

Abstract
In addition to dopaminergic and motor deficits, patients with Parkinson’s disease (PD) suffer from non-motor symptoms, including early cognitive and social impairment, that do not respond well to dopaminergic therapy. Cholinergic deficits may contribute to these problems, but cholinesterase inhibitors have limited efficacy. Mice over-expressing α-synuclein, a protein critically associated with PD, show deficits in cognitive and social interaction tests, as well as a decrease in cortical acetylcholine. We have evaluated the effects of chronic administration of nicotine in mice over-expressing wild type human α-synuclein under the Thy1-promoter (Thy1-aSyn mice). Nicotine was administered subcutaneously by osmotic minipump for 6 months from 2 to 8 months of age at 0.4 mg/kg/h and 2.0 mg/kg/h. The higher dose was toxic in the Thy1-aSyn mice, but the low dose was well tolerated and both doses ameliorated cognitive impairment in Y-maze performance after 5 months of treatment. In a separate cohort of Thy1-aSyn mice, nicotine was administered at the lower dose for one month beginning at 5 months of age. This treatment partially eliminated the cognitive deficit in novel object recognition and social impairment. In contrast, chronic nicotine did not improve motor deficits after 2, 4 or 6 months of treatment, nor modified α-synuclein aggregation, tyrosine hydroxylase immunostaining, synaptic and dendritic markers, or microglial activation in Thy1-aSyn mice. These results suggest that cognitive and social impairment in synucleinopathies like PD may result from deficits in cholinergic neurotransmission and may benefit from chronic administration of nicotinic agonists.
Keywords: Parkinson’s disease, mouse model, α-synuclein, nicotine, motor deficits, cognitive deficits, social impairment
Introduction
In patients with Parkinson’s disease (PD), neurodegeneration primarily affects dopaminergic neurons of the substantia nigra (SN) of the midbrain, leading to dopamine loss in the striatum and dysregulation of neuronal circuits in the basal ganglia. This dopaminergic deficit is the primary cause of the characteristic motor symptoms of the disease, bradykinesia, resting tremor, and rigidity. However, the symptoms of PD extend beyond motor dysfunction and also involve non-motor aspects that include cognitive deficits, social impairment, sleep disturbances, and sensory and autonomic dysfunction (Anderson et al., 2013; Bassetti, 2011; Chaudhuri et al., 2011; Wolters, 2009). The mechanisms of these non-motor symptoms of PD are only partially understood. Dopamine depletion may play a role in non-motor deficits; but their early appearance, often before motor deficits, and their poor response to dopaminergic treatments suggest additional mechanisms (Abbott et al., 2007; Postuma et al., 2012; Ross et al., 2006).
α-Synuclein has emerged as a potential pathogenic factor in PD based on genetic evidence that mutation or gene duplication in the SNCA gene cause familial forms of PD, and polymorphisms are strongly associated with sporadic PD (Edwards et al., 2010; Kruger et al., 1998; Polymeropoulos et al., 1997; Singleton et al., 2003). Furthermore, α-synuclein is a component of Lewy bodies, a hallmark of PD, and forms aggregates that are broadly distributed in the central and peripheral nervous system (Bloch et al., 2006; Braak et al., 2006; Spillantini and Goedert, 2000; Spillantini et al., 1997). The hypothesis that α-synuclein pathology may contribute to non-motor deficits in PD is supported by experimental evidence that mouse models of α-synuclein over-expression display deficits in behaviors also affected in early PD (Chesselet et al., 2012; Hatami and Chesselet, 2015; Magen and Chesselet, 2011). This has been extensively characterized in mice over-expressing α-synuclein under the Thy1 promoter developed by Eliezer Masliah (Thy1-aSyn mice; line 61) (Rockenstein et al., 2002). These mice express human wild-type α-synuclein at 2-4-fold higher levels than endogenous α-synuclein and exhibit broadly distributed α-synuclein aggregates throughout the brain. They also exhibit extensive behavioral deficits in many domains affected in pre-manifest PD, including cognition, sleep, social interaction and autonomic function, as well as inflammation, mitochondrial dysfunction, and later loss of striatal dopamine. Thus, α-synuclein line 61 mice recapitulate many aspects of sporadic PD (Chesselet et al., 2012; Magen et al., 2015; McDowell et al., 2014; Subramaniam et al., 2014).
In previous studies (Magen et al., 2012), we have detected a decrease in acetylcholine in the cortex of Thy1-aSyn mice at an age when cognitive deficits were present, suggesting that α-synuclein pathology is sufficient to induce acetylcholine deficits analogous to those reported in PD patients (Pfeiffer et al., 2014; Yang et al., 2016; Zarow et al., 2003). Cholinesterase inhibitors are clinically used for cognitive deficits in PD patients, on the assumption that the inhibitors primarily exert any therapeutic effects by inhibiting the hydrolysis of acetylcholine to choline; but these drugs show limited efficacy (Emre et al., 2004; Grace et al., 2009; Hanagasi et al., 2017; Mamikonyan et al., 2015; Wang et al., 2015). Stimulating nicotinic receptors may be a better approach to treat cognitive disorders in a variety of disorders (Gee et al., 2017; Harris et al., 2004; Preskorn et al., 2014; Wallace and Porter, 2011). However, there is little preclinical information on the ability of nicotine to improve deficits due to α-synuclein pathology as present in PD or other synucleinopathies.
In the current study we use the Thy1-aSyn mouse model to test the hypothesis that chronic nicotine administration could improve deficits in cognition and social behavior induced by α-synuclein pathology. The aim of this study was to provide preclinical data to support or invalidate the development of nicotinic agents to treat aspects of PD. We administered nicotine subcutaneously by osmotic minipump either for either 6 months (2-8 months of age) or one month (5-6 months of age) to male Thy1-aSyn mice and measured their performance in tests of cognition and social interaction. In addition, because previous studies in toxin-induced animal models of dopamine degeneration have proposed a protective role of nicotine in PD (Costa et al., 2001; Picciotto and Zoli, 2008; Quik et al., 2012; Ryan et al., 2001; Toulorge et al., 2011), we assessed the effects of nicotine administration on early motor deficits and α-synuclein pathology in these mice. The results show that chronic nicotine administration improves cognitive and social impairments without affecting motor deficits or α-synuclein aggregates in Thy1-aSyn mice.
Materials and Methods
Animals
Mice over-expressing human, full length, wild type (Wt) α-synuclein under the murine Thy1-promoter (Thy1-aSyn mice) were developed at the laboratory of Dr. E. Masliah (University of California, San Diego) (‘line 61” of (Rockenstein et al., 2002). Thy1-aSyn mice were maintained on a hybrid C57BL6/DBA2 background as described previously (Fleming et al., 2004; Rockenstein et al., 2002). The genotypes of all Thy1-aSyn and Wt mice were determined at 1 month of age and confirmed at the end of the experiment by polymerase chain reaction (PCR) amplification analysis of tail DNA. We studied male mice to maintain gene dosage, because the transgene is inserted in the X chromosome in this line. Transgenics and their Wt littermates were distributed among the study groups to avoid any litter effect, and the group sizes were determined based on our power analysis (Chesselet et al., 2012). Animals were socially housed ≤ 4 in each cage and maintained on a reverse light/dark cycle to facilitate behavioral analysis. Mice were housed on standard bedding (shredded wood) and material for nest-building (Nestlets, Ancare, NY, USA) was provided. Mice had ad libitum access to water and standard rodent diet (NIH-31 modified 7013, Harlan). Room temperature in the mouse holding room was 23 ± 2°C and relative humidity was ~60%; values were recorded during the daily animal check. Animal care complied with the United States Public Health Service Guide for the Care and Use of Laboratory Animals and all experimental procedures were approved by the UCLA Institutional Care and Use Committee.
Nicotine administration: first (“six-month”) cohort
Two separate cohorts of male mice were used (Fig. 1). A first cohort was treated with nicotine for 6 months. Thy1-aSyn mice were divided into three groups (minimum of n=20 per group) and treated with vehicle (saline; Teknova, USA; #S5819) supplemented with either zero, 0.4, or 2.0 mg/kg/h nicotine (calculated as the free base, prepared from nicotine tartrate, Sigma, USA; #N5260). A fourth group, Wt littermates (minimum of n=20), were treated with vehicle plus zero nicotine, serving as additional controls. Treatment began at 2 months of age. Solutions could be infused subcutaneously for one month using implanted osmotic minipumps (Alzet, USA; Model 2004). Briefly, vehicle, with or without nicotine, was loaded into the minipump and maintained at 37°C for 48 h before the implantation surgery. For the initial implant surgeries (at 2 months), mice were anesthetized with isoflurane. A subcutaneous pocket was created in the dorsal thoracic area and received the minipump. Mice treated with the higher nicotine dose (2.0 mg/kg/h) lost weight after this surgery; 50% died after one week. Because the toxicity in the high dose nicotine (2.0 mg/kg/h) group might have been due to the combined toxic effects of isoflurane and a higher dose of nicotine, we used pentobarbital instead of isoflurane for induction of anesthesia for subsequent surgeries in this cohort. Briefly, anesthesia was induced with pentobarbital (6.6 mg/kg, i.p.) and a surgical level of anesthesia was maintained with isoflurane (0.5-1.5% in O2). The use of combined pentobarbital and isoflurane markedly reduced the mortality rate and improved weight gain in the higher dose group. At one month intervals, fresh osmotic mini-pumps were loaded with nicotine solutions adjusted for body weight, and were implanted at the same subcutaneous pocket.
