions or psychological strain71,80,96, which are events that stimulate the sympathetic release of epinephrine known to sensitize nociceptors97,98 and promote inflammation99–107. Thus, repeated environmental exposures to injury, inflammation, and stress in genetically-predisposed individuals results in the transition from acute pain to chronic pain, in part through neuroinflammation (Fig. 2). Of note, when a challenge of lipopolysaccharide (LPS) is preceded by a surgical procedure, it has been shown that this early priming of spinal microglial cells increases the duration and intensity of pain through contributions to neuroinflammation108.
Glial activation as a primary feature of neuroinflammation and a driver of chronic pain
Glial activation, gliosis, and gliopathy after painful injuries
Neuroinflammation is characterized by activation of peripheral glia including Schwann cells in the nerve and satellite glial cells in the DRG and trigeminal ganglia, and central glia including microglia, astrocytes, and oligodendrocytes in the spinal cord and brain109,110. In this review we focus on central glia, especially microglia and astrocytes and the mechanisms by which glia-produced mediators modulate synaptic plasticity and central sensitization. The previous decade has seen an exponential increase in literature documenting the role of microglia and astrocytes in the pathogenesis of chronic pain, and “glial activation” is emerging as a powerful mechanism underlying pathogenesis of chronic pain111–117, and chronic pain may also manifest as a “gliopathy”110.
After painful injuries, there are different activation states of microglia and astrocytes. The morphological activation (glial reaction or gliosis) is the most studied activation state, although this type of glial activation may not directly cause pain110. Glial reaction is characterized by increased expression of microglial markers such as CD11b, IBA1, and CX3CR1 and astroglial markers GFAP (glial fibrillary acid protein), as well as morphological changes such as hypertrophy or process retraction/extension of microglia and astrocytes. Microglial reaction in the spinal cord is very rapid and dramatic, whereas astrocyte reaction in the spinal cord is more persistent and occurs in more painful conditions (Fig. 3, A–F) 116,118. Subcutaneous formalin injection into a hind paw of rat or mouse is probably the most used animal model of inflammatory pain, which lasts for less than one hour. Of interest, in 1999 Maixner and coworkers showed that subcutaneous formalin also caused a robust microglial reaction (labeled with CD11b) in the spinal cord days after the injection. This microglial reaction from formalin-induced nerve injury is associated with the development of mechanical allodynia119. This is one of the earliest reports to support a possible role of spinal microglia in pain regulation. Functional imaging also reveals glial activation in patients with chronic pain120. In 2003, Eisenach and coworkers showed that incision and nerve injury causes upregulation of cyclooxygenase-1 (COX-1) in spinal glial cells, which is important for the development of postoperative pain and neuropathic pain121,122.
Figure 3.

Distinct and time-dependent activation of microglia and astrocytes in the spinal cord after nerve injury. (A, B) Microglia activation revealed by increased CX3CR1 expression in the spinal cord 10 days (A) and 21 days (B) after nerve injury in Cx3cr1-GFP mice. Scale, 100 μm. (C) Phosphorylation of p38 MAP kinase (P-p38) in CD11b+ microglia in the spinal cord dorsal horn 7 days after nerve injury. Scale, 20 μm. (D, E) Astroctye activation revealed by increased GFAP expression in the spinal cord 10 days (D) and 21 days (E) after nerve injury in mice. Scale, 100 μm. (F) Expression of Cx43 in GFAP+ astrocytes in the spinal cord dorsal horn 21 days after nerve injury. Scale, 20 μm. All the images have not been published.
Signaling mechanisms in glial regulation of allodynia and hyperalgesia
Several neuromodulators such as ATP, chemokines (CX3CL1, CCL2, CXCL13), and neuropeptides (SP and CGRP), as well as colony-stimulating factor 1 (CSF1) are involved in glial activation after painful insults110,123,124 (Table-2). The up-regulations of glia-specific receptors and channels are functionally correlated with pain hypersensitivity. ATP modulates glial activation via stimulation of ionotropic P2X receptors and metabotropic P2Y receptors125,126. Peripheral nerve injury increases the expression of ATP P2Y receptors (P2X4, P2X7, and P2Y12) in spinal microglia, and each up-regulation was implicated in neuropathic pain sensitization (mechanical allodynia) 127,128. Accumulating evidence suggests that after tissue and nerve injury ATP is generated from different cell types including astrocytes, neurons, and microglia. Astrocyte-expressing hemichannels connexin-43 are permeable to ATP129. Glucocorticoids induce ATP release from spinal astrocytes, leading to microglial activation and diurnal exacerbation of allodynia130. Vesicular nucleotide transporter regulates ATP release from spinal cord neurons after nerve injury131. Microglial pannexin-1 channel was also shown to facilitate ATP release in neuropathic pain132. Nerve injury results in cleavage and activation of chemokine CX3CL1/fractalkine by protease cathepsin S, leading to microglia activation through stimulation of CX3CR1 receptor115. CX3CR1, one of best-known markers of microglia, is strongly up-regulated after nerve injury, as revealed in Cx3cr1-GFP mice (Fig. 3A,B). This microglial receptor is also critical for the development of neuropathic pain symptoms, since mechanical allodynia after nerve injury is reduced after spinal administration of the CX3CR1 neutralizing antibody and abrogated in Cx3cr1 knockout mice115,133,134. Following chronic constriction injury (CCI) of the sciatic nerve, CX3CR1 up-regulation peaks within 10 days. In the late phase of mechanical allodynia (>3 weeks), microglial CX3CR1 expression markedly declines (Fig. 3A,B). Microglia may play a role in maintaining persistent hyperalgesia and allodynia after bone cancer135. Orthopedic surgery such as bone fracture also results in nerve injury and microglial activation in the spinal cord. It was proposed that spinal microglial activation also contribute to postoperative cognitive dysfunction such as delirium136. Substance P signaling from C-fiber afferent terminals in the spinal cord results in microglia activation and central sensitization after bone fracture137.
Table 2.
Signal transduction in spinal cord microglia and astrocytes after tissue and nerve injury.
Painful tissue and nerve injuries induce release of glial activators, which in turn bind their respective receptors on microglia and astrocytes. Upon activation, the glial receptors cause intracellular signal transduction and activation of protein kinases (phosphorylation of MAP kinase and SRC kinase), leading to increased synthesis and release of glial mediators that can produce central sensitization and hyperalgesia and allodynia.
| Microglia | Astrocytes |
|---|---|
| Activators | Activators |
| ATP | ATP |
| CX3CL1 | TNF |
| CSF1 | CXCL13 |
| LPS/HMGB1 | LPS/HMGB1 |
| CASP6 | MMP-2 |
|
| |
| Receptors | Receptors |
| P2X4, P2X7, P2Y12 | P2X/P2Y |
| CX3CR1 | TNFR1 |
| CSF1R | CXCR5 |
| TLR4 | TLR4 |
| C5aR | Cx43 |
|
| |
| Intracellular signaling | Intracellular signaling |
| P-P38, P-ERK, P-SRC | P-JNK, P-ERK |
|
| |
| Mediators | Mediators |
| TNF | CCL2, CXCL1 |
| IL-1β, IL-18 | TSP1, TSP4 |
| BDNF | bFGF, IL-1β |
Abbreviations: bFGF-2, basic fibroblast growth factor 2; CASP6, caspase 6, CSF1; colony stimulating factor 1; CXCL13, chemokine (C-X-C motif) ligand 13, Cx43, connexin-43; C5aR, complement C5 receptor; HMGB1, high motility group box protein 1; LPS, lipopolysaccharide; MMP-2, matrix metalloprotease-2; TLR4, toll-like receptor 4; TNF, tumor necrosis factor (alpha); TNFR, TNF receptor; TSP-1, thromspondin-1; TSP-4, thromspondin-4.
After painful insults, gliopathy is also characterized by dysfunction of astrocytes, such as downregulations of the glutamate transporters (GLT-1 and GLAST) in spinal cord astrocytes, resulting in glutamate accumulation in synaptic clefts that causes neuronal hyperactivity110,138. Connexin-43 (Cx43) is a critical astrocytic signaling molecule that controls the release of astroglial mediators including glutamate and ATP139. Of note, Cx43 upregulation in spinal cord astrocytes is sustained after spinal cord injury and nerve injury (Fig. 3F) and contributory to the development and maintenance of mechanical allodynia140,141. Additionally, chronic pain-associated gliopathy could manifest as a functional switch of Cx43 from gap junction communication to hemichannel regulation, so that astrocytes become “leaky” during this switch, resulting in increased secretion of cytokines (IL-1β) and chemokines (CCL2, CXCL1)141,142. Chemokines regulate bidirectional interactions of neurons and glial cells143. Astrocytes not only produce chemokines that can “talk to” neurons by modulating neuronal activity but also “listen to” neurons by responding to chemokines (e.g., CXCL13) derived from neurons124. Astrocytes also release thrombospondin 4 (TSP4) to modulate synapse formation, synaptic plasticity, and behavioral hypersensitivity 144(Table-2).
A critical step of glial activation in persistent pain is the activation of intracellular signaling pathways especially the mitogen-activated protein kinase (MAPK) pathways. There are three major members in the MAPK family: ERK (extracellular signal-regulated kinase 1 and 2), p38, and JNK (c-Jun N-terminal kinase) is a 145. Phosphorylation of p38 (P-p38) in spinal microglia occurs in different pain conditions after surgery, nerve injury (Fig. 3C), and opioid tolerance and results in increased synthesis and release of microglial mediators (TNF, IL-1β, and BDNF) and pain hypersensitivity134,135,146–149. Inflammation or nerve injury also activates JNK in astrocytes 150,151, leading to increased secretion of CCL2 and CXCL1 and enhanced pain states141,152. Nerve injury also causes sequential activation of ERK in microglia (early phase) and astrocytes (late phase)145, indicating distinct involvement of these two glial cell types in chronic pain induction and maintenance.
Glia activation in the brain after painful injuries
Accumulating evidence also suggests a role of glial activation, revealed in different brain regions, in regulating neuroinflammation and pain sensitivity. Sciatic nerve ligation induces astrocyte activation in the S1 sensory cortex, which is associated with upregulation of metabotropic glutamate receptor 5. Activation of this glutamate receptor subtype in astroglia induces spontaneous somatic Ca2+ transients and secretion of thrombospondin 1 (TSP1) from astrocytes, leading to new synapse formation and mechanical allodynia153. Toll-like receptor 4 (TLR4) plays a critical role in glial activation and neuroinflammation in the spinal cord, as well as hyperalgesia and allodynia154,155. TLR4 also contributes to neuroinflammation in the prefrontal cortex and visceral pain after chronic stress. Increased expression of TLR4 is associated enhanced glia activation in the prefrontal cortex and increased levels of proinflammatory cytokines. Administration of a TLR4 specific antagonist TAK-242 in the prefrontal cortex is sufficient to attenuate visceral hypersensitivity156. Peripheral nerve injury causes microglia activation within the mesolimbic reward circuitry, leading to a disruption of dopaminergic signaling and reward behavior157. Furthermore, nerve injury causes activation of microglia and astrocytes in the anterior cingulate cortex, and administration of microglial inhibitor minocycline in this brain region inhibited mechanical allodynia 153. However, earlier studies showed that nerve injury did not cause microgliosis in the anterior cingulate cortex and that long-term synaptic plasticity (long-term potentiation) was not altered by minocycline158,159. Future studies are warranted to investigate how microglia and astrocytes regulate different forms of synaptic plasticity in different brain regions following painful insults in the peripheral tissues and the central nervous system.