Figure 1. Schematic illustration of the timeline in all experiments.
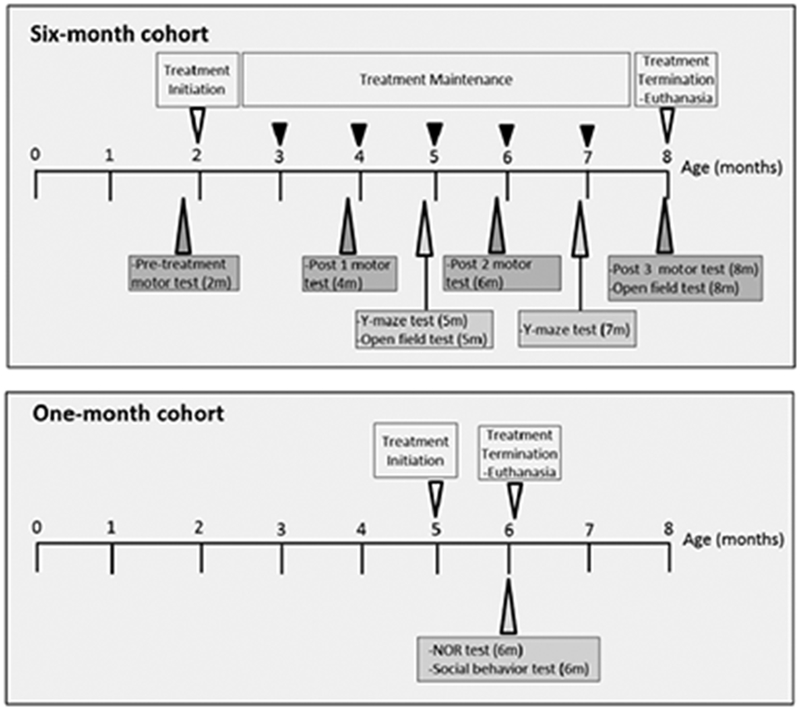
Treatment initiation- osmotic mini-pump implantation subcutaneously for one month; Treatment maintenance- reimplantation of osmotic mini-pump in the same subcutaneous pocket with vehicle or nicotine adjusted for body weight; motor test – challenging beam test and vertical pole test; Y-maze test - Y-maze spontaneous alternations test; NOR test- novel object recognition test; m – months.
For this cohort, we intended to treat for 6 months. The full time course was achieved for the lower nicotine dose of 0.4 mg/kg/h. However, Thy1-aSyn mice treated with the higher nicotine dose, 2.0 mg/kg/h nicotine, died at an unexpectedly high rate: 50% mortality was observed in this group after four months of nicotine administration. We minimized pain and suffering by euthanizing the remaining members of the Thy1-aSyn higher dose group at 6 months of age (4 months of treatment). It must be noted that a few mice (n=3) in the higher dose group completed full time course of treatment before a decision was made to shorten their treatment.
Nicotine administration: second (“one-month”) cohort
Experience gained with the first cohort allowed us to minimize animal use, pain, and suffering in a second cohort. Treatment began at 5 months of age. We used the pentobarbital-isoflurane anesthesia protocol. Thy1-aSyn mice and their Wt littermates were divided into two groups each (minimum of n=20 per group) and received either vehicle or the lower nicotine dose, 0.4 mg/kg/h. Each mouse received a single minipump, for a single month of treatment.
For both the first (six-month) and the second (one-month) cohort, general health was monitored daily. Body weights were monitored and recorded daily for a week after implant surgery and twice weekly thereafter. Mice receiving the lower nicotine dose (0.4 mg/kg/h) showed no differences in general health compared to mice administered with vehicle. Above, we summarize health issues in the first cohort, exposed to 2.0 mg/kg/h nicotine.
Behavioral testing
All behavioral testing was performed (Fig. 1) and analyzed by investigators without knowledge of genotype and drug treatment. Body weights were recorded before the behavioral tests to control for any confounding effects of differences in weight on measures of activity. Motoric behavior was tested in only the six-month treatment cohort, once before treatment at 2 months (pre-treatment) and then at 4 (Post 1), 6 (Post 2) and 8 months (Post 3) of age, on the challenging beam and the vertical pole as described previously (Fleming et al., 2004). Briefly, for challenging beam test, each mouse was trained on a narrowing horizontal beam for two days and tested on third day using the same narrowing horizontal beam but with grids placed on it for 5 trials. For the vertical pole test, each mouse was trained for two days to turn and climbdown the vertical pole without falling, and tested on third day for 5 trials. Each mouse was also tested for general locomotor activity in the open field (15 min) at 5 months of age and 8 months of age as described previously (Lam et al., 2011; Wang et al., 2012).
Cognitive behavior testing was performed in the six-month treatment cohort mice at ages 5 months and 7 months (Y-maze spontaneous alternations). In one-month treatment cohort novel object recognition and social behavior tests was performed at 6 months of age.
Y-maze spontaneous alteration and novel object recognition were assessed as described previously (Magen et al., 2012). Briefly, mice were allowed to explore the Y-maze for 7 min, while the visits to each arm were counted. Successive visits to the three different arms were considered as an alternation and percentage of spontaneous alternations were calculated as alternations/(arm entries-2)*100 – 50% to set chance level at zero. Mice which had < 7 visits in the 7-minute tested period were excluded from analysis as non-performers, slightly modified from (Lelong-Boulouard et al., 2006), who set the exclusion criteria at 5 visits for 5 min.
For novel object recognition (NOR) test, mice were first habituated to the cylindrical tank for 5 min the day prior to the testing day. On the testing day mice were exposed to two identical objects (pig or wolf figurines) for 10 min and after a 30-40 min interim interval one of the objects was replaced with a novel object. During exploration, the mice were videotaped, and later analyzed in a blinded fashion. The objects were balanced among different mice to avoid any object preference bias. Mice with total exploration time of < 7 s, indicating low interest in the objects, were excluded from all further analyses (de Bruin and Pouzet, 2006). The discrimination index was defined as DI = (N − F) / (N + F), where N and F represents total time spent exploring the novel and familiar objects, respectively.
The social approach task was performed in only the one-month treatment cohort at 6 months of age as described before (Magen et al., 2015). Briefly, mice were first habituated to the center chamber of a three-chamber Perspex® apparatus with the two side chambers closed, for 10 min on the testing day (phase I). Thereafter, the doors connecting the chambers were opened and the mice were now allowed to explore all three chambers for 10 min (phase II). In the final phase of the test (phase III), an empty 1 L glass beaker was placed in one of the side chambers whereas the same beaker containing a novel mouse from the 129Sv strain was placed in the other side chamber. Locations of empty beaker and beaker containing a mouse were balanced to avoid potential confounding effect of baseline side preference. Mice were allowed to explore all three chambers as in phase II. The time spent in the side chambers in phase III was recorded during testing, while the sniffing times at the empty beaker and at the beaker containing the mouse were measured from video recordings by a blinded observer.
Euthanasia and immunohistology
The measurement of pathological endpoints by immunohistology was performed in only the six-month treatment cohort. After the treatment phase, 50% of the mice (n=10) from each group were anesthetized with pentobarbital, perfused shortly with PBS and then with 4% paraformaldehyde. Their brains were processed for histological analysis. Because the nicotine treatment in the higher dose group was terminated early (at 6 months of age), the immunohistology results for this group were obtained in 6-month old mice.
Brain sections from the Wt and each transgenic group were reacted for α-synuclein with a mouse (anti-human and anti-mouse) α-synuclein monoclonal antibody (BD Transduction Laboratories, USA, 610787) at 1:250 dilution, for tyrosine hydroxylase with rabbit monoclonal anti-TH antibody (Millipore, USA; AB152) at 1:600 dilution, for synaptophysin with mouse monoclonal anti-synaptophysin antibody (Millipore, USA; MAB5258) at 1:2000 dilution, and for microtubule-associated protein 2 (MAP2) with rabbit polyclonal anti-MAP2 antibody (Millipore, USA; AB5622 at 1:500 dilution). Secondary antibodies conjugated with Alexa 488 or Alexa 594 were used for immunofluorescence labeling and quantified as described previously (Richter et al., 2014a). For the assessment of α-synuclein aggregates, the sections were treated with proteinase K and quantified as described previously (Fernagut et al., 2007; Fleming et al., 2011; Richter et al., 2014a). Microglial activation was quantified in SN using rabbit polyclonal antibody against ionized calcium binding adaptor molecule (anti-Iba-1) (Wako, Osaka, Japan, #019-19741, at 1:500 dilution) as described (Watson et al., 2012). All analyses were performed by investigators unaware of genotype and treatment groups.