Sex dimorphism in glial regulation of allodynia and hyperalgesia
Chronic pain such as chronic orofacial pain associated with TMD occurs more frequently in women160,161. In 1993 Maixner and coworker reported gender differences in pain and cardiovascular responses to forearm ischemia in humans162. Paradoxically, the majority, if not all, pain-related studies were conducted in male animals 163. However, it was fortunate to test males because spinal microglia play little or no role in regulating inflammatory and neuropathic pain primarily in female rodents164–167 (Fig 4). Mogil and coworkers demonstrated that spinal TLR4, an important receptor for microglia activation, regulates hyperalgesia and allodynia resulting from inflammation or nerve injury exclusively in male mice164. Of note, morphological activation and proliferation of spinal microglia are identical in males and females after nerve injury. Nerve injury-evoked mechanical allodynia is also equivalent in both sexes during the tested times165,166. However, mechanical allodynia after nerve injury was exclusively attenuated in male mice after intrathecal injection of microglial inhibitor (minocycline), microglial toxin, or P2X4 blocker, or following special deletion of Bdnf in microglia165. Furthermore, nerve injury activates p38 in spinal microglia of male but not female mice; and in agreement, spinal administration of p38 inhibitor reduces neuropathic pain only in male mice166. Caspase-6 is a microglial activator and released from axonal terminal in the spinal cord following tissue and nerve injury, which can act on microglia to release TNF168. Sex dimorphism was also revealed in caspase-6-mediated microglial signaling, and caspase-6 regulates neuropathic pain exclusively in males167. It appears some of male-specific microglial responses require testosterone, as minocycline reduces allodynia in testosterone-treated females but not in castrated males. In female rodents, the role of microglia in neuropathic pain appears to be replaced by T cells165.
Figure 4.

Schematic illustration of local and remote central sensitization induced by glial activation and neuroinflammation in the spinal cord. Activation of spinal microglia and astrocytes by painful insults results in secretion of glial mediators such as TNF, IL-1β, CCL2, CXCL1, BDNF, D-serine, which can act as neuromodulators to induce local central sensitization in surrounding excitatory synapses (facilitation) and inhibitory synapses (dis-inhibition). During neuroinflammation these glial mediators are also present in the CSF and affect synapses in different spinal segments to cause remote central sensitization and extra-territorial and widespread pain beyond the initial injury site.
Central sensitization controls augmentation and spread of pain hypersensitivity
Term development
Central sensitization is a powerful phenomenon in the pain field. As a key mechanism of chronic pain, it also guides clinical treatment for conditions associated with widespread pain169,170. In this review, we highlight the role of glial cells and neuroinflammation in promoting central sensitization and widespread chronic pain. In 1983 Woolf presented evidence for a central component of post-injury pain hypersensitivity36. The International Association for the Study of Pain (IASP) describes central sensitization as increased responsiveness of nociceptive neurons in the central nervous system to their normal or subthreshold afferent input171. Central sensitization may also include conditions like increased central responsiveness due to dysfunction of endogenous pain control systems, regardless if there is functional change of peripheral neurons. In 2003, Ji and Woolf defined central sensitization as the increased synaptic efficacy established in somatosensory neurons in the dorsal horn of the spinal cord following intense peripheral noxious stimuli, tissue injury, or nerve damage. This heightened synaptic transmission results in reduction in pain threshold, an amplification of pain responses and a spread of pain sensitivity to non-injured areas6. In 2009, Latremoliere and Woolf described central sensitization as an enhancement in the function of neurons and circuits in nociceptive pathways caused by increases in membrane excitability and synaptic efficacy as well as to reduced inhibition and is a manifestation of the remarkable plasticity of the somatosensory nervous system in response to activity, inflammation, and neural injury. In this review, the authors highlighted disinhibition (reduced inhibition) and also emphasized that the net effect of central sensitization is to recruit previously subthreshold synaptic inputs to nociceptive neurons, generating an increased or augmented action potential output: a state of facilitation, potentiation, augmentation, or amplification38.
Mechanisms of central sensitization
As shown in Fig. 5, activation of NMDA receptor (NMDAR) is an essential step in initiating and maintaining the central sensitization and pain hypersensitivity after tissue and nerve injury172,173. Glutamate is a primary excitatory neurotransmitter in the pain pathway. Under normal circumstances NMDAR channel is blocked by Mg2+ ions, but this blockade is removed by membrane de-polarization following activation of nociceptive primary afferents. Activation of NMDAR boosts synaptic efficacy and causes Ca2+ influx, which can activate intracellular signaling pathways that initiate and maintain central sensitization6,38. In particular, tissue and nerve injury increases the expression of the NMDAR-NR2B (GluN2B) subunit, which regulates spinal synaptic plasticity in persistent pain conditions together with NR1 subunit174. Interestingly, NR2B/GluN2B receptor activity and surface expression in spinal cord dorsal horn (SCDH) neurons is negatively regulated by β-arrestin 2175, a scaffold protein that was traditionally known as an inhibitor of GPCRs. Deficiency of β-arrestin 2 results in enhanced acute opioid analgesia, produced by morphine and DAMGO, a selective μ opioid receptor agonist175,176. Paradoxically, DAMGO-induced hyperalgesia is also potentiated after β-arrestin 2 deficiency, as a result of enhanced surface and synapse expression of GluN2B that results in hyperactivity of the receptor. Loss of β-arrestin 2 also leads to a prolongation of inflammatory and neuropathic pain, as these pain conditions critically depend on GluN2B175. It appears that for the resolution of persistent pain, it is more important for β-arrestin 2 to regulate NMDA receptors via ERK signaling pathway (Fig. 5). Furthermore, surface trafficking of AMPA receptor (AMPAR), especially calcium-permeable subunit (GluR1/GluR-A), play a critical role in spinal cord synaptic plasticity and pain hypersensitivity after tissue injury177,178. The scaffold protein Homer1a operates in a negative feedback loop to regulate the calcium signaling and excitability of the spinal cord pain pathway in an activity-dependent manner 179. Given a critical role of AMPAR in direct control of excitatory synaptic transmission in the pain circuits, NMDAR-independent central sensitization may also exist.
Figure 5.

Molecular mechanisms of central sensitization in first-order excitatory synapses in the spinal cord dorsal horn pain circuit and induction of central sensitization by proinflammatory cytokines and chemokines (e.g., TNF, IL-1β, CCL2, CXCL1) that are produced by glial cells. At presynaptic sites, i.e. central terminals of nociceptive primary afferents, activation of receptors of cytokine and chemokine receptors results in phosphorylation and activation of ERK and p38 (P-ERK, P-p38), leading to glutamate (Glu) release from synaptic vesicles, via activation of ion channels TRPV1, Nav1.7, and Nav1.8. At postsynaptic sties, increased release of neurotransmitters (e.g., Glu) also induces P-ERK, which can induce central sensitization by positive modulation of NMDA receptor (NMDAR, Step-1). AMPA receptor (AMPAR, Step-2) and negative modulation of potassium channel subunit Kv4.2 (Step-3). P-ERK also maintains central sensitization via inducing CREB phosphorylation (P-CREB, Step-4). Opioids such as morphine inhibit neurotransmitter release via mu opioid receptors (MOR) and N-type calcium channels. The scaffold protein β-arrestin-2 (βarr2) inhibits MOR signaling by desensitization and degradation of GPCRs, leading to enhanced acute opioid analgesia in βarr2 knockout mice. Paradoxically, βarr2 also inhibits NMDAR and ERK signaling, leading to a transition from acute pain to chronic pain175.
Activation of intracellular pathways by protein kinases, such as protein kinase A (PKA), protein kinase C (PKC), Ca2+/calmodulin-dependent kinase-II (CaMKII), Src (a tyrosine kinase encoded by sacoma oncogene), and extracellular signal-regulated kinases (ERK, including ERK1 and ERK2) are important for the generation of central sensitization6,37,180. Notably, activation of ERK via phosphorylation of ERK (pERK) in spinal cord dorsal horn (SCDH) neurons is nociceptive-specific and serves as a marker of central sensitization181–183. pERK is a common pathway following the activation of various ionotropic and metabotropic receptors and protein kinases (PKA, PKC, and Src) and also a downstream event of Ca2+ signaling184–186. pERK induces central sensitization via rapid post-translational regulation, such as suppression of potassium channel Kv4.2 activity, leading to hyperactivity of SCDH187,188. pERK also contributes to rapid upregulation of NMDAR (GluN2B) function in SCDH neurons in response to inflammatory mediators152,189. Furthermore, translocation of pERK to the nuclei of SCDH neurons activates the transcription factor cAMP response element binding protein (CREB), leading to increased expression of pronociceptive genes encoding for c-Fos, NK-1, and prodynorphin184,190.
Does central sensitization require peripheral input?
Historically, it has been believed that the central sensitization to noxious stimuli requires a sustained, intense and repeated applications of the stimulus. More recently, it has become apparent that persistent peripheral nociceptive input may not be required to elicit central sensitization, because central sensitization can result from changes in the properties of neurons in the central nervous system that appear to be independent of peripheral input38. Central sensitization produces pain hypersensitivity by changing the sensory responses elicited by normal inputs, including subthreshold innocuous tactile stimulation. For example, spinal cord disinhibition by intrathecal injection of GABA and glycine receptor antagonists is sufficient to induce central sensitization and mechanical allodynia via disinhibition of inhibitory signaling and subsequent activation of excitatory signaling that is mediated by NMDAR191. Dis-inhibition of GABAergic and glycinergic synaptic transmission in the spinal cord pain circuitry is critical to the generation of chronic pain86–89. Gate control theory describes a tonic inhibition in the spinal cord pain circuit via inhibitory neurons192. A feed-forward spinal cord glycinergic neural circuit in the laminae II-III dorsal horn gates mechanical allodynia, and nerve injury impairs glycinergic synaptic transmission and opens the gate to elicit mechanical allodynia86.
It is noteworthy that some central etiologies/injuries such as spinal cord injury, traumatic brain injury, multiple sclerosis may cause neuroinflammation, central sensitization and chronic pain without a peripheral insult193 (Fig. 2). Therefore, it is important for future studies to examine neuroinflammation in the brain, including those regions that do not receive input from primary afferents. Spinal cord injury is sufficient to produce hyperexcitability in primary sensory neurons194, via possible retrograde signaling from the dorsal root reflex. Neuroinflammation in the spinal cord may also regulate gene expression of primary sensory neurons via diffusible inflammatory mediators that can reach to DRGs. Thus, there could be bi-directional interactions between peripheral sensitization and central sensitization. Central sensitization is not only secondary to peripheral sensitization but may, in turn, regulate peripheral sensitization (Fig. 4).
Neuroinflammation drives central sensitization and widespread pain via glia-produced cytokines and chemokines
An important step forward in revealing the role of central sensitization in widespread chronic pain is to demonstrate direct involvement of cytokines and chemokines (small cytokines) in the induction and maintenance of central sensitization152,195–197. In 2001 Samad et al. showed that spinal IL-1β contributes to central sensitization and inflammatory pain hypersensitivity via transcriptional regulation that causes up-regulations of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2)195. Notably, unilateral inflammation in the hindpaw of rats caused widespread and bilateral increase of COX-2 in the spinal cord and brain, due to possible increase of IL-1β level in the CSF195. This may partially explain widespread pain in some chronic pain conditions (Fig. 4). However, contribution of the spinal COX/PGE2 pathway to central sensitization is not supported by a clinical trial in post-operative pain198. Despite a critical contribution of PGE2 to peripheral sensitization, the involvement of this important inflammatory mediator in regulating central sensitization is not well studied but see199. In 2008, Kawasaki et al. demonstrated a direct induction of central sensitization by the pro-inflammatory cytokines TNF, IL-1β, and IL-6. These cytokines elicit very rapid increases (within 1 minute) in excitatory synaptic transmission on spinal cord neurons196. In support of this direct modulation, TNF, IL-1β, and IL-6 rapidly modulate the function of neurotransmitter receptors such as AMPAR, NMDAR, GlyR, and GABAR, which results in enhanced excitatory synaptic transmission and suppressed inhibitory synaptic transmission in the spinal pain circuit196. In agreement with this finding, intrathecal injection of TNF, IL-1β, or IL-6 elicits rapid pain hypersensitivity in naive animals196. Because these cytokines are elevated and circulating in the CSF in chronic pain conditions200,201, they are possible mediators of widespread pain, as a result of widespread central sensitization (Fig. 4).
TNF can be produced by microglia, astrocytes, and even DRG primary sensory neurons202,203. However, in the spinal cord TNF is primarily produced by microglia, as indicated by single-cell analysis in microglia, astrocytes, and neurons168. Electrophysiological analysis reveals that TNF increases glutamate release in TRPV1+ C-fiber terminals, leading to enhanced excitatory synaptic transmission in lamina IIo excitatory SCDH interneurons204. These lamina IIo interneurons synapse to lamina I projection neurons to form a pain circuit and potentiate pathological pain 85, 86. TNF also increases NMDA currents in IIo excitatory interneurons via ERK activation189. Additionally, TNF inhibits spontaneous action potentials in GABAergic neurons in the dorsal horn205. Both type I and type II rector of TNF (TNFR1 and TNFR2) are involved in behavioral manifestations of central sensitization following intrathecal TNF treatment or during formalin-induced 2nd phase pain206. Notably, caspase-6 triggers TNF release from microglia to elicit central sensitization via TNF receptor signaling168.
IL-1β is expressed by both microglia and astrocytes in the spinal cord 112,149,207. IL-18, a highly-related family member of IL-1β, is induced in microglia by nerve injury and bone cancer135,208. Caspase-1 cleaves and activates IL-1β and IL-18 and acts as a key component of inflammasome, which contributes to the pathogenesis of chronic pain209. In addition, matrix metalloproteases MMP-9 and MMP-2 have been implicated in IL-1β cleavage and activation and regulation of glial activation in early vs. late-phase of neuropathic pain210. IL-1β induces central sensitization via both presynaptic modulation (increasing glutamate release)196 and post-synaptic regulation (phosphorylation of NMDAR211,212 and enhancement of NMDA current196). Endogenous IL-1β also potentiates presynaptic NMDAR function in neuropathic pain213. Furthermore, IL-1β suppresses inhibitory synaptic transmission (IPSCs) and GABA and glycine-induced currents in spinal lamina IIo neurons196. IL-18 also causes hyperactivity of spinal wide dynamic range (WDR) neurons following mechanical stimuli in vivo135. Thus, cytokines regulate central sensitization through multiple mechanisms that involve presynaptic and post-synaptic modulation as well as excitatory and inhibitory synaptic transmission modulation.
Astrocyte-produced chemokines CCL2 and CXCL1 also play an important role in central sensitization and chronic pain141,152,214. CCR2, the major receptor for CCL2, is expressed in neurons including DRG and SCDH neurons152,215,216. Bath application of CCL2 to spinal cord slices induces rapid ERK activation and causes ERK-dependent potentiation of NMDA currents in dorsal horn neurons via the activation of CCR2 receptors152,216. Intrathecal injection of CXCL1 induced ERK and CREB activation in spinal neurons via the stimulation of CXCR2 receptors214, which is epigenetically regulated after injury217. Notably, late-phase neuropathic pain and synaptic plasticity (EPSC increase) in the spinal cord pain circuit, 3 weeks after nerve injury, is transiently reversed by the CXCR2 antagonist SB225002. These results suggest an active role of CXCL1/CXCR2 in the maintenance of central sensitization141.
Glial cells also produce growth factors such as brain-derived growth factor (BDNF) and basic fibroblast growth factor (bFGF, also called FGF-2) to enhance central sensitization and chronic pain218,219. BDNF release from central terminals of primary afferents elicits central sensitization via activation of ERK and potentiation of NMDAR184,220–222. BDNF signaling in spinal microglia also facilitates central sensitization, neuropathic pain, and morphine tolerance218,223. Exposure of spinal lamina I projection neurons to BDNF results in a depolarizing shift in the anion reversal potential, causing disinhibition of GABAergic system, a key regulatory mechanism in chronic pain218,223. BDNF also modulates excitatory synaptic transmission in SCDH lamina I neurons via activation of protein kinase Fyn180.
Long-term potentiation, central sensitization, and widespread pain
Spinal cord LTP (sLTP) is an important form of synaptic plasticity and a unique form of central sensitization in chronic pain224,225. sLTP of C-fiber-evoked field potentials is typically induced by high-frequency tetanic stimulation of the sciatic nerve226. sLTP is also induced by nerve injury and opioid withdrawal227–229. There are striking similarities between sLTP and central sensitization, and both show the critical requirements of NMDAR and involvement of key signaling transduction pathways including the PKC, ERK, and Src, as well as dependence of protein synthesis and gene transcription38,181,230. However, maintenance of late phase LTP (>4 h) may require additional mechanisms226. It remains to be determined if there is persistent spinal LTP months after painful insults, due to technical difficulty to follow LTP over time.
Several lines of evidence suggest an essential role of neuroinflammation in inducing and sustaining sLTP. First, spinal TNF is both sufficient and required for the induction of sLTP via both TNFR1 and TNFR2204,231. Caspase-6 also contributes to the induction and maintenance of sLTP via TNF-signaling168. Second, IL-1β triggers sLTP not only in excitatory synapses232 but also in glycinergic synapses on GABAergic neurons in spinal cord slices233, serving as another example of cytokine-induced disinhibition. Third, activation of spinal microglial CX3CR1 via CX3CL1/fractalkine is sufficient to elicit sLTP 234. Finally, the chemokine receptor CCR2 is required for the maintenance of sLTP216.
Recently, Sandkuhler and coworkers demonstrated a gliogenic sLTP that can spread widely in nociceptive pathways235. A fundamental feature of LTP induction in the brain is the requirement for coincident pre- and postsynaptic activity, which is important to restrict LTP expression to activated synapses only (homosynaptic LTP) as well as to define the input specificity of LTP. Gliogenic sLTP can travel long distances via CSF, because this LTP can be induced by glial activation and diffusible messengers, such as d-serine and TNF235. Strikingly, transfer of spinal CSF from a donor animal displaying LTP is able to induce LTP in a naïve receiver animal235. Therefore, this diffusible sLTP affects susceptible synapses at remote sites. Collectively, gliogenic LTP, as well as other forms of diffusible and glial-mediated central sensitization may underlie widespread pain in chronic pain patients, especially in those with overlapping chronic pain conditions (Fig. 6).
Figure 6.

Non-pharmacological approaches that can control neuroinflammation and produce multiple beneficial effects including prevention and resolution of chronic pain, prevention of neurodegeneration, and repair of cognitive function deficits. Abbreviations: DHA, docosahexaenoic acid; EA, electroacupuncture; EPA, eicosapentaenoic acid; VNS, vagus nerve stimulation; SPMs, specialized pro-resolution mediators; TENS, transcutaneous electrical nerve stimulation; TMS, transcranial magnetic stimulation.
Synaptic plasticity and LTP have also been investigated in the brain, such as anterior cingulate cortex236, amygdala237, and hippocampus238 after tissue and nerve injury. Further investigation is warranted to define the role of neuroinflammation and glial activation in these pain-associated synaptic changes in the brain.
Clinical trials and translational gap
The current levels of enthusiasm and interest in the development of new treatments that specifically target neuroinflammation and glial activation may be tempered by the somewhat disappointing results of prior clinical trials. Although animal models, as reviewed in the previous sections, demonstrated promising results in the reduction of neuropathic pain, glial activation, and neuroinflammation when treated by the glial modulator propentofylline239,240, the same compound failed to provide beneficial reductions in neuropathic pain when administered to patients suffering from post-herpetic neuralgia241. Despite its efficacy in reducing postoperative pain in animal model, intrathecal injection of cyclooxygenase inhibitor such as ketorolac did not improve acute or chronic pain after hip arthroplastry121,198. While the clinical trial does not support a central role of COX/PGE2 pathway in clinical pain, cytokines and chemokines can independently regulate central sensitization via direct actions on nociceptive neurons (Fig. 5). Thus, future clinical studies are still needed to test the effects of spinal inhibition of cytokines/chemokines in clinical pain. Neuropathic pain patients demonstrate tolerance to p38 inhibitors including dilmapimod and losmapimod242,243, but these drugs have inconsistent levels of efficacy. An exploratory trial of dilmapimod significantly inhibited nerve injury-induced neuropathic pain 242, however losmapimod did not demonstrate any significant analgesic effect in comparison to placebo controls243. Intriguingly, acute postsurgical dental pain was significantly reduced when treated with another p38 inhibitor, specifically the novel p38α MAPK inhibitor SCIO-469.244.
The use of cytokine inhibitors and glial modulators in clinical trials has shown some encouraging results. Intractable discogenic lower back pain patients showed up to 8 weeks of pain relief when given a single intradiscal treatment of entanercept, a TNF inhibitor 245. Additionally, lumbar disc herniation patients who received entanercept via transforaminal epidural injections had up to 26 weeks of pain relief following two injections in a randomized, double-blind, and placebo-controlled trial246. Another cytokine inhibitor, the IL-1 trap rilonacept, is well tolerated by patients, and in a proof-of-concept study, treatment with the drug showed pain relief for a small group of patients being treated for chronic refractory gouty arthritis247. By contrast, subcutaneous inhibition of IL-1β with anakinra has no beneficial effect on chronic fatigue syndrome CFS248. Microglial activation can be blocked in vitro by low doses of naltrexone 249, and a pilot trial also showed that treatment with the drug can help to reduce symptoms related to fibromyalgia.250.
Certain limitations and concerns regarding the design of the mentioned trials need to be addressed. The first concern regards lack of neuroinflammation analysis by biomarkers, as inhibition of in vivo glial responses in the propentyofylline study notably had no biomarker validation241. A second concern is the lack of validation regarding whether central sites, including the spinal cord, received exposure to systemically-administered drugs. For instance, it was noted by the authors of the losmapimod study, that the lack of response may be a result of central sites having a lack of adequate exposure to the drug 243. In the majority of preclinical studies, inhibitors of glial cells, MAP kinases, cytokines, and chemokines are given via intrathecal administration, which ensures direct exposure of central sites to the drug being studied110. The third concern is the absence of analysis of sex-dependent effects of treatments in previous studies of p38 and glial inhibitors in light of emerging evidence of sex-dimorphism in microglial and p38 signaling and T cell signaling in inflammatory and neuropathic pain165,166. Fourth, the timing of administration appears to be a critical component of a treatment’s efficacy. During the acute phase of pain, inhibitors of microglia, p38 MAP kinase, and TNF-α show stronger efficacy, with more partial effects in the chronic phase according to preclinical studies110,251. In line with this point, the effective results of the p38 inhibitor SCIO-46 trial were demonstrated in dental patients during the acute phase of postsurgical pain244. It should be noted however, that the intrathecal administration of the IL-1 receptor antagonist anakinra at 16 weeks post-bone fracture in rodents can reduce chronic postoperative pain58. Finally, the mechanisms of the tested drugs need to be further elucidated. Propentofylline, although a glial modulator, also acts as a phopsphodiesterase inhibitor as well as an adenosine uptake inhibitor. Thus, the administration of this drug may alter cAMP and adenosine levels in both glial and non-glial cells252.
Animal models are of exceeding importance in the understanding of the mechanisms of pain as well as testing for newly developed therapeutics. However, animal models cannot reproduce all key clinical symptoms of pain, and thus the gap in translation between preclinical studies in rodents and clinical studies in humans is a major topic in pain research discussed in numerous review articles253,254. With notion to methods of measurement, in animal studies pain is measured by application of von Frey filaments to cause reflex pain with subsequent quantification of paw withdrawal thresholds, whereas most clinical studies measure pain based on the visual pain scale. Additionally, the time course of pain is very different between animals and humans, with the duration of pain in preclinical studies lasting normally from days to weeks while on the other hand clinical patients with chronic pain suffer from their symptoms often from months to years. Thus, developing animal models that accurately reflect the various genetic, sex-dependent, psychological, and environmental factors and sequelae of the development and maintenance of chronic pain proves to pose a continuing challenge. Although clinically effective treatments do show efficacy in animal models thus far, increasing efforts to increase the translatability of preclinical and clinical studies will prove important, for instance measurement of spontaneous pain in animal models and quantitative sensory testing in human patients59,255–258.
Alternative treatments that can modulate neuroinflammation
Although direct targeting of neuroinflammation via inhibitors of cytokines, chemokines, and MAP kinases could be effective, these drugs may also produce side effects such as infection after long-term treatment and impair the resolution of inflammation39. In this review, we highlight the alternative approaches that can control excessive neuroinflammation, including specialized pro-resolution mediators, cell therapies, and neuromodulation (Fig. 6).