Statistics
Statistical analysis was performed with SigmaPlot software (v. 12.0, Systat Inc., USA). For the analysis of the 6-month treatment cohort: (1) two-tailed Student’s t-test (when data were homoscedastic) or Mann Whitney U-test (when data were heteroscedastic) were used to compare Wt-vehicle and Thy1-aSyn-vehicle groups for genotype differences, (2) Analysis of variance (ANOVA) was used to compare the nicotine treatment effect in Thy1-aSyn groups, followed when appropriate with post hoc Bonferroni t-tests versus control (Thy1-aSyn-vehicle) group. For the analysis of 1-month treatment cohort, two-way ANOVA followed by post hoc Bonferroni t-tests were used. Data were tested for equal variances prior to ANOVA. In addition, for cognitive tests the data were compared to chance level (zero) by using one-sample t-test. A paired t-test was used to compare the effect on same groups at different timepoints. Outliers in all the analysis were excluded with Grubb’s outlier test. The investigator was blinded to the groups when assessing the outcome of the experiments.
Results
Nicotine effects on Y-maze alternation
Spontaneous Y-maze alternation test was performed at 5 and 7 months of age (3 and 5 months of nicotine or vehicle administration, respectively). The Thy1-aSyn-vehicle group performed significantly worse at 7 than at 5 months (paired t-test, t20=2.18, p=0.04) (Fig. 2). No significant performance deficit was noted in the Wt-vehicle mice. These data indicate the development of cognitive deficits with age in the Thy1-aSyn mice, consistent with previous results (Magen et al., 2012).
Figure 2. Nicotine treatment improves cognitive defects in Y-maze spontaneous alternation test at 7 months of age.
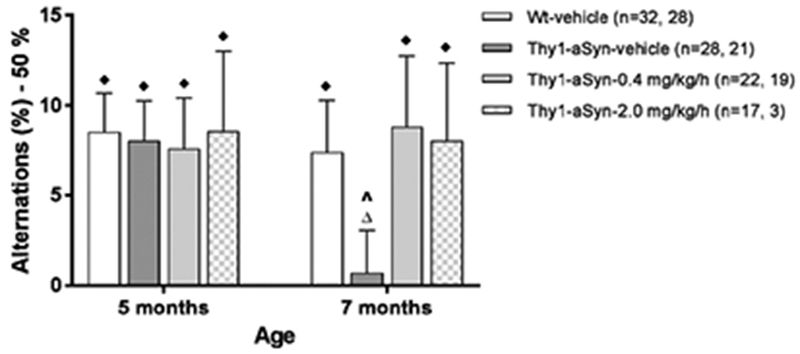
One-sample t test on percent alternations compared to chance level of zero was performed. Thy1-aSyn mice treated with vehicle showed decreased percentage of alternations at 7 months, but not at 5 months. Nicotine treatment at 0.4 mg/kg/h for 5 months (7 months of age) reversed the deficit. Data are represented as the mean ± SEM. ♦p<0.05, ∆not significant vs. chance level zero compared by one sample t test. A paired t test was also used to compare each group at 5 vs 7 months. ^p<0.05 compared to respective group at previous age.
Because several mice died between 5 and 7 months of age, we analyzed the data at each time point separately. At 5 months of age (Fig. 2), all groups displayed significantly more alternations than the chance level of zero percent (p<0.05, one-sample t-test), indicating intact alternation behavior and spatial memory. At 7 months of age, Wt-vehicle mice but not Thy1-aSyn-vehicle mice maintained this significantly higher than zero alternation percentage (one-sample t-test, p=0.02 and 0.77, respectively), indicating that Y-maze performance was impaired in Thy1-aSyn mice, as previously reported for that age (Magen et al., 2012). In contrast, Thy1-aSyn mice administered 0.4 mg/kg/h nicotine performed above chance (p=0.04), at the same level as Wt-vehicle mice, indicating that nicotine substantially reverses this impairment (Fig. 2). The total number of entries did not differ between the groups at any time point, indicating a similar level of activity (data not shown). The data indicate that 5 months of nicotine administration at 0.4 mg/kg/h improve cognitive performance, as tested by Y-maze alternation.
Thy1-aSyn mice treated with 2.0 mg/kg/h nicotine also displayed significantly higher than zero alternation percentage at 7 months. However, only three mice completed the full course of 6 months treatment (see methods).
Previous studies on Thy1-aSyn mice showed small deficits in the Y-maze at 5-6 months of age (Magen et al., 2012) but the magnitude of the deficit was much smaller than at 7-9 months of age, when an effect was also observed in the six-months treatment cohort in the present study. Similarly, in the one-month treatment cohort, ANOVA revealed no main effect of either genotype (F1,69=0.715, p=0.4) or 0.4 mg/kg/h nicotine treatment (F1,69=0.43, p=0.51), and also no interaction effect (F1,69=0.035, p=0.85). The percentage of spontaneous alternations for the groups are as follows: Wt-vehicle: 3.43±3.21%, n=19; Thy1-aSyn-vehicle: 6.92±3.92%, n=13; Wt-0.4 mg/kg/h nicotine: 1.84±3.38%, n=18; Thy1-aSyn-0.4 mg/kg/h nicotine: 4.07±3.15%, n=21 (Mean ± SEM).
Nicotine effects on NOR
In contrast to previous results obtained in naïve mice (Magen et al., 2012), we observed no deficit in NOR performed in the six-month treatment cohort at 7 months of age (after 5 months of nicotine or vehicle administration). The groups had discrimination index values as follows: Wt-vehicle: 0.1±0.08, n=18; Thy1-aSyn-vehicle: 0.08±0.08, n=18; Thy1-aSyn-0.4 mg/kg/h nicotine: 0.11±0.08, n=15; Thy1-aSyn-2.0 mg/kg/h nicotine: 0.29±0.21, n=3 (Mean ± SEM). Total exploration times of the objects did not differ between the groups, consistent with previous data (Magen et al., 2012). It is possible that the repeated surgeries and behavioral testing have led to altered performance in both Wt/vehicle and the Thy1-aSyn/vehicle groups, which were much lower than those previously observed in naïve mice (Magen et al., 2012) (and see below), thus impairing detection of a deficit in the transgenics.
In the one-month cohort tested for NOR, we administered the lower nicotine dose (0.4 mg/kg/h) to 5-month-old mice for only one month. In contrast to the 6-month cohort, which received multiple surgeries and behavioral testing, vehicle-treated Thy1-aSyn mice in this one-month cohort showed a significant deficit in NOR at 6 months of age compared to vehicle-treated Wt littermates, and this deficit was reversed in mice that received 0.4 mg/kg/h of nicotine (Fig. 3). The groups displayed DI values as follows (Mean ± SEM): Wt-vehicle: 0.28±0.11, n=17; Thy1-aSyn-vehicle: 0.00±0.09, n=12; Wt-0.4 mg/kg/h nicotine: 0.11±0.09, n=17; Thy1-aSyn-0.4 mg/kg/h nicotine: 0.23±0.07, n=19. Two-way ANOVA revealed no main effect of genotype (F1,59=0.677, p=0.414) or of treatment (F1,59=0.103, p=0.750) but showed an interaction effect (F1,59=4.35, p=0.042). The Wt-vehicle mice, but not the Thy1-aSyn-vehicle mice, displayed discrimination indices significantly higher than zero (p=0.027 vs p>0.99, respectively, one-sample t-test). In contrast, Thy1-aSyn/0.4 mg/kg/h nicotine mice performed significantly higher than zero (p=0.006) (Fig. 3). Thus, NOR performance was impaired in Thy1-aSyn mice, extending previous data at the earlier time of 4-5 months (Magen et al., 2012). Importantly, the present data show that treatment with 0.4 mg/kg/h nicotine for one month completely reversed this impairment. Unexpectedly, however, nicotine impaired performance in Wt mice, which no longer performed above chance level (p=0.25).
Figure 3. Nicotine treatment improves cognitive defects in novel object recognition test in Thy1-aSyn mice at 6 months of age.
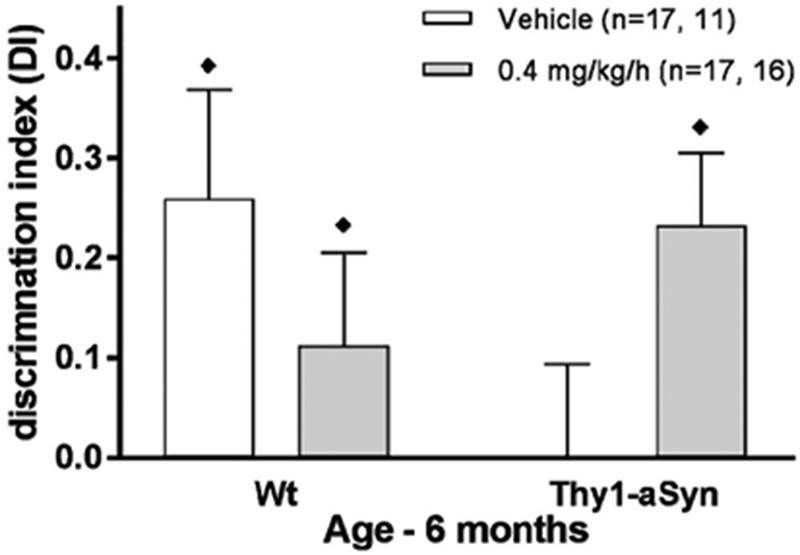
Thy1-aSyn mice treated with vehicle showed decreased discrimination index at 6 months of age. Nicotine treatment at 0.4 mg/kg/h for one month abolished this impairment. Data are presented as the mean ± SEM. ♦p<0.05 vs. chance level (zero) compared by one-sample t test.