Specialized pro-resolution mediators (SPMs), including lipoxins, resolvins, protectins, and maresins are biosynthesized from polyunsaturated fatty acids, including omega-3 fatty acids docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), two major components of fish oil. SPMs have multiple beneficial actions for treating inflammation-associated disease conditions259,260. Lipoxin A4, resolvin D1, resolvin E1, resolvin D2, neuroprotectin D1, and maresin 1, at very low doses (10–500, ng range), reduced inflammatory pain, postoperative pain, and neuropathic pain in animal models189,204,260–265. In the hippocampus, treatment with resolvin D1 prevented LTP impairment by reducing pro-inflammatory cytokines after surgery266. The benefits of these SPMs include high potency, wide safety profile, and multiple mechanisms of action including but not limited to control of inflammation in peripheral tissues, control of neuroinflammation in the PNS and CNS, resolution of synaptic plasticity, modulation of TRPA1 and TRPV1 activities, and protection against nerve injury39,228,260,266. Spinal administration of neuroprotectin D1, even 2 hours after LTP induction, is also effective in reversing LTP in the intact spinal cord204,263. SPMs are especially effective in preventing neuroinflammation, postoperative pain, and neuropathic pain after nerve injury and surgery, although post-treatment of SPMs also exhibit transient analgesic effects. Notably, thoracotomies produce a high incidence of chronic postoperative pain by nerve compression267. Strichartz and coworkers found that intrathecal and perioperative treatment with resolvin D1 and resolvin D2 effectively prevents postoperative pain in a rat model of chronic post-thoracotomy265.
Cell therapies are emerging treatments for chronic pain and neurodegenerative conditions. Implantation of bone marrow stem cells produces long-term pain relief268,269. Bone marrow stromal cells or bone marrow stem cells (BMSCs) promote tissue regeneration and tissue repair by secreting growth factors and control inflammation and neuroinflammation by secreting anti-inflammatory mediators such as transforming growth factor-beta1 (TGF-β1)268. A single intrathecal injection of BMSCs not only inhibits nerve injury-induced neuropathic pain for many weeks via secretion of TGF-β1, but also blocks nerve injury-induced neuroinflammation (glial activation and cytokine/chemokine up-regulation) in DRG and spinal cord tissues268. This paracrine modulation of neuroinflammation by BMSCs is very different from typical cell replacement strategies, such as implantation of forebrain GABAergic precursor cells into the spinal cord to generate functional GABAergic neurons for chronic pain control270. Furthermore, subcutaneous treatment with human umbilical cord blood-derived multipotent stem cells reduced peripheral neuropathic pain in rats and inhibited spinal MMP-9 and MMP-2 expression after nerve injury271. Macrophages and T cells also play a role in the resolution of pain. Pro-resolution macrophages (M2-like) may inhibit neuroinflammation and chronic pain by secreting SPMs and anti-inflammatory cytokines such as IL-1026,272. Adoptive transfer of regulatory T cells (Treg) reduced neuropathic pain, while CD8+ cytotoxic T cells increased neuropathic pain after chemotherapy following intrathecal injection30. However, adoptive transfer of CD8+ T cells via systemic route was also shown to resolve chemotherapy-induced neuropathic pain by increasing IL-10 receptor expression in DRG neurons273. Autologous conditioned serum (ACS) is prepared from whole blood, which is incubated with glass beads to initiate monocyte activation. ACS contains increased levels of IL-1 receptor antagonist IL-1ra as well as the anti-inflammatory cytokines IL-4 and IL-10 and demonstrates efficacy in relieving pain in patients with knee osteoarthritis 274,275. Platelet-rich plasma (PRP) was also shown to reduce clinical pain in knee osteoarthritis via possible modulation of inflammatory responses276.
Neuromodulation via electrical and magnetic stimulation, such as spinal cord stimulation (SCS), deep brain stimulation (DBS), transcranial magnetic stimulation (TMS), transcutaneous electrical nerve stimulation (TENS), vagus nerve stimulation (VNS), auricular stimulation, and DRG stimulation, as well as acupuncture including electroacupuncture (EA), has been used to provide pain relief in patients and animals277,278 via activation of specific neural pathways279,280, suppression of nociceptive neuron activities (e.g. wide-dynamic neurons and projection neurons in the spinal cord281,282), and release of pain suppressing neurotransmitters and neuromodulators283. However, transient modulation of neuronal activity in the pain circuits during stimulation cannot explain the long-term benefits of neuromodulation. Increasing evidence suggests that neuromodulation such as vagus nerve stimulation can powerfully regulate inflammation284,285. While acupuncture elicits transient analgesia via releasing opioid peptides and adenosine283,286, acupuncture and sciatic nerve activation also regulates inflammation and sepsis through vagus nerve activation and dopamine release287. In 1984, Maixner and Randich demonstrated an antinociceptive role of vagal stimulation288. Notably, acupuncture also causes neuronal activation in the nucleus of solitary tract to mediate vagal responses289,290. In a rodent model of inflammatory muscle pain, acutpuncture elecits anti-inflammatory effects via IL-10 release291. The identification of the “inflammatory reflex” reveals a neural circuit capable of providing information in real-time to the brain about the body’s inflammatory status, allowing rapid neural regulatory responses via the vagus nerve292. This has clear implications in developing novel bioelectronic technologies to treat pain and improve postoperative outcomes in vulnerable patients285.
Concluding remarks and future directions
Neuroinflammation resulting from neuro-glial and neuro-immune interactions not only serves as a driving force for chronic pain, but is also implicated in other neurological and psychiatric diseases such as Alzheimer’s disease, Parkinson disease, multiple sclerosis, autism, major depression, and schizophrenia39. Chronic pain is commonly associated with depression, anxiety, and sleep disorders, and the prevalence of chronic pain is especially high in the rapidly growing populations of aged people and females with overlapping chronic pain conditions. Thus, targeting excessive neuroinflammation will help to alleviate chronic pain and control the progression of neurological and psychiatric diseases in aged as well as younger populations. Mechanistically, neuroinflammation drives widespread chronic pain via central sensitization, which can be induced and maintained by cytokines, chemokines, and other glia-produced mediators circulated in the CSF. Therefore, increasing the precision with which drugs can target neuroinflammation in the CNS, namely by increasing access to the spinal cord and brain, and developing the ability to accurately measure evolving neuroinflammatory changes in the CNS particularly in the CSF will be of great importance. By developing specific neuroinflammatory profiles, their creation may also reveal novel biomarkers and means to identify chronic pain states. For instance, a recent study utilizing functional imaging of patients with chronic low-back pain revealed glial activation in the brain120. In line with the use of an analgesic marker of glial activation, translocator protein (TSPO)293, the development of additional novel radioligands to characterize different activation states of glial cells in the brain, as well as in spinal cord, DRG, and peripheral nerves is of pressing need. Specialized pro-resolution mediators (SPMs) including resolvins and protectins, as well as bone marrow stem cells and neuromodulation can serve as alternative approaches to providing effective control of neuroinflammation with perhaps less side effects than more directly targeted neuroinflammation treatments including cytokine, chemokine, and MAP kinase inhibitors. With the high incidence of postoperative pain following trauma and nerve injury resulting from surgeries267 and the worsening opioid crisis in the United States294, the development of effective non-opioid treatments for the prevention and resolution of neuroinflammation and postoperative pain is of utmost importance for clinical practice and health care. The benefits of developing novel treatments may extend further towards improving cognitive function in postoperative patients, a topic that will be addressed by the authors in future reviews.
Acknowledgments
Supported in part by grants of R01DE17794 and R01DE22743, and R01NS87988 to RRJ from the National Institutes of Health, Bethesda, USA
Footnotes
Conflict of interest: The authors have no financial interests in this study.
References
- 1.Pizzo PA, Clark NM. Alleviating suffering 101--pain relief in the United States. N Engl J Med. 2012;366:197–199. doi: 10.1056/NEJMp1109084. [DOI] [PubMed] [Google Scholar]
- 2.Gereau RWt, Sluka KA, Maixner W, Savage SR, Price TJ, Murinson BB, Sullivan MD, Fillingim RB. A pain research agenda for the 21st century. J Pain. 2014;15:1203–14. doi: 10.1016/j.jpain.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest. 2010;120:3779–3787. doi: 10.1172/JCI43766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo C, Kuner T, Kuner R. Synaptic plasticity in pathological pain. Trends Neurosci. 2014;37:343–355. doi: 10.1016/j.tins.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawes JM, Calvo M, Perkins JR, Paterson KJ, Kiesewetter H, Hobbs C, Kaan TK, Orengo C, Bennett DL, McMahon SB. CXCL5 Mediates UVB Irradiation-Induced Pain. Sci Transl Med. 2011;3:90ra60. doi: 10.1126/scitranslmed.3002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angst MS, Clark JD, Carvalho B, Tingle M, Schmelz M, Yeomans DC. Cytokine profile in human skin in response to experimental inflammation, noxious stimulation, and administration of a COX-inhibitor: a microdialysis study. Pain. 2008;139:15–27. doi: 10.1016/j.pain.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 13.Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain. 2011;152:2802–12. doi: 10.1016/j.pain.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan HL, Liu BL, Lin W, Zhang YQ. Modulation of Nav1.8 by Lysophosphatidic Acid in the Induction of Bone Cancer Pain. Neurosci Bull. 2016;32:445–54. doi: 10.1007/s12264-016-0060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SE, Coste B, Chadha A, Cook B, Patapoutian A. The role of Drosophila Piezo in mechanical nociception. Nature. 2012;483:209–212. doi: 10.1038/nature10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu XZ. Demystifying Mechanosensitive Piezo Ion Channels. Neurosci Bull. 2016;32:307–9. doi: 10.1007/s12264-016-0033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ, Ji RR. Extracellular MicroRNAs Activate Nociceptor Neurons to Elicit Pain via TLR7 and TRPA1. Neuron. 2014;82:47–54. doi: 10.1016/j.neuron.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 20.Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393–2401. doi: 10.1172/JCI25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woolf CJ, Allchorne A, Safieh-Garabedian B, Poole S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol. 1997;121:417–424. doi: 10.1038/sj.bjp.0701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schafers M, Sorkin LS, Sommer C. Intramuscular injection of tumor necrosis factor-alpha induces muscle hyperalgesia in rats. Pain. 2003;104:579–588. doi: 10.1016/S0304-3959(03)00115-5. [DOI] [PubMed] [Google Scholar]
- 23.Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sommer C, Schmidt C, George A. Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp Neurol. 1998;151:138–142. doi: 10.1006/exnr.1998.6797. [DOI] [PubMed] [Google Scholar]