Nicotine effects on social behavior
In our previous studies, we detected a deficit in a social approach task in Thy1-aSyn mice, suggesting a deficit akin to the impairment of theory of mind described in PD patients (Magen et al., 2015). This task was performed in the one-month nicotine treatment cohort at 6 months of age. As described in methods, we compared sniffing times at an empty beaker versus a beaker containing a mouse from another genetic background after appropriate habituation. Results in Wt mice revealed the expected strong preference for the beaker containing a mouse, whereas this preference was decreased in Thy1-aSyn mice receiving vehicle, confirming our previous data (Fig. 4). Repeated measure ANOVA with group as between-subject factor and variable (i.e. empty beaker vs. beaker with a mouse) as a within-subject factor revealed no main effect of group (F3,148=2.164, p=0.1) or interaction effect between group and variable (F3,148=0.603, p=0.615) but did reveal an effect for the sniffed item (F1,150=41.76, p<0.001). The two Wt groups showed a significant preference for exploring a beaker with a novel mouse over exploring an empty beaker (p<0.001), whereas the Thy1-aSyn/vehicle group showed no such preference (Bonferroni post-hoc test). Furthermore, Thy1-aSyn-vehicle mice sniffed a beaker with a novel mouse to a lesser extent than Wt/vehicle mice (p=0.044). Treatment with 0.4 mg/kg/h nicotine completely restored the preference for the mouse versus empty beaker in Thy1-aSyn mice (p<0.001). Comparing chamber time between the groups revealed no main effects (p>0.05) indicating that the mice had no preference for either chamber, an important control. The results confirm our previous findings (Magen et al., 2015) that Thy1-aSyn mice have a decreased interest in social stimuli compared to Wt mice while they explore a neutral, inanimate object to the same extent, indicating social cognition impairment rather than general exploratory deficit. This impairment was reversed by 0.4 mg/kg/h nicotine administration.
Figure 4. Nicotine treatment for one month improves social impairment in Thy1-aSyn mice tested at 6 months of age.
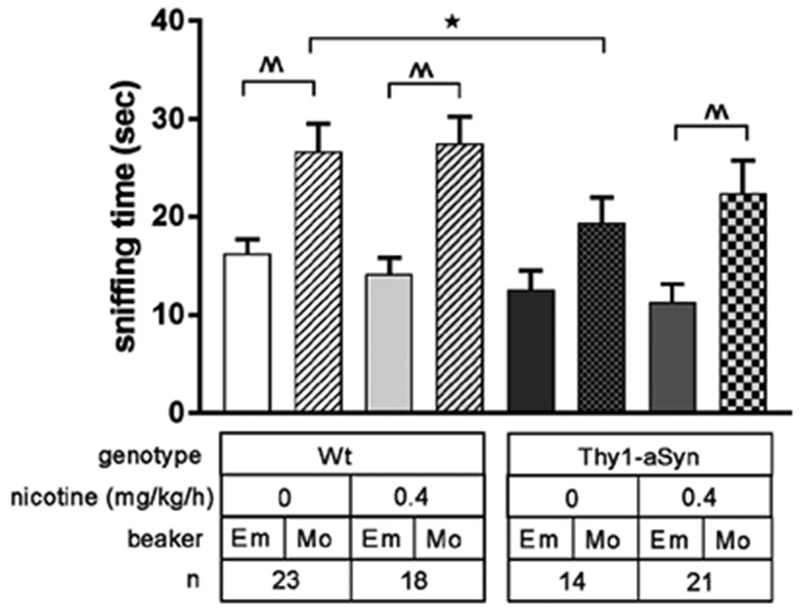
Thy1-aSyn mice treated with vehicle sniffed a beaker with a novel mouse (Mo) for less time than Wt mice treated with vehicle. Vehicle treated Thy1-aSyn mice did not show a preference for exploring a novel mouse (Mo) over an empty beaker (Em), but nicotine administration at 0.4 mg/kg/h for one month restored the interest in a novel mouse. Data are presented as mean ± SEM. Two-way repeated measures ANOVA followed by post hoc Bonferroni t test. *p<0.05, Thy1-aSyn-vehicle compared to Wt-vehicle; ^^p<0.01 compared within the group (empty beaker vs. mouse beaker).
Chronic nicotine treatment did not improve motor deficits in challenging beam and vertical pole tests
To evaluate the effect of nicotine treatment on α-synuclein-induced motor deficits, we tested the mice in the challenging beam and vertical pole tests at four time points including pre-treatment time point (2 months), and at 3 time points during nicotine administration called Post 1 (4 months), Post 2 (6 months) and Post 3 (8 months). Motor deficits in these challenging tests are robust and appear as early as 2 months in the Thy1-aSyn mice (Chesselet et al., 2012; Fleming et al., 2004). These deficits are distinct from parkinsonism, which occurs at much older ages when the Thy1-aSyn mice begin to show decreased striatal dopamine (Lam et al., 2011). They reflect impairment in nigrostriatal (Lam et al., 2011) and/or extranigral (Watson et al., 2009; Wu et al., 2010) circuits resulting from α-synuclein overexpression. Deficits in the challenging beam were confirmed in this study in 2-month-old transgenic mice, which show a large increase in number of errors per step (Fig. 5A) (Fleming et al., 2004). As previously reported, this motor deficit progressed between 2 and 8 months of age in the Thy1-aSyn mice (Fig. 5B).
Figure 5. Nicotine treatment does not improve motor performance in the challenging beam test.
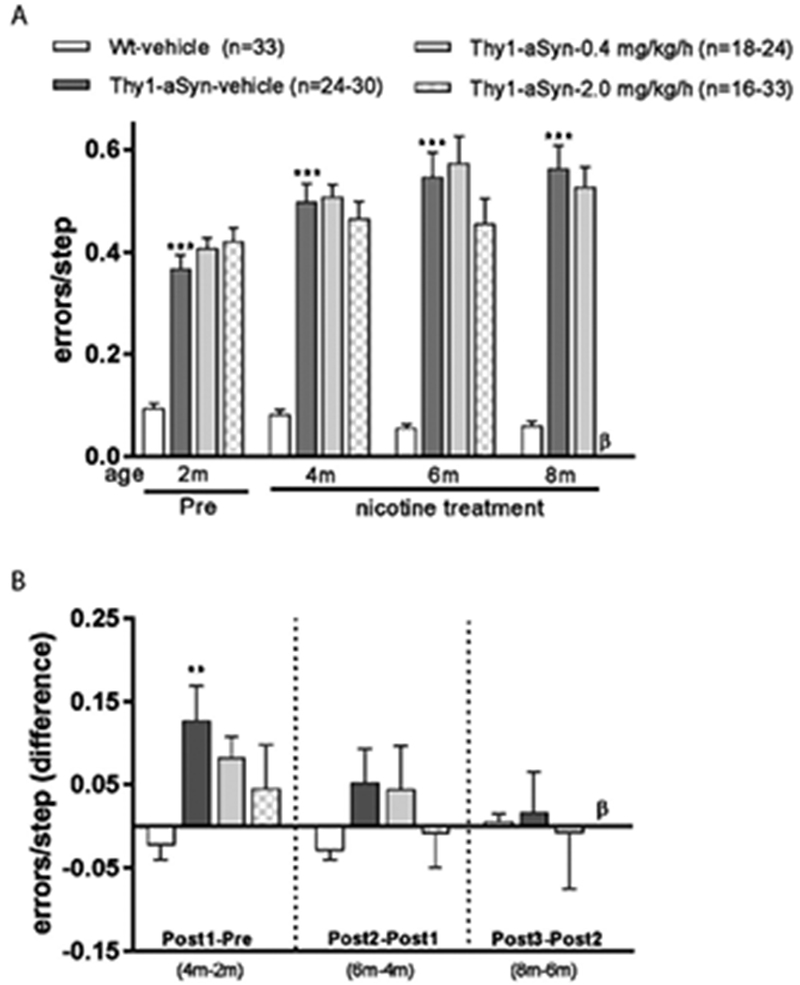
Thy1-aSyn mice treated with vehicle showed increased errors per step (A) and a progression of the defect from 2 months to 4 months of age (B) compared to Wt-vehicle treated mice. Nicotine treatment at both doses did not improve this defect initially (A) but partially inhibited the progression of this defect at 4 months of age (B). Data are presented as mean ± SEM; Student’s t test or the Mann Whitney U-test (see Methods) was performed to compare Wt-vehicle vs. Thy1-aSyn-vehicle within each time point, ***p<0.001, **p<0.01. One-way ANOVA followed by a Bonferroni t test was performed to compare the effect of treatment in Thy1-aSyn mice. β, Thy1-aSyn-nicotine-2.0 mg/kg/h group was euthanized before 8 months of age (see Methods).