- 25.Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354:572–577. doi: 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. 2012;15:1063–1067. doi: 10.1038/nn.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu T, Ji RR. New insights into the mechanisms of itch: are pain and itch controlled by distinct mechanisms? Pflugers Arch. 2013 doi: 10.1007/s00424-013-1284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu T, Berta T, Xu ZZ, Park CK, Zhang L, Lu N, Liu Q, Liu Y, Gao YJ, Liu YC, Ma Q, Dong X, Ji RR. TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest. 2012;122:2195–2207. doi: 10.1172/JCI45414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu XJ, Zhang Y, Liu T, Xu ZZ, Park CK, Berta T, Jiang D, Ji RR. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014 doi: 10.1038/cr.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talbot S, Foster SL, Woolf CJ. Neuroimmunity: Physiology and Pathology. Annu Rev Immunol. 2016;34:421–47. doi: 10.1146/annurev-immunol-041015-055340. [DOI] [PubMed] [Google Scholar]
- 32.Liu XJ, Liu T, Chen G, Wang B, Yu XL, Yin C, Ji RR. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep. 2016;6:28188. doi: 10.1038/srep28188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, Dougherty PM. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2013;14:1031–44. doi: 10.1016/j.jpain.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei T, Li WW, Guo TZ, Zhao R, Wang L, Clark DJ, Oaklander AL, Schmelz M, Kingery WS. Post-junctional facilitation of Substance P signaling in a tibia fracture rat model of complex regional pain syndrome type I. Pain. 2009;144:278–86. doi: 10.1016/j.pain.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiu IM, Heesters BA, Ghasemlou N, von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, Bubeck WJ, Hwang SW, Carroll MC, Woolf CJ. Bacteria activate sensory neurons that modulate pain and inflammation. Nature. 2013;501:52–57. doi: 10.1038/nature12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 37.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 38.Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014;13:533–548. doi: 10.1038/nrd4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 41.Culley DJ, Snayd M, Baxter MG, Xie Z, Lee IH, Rudolph J, Inouye SK, Marcantonio ER, Crosby G. Systemic inflammation impairs attention and cognitive flexibility but not associative learning in aged rats: possible implications for delirium. Front Aging Neurosci. 2014;6:107. doi: 10.3389/fnagi.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiu IM. Infection, Pain, and Itch. Neurosci Bull. 2017 doi: 10.1007/s12264-017-0098-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 45.Sisignano M, Baron R, Scholich K, Geisslinger G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat Rev Neurol. 2014;10:694–707. doi: 10.1038/nrneurol.2014.211. [DOI] [PubMed] [Google Scholar]
- 46.Huang W, Calvo M, Karu K, Olausen HR, Bathgate G, Okuse K, Bennett DL, Rice AS. A clinically relevant rodent model of the HIV antiretroviral drug stavudine induced painful peripheral neuropathy. Pain. 2013;154:560–575. doi: 10.1016/j.pain.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 47.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 48.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 49.Uceyler N, Gobel K, Meuth SG, Ortler S, Stoll G, Sommer C, Wiendl H, Kleinschnitz C. Deficiency of the negative immune regulator B7-H1 enhances inflammation and neuropathic pain after chronic constriction injury of mouse sciatic nerve. Exp Neurol. 2010;222:153–160. doi: 10.1016/j.expneurol.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 50.Chen GKY, Ji RR. PD-L1 inhibits acute and chronic pain and suppresses nociceptor activitity via PD-1. Nature Neuroscience. 2017 In press. [Google Scholar]
- 51.Rees H, Sluka KA, Westlund KN, Willis WD. Do dorsal root reflexes augment peripheral inflammation? Neuroreport. 1994;5:821–4. doi: 10.1097/00001756-199403000-00021. [DOI] [PubMed] [Google Scholar]
- 52.Sluka KA, Willis WD, Westlund KN. Central Control of Peripheral Joint Inflammation and Heat Hyperalgesia. Prog Pain Res Manag. 1994;2:359–371. [PMC free article] [PubMed] [Google Scholar]
- 53.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 54.Christianson CA, Dumlao DS, Stokes JA, Dennis EA, Svensson CI, Corr M, Yaksh TL. Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain. 2011;152:2881–2891. doi: 10.1016/j.pain.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 2012;32:10833–10840. doi: 10.1523/JNEUROSCI.5628-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sluka KA, Clauw DJ. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience. 2016;338:114–129. doi: 10.1016/j.neuroscience.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uceyler N, Zeller D, Kahn AK, Kewenig S, Kittel-Schneider S, Schmid A, Casanova-Molla J, Reiners K, Sommer C. Small fibre pathology in patients with fibromyalgia syndrome. Brain. 2013;136:1857–1867. doi: 10.1093/brain/awt053. [DOI] [PubMed] [Google Scholar]
- 58.Wei T, Guo TZ, Li WW, Kingery WS, Clark JD. Acute versus chronic phase mechanisms in a rat model of CRPS. J Neuroinflammation. 2016;13:14. doi: 10.1186/s12974-015-0472-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maixner W, Fillingim RB, Williams DA, Smith SB, Slade GD. Overlapping Chronic Pain Conditions: Implications for Diagnosis and Classification. J Pain. 2016;17:T93–t107. doi: 10.1016/j.jpain.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ciszek BP, Khan AA, Dang H, Slade GD, Smith S, Bair E, Maixner W, Zolnoun D, Nackley AG. MicroRNA expression profiles differentiate chronic pain condition subtypes. Transl Res. 2015;166:706–720. e11. doi: 10.1016/j.trsl.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torpy DJ, Papanicolaou DA, Lotsikas AJ, Wilder RL, Chrousos GP, Pillemer SR. Responses of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis to interleukin-6: a pilot study in fibromyalgia. Arthritis Rheum. 2000;43:872–80. doi: 10.1002/1529-0131(200004)43:4<872::AID-ANR19>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 62.Evaskus DS, Laskin DM. A biochemical measure of stress in patients with myofascial pain-dysfunction syndrome. J Dent Res. 1972;51:1464–6. doi: 10.1177/00220345720510053501. [DOI] [PubMed] [Google Scholar]
- 63.Perry F, Heller PH, Kamiya J, Levine JD. Altered autonomic function in patients with arthritis or with chronic myofascial pain. Pain. 1989;39:77–84. doi: 10.1016/0304-3959(89)90177-2. [DOI] [PubMed] [Google Scholar]
- 64.Bote ME, Garcia JJ, Hinchado MD, Ortega E. An exploratory study of the effect of regular aquatic exercise on the function of neutrophils from women with fibromyalgia: role of IL-8 and noradrenaline. Brain Behav Immun. 2014;39:107–12. doi: 10.1016/j.bbi.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 65.Marbach JJ, Levitt M. Erythrocyte catechol-O-methyltransferase activity in facial pain patients. J Dent Res. 1976;55:711. doi: 10.1177/00220345760550043801. [DOI] [PubMed] [Google Scholar]
- 66.Smith SB, Reenila I, Mannisto PT, Slade GD, Maixner W, Diatchenko L, Nackley AG. Epistasis Between Polymorphisms in COMT, ESR1, and GCH1 Influences COMT Enzyme Activity and Pain. Pain. 2014 doi: 10.1016/j.pain.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 68.Hartung JE, Ciszek BP, Nackley AG. beta2- and beta3-adrenergic receptors drive COMT-dependent pain by increasing production of nitric oxide and cytokines. Pain. 2014;155:1346–55. doi: 10.1016/j.pain.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–10. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- 70.Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–3. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 71.Slade GD, Sanders AE, Ohrbach R, Bair E, Maixner W, Greenspan JD, Fillingim RB, Smith S, Diatchenko L. COMT Diplotype Amplifies Effect of Stress on Risk of Temporomandibular Pain. J Dent Res. 2015;94:1187–95. doi: 10.1177/0022034515595043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith SB, Reenila I, Mannisto PT, Slade GD, Maixner W, Diatchenko L, Nackley AG. Epistasis between polymorphisms in COMT, ESR1, and GCH1 influences COMT enzyme activity and pain. Pain. 2014;155:2390–9. doi: 10.1016/j.pain.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barbosa FR, Matsuda JB, Mazucato M, de Castro Franca S, Zingaretti SM, da Silva LM, Martinez-Rossi NM, Junior MF, Marins M, Fachin AL. Influence of catechol-O-methyltransferase (COMT) gene polymorphisms in pain sensibility of Brazilian fibromialgia patients. Rheumatol Int. 2012;32:427–30. doi: 10.1007/s00296-010-1659-z. [DOI] [PubMed] [Google Scholar]
- 74.Cohen H, Neumann L, Glazer Y, Ebstein RP, Buskila D. The relationship between a common catechol-O-methyltransferase (COMT) polymorphism val(158) met and fibromyalgia. Clin Exp Rheumatol. 2009;27:S51–6. [PubMed] [Google Scholar]
- 75.Gursoy S, Erdal E, Herken H, Madenci E, Alasehirli B, Erdal N. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23:104–7. doi: 10.1007/s00296-002-0260-5. [DOI] [PubMed] [Google Scholar]
- 76.Matsuda JB, Barbosa FR, Morel LJ, de Franca SC, Zingaretti SM, da Silva LM, Pereira AM, Marins M, Fachin AL. Serotonin receptor (5-HT 2A) and catechol-O-methyltransferase (COMT) gene polymorphisms: triggers of fibromyalgia? Rev Bras Reumatol. 2010;50:141–9. [PubMed] [Google Scholar]
- 77.Vargas-Alarcon G, Fragoso JM, Cruz-Robles D, Vargas A, Vargas A, Lao-Villadoniga JI, Garcia-Fructuoso F, Ramos-Kuri M, Hernandez F, Springall R, Bojalil R, Vallejo M, Martinez-Lavin M. Catechol-O-methyltransferase gene haplotypes in Mexican and Spanish patients with fibromyalgia. Arthritis Res Ther. 2007;9:R110. doi: 10.1186/ar2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14:135–43. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 79.Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–3. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- 80.McLean SA, Diatchenko L, Lee YM, Swor RA, Domeier RM, Jones JS, Jones CW, Reed C, Harris RE, Maixner W, Clauw DJ, Liberzon I. Catechol O-methyltransferase haplotype predicts immediate musculoskeletal neck pain and psychological symptoms after motor vehicle collision. J Pain. 2011;12:101–7. doi: 10.1016/j.jpain.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meloto CB, Bortsov AV, Bair E, Helgeson E, Ostrom C, Smith SB, Dubner R, Slade GD, Fillingim RB, Greenspan JD, Ohrbach R, Maixner W, McLean SA, Diatchenko L. Modification of COMT-dependent pain sensitivity by psychological stress and sex. Pain. 2016;157:858–67. doi: 10.1097/j.pain.0000000000000449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kline RHt, Exposto FG, O’Buckley SC, Westlund KN, Nackley AG. Catechol-O-methyltransferase inhibition alters pain and anxiety-related volitional behaviors through activation of beta-adrenergic receptors in the rat. Neuroscience. 2015;290:561–9. doi: 10.1016/j.neuroscience.2015.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both beta2- and beta3-adrenergic receptors. Pain. 2007;128:199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ciszek BP, O’Buckley SC, Nackley AG. Persistent Catechol-O-methyltransferase-dependent Pain Is Initiated by Peripheral beta-Adrenergic Receptors. Anesthesiology. 2016;124:1122–35. doi: 10.1097/ALN.0000000000001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hartung JE, BC, O’Buckley SC, Nackley A. Distinct mechanisms underlie the development and maintenance of COMT-dependent pain. APS 35th Annual Scientific Meeting; Austin, TX: American Pain Society; 2016. [Google Scholar]
- 86.Hartung J, Ciszek B, O’Buckley S, Nackley A. (374) Distinct mechanisms underlie the development and maintenance of COMT-dependent pain. J Pain. 2016;17:S68. [Google Scholar]
- 87.Bote ME, Garcia JJ, Hinchado MD, Ortega E. Inflammatory/stress feedback dysregulation in women with fibromyalgia. Neuroimmunomodulation. 2012;19:343–51. doi: 10.1159/000341664. [DOI] [PubMed] [Google Scholar]
- 88.Larson AA, Giovengo SL, Russell IJ, Michalek JE. Changes in the concentrations of amino acids in the cerebrospinal fluid that correlate with pain in patients with fibromyalgia- implications for nitric oxide pathways. Pain. 2000;87:201–211. doi: 10.1016/S0304-3959(00)00284-0. [DOI] [PubMed] [Google Scholar]
- 89.Shafer D, Assael L, White LB, Rossomando EF. Tumor Necrosis Factor-a as a biochemical marker of pain and outcome in temporomandibular joints with internal derangements. J Oral Maxillofac Surg. 1994;52:786–791. doi: 10.1016/0278-2391(94)90217-8. [DOI] [PubMed] [Google Scholar]
- 90.Kubota E, Kubota T, Matsumoto J, Shibata T, Murakami KI. Synovial fluid cytokines and proteinases as markers of temporomandibular joint disease. J Oral Maxillofac Surg. 1998;56:192–198. doi: 10.1016/s0278-2391(98)90868-0. [DOI] [PubMed] [Google Scholar]
- 91.Fan W, Huang F, Wu Z, Zhu X, Li D, He H. The role of nitric oxide in orofacial pain. Nitric Oxide. 2012;26:32–7. doi: 10.1016/j.niox.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Ferrari LF, Araldi D, Levine JD. Distinct terminal and cell body mechanisms in the nociceptor mediate hyperalgesic priming. J Neurosci. 2015;35:6107–16. doi: 10.1523/JNEUROSCI.5085-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang H, Heijnen CJ, Eijkelkamp N, Garza Carbajal A, Schedlowski M, Kelley KW, Dantzer R, Kavelaars A. GRK2 in sensory neurons regulates epinephrine-induced signalling and duration of mechanical hyperalgesia. Pain. 2011;152:1649–58. doi: 10.1016/j.pain.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 94.Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eijkelkamp N, Wang H, Garza-Carbajal A, Willemen HL, Zwartkruis FJ, Wood JN, Dantzer R, Kelley KW, Heijnen CJ, Kavelaars A. Low nociceptor GRK2 prolongs prostaglandin E2 hyperalgesia via biased cAMP signaling to Epac/Rap1, protein kinase Cepsilon, and MEK/ERK. J Neurosci. 2010;30:12806–12815. doi: 10.1523/JNEUROSCI.3142-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McLean SA. The potential contribution of stress systems to the transition to chronic whiplash-associated disorders. Spine (Phila Pa 1976) 2011;36:S226–32. doi: 10.1097/BRS.0b013e3182387fb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khasar SG, Lin Y-H, Martin A, Dadgar J, McMahon T, Wang D, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ji RR. Mitogen-activated protein kinases as potential targets for pain killers. Curr Opin Investig Drugs. 2004;5:71–75. [PubMed] [Google Scholar]
- 99.Akimoto Y, Horinouchi T, Shibano M, Matsushita M, Yamashita Y, Okamoto T, Yamaki F, Tanaka Y, Koike K. Nitric oxide (NO) primarily accounts for endothelium dependent component of B-adrenoceptor-activated smooth muscle relaxation of mouse aorta in response to isoprenaline. J Smooth Muscle Res. 2002;38:87–99. doi: 10.1540/jsmr.38.87. [DOI] [PubMed] [Google Scholar]
- 100.Canova NK, Lincova D, Kmonickova E, Kamenikova L, Farghali H. Nitric oxide production from rat adipocytes is modulated by β3-adrenergic receptor agonists and is involved in a cyclic AMP-dependent lipolysis in adipocytes. Nitric Oxide. 2006;14:200–11. doi: 10.1016/j.niox.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 101.Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of β-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009:297. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- 102.Frost RA, Nystrom GJ, Lang CH. Epinephrine stimulates IL-6 expression in skeletal muscle and C2C12 myoblasts: role of c-Jun NH2-terminal kinase and histone deacetylase activity. Am J Physiol Endocrinol Metab. 2004:E809–E817. doi: 10.1152/ajpendo.00560.2003. [DOI] [PubMed] [Google Scholar]
- 103.Furlan C, Sterin-Borda L, Borda E. Activation of β3 adrenergic receptor decreases DNA synthesis in human skin fibroblasts via cyclic GMP/nitric oxide pathway. Cell Physiol Biochem. 2005;18:175–182. doi: 10.1159/000089843. [DOI] [PubMed] [Google Scholar]
- 104.Hodges GJ, Kosiba WA, Zhao K, Johnson JM. The involvement of norepinephrine, neuropeptide Y, and nitric oxide in the cutaneous vasodilator response to local heating in humans. J Appl Physiol. 2008;105:233–40. doi: 10.1152/japplphysiol.90412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maimone D, Cioni C, Rosa S, Macchia G, Aloisi F, Annunziata P. Norepinephrine and vasoactive intestinal peptide induce IL-6 secretion by astrocytes: synergism with IL-1 beta and TNF alpha. J Neuroimmunol. 1993;47:73–81. doi: 10.1016/0165-5728(93)90286-8. [DOI] [PubMed] [Google Scholar]
- 106.Tchivileva IE, Tan KS, Gambarian M, Nackley AG, Medvedev AV, Romanov S, Flood PM, Maixner W, Makarov SS, Diatchenko L. Signaling pathways mediating β3-adrenergic receptor-induced production of interleukin-6 in adipocytes. Mol Immunol. 2009;46:2256–66. doi: 10.1016/j.molimm.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. β2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007;19:251–60. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 108.Hains LE, Loram LC, Weiseler JL, Frank MG, Bloss EB, Sholar P, Taylor FR, Harrison JA, Martin TJ, Eisenach JC, Maier SF, Watkins LR. Pain intensity and duration can be enhanced by prior challenge: initial evidence suggestive of a role of microglial priming. J Pain. 2010;11:1004–1014. doi: 10.1016/j.jpain.2010.01.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov. 2005;4:834–844. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ji RR, Berta T, Nedergaard M. Glia and pain: Is chronic pain a gliopathy? Pain. 2013 doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 112.Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 115.Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U SA. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- 117.Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Fabisiak TJ, Anderson ND, Cheng K, Greene LI, Berkelhammer D, Zhang Y, Ellis AL, Yin HH, Campeau S, Rice KC, Roth BL, Maier SF, Watkins LR. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1602070113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010;7:482–493. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fu KY, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999;825:59–67. doi: 10.1016/s0006-8993(99)01186-5. [DOI] [PubMed] [Google Scholar]
- 120.Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, Riley M, Wasan AD, Zurcher NR, Albrecht DS, Vangel MG, Rosen BR, Napadow V, Hooker JM. Evidence for brain glial activation in chronic pain patients. Brain. 2015;138:604–615. doi: 10.1093/brain/awu377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu X, Conklin D, Eisenach JC. Cyclooxygenase-1 in the spinal cord plays an important role in postoperative pain. Pain. 2003;104:15–23. doi: 10.1016/s0304-3959(02)00465-7. [DOI] [PubMed] [Google Scholar]
- 122.Zhu X, Eisenach JC. Cyclooxygenase-1 in the spinal cord is altered after peripheral nerve injury. Anesthesiology. 2003;99:1175–1179. doi: 10.1097/00000542-200311000-00026. [DOI] [PubMed] [Google Scholar]
- 123.Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. 2016;19:94–101. doi: 10.1038/nn.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, Zhang WW, Wu XB, Berta T, Ji RR, Gao YJ. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126:745–61. doi: 10.1172/JCI81950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu Y, Willcockson HH, Maixner W, Light AR. Suramin inhibits spinal cord microglia activation and long-term hyperalgesia induced by formalin injection. J Pain. 2004;5:48–55. doi: 10.1016/j.jpain.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 126.Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 127.Tsuda M, Tozaki-Saitoh H, Inoue K. Pain and purinergic signaling. Brain Res Rev. 2010;63:222–232. doi: 10.1016/j.brainresrev.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 128.Trang T, Beggs S, Salter MW. ATP receptors gate microglia signaling in neuropathic pain. Exp Neurol. 2012;234:354–361. doi: 10.1016/j.expneurol.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M. Connexin 43 hemichannels are permeable to ATP. J Neurosci. 2008;28:4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koyanagi S, Kusunose N, Taniguchi M, Akamine T, Kanado Y, Ozono Y, Masuda T, Kohro Y, Matsunaga N, Tsuda M, Salter MW, Inoue K, Ohdo S. Glucocorticoid regulation of ATP release from spinal astrocytes underlies diurnal exacerbation of neuropathic mechanical allodynia. Nat Commun. 2016;7:13102. doi: 10.1038/ncomms13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Masuda T, Ozono Y, Mikuriya S, Kohro Y, Tozaki-Saitoh H, Iwatsuki K, Uneyama H, Ichikawa R, Salter MW, Tsuda M, Inoue K. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat Commun. 2016;7:12529. doi: 10.1038/ncomms12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, Mousseau M, Shankara JV, Stemkowski PL, Baimoukhametova D, Bains JS, Antle MC, Zamponi GW, Cahill CM, Borgland SL, De Koninck Y, Trang T. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat Med. 2017;23:355–360. doi: 10.1038/nm.4281. [DOI] [PubMed] [Google Scholar]
- 133.Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- 134.Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang Y, Li H, Li TT, Luo H, Gu XY, Lu N, Ji RR, Zhang YQ. Delayed Activation of Spinal Microglia Contributes to the Maintenance of Bone Cancer Pain in Female Wistar Rats via P2X7 Receptor and IL-18. J Neurosci. 2015;35:7950–7963. doi: 10.1523/JNEUROSCI.5250-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang MD, Barde S, Yang T, Lei B, Eriksson LI, Mathew JP, Andreska T, Akassoglou K, Harkany T, Hokfelt TG, Terrando N. Orthopedic surgery modulates neuropeptides and BDNF expression at the spinal and hippocampal levels. Proc Natl Acad Sci U S A. 2016;113:E6686–E6695. doi: 10.1073/pnas.1614017113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li WW, Guo TZ, Shi X, Sun Y, Wei T, Clark DJ, Kingery WS. Substance P spinal signaling induces glial activation and nociceptive sensitization after fracture. Neuroscience. 2015;310:73–90. doi: 10.1016/j.neuroscience.2015.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cotrina ML, Lin JH, Lopez-Garcia JC, Naus CC, Nedergaard M. ATP-mediated glia signaling. J Neurosci. 2000;20:2835–2844. doi: 10.1523/JNEUROSCI.20-08-02835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR, Nedergaard M. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia. 2012;60:1660–1670. doi: 10.1002/glia.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 2014;137:2193–2209. doi: 10.1093/brain/awu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev. 2009;60:57–64. doi: 10.1016/j.brainresrev.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim DS, Li KW, Boroujerdi A, Peter YY, Zhou CY, Deng P, Park J, Zhang X, Lee J, Corpe M, Sharp K, Steward O, Eroglu C, Barres B, Zaucke F, Xu ZC, Luo ZD. Thrombospondin-4 Contributes to Spinal Sensitization and Neuropathic Pain States. J Neurosci. 2012;32:8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ji RR, Gereau RW, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29:3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J Neurosci. 2010;30:573–582. doi: 10.1523/JNEUROSCI.3295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain. 2010;148:309–319. doi: 10.1016/j.pain.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim SK, Hayashi H, Ishikawa T, Shibata K, Shigetomi E, Shinozaki Y, Inada H, Roh SE, Kim SJ, Lee G, Bae H, Moorhouse AJ, Mikoshiba K, Fukazawa Y, Koizumi S, Nabekura J. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest. 2016;126:1983–97. doi: 10.1172/JCI82859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. 2005 doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu T, Han Q, Chen G, Huang Y, Zhao LX, Berta T, Gao YJ, Ji RR. Toll-like receptor 4 contributes to chronic itch, alloknesis, and spinal astrocyte activation in male mice. Pain. 2016;157:806–17. doi: 10.1097/j.pain.0000000000000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tramullas M, Finger BC, Moloney RD, Golubeva AV, Moloney G, Dinan TG, Cryan JF. Toll-like receptor 4 regulates chronic stress-induced visceral pain in mice. Biol Psychiatry. 2014;76:340–8. doi: 10.1016/j.biopsych.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 157.Taylor AM, Castonguay A, Taylor AJ, Murphy NP, Ghogha A, Cook C, Xue L, Olmstead MC, De Koninck Y, Evans CJ, Cahill CM. Microglia disrupt mesolimbic reward circuitry in chronic pain. J Neurosci. 2015;35:8442–50. doi: 10.1523/JNEUROSCI.4036-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang F, Vadakkan KI, Kim SS, Wu LJ, Shang Y, Zhuo M. Selective activation of microglia in spinal cord but not higher cortical regions following nerve injury in adult mouse. Mol Pain. 2008;4:15. doi: 10.1186/1744-8069-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tsuda M, Koga K, Chen T, Zhuo M. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. J Neurochem. 2017;141:486–498. doi: 10.1111/jnc.14001. [DOI] [PubMed] [Google Scholar]