Administration of neither the lower (0.4 mg/kg/h, 6 months) nor the higher (2.0 mg/kg/h, 4 months) nicotine dose, improved this motor deficit (Fig. 5A). To evaluate the effect of nicotine on the progression of beam deficits, we calculated the difference between the numbers of errors per step at each time point for each mouse. Thy1-aSyn mice show a significant worsening at 4 months compared to 2 months of age; this was not significantly affected by nicotine (Fig. 5B), despite the good power conferred by using relatively large groups of mice. The deficit on the beam leveled off at later time points; and again, nicotine had no effect. Thus, nicotine administration did not significantly improve deficits on the challenging beam when administered after this motor deficit was already established.
In the vertical pole, a test of balance and coordination, Thy1-aSyn mice take longer than Wt mice to turn around and descend the pole (Fig. 6A) (Fleming et al., 2004). Nicotine treatment, 0.4 mg/kg/h (for 6 months) and 2.0 mg/kg/h (for 4 months), did not improve this motor deficit at any time point tested (Fig. 6A). About 50% of the Thy1-aSyn mice were able to perform this task within the time limit at 2 months of age but only 25% at 4 months and almost none at later ages. Nicotine did not affect the number of mice able to perform the task (Fig. 6B). These results indicate that chronic nicotine administration failed to improve the progressive motor deficits observed in Thy1-aSyn mice, even after 6 months of continuous treatment.
Figure 6. Nicotine treatment does not improve motor performance in the vertical pole test.
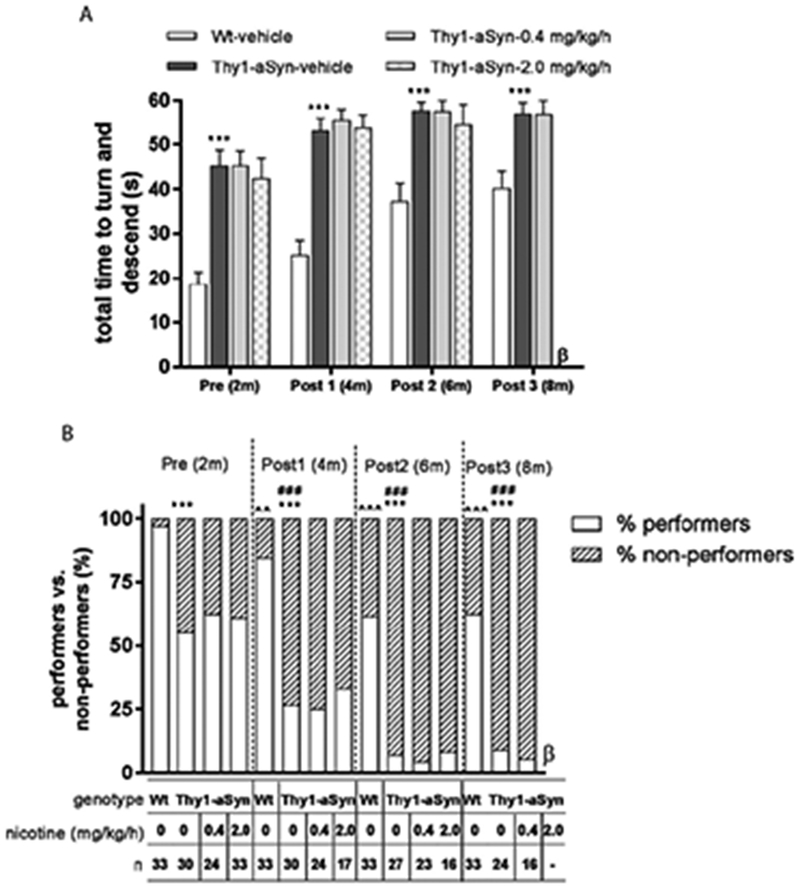
(A) Thy1-aSyn mice treated with vehicle showed increased total time to turn and descend compared to Wt-vehicle treated mice at the ages mentioned. Nicotine treatment did not improve this defect. Data are represented as the mean ± SEM; Student’s t test or the Mann Whitney U-test (see Methods) was performed to compare Wt-vehicle vs. Thy1-aSyn vehicle within each time point, ***p<0.001. One-way ANOVA followed by a Bonferroni t test was performed to compare the effect of treatment in Thy1-aSyn mice. (B) The percentage of mice performing the pole test was decreased by 6 months of age and neared zero in Thy1-aSyn mice. Data are presented as percentage performers vs. non-performers. Chi-square test was performed, ***p<0.001 compared with Wt mice treated with vehicle within the time point tested, ^^p<0.01, ^^^p<0.001 vs. Wt-vehicle group at pre-treatment, ###p<0.001 vs. Thy1-vehicle group at pre-treatment. β = Thy1-aSyn-nicotine-2.0 mg/kg/h group was euthanized before 8 months of age (see Methods).
Chronic nicotine treatment did not alter hyperactivity in the open field
Locomotor activity in open field was examined at 5 months (3 months of nicotine treatment) and 8 months (6 months of nicotine treatment) of age. We measured move episodes, total move time, move time per episode, total distance traveled, distance traveled per episode, movement velocity, center entries, and rearings. Previous studies by our group showed hyperactivity in relatively young (4-7 months of age) Thy1-aSyn mice (Lam et al., 2011; Wang et al., 2012). This observation was confirmed in the present cohort (Table 1): Thy1-aSyn mice receiving vehicle showed a decrease in move episodes (p<0.01) and an increase in move time per episode (p<0.001), as well as the other parameters measured, indicating hyperactivity compared to Wt vehicle treated mice at both time points. Several lines of transgenic mice over-expressing α-synuclein show hyperactivity at early ages (Chesselet et al., 2012) and we have previously attributed this hyperactivity to increased extracellular dopamine in the striatum of young Thy1-aSyn mice (Lam et al., 2011). Administration of nicotine at two doses, 0.4 mg/kg/h and 2.0 mg/kg/h, for 3 months and 6 months produced no change in hyperactivity of the mice (Table 1).
Table 1. Nicotine treatment did not alter open field hyperactivity in Thy1-aSyn mice.
Open-field test parameters showed hyperactivity of Thy1-aSyn mice at both 5 and 8 months of age. Data are presented as the mean ± SEM. Student’s t test or the Mann- Whitney U-test (if unequal variances exist) was performed to compare Wt-vehicle vs. Thy1-aSyn vehicle within each time point, *p<0.05, **p<0.01, ***p<0.001. One-way ANOVA followed by a Bonferroni t test was performed to compare the effect of treatment in Thy1-aSyn mice. β = Thy1-aSyn-nicotine-2.0 mg/kg/h group was not included in the statistical analysis at 8 months of age, because mice in this group were euthanized at 6 months of age (see Methods).
| Genotype | Wt | Thy1-aSyn | ||
|---|---|---|---|---|
|
| ||||
| Number of mice tested | ||||
| at age 5 months | 34 | 30 | 25 | 18 |
| at age 8 months | 32 | 25 | 18 | β |
|
| ||||
| Nicotine, mg/kg/h | 0 | 0 | 0.4 | 2.0 |
|
| ||||
| Move episodes (number) | ||||
| 5 months | 292 ± 4 | 267 ± 6*** | 252 ± 9 | 233 ± 14 |
| 8 months | 266 ± 5 | 237 ± 10* | 263 ± 6 | β |
| Move time (seconds) | ||||
| 5 months | 517 ± 14 | 595 ± 15*** | 579 ± 24 | 605 ± 27 |
| 8 months | 442 ± 18 | 569 ± 30*** | 567 ± 23 | β |
| Move time/episode (seconds) | ||||
| 5 months | 1.79 ± 0.06 | 2.28 ± 0.10*** | 2.29 ± 0.11 | 2.63 ± 0.23 |
| 8 months | 1.66 ± 0.07 | 2.48 ± 0.17*** | 2.12 ± 0.09 | β |
| Distance (cm) | ||||
| 5 months | 1860 ± 91 | 2540 ± 160** | 2550 ± 230 | 2700 ± 340 |
| 8 months | 1560 ± 115 | 2780 ± 270*** | 2630 ± 250 | β |
| Distance/episode (cm) | ||||
| 5 months | 6.52 ± 0.38 | 9.19 ± 0.76*** | 10.26 ± 1.03 | 12.49 ± 2.27 |
| 8 months | 5.89 ± 0.45 | 11.18 ± 1.06*** | 8.74 ± 0.56 | β |
| Velocity (cm/second) | ||||
| 5 months | 3.55 ± 0.11 | 4.05 ± 0.15* | 4.41 ± 0.22 | 4.54 ± 0.32 |
| 8 months | 3.42 ± 0.11 | 4.71 ± 0.28*** | 4.51 ± 0.28 | β |
| Center entries (number) | ||||
| 5 months | 66.8 ± 3.7 | 84.2 ± 5.1* | 82.0 ± 7.8 | 87.9 ± 10.5 |
| 8 months | 57.0 ± 4.4 | 86.0 ± 8.5** | 86.6 ± 5.9 | β |
| Rears (number) | ||||
| 5 months | 64.9 ± 5.4 | 79.7 ± 9.5 | 77.0 ± 9.9 | 69.2 ± 11.2 |
| 8 months | 39.8 ± 5.6 | 63.8 ± 11.5 | 55.8 ± 8.6 | β |
Lack of chronic nicotine effects on histological parameters in Thy1-aSyn mice
A hallmark of sporadic PD is the accumulation of Lewy bodies or Lewy neurites, containing α-synuclein. Similar to other transgenic models, Thy1-aSyn mice do not show mature fibrillar structures identical to Lewy bodies but do show proteinase K-resistant aggregates of α-synuclein. Although the role of α-synuclein aggregates in neurotoxicity remains unclear, decreased α-synuclein levels have been associated with functional improvement in the model studied here (Richter et al. 2014). To assess the effects of nicotine treatment on the α-synuclein levels in brain, we measure α-synuclein immunofluorescence in the substantia nigra (SN). Regional measurement of the immunofluorescence intensity in the SN showed a significant increase in α-synuclein levels in Thy1-aSyn mice in the SN pars compacta. Nicotine treatment at 0.4 mg/kg/h (for 6 months) or at 2.0 mg/kg/h (for 4 months) did not alter the levels of α-synuclein in SN of Thy1-aSyn mice. Similarly, nicotine treatment did not modify the α-synuclein aggregate load in Thy1-aSyn mice (Table 2).