- 160.Horst OV, Cunha-Cruz J, Zhou L, Manning W, Mancl L, DeRouen TA. Prevalence of pain in the orofacial regions in patients visiting general dentists in the Northwest Practice-based REsearch Collaborative in Evidence-based DENTistry research network. J Am Dent Assoc. 2015;146:721–8. e3. doi: 10.1016/j.adaj.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Slade GD, Fillingim RB, Sanders AE, Bair E, Greenspan JD, Ohrbach R, Dubner R, Diatchenko L, Smith SB, Knott C, Maixner W. Summary of findings from the OPPERA prospective cohort study of incidence of first-onset temporomandibular disorder: implications and future directions. J Pain. 2013;14:T116–24. doi: 10.1016/j.jpain.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Maixner W, Humphrey C. Gender differences in pain and cardiovascular responses to forearm ischemia. Clin J Pain. 1993;9:16–25. doi: 10.1097/00002508-199303000-00003. [DOI] [PubMed] [Google Scholar]
- 163.Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci. 2012;13:859–866. doi: 10.1038/nrn3360. [DOI] [PubMed] [Google Scholar]
- 164.Sorge RE, Lacroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin JS, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. 2011;31:15450–15454. doi: 10.1523/JNEUROSCI.3859-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18:1081–1083. doi: 10.1038/nn.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, Van de Ven T, Laufer S, Ji RR. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav Immun. 2016;55:70–81. doi: 10.1016/j.bbi.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Berta T, Qadri YJ, Chen G, Ji RR. Microglial Signaling in Chronic Pain with a Special Focus on Caspase 6, p38 MAP Kinase, and Sex Dependence. J Dent Res. 2016 doi: 10.1177/0022034516653604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lu N, Liu YC, Ji RR. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest. 2014;124:1173–1186. doi: 10.1172/JCI72230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Clauw DJ. Diagnosing and treating chronic musculoskeletal pain based on the underlying mechanism(s) Best Pract Res Clin Rheumatol. 2015;29:6–19. doi: 10.1016/j.berh.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 170.Yunus MB. Editorial review: an update on central sensitivity syndromes and the issues of nosology and psychobiology. Curr Rheumatol Rev. 2015;11:70–85. doi: 10.2174/157339711102150702112236. [DOI] [PubMed] [Google Scholar]
- 171.Loeser JD, Treede RD. The Kyoto protocol of IASP Basic Pain Terminology. Pain. 2008;137:473–7. doi: 10.1016/j.pain.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 172.Ren K, Hylden JL, Williams GM, Ruda MA, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain. 1992;50:331–344. doi: 10.1016/0304-3959(92)90039-E. [DOI] [PubMed] [Google Scholar]
- 173.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 174.Liu XJ, Gingrich JR, Vargas-Caballero M, Dong YN, Sengar A, Beggs S, Wang SH, Ding HK, Frankland PW, Salter MW. Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat Med. 2008;14:1325–1332. doi: 10.1038/nm.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen G, Xie RG, Gao YJ, Xu ZZ, Zhao LX, Bang S, Berta T, Park CK, Lay M, Chen W, Ji RR. beta-arrestin-2 regulates NMDA receptor function in spinal lamina II neurons and duration of persistent pain. Nat Commun. 2016;7:12531. doi: 10.1038/ncomms12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 177.Hartmann B, Ahmadi S, Heppenstall PA, Lewin GR, Schott C, Borchardt T, Seeburg PH, Zeilhofer HU, Sprengel R, Kuner R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–50. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 178.Tao YX. AMPA receptor trafficking in inflammation-induced dorsal horn central sensitization. Neurosci Bull. 2012;28:111–20. doi: 10.1007/s12264-012-1204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tappe A, Klugmann M, Luo C, Hirlinger D, Agarwal N, Benrath J, Ehrengruber MU, During MJ, Kuner R. Synaptic scaffolding protein Homer1a protects against chronic inflammatory pain. Nat Med. 2006;12:677–681. doi: 10.1038/nm1406. [DOI] [PubMed] [Google Scholar]
- 180.Zhang WB, Ross PJ, Tu Y, Wang Y, Beggs S, Sengar AS, Ellis J, Salter MW. Fyn Kinase regulates GluN2B subunit-dominant NMDA receptors in human induced pluripotent stem cell-derived neurons. Sci Rep. 2016;6:23837. doi: 10.1038/srep23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 182.Karim F, Wang CC, Gereau RW. Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der MC, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hu HJ, Gereau RW. ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol. 2003;90:1680–1688. doi: 10.1152/jn.00341.2003. [DOI] [PubMed] [Google Scholar]
- 186.Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hu HJ, Glauner KS, Gereau RW. ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J Neurophysiol. 2003;90:1671–1679. doi: 10.1152/jn.00340.2003. [DOI] [PubMed] [Google Scholar]
- 188.Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW. The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 189.Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, Serhan CN, Ji RR. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med. 2010;16:592–7. 1p. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- 192.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 193.Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yang Q, Wu Z, Hadden JK, Odem MA, Zuo Y, Crook RJ, Frost JA, Walters ET. Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci. 2014;34:10765–9. doi: 10.1523/JNEUROSCI.5316-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 196.Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhou LJ, Liu XG. Glial Activation, A Common Mechanism Underlying Spinal Synaptic Plasticity? Neurosci Bull. 2016 doi: 10.1007/s12264-016-0091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wang L, Bauer M, Curry R, Larsson A, Sessler DI, Eisenach JC. Intrathecal ketorolac does not improve acute or chronic pain after hip arthroplasty: a randomized controlled trial. J Anesth. 2014;28:790–3. doi: 10.1007/s00540-014-1798-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci. 2002;5:34–40. doi: 10.1038/nn778. [DOI] [PubMed] [Google Scholar]
- 200.DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- 201.Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schafers M, Geis C, Svensson CI, Luo ZD, Sommer C. Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003;17:791–804. doi: 10.1046/j.1460-9568.2003.02504.x. [DOI] [PubMed] [Google Scholar]
- 203.Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006;123:306–321. doi: 10.1016/j.pain.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 204.Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, Ji RR. Resolving TRPV1- and TNF-a-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci. 2011;31:15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J Neurosci. 2010;30:12844–12855. doi: 10.1523/JNEUROSCI.2437-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain. 2011;152:419–427. doi: 10.1016/j.pain.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, Lao L. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118:125–136. doi: 10.1016/j.pain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 208.Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci. 2008;28:12775–12787. doi: 10.1523/JNEUROSCI.3512-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li WW, Guo TZ, Liang D, Shi X, Wei T, Kingery WS, Clark JD. The NALP1 inflammasome controls cytokine production and nociception in a rat fracture model of complex regional pain syndrome. Pain. 2009;147:277–286. doi: 10.1016/j.pain.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135:232–239. doi: 10.1016/j.pain.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yan X, Weng HR. Endogenous interleukin-1beta in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic NMDA receptors. J Biol Chem. 2013 doi: 10.1074/jbc.M113.495465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–2197. doi: 10.1016/j.pain.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, LaMotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U SA. 2005;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Xie RG, Gao YJ, Park CK, Lu N, Luo C, Wang WT, Wu SX, Ji RR. Spinal CCL2 Promotes Central Sensitization, Long-Term Potentiation, and Inflammatory Pain via CCR2: Further Insights into Molecular, Synaptic, and Cellular Mechanisms. Neurosci Bull. 2017 doi: 10.1007/s12264-017-0106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sun Y, Sahbaie P, Liang DY, Li WW, Li XQ, Shi XY, Clark JD. Epigenetic regulation of spinal CXCR2 signaling in incisional hypersensitivity in mice. Anesthesiology. 2013;119:1198–208. doi: 10.1097/ALN.0b013e31829ce340. [DOI] [PubMed] [Google Scholar]
- 218.Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 219.Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–269. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Slack SE, Pezet S, McMahon SB, Thompson SW, Malcangio M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20:1769–1778. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- 223.Ferrini F, Trang T, Mattioli TA, Laffray S, Del’guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De KY. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013 doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sandkuhler J. Learning and memory in pain pathways. Pain. 2000;88:113–118. doi: 10.1016/S0304-3959(00)00424-3. [DOI] [PubMed] [Google Scholar]
- 225.Sandkuhler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- 226.Ruscheweyh R, Wilder-Smith O, Drdla R, Liu XG, Sandkuhler J. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Mol Pain. 2011;7:20. doi: 10.1186/1744-8069-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang HM, Zhou LJ, Hu XD, Hu NW, Zhang T, Liu XG. Acute nerve injury induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn of intact rat. Sheng Li Xue Bao. 2004;56:591–596. [PubMed] [Google Scholar]
- 228.Xu ZZ, Liu XJ, Berta T, Park CK, Lu N, Serhan CN, Ji RR. Neuroprotectin/Protectin D1 protects neuropathic pain in mice after nerve trauma. Ann Neurol. 2013 doi: 10.1002/ana.23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Drdla R, Gassner M, Gingl E, Sandkuhler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. doi: 10.1126/science.1171759. [DOI] [PubMed] [Google Scholar]
- 230.Bonin RP, De Koninck Y. A spinal analog of memory reconsolidation enables reversal of hyperalgesia. Nat Neurosci. 2014;17:1043–5. doi: 10.1038/nn.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu YL, Zhou LJ, Hu NW, Xu JT, Wu CY, Zhang T, Li YY, Liu XG. Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52:708–715. doi: 10.1016/j.neuropharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 232.Gruber-Schoffnegger D, Drdla-Schutting R, Honigsperger C, Wunderbaldinger G, Gassner M, Sandkuhler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-alpha and IL-1beta is mediated by glial cells. J Neurosci. 2013;33:6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Chirila AM, Brown TE, Bishop RA, Bellono NW, Pucci FG, Kauer JA. Long-term potentiation of glycinergic synapses triggered by interleukin 1beta. Proc Natl Acad Sci U SA. 2014;111:8263–8268. doi: 10.1073/pnas.1401013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkuhler J. Selective activation of microglia facilitates synaptic strength. J Neurosci. 2015;35:4552–70. doi: 10.1523/JNEUROSCI.2061-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kronschlager MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkuhler J. Gliogenic LTP spreads widely in nociceptive pathways. Science. 2016;354:1144–1148. doi: 10.1126/science.aah5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bliss TV, Collingridge GL, Kaang BK, Zhuo M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci. 2016;17:485–96. doi: 10.1038/nrn.2016.68. [DOI] [PubMed] [Google Scholar]
- 237.Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol. 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG. TNF-alpha Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J Neurosci. 2017;37:871–881. doi: 10.1523/JNEUROSCI.2235-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001;297:1210–1217. [PubMed] [Google Scholar]
- 240.Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain. 2003;104:655–664. doi: 10.1016/S0304-3959(03)00138-6. [DOI] [PubMed] [Google Scholar]