Table 2. Nicotine treatment for 6 months did not alter histological parameters in SN and striatum.
Thy1-aSyn mice showed increased α-synuclein levels overall in SN (SNc, SN pars compacta; SNr, SN pars reticulata) compared to Wt mice treated with vehicle; and nicotine treatment did not alter these levels. The percentage area occupied by the proteinase K-resistant aggregates in SN subregions of Thy1-aSyn mice was not altered by nicotine treatment for 6 months. Other parameters show μm2 / section. TH immunofluorescence was not altered in Thy1-aSyn mice compared to Wt mice treated with vehicle in both SN and striatum. Nicotine treatment for 6 months did not change nigral or striatal tyrosine TH immunofluorescence compared to vehicle-treated Thy1-aSyn mice. Thy1-aSyn mice showed no change in striatal synaptophysin or MAP2 (microtubule-associated protein 2) immunofluorescence compared to vehicle-treated Wt mice. Nicotine treatment for 6 months did not alter striatal synaptophysin or MAP2 immunofluorescence at several levels (rostral, medial and caudal) compared to vehicle-treated Thy1-aSyn mice. Data are presented as the mean ± SEM; Wt-vehicle (n=8-10), Thy1-aSyn-vehicle (n=8-10), Thy1-aSyn-0.4 mg/kg/h (n=8-10), Thy1-aSyn-2.0 mg/kg/h (n=8-10); repeated measures ANOVA followed by a post hoc Bonferroni t test. *p<0.05, compared to Wt vehicle group within the brain sub-region. ^p<0.05, Thy1-aSyn-0.4 mg/kg/h group vs Thy1-aSyn-2.0 mg/kg/h group within the brain sub-region. β = Thy1-aSyn-nicotine-2.0 mg/kg/h mice were euthanized at 6 months of age (see Methods) and their brains were processed for histology.
| Immunohistological measurements | Genotype | Nicotine mg/kg/h | Brain Region (with sub-regions) | |||
|---|---|---|---|---|---|---|
|
| ||||||
|
SN
|
||||||
| SNc | SNr | |||||
|
| ||||||
| α-synuclein (immunofluorescence) | Wt | 0 | 18900 ± 2100 | 29900 ± 2400 | ||
| Thy1-aSyn | 0 | 30100 ± 2400* | 35000 ± 2600 | |||
| Thy1-aSyn | 0.4 | 28700 ± 3200* | 33400 ± 3500 | |||
| Thy1-aSyn | 2.0 β | 26900 ± 2700* | 31500 ± 2400 | |||
|
| ||||||
| α-synuclein aggregates (proteinase K-resistant) | Dorsolateral | Dorsomedial | Ventrolateral | Ventromedia | ||
|
|
||||||
| Thy1-aSyn | 0 | 1.16±0.10 | 0.99±0.07 | 1.11±0.10 | 1.18±0.08 | |
| Thy1-aSyn | 0.4 | 1.14±0.05 | 1.03±0.07 | 1.18±0.08 | 1.17±0.07 | |
| Thy1-aSyn | 2.0 β | 1.21±0.08 | 1.06±0.04 | 1.35±0.09 | 1.20±0.07 | |
|
| ||||||
| TH (immunofluorescence) | SNc | SNr | ||||
|
|
||||||
| Wt | 0 | 13400 ± 880 | 7570 ± 450 | |||
| Thy1-aSyn | 0 | 13500 ± 610 | 7380 ± 370 | |||
| Thy1-aSyn | 0.4 | 15300 ±1300 | 7570 ± 550 | |||
| Thy1-aSyn | 2.0 β | 12700 ± 840^ | 6870 ± 400 | |||
|
| ||||||
|
striatum
|
||||||
| Rostral | Medial | Caudal | ||||
|
| ||||||
| TH (immunofluorescence) | Wt | 0 | 23000 ± 1200 | 19270 ± 1310 | 19100 ± 930 | |
| Thy1-aSyn | 0 | 22400 ± 1400 | 18090 ± 1140 | 17110 ± 930 | ||
| Thy1-aSyn | 0.4 | 23800 ± 1100 | 19740 ± 1360 | 19100 ±1100 | ||
| Thy1-aSyn | 2.0 β | 20200 ± 1500^ | 17140 ± 890 | 16400 ± 490 | ||
|
| ||||||
| Synaptophysin (immunofluorescence) | Wt | 0 | 47200 ± 1700 | 33200 ± 1500 | 33400 ± 1900 | |
| Thy1-aSyn | 0 | 47300 ± 2200 | 30700 ± 1700 | 29700 ± 1300 | ||
| Thy1-aSyn | 0.4 | 46100 ±1500 | 31200 ± 1500 | 31600 ± 1200 | ||
| Thy1-aSyn | 2.0 β | 52700 ±1800 | 34300 ± 1700 | 33200 ± 1600 | ||
|
| ||||||
| MAP2 (immunofluorescence) | Wt | 0 | 33600 ± 1700 | 29500 ± 2500 | 30100 ± 2700 | |
| Thy1-aSyn | 0 | 38200 ± 2000 | 30200 ± 2100 | 30600 ± 1700 | ||
| Thy1-aSyn | 0.4 | 33300 ± 2100 | 27900 ± 1000 | 28900 ± 1000 | ||
| Thy1-aSyn | 2.0 β | 34800 ± 2200 | 30700 ± 2500 | 30600 ± 2000 | ||
To ensure that nicotine administration did not have detrimental effects on synaptic and dendritic markers in the striatum, we analyzed the levels of synaptophysin (a major presynaptic vesicle protein) and MAP2 (a dendrite-specific microtubule-associated protein) by immunohistochemistry in 8-month-old Thy1-aSyn mice (6 months after the start of treatment). Confirming previous results, vehicle-treated Thy1-aSyn mice showed unchanged synaptophysin or MAP2 levels compared to Wt mice, and nicotine did not affect these parameters (Table 2).
Confirming our previous observation that tyrosine hydroxylase (TH) loss occurs beginning at 14 months of age in Thy1-aSyn mice (Lam et al., 2011), we found unchanged TH immunoreactivity in Thy1-aSyn mice compared to Wt mice; and nicotine treatment did not affect TH levels in the SN or striatum (Table 2), indicating a lack of toxicity on dopaminergic terminals.
To determine the effect of nicotine administration on inflammation in Thy1-aSyn mice, we measured the levels of activated microglia in SN of mice using Iba-1, a microglial marker. Based on our previous observations, resting microglia are 1 to 4 μm in diameter whereas activated microglia are >6 μm in diameter. We previously observed a microglial activation in 5-6 months old Thy1-aSyn mice together with an increased production of pro-inflammatory cytokines (Watson et al., 2012). Wt mice had a markedly lower level of microglial activation in this study compared to our previous studies (Richter et al., 2014a; Watson et al., 2012), suggesting an overall lower level of inflammation in the present cohort, possibly related to general improvements in husbandry. In contrast to our previous observations in younger, 5-6 month-old mice (Watson et al., 2012), we did not observe any microglial activation in vehicle-treated 8-month-old Thy1-aSyn mice compared to Wt mice (Fig. 7A), consistent with our findings that α-synuclein over-expressing mice have only transient microglial activation (Watson et al., 2012). Nicotine administration induced no change in microglial activation in Thy1-aSyn mice (Fig. 7B, 7C).
Figure 7. Measurement of microglial activation by IBA-1 staining in SN.