- 241.Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol. 2012;234:340–350. doi: 10.1016/j.expneurol.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 242.Anand P, Shenoy R, Palmer JE, Baines AJ, Lai RY, Robertson J, Bird N, Ostenfeld T, Chizh BA. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur J Pain. 2011;15:1040–1048. doi: 10.1016/j.ejpain.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 243.Ostenfeld T, Krishen A, Lai RY, Bullman J, Baines AJ, Green J, Anand P, Kelly M. Analgesic efficacy and safety of the novel p38 MAP kinase inhibitor, losmapimod, in patients with neuropathic pain following peripheral nerve injury: a double-blind, placebo-controlled study. Eur J Pain. 2013;17:844–857. doi: 10.1002/j.1532-2149.2012.00256.x. [DOI] [PubMed] [Google Scholar]
- 244.Tong SE, Daniels SE, Black P, Chang S, Protter A, Desjardins PJ. Novel p38alpha mitogen-activated protein kinase inhibitor shows analgesic efficacy in acute postsurgical dental pain. J Clin Pharmacol. 2012;52:717–728. doi: 10.1177/0091270011405496. [DOI] [PubMed] [Google Scholar]
- 245.Sainoh T, Orita S, Miyagi M, Inoue G, Kamoda H, Ishikawa T, Yamauchi K, Suzuki M, Sakuma Y, Kubota G, Oikawa Y, Inage K, Sato J, Nakata Y, Nakamura J, Aoki Y, Toyone T, Takahashi K, Ohtori S. Single Intradiscal Administration of the Tumor Necrosis Factor-Alpha Inhibitor, Etanercept, for Patients with Discogenic Low Back Pain. Pain Med. 2016;17:40–5. doi: 10.1111/pme.12892. [DOI] [PubMed] [Google Scholar]
- 246.Freeman BJ, Ludbrook GL, Hall S, Cousins M, Mitchell B, Jaros M, Wyand M, Gorman JR. Randomized, double-blind, placebo-controlled, trial of transforaminal epidural etanercept for the treatment of symptomatic lumbar disc herniation. Spine (Phila Pa 1976) 2013;38:1986–94. doi: 10.1097/01.brs.0000435140.61593.4c. [DOI] [PubMed] [Google Scholar]
- 247.Terkeltaub R, Sundy JS, Schumacher HR, Murphy F, Bookbinder S, Biedermann S, Wu R, Mellis S, Radin A. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68:1613–7. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Roerink ME, Bredie SJH, Heijnen M, Dinarello CA, Knoop H, Van der Meer JWM. Cytokine Inhibition in Patients With Chronic Fatigue Syndrome: A Randomized Trial. Ann Intern Med. 2017;166:557–564. doi: 10.7326/M16-2391. [DOI] [PubMed] [Google Scholar]
- 249.Wang X, Zhang Y, Peng Y, Hutchinson MR, Rice KC, Yin H, Watkins LR. Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br J Pharmacol. 2016;173:856–69. doi: 10.1111/bph.13394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Younger J, Mackey S. Fibromyalgia symptoms are reduced by low-dose naltrexone: a pilot study. Pain Med. 2009;10:663–72. doi: 10.1111/j.1526-4637.2009.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Echeverry S, Shi XQ, Yang M, Huang H, Wu Y, Lorenzo LE, Perez-Sanchez J, Bonin RP, De Koninck Y, Zhang J. Spinal microglia are required for long-term maintenance of neuropathic pain. Pain. 2017;158:1792–1801. doi: 10.1097/j.pain.0000000000000982. [DOI] [PubMed] [Google Scholar]
- 252.Frampton M, Harvey RJ, Kirchner V. Propentofylline for dementia. Cochrane. Database Syst Rev. 2003:CD002853. doi: 10.1002/14651858.CD002853. [DOI] [PubMed] [Google Scholar]
- 253.Yekkirala AS, Roberson DP, Bean BP, Woolf CJ. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov. 2017;16:545–564. doi: 10.1038/nrd.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci. 2009;10:283–294. doi: 10.1038/nrn2606. [DOI] [PubMed] [Google Scholar]
- 255.King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, Matsumiya L, Sorge RE, Sotocinal SG, Tabaka JM, Wong D, van den Maagdenberg AM, Ferrari MD, Craig KD, Mogil JS. Coding of facial expressions of pain in the laboratory mouse. Nat Methods. 2010;7:447–449. doi: 10.1038/nmeth.1455. [DOI] [PubMed] [Google Scholar]
- 257.Rosenberg MB, Carroll FI, Negus SS. Effects of monoamine reuptake inhibitors in assays of acute pain-stimulated and pain-depressed behavior in rats. J Pain. 2013;14:246–259. doi: 10.1016/j.jpain.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Cobos EJ, Ghasemlou N, Araldi D, Segal D, Duong K, Woolf CJ. Inflammation-induced decrease in voluntary wheel running in mice: a nonreflexive test for evaluating inflammatory pain and analgesia. Pain. 2012;153:876–884. doi: 10.1016/j.pain.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011 doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Huang L, Wang CF, Serhan CN, Strichartz G. Enduring prevention and transient reduction of postoperative pain by intrathecal resolvin D1. Pain. 2011;152:557–565. doi: 10.1016/j.pain.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M, Petasis NA. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012;26:1755–1765. doi: 10.1096/fj.11-201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Park CK, Xu ZZ, Liu T, Lu N, Serhan CN, Ji RR. Resolvin d2 is a potent endogenous inhibitor for transient receptor potential subtype v1/a1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin d1, d2, and e1. J Neurosci. 2011;31:18433–18438. doi: 10.1523/JNEUROSCI.4192-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Svensson CI, Zattoni M, Serhan CN. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med. 2007;204:245–252. doi: 10.1084/jem.20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wang JC, Strichartz GR. Prevention of Chronic Post-Thoracotomy Pain in Rats By Intrathecal Resolvin D1 and D2: Effectiveness of Perioperative and Delayed Drug Delivery. J Pain. 2017 doi: 10.1016/j.jpain.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Terrando N, Gomez-Galan M, Yang T, Carlstrom M, Gustavsson D, Harding RE, Lindskog M, Eriksson LI. Aspirin-triggered resolvin D1 prevents surgery-induced cognitive decline. FASEB J. 2013;27:3564–71. doi: 10.1096/fj.13-230276. [DOI] [PubMed] [Google Scholar]
- 267.Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367:1618–1625. doi: 10.1016/S0140-6736(06)68700-X. [DOI] [PubMed] [Google Scholar]
- 268.Chen G, Park CK, Xie RG, Ji RR. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-beta secretion. J Clin Invest. 2015;125:3226–3240. doi: 10.1172/JCI80883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Guo W, Wang H, Zou S, Gu M, Watanabe M, Wei F, Dubner R, Huang GT, Ren K. Bone marrow stromal cells produce long-term pain relief in rat models of persistent pain. Stem Cells. 2011;29:1294–1303. doi: 10.1002/stem.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Braz JM, Sharif-Naeini R, Vogt D, Kriegstein A, Alvarez-Buylla A, Rubenstein JL, Basbaum AI. Forebrain GABAergic neuron precursors integrate into adult spinal cord and reduce injury-induced neuropathic pain. Neuron. 2012;74:663–675. doi: 10.1016/j.neuron.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Lee MJ, Yoon TG, Kang M, Kim HJ, Kang KS. Effect of subcutaneous treatment with human umbilical cord blood-derived multipotent stem cells on peripheral neuropathic pain in rats. Korean J Physiol Pharmacol. 2017;21:153–160. doi: 10.4196/kjpp.2017.21.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Willemen HL, Eijkelkamp N, Garza Carbajal A, Wang H, Mack M, Zijlstra J, Heijnen CJ, Kavelaars A. Monocytes/Macrophages control resolution of transient inflammatory pain. J Pain. 2014;15:496–506. doi: 10.1016/j.jpain.2014.01.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Li Y, Dougherty PM, Heijnen CJ, Kavelaars A. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J Neurosci. 2016;36:11074–11083. doi: 10.1523/JNEUROSCI.3708-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Barreto A, Braun TR. A method to induce Interleukin-1 Receptor Antagonist Protein from autologous whole blood. Cytokine. 2016;81:137–41. doi: 10.1016/j.cyto.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 275.Meijer H, Reinecke J, Becker C, Tholen G, Wehling P. The production of anti-inflammatory cytokines in whole blood by physico-chemical induction. Inflamm Res. 2003;52:404–7. doi: 10.1007/s00011-003-1197-1. [DOI] [PubMed] [Google Scholar]
- 276.Shen L, Yuan T, Chen S, Xie X, Zhang C. The temporal effect of platelet-rich plasma on pain and physical function in the treatment of knee osteoarthritis: systematic review and meta-analysis of randomized controlled trials. J Orthop Surg Res. 2017;12:16. doi: 10.1186/s13018-017-0521-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Moreno-Duarte I, Morse LR, Alam M, Bikson M, Zafonte R, Fregni F. Targeted therapies using electrical and magnetic neural stimulation for the treatment of chronic pain in spinal cord injury. Neuroimage. 2014;85(Pt 3):1003–13. doi: 10.1016/j.neuroimage.2013.05.097. [DOI] [PubMed] [Google Scholar]
- 278.Shamji MF, De Vos C, Sharan A. The Advancing Role of Neuromodulation for the Management of Chronic Treatment-Refractory Pain. Neurosurgery. 2017;80:S108–S113. doi: 10.1093/neuros/nyw047. [DOI] [PubMed] [Google Scholar]
- 279.Pawela CP, Kramer JM, Hogan QH. Dorsal root ganglion stimulation attenuates the BOLD signal response to noxious sensory input in specific brain regions: Insights into a possible mechanism for analgesia. Neuroimage. 2017;147:10–18. doi: 10.1016/j.neuroimage.2016.11.046. [DOI] [PubMed] [Google Scholar]
- 280.Ji RR, Schlaepfer TE, Aizenman CD, Epstein CM, Qiu D, Huang JC, Rupp F. Repetitive transcranial magnetic stimulation activates specific regions in rat brain. Proc Natl Acad Sci U SA. 1998;95:15635–15640. doi: 10.1073/pnas.95.26.15635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Yang F, Zhang T, Tiwari V, Shu B, Zhang C, Wang Y, Vera-Portocarrero LP, Raja SN, Guan Y. Effects of Combined Electrical Stimulation of the Dorsal Column and Dorsal Roots on Wide-Dynamic-Range Neuronal Activity in Nerve-Injured Rats. Neuromodulation. 2015;18:592–7. doi: 10.1111/ner.12341. discussion 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Zhang TC, Janik JJ, Peters RV, Chen G, Ji RR, Grill WM. Spinal sensory projection neuron responses to spinal cord stimulation are mediated by circuits beyond gate control. J Neurophysiol. 2015;114:284–300. doi: 10.1152/jn.00147.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Han JS. Acupuncture: neuropeptide release produced by electrical stimulation of different frequencies. Trends Neurosci. 2003;26:17–22. doi: 10.1016/s0166-2236(02)00006-1. [DOI] [PubMed] [Google Scholar]
- 284.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–62. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 285.Steinberg BE, Sundman E, Terrando N, Eriksson LI, Olofsson PS. Neural Control of Inflammation: Implications for Perioperative and Critical Care. Anesthesiology. 2016;124:1174–89. doi: 10.1097/ALN.0000000000001083. [DOI] [PubMed] [Google Scholar]
- 286.Goldman N, Chen M, Fujita T, Xu Q, Peng W, Liu W, Jensen TK, Pei Y, Wang F, Han X, Chen JF, Schnermann J, Takano T, Bekar L, Tieu K, Nedergaard M. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat Neurosci. 2010;13:883–888. doi: 10.1038/nn.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Torres-Rosas R, Yehia G, Pena G, Mishra P, del Rocio Thompson-Bonilla M, Moreno-Eutimio MA, Arriaga-Pizano LA, Isibasi A, Ulloa L. Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat Med. 2014;20:291–5. doi: 10.1038/nm.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Maixner W, Randich A. Role of the right vagal nerve trunk in antinociception. Brain Res. 1984;298:374–7. doi: 10.1016/0006-8993(84)91441-0. [DOI] [PubMed] [Google Scholar]
- 289.Fang JF, Du JY, Shao XM, Fang JQ, Liu Z. Effect of Electroacupuncture on the NTS is modulated primarily by acupuncture point selection and stimulation frequency in normal rats. BMC Complement Altern Med. 2017;17:182. doi: 10.1186/s12906-017-1690-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lim HD, Kim MH, Lee CY, Namgung U. Anti-Inflammatory Effects of Acupuncture Stimulation via the Vagus Nerve. PLoS One. 2016;11:e0151882. doi: 10.1371/journal.pone.0151882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.da Silva MD, Bobinski F, Sato KL, Kolker SJ, Sluka KA, Santos AR. IL-10 cytokine released from M2 macrophages is crucial for analgesic and anti-inflammatory effects of acupuncture in a model of inflammatory muscle pain. Mol Neurobiol. 2015;51:19–31. doi: 10.1007/s12035-014-8790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–9. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 293.Wei XH, Wei X, Chen FY, Zang Y, Xin WJ, Pang RP, Chen Y, Wang J, Li YY, Shen KF, Zhou LJ, Liu XG. The upregulation of translocator protein (18 kDa) promotes recovery from neuropathic pain in rats. J Neurosci. 2013;33:1540–51. doi: 10.1523/JNEUROSCI.0324-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Volkow ND, Collins FS. The Role of Science in Addressing the Opioid Crisis. N Engl J Med. 2017;377:391–394. doi: 10.1056/NEJMsr1706626. [DOI] [PubMed] [Google Scholar]