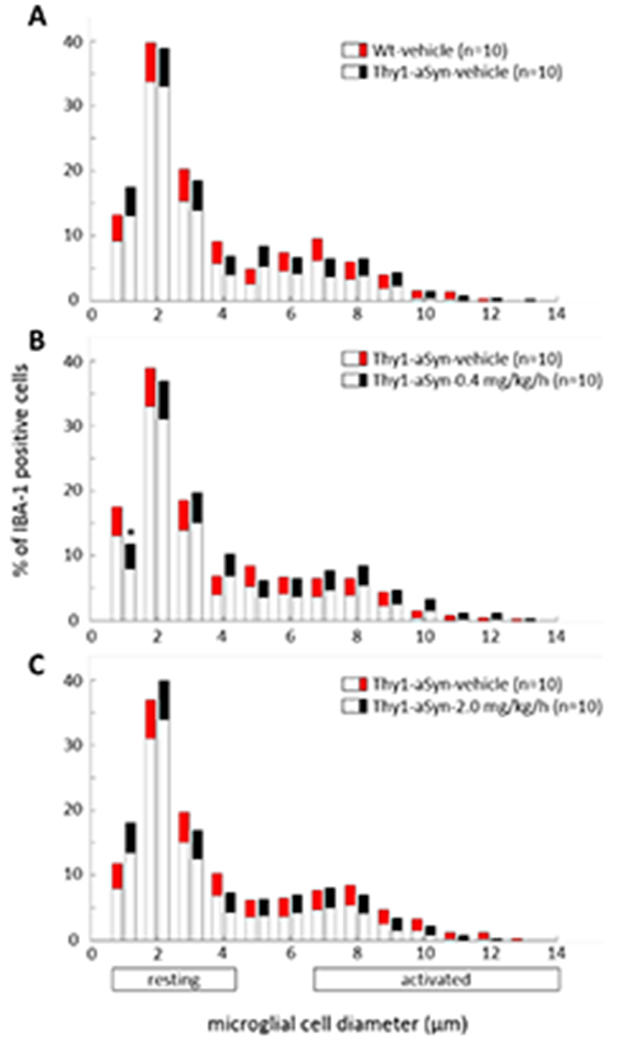
Thy1-aSyn-vehicle mice showed no change in Iba-1 staining of activated microglia compared to Wt-vehicle mice (A). Nicotine treatment at both doses (0.4 mg/kg/h and 2.0 mg/kg/h) did not alter microglial activation compared to Thy1-aSyn-vehicle mice (B, C). Data are presented as the mean ± 95% CI. Significance is represented if confidence intervals (CI) fall outside 95%, calculated by bootstrapping (*p<0.05). Thy1-aSyn-nicotine-2.0 mg/kg/h mice were euthanized at 6 months of age (see Methods) and their brains were processed for histology.
Discussion
In the present study we investigated the effects of sustained administration of two doses of nicotine for six months on tests of motor and cognitive function, as well histological markers, in a genetic model of pre-manifest PD (Chesselet et al., 2012). We further examined the effects of a sustained administration of the safer nicotine dose for one month on cognitive and social behaviors in these mice. Chronic nicotine administration improved synuclein-induced cognitive and social impairments in young Thy1-aSyn mice, but benefited neither early motor deficits, nor alterations in the levels of overexpressed human α-synuclein protein, nor its proteinase-K resistant aggregates in the SN.
Pre-manifest deficits in Thy1-aSyn mice
We conducted our study in Thy1-aSyn mice (line 61)(Rockenstein et al., 2002) because these mice offer a well characterized model of pre-manifest PD that has now been used in several laboratories for preclinical studies of potential disease modifying treatment (Lee et al., 2011; Magen et al., 2014; Richter et al., 2014a; Richter et al., 2014b; Wrasidlo et al., 2016). Beginning at an early age, these mice exhibit cognitive decline (Magen et al., 2012) and other non-motor deficits (Chesselet et al., 2012; Fleming et al., 2008; McDowell et al., 2014; Subramaniam et al., 2014), reminiscent of defects observed in the pre-manifest phase of PD. The Thy1-aSyn mice show hyperactivity when young, and elevated extracellular dopamine at 6 months of age (Lam et al., 2011). Interestingly, elevated striatal dopamine occurs in vivo in pre-symptomatic human carriers of LRRK2 mutations, a cause of familial PD (Sossi et al., 2010). In contrast, the Thy1-aSyn mice of line 61 develop parkinsonism with loss of striatal dopamine and L-dopa-responsive deficits at a relatively late age, 14 months (Lam et al., 2011).
We note previous reports that the phenotype of this model differs in some respects from other α-synuclein over-expressing lines constructed with portions of the same mouse Thy-1 promoter, and with various α-synuclein mutants. These differences include both the cell types that show abnormalities and the behavior. Phenotypic differences among these strains may arise from the pathophysiology of the mutant, the detailed sequence of the promoter fragment used, the insertion locus, and the host mouse strain (Chesselet and Richter, 2011) (http://jaxmice.jax.org/strain/017682.html).
The present study confirms previous findings that the Thy1-aSyn mice show deficits in cognitive function (Magen et al., 2012), accompanied neither by striatal dopamine loss (Lam et al., 2011) nor by changes in TH immunoreactivity (Fernagut et al., 2007). Performance on several of these cognitive tests depends on the integrity of cortical neurotransmission involving acetylcholine. Such systems are typically termed “cholinergic”, typically meaning actions of acetylcholine on either nicotinic acetylcholine receptors (nAChRs) or muscarinic receptors. In some brain regions, nicotinic α7 receptors may also be activated by circulating levels of choline itself, another eponymous cholinergic transmitter. As one example of a muscarinic cognitive effect, spontaneous alternations in Y-maze and NOR are impaired by the administration of a muscarinic antagonist, scopolamine (Barker and Warburton, 2009; Botton et al., 2010; Wall and Messier, 2002). Interestingly, the levels of cortical acetylcholine were decreased at 6 months of age in Thy1-aSyn mice (Magen et al., 2012) suggesting that the deficits in Y-maze and NOR observed in our study may be due to cholinergic deficits. It is important to note that cholinergic systems in PD patients can be impaired by Lewy body pathology (Braak et al., 2003) and cholinergic cell loss (Zarow et al., 2003) in the basal nucleus of Meynert. PD patients display reduced nAChRs and cholinergic denervation (Bohnen and Albin, 2009; Bohnen et al., 2015; Fujita et al., 2006; Meyer et al., 2009), and this may be associated with mild cognitive and depressive symptoms. Also, up to 50% reduction in the nAChR levels are reported in PD patients especially in frontal and temporal areas that are involved in memory, learning and stimulus-seeking behaviors (Hershey and Perlmutter, 2014; Quik and Jeyarasasingam, 2000). In Thy1-aSyn mice, human α-synuclein accumulates in cholinergic neurons of the basal nucleus of Meynert as early as 5 months of age (Magen et al., 2012).
Brain cholinergic systems are vital for various cognitive functions including memory, learning and attention (Everitt and Robbins, 1997), involving several regions of the brain, in both primates and rodents. nAChRs occur widely in neocortical and subcortical areas (Perry et al., 1992; Poorthuis et al., 2009). Evidence indicate that there is significant reduction of cortical nAChRs in post-mortem brains of both demented and non-demented PD patients (Oishi et al., 2007; Pimlott et al., 2004; Rinne et al., 1991). Previous studies from our group implies that chronic nicotine exposure upregulates some nAChRs (Henderson and Lester, 2015; Lester et al., 2009). Upregulated nAChRs include α4β2* and α6* (*indicating other subunits may be present) (Henderson et al., 2014; Lester et al., 2009; Srinivasan et al., 2014). Previous evidence for a decrease in choline acetyl transferase in Thy1-aSyn mice (Magen et al., 2012) together with the current profound effect of chronic nicotine administration on cognitive deficits, at a time when no dopaminergic deficit can be detected in this model, supports the hypothesis that the defects in cholinergic rather than dopaminergic systems in the Thy1-aSyn mice might have caused the cognitive impairment. Future studies should be directed at determining whether this impairment was improved by chronic nicotine treatment via upregulation of nAChRs in addition to direct stimulation of these receptors, as suggested by our previous work (Henderson and Lester, 2015; Henderson et al., 2014; Lester et al., 2009; Srinivasan et al., 2014).
In addition to cognitive deficits, Thy1-aSyn mice display impairments in social behaviors (Magen et al. 2015), and these were improved by nicotine administration. Impaired social behavior in humans is characterized by deficits in non-verbal expression and in social approach, abnormalities in social use or understanding of language, or markedly diminished peer relationships (Moy et al., 2004). Social interactions of PD patients may be affected by disrupted social connectedness (Soleimani et al., 2014); PD patients with mild cognitive deficits showed impairment in social behavior (Anderson et al., 2013). As mentioned, nicotine and other nicotinic agonists activate several nAChR subtypes which are involved in social behavior in monkeys and rodents (Bitner et al., 2010; Boess et al., 2013; Feuerbach et al., 2009; Henderson and Lester, 2015; Salas et al., 2013; Sarter et al., 2009; Timmermann et al., 2012). Chronic nicotine treatment also benefited social behavior in G72Tg mice, a possible mouse model of schizophrenia (Hambsch et al., 2014). Hence, the observed improvement in social impairment by nicotine in Thy1-aSyn mice might be related to nAChR upregulation.
Previously reported early α-synuclein-induced motor deficits and hyperactivity (Fleming et al., 2004; Lam et al., 2011) were also observed in the current study. These highly reproducible phenotypes confer good power to detect drug effects (Chesselet et al., 2012), rendering this mouse model reliable and robust for pre-clinical drug efficacy studies (Fleming et al., 2011; Magen et al., 2014; Richter et al., 2014a; Richter et al., 2014b). However, chronic nicotine administration did not ameliorate the motor deficits or hyperactivity phenotype in the Thy1-aSyn mice. This is unlikely due to the severity of the deficit, because other compounds improve these deficits in mice of approximately the same age as in the present study (Richter et al., 2014b; Wrasidlo et al., 2016). The use of chronic administration of nicotine in this study adds to previous evidence obtained with more acute administration of nicotine or nAChR agonists that also did not improve motor deficits in models of PD as reviewed in (Quik et al., 2015).
Similarly, our results showed that nicotine administration did not alter the levels of α-synuclein in the SN and striatum of Thy1-aSyn mice, nor the level of proteinase K resistant α-synuclein aggregates in SN. This suggests that in our experimental conditions, nicotine did not markedly modify the production, clearance or aggregation of human α-synuclein in vivo. In vitro, nicotine, at concentrations at least 100-fold greater than those produced in mouse brain by our treatments (Nashmi et al., 2007) prevented the fibrillation of purified α-synuclein (Hong et al., 2009; Ono et al., 2007). These effects of nicotine on purified α-synuclein are irrelevant to any form of human nicotine use. Together our findings highlight the inability of chronic nicotine treatment to improve early α-synuclein-induced motor deficits or pathology in a pre-manifest mouse model of PD, thus providing no support for a neuroprotective effect of nicotine.
Many case-control epidemiological studies demonstrate an inverse relationship between smoking and PD risk (Gorell et al., 1999; Tanner et al., 2002), and the risk of PD in someone who has ever smoked is about half of that of a non-smoker (Checkoway et al., 2002; Sugita et al., 2001). Nicotine has been a strong candidate for tobacco constituent responsible for the apparent neuroprotective effect of smoking, because in animal models, nicotine has neuroprotective effects against toxin-induced loss of nigrostriatal dopaminergic neurons (Costa et al., 2001; Khwaja et al., 2007; Quik et al., 2013; Quik et al., 2012). Nicotine may protect dopaminergic neurons against apoptosis by decreasing endoplasmic reticulum stress and the unfolded protein response (Srinivasan et al., 2016). However, toxin-induced models may lack predictive value for neuroprotection because they do not reproduce the pathophysiology that leads to widespread neuronal dysfunction and dopaminergic cell loss in PD. Furthermore, recent epidemiological studies question the interpretation of the relationship between PD and smoking (Hershey and Perlmutter, 2014; Ritz et al., 2014). Based on evidence that individuals in early stages of PD quit smoking much more easily than controls, Ritz and colleagues (2014) suggested that the lower number of smokers in the PD group occurs because nicotine loses its reinforcing properties in early PD, rather than nicotine protecting the neurons. Accordingly, the neuroprotective properties of nicotine in PD remain unproven. Previous clinical trials have failed to show any beneficial effect of short-term nicotine administration in PD patients (Clemens et al., 1995; Vieregge et al., 2001). Chronic nicotine treatment in PD patients has been confirmed to be safe and tolerable (Villafane et al., 2007) but its neuroprotective effect awaits the results of ongoing clinical trials that are examining transdermal nicotine patches (NCT01560754) in PD. Varenicline, a nicotinic agonist, is also currently under clinical trials (NCT02473562, NCT02933372, NCT01341080) for PD therapy.
Conclusion
Our results suggest that chronic administration of nAChR agonists may improve cognitive and social deficits, debilitating non-motor aspects of early PD. Although not supported by the results of this study, this does not exclude a possible neuroprotective effect of nicotine on dopaminergic neurons. Even though the ultimate goal is to stop or reverse the course of neurodegenerative diseases, improvement of symptoms such as cognitive or social deficits can have a very positive effect on patients’ quality of life.
Highlights.
Chronic nicotine treatment for five months reverses cognitive deficits in Y-maze spontaneous test in Thy1-aSyn mice.
Chronic nicotine treatment for one month improves cognitive defects in novel object recognition test and social impairment in Thy1-aSyn mice.
Chronic nicotine treatment for six months did not alter early motor deficits nor modified α-synuclein pathology, tyrosine hydroxylase immunostaining, or microglial activation in Thy1-aSyn mice.
Chronic administration of nicotinic acetylcholine receptor (nAChR) agonists may improve cognitive and social deficits, the debilitating non-motor aspects of early PD.
Acknowledgements
This work was supported by the Michael J. Fox Foundation (MJFF), by the Caltech-UCLA Joint Center for Translational Medicine (JCTM), by NIH grant AG-033954, by gifts to the Center for the Study of Parkinson’s disease at UCLA, and by gifts from Louis and Janet Fletcher at Caltech. We thank Dr. Franziska Richter and Sheri McKinney for help in designing the study. We also thank undergraduate students Bansi Patel, Jacky Kwong, Sean Campeau and Diana Dinh for their help with behavior rating and histology. MFC has received honoraria (for service as a reviewer) and travel reimbursement from MJFF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abbott RD et al. Bowel movement frequency in late-life and incidental Lewy bodies Mov Disord 2007. 22 1581–6 [DOI] [PubMed] [Google Scholar]
- Anderson RJ et al. Social problem solving, social cognition, and mild cognitive impairment in Parkinson’s disease Behav Neurosci 2013. 127 184–92 [DOI] [PubMed] [Google Scholar]
- Barker GR Warburton EC Critical role of the cholinergic system for object-in-place associative recognition memory Learn Mem 2009. 16 8–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti CL Nonmotor disturbances in Parkinson’s disease Neurodegener Dis 2011. 8 95–108 [DOI] [PubMed] [Google Scholar]
- Bitner RS et al. In vivo pharmacological characterization of a novel selective α7 neuronal nicotinic acetylcholine receptor agonist ABT-107: preclinical considerations in Alzheimer’s disease J Pharmacol Exp Ther 2010. 334 875–86 [DOI] [PubMed] [Google Scholar]
- Bloch A et al. α-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects Neuropathol Appl Neurobiol 2006. 32 284–95 [DOI] [PubMed] [Google Scholar]
- Boess FG et al. Pharmacological and behavioral profile of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-6-chinolincarboxamide (EVP-5141), a novel α7 nicotinic acetylcholine receptor agonist/serotonin 5-HT3 receptor antagonist Psychopharmacology (Berl) 2013. 227 1–17 [DOI] [PubMed] [Google Scholar]
- Bohnen NI Albin RL Cholinergic denervation occurs early in Parkinson disease Neurology 2009. 73 256–7 [DOI] [PubMed] [Google Scholar]
- Bohnen NI et al. Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects JAMA Neurol 2015. 72 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botton PH et al. Caffeine prevents disruption of memory consolidation in the inhibitory avoidance and novel object recognition tasks by scopolamine in adult mice Behav Brain Res 2010. 214 254–9 [DOI] [PubMed] [Google Scholar]
- Braak H et al. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology Neurosci Lett 2006. 396 67–72 [DOI] [PubMed] [Google Scholar]
- Braak H et al. Staging of brain pathology related to sporadic Parkinson’s disease Neurobiol Aging 2003. 24 197–211 [DOI] [PubMed] [Google Scholar]
- Chaudhuri KR et al. Parkinson’s disease: the non-motor issues Parkinsonism Relat Disord 2011. 17 717–23 [DOI] [PubMed] [Google Scholar]
- Checkoway H et al. Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake Am J Epidemiol 2002. 155 732–8 [DOI] [PubMed] [Google Scholar]
- Chesselet MF Richter F Modelling of Parkinson’s disease in mice Lancet Neurol 2011. 10 1108–18 [DOI] [PubMed] [Google Scholar]
- Chesselet MF et al. A progressive mouse model of Parkinson’s disease: the Thy1-aSyn (“Line 61”) mice Neurotherapeutics 2012. 9 297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens P et al. The short-term effect of nicotine chewing gum in patients with Parkinson’s disease Psychopharmacology (Berl) 1995. 117 253–6 [DOI] [PubMed] [Google Scholar]
- Costa G et al. Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra Brain Res 2001. 888 336–342 [DOI] [PubMed] [Google Scholar]
- de Bruin N Pouzet B Beneficial effects of galantamine on performance in the object recognition task in Swiss mice: deficits induced by scopolamine and by prolonging the retention interval Pharmacol Biochem Behav 2006. 85 253–60 [DOI] [PubMed] [Google Scholar]
- Edwards TL et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease Ann Hum Genet 2010. 74 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre M et al. Rivastigmine for dementia associated with Parkinson’s disease N Engl J Med 2004. 351 2509–18 [DOI] [PubMed] [Google Scholar]
- Everitt BJ Robbins TW Central cholinergic systems and cognition Annu Rev Psychol 1997. 48 649–84 [DOI] [PubMed] [Google Scholar]
- Fernagut PO et al. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of α-synuclein over-expression Synapse 2007. 61 991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerbach D et al. The selective nicotinic acetylcholine receptor α7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain Neuropharmacology 2009. 56 254–63 [DOI] [PubMed] [Google Scholar]
- Fleming SM et al. A pilot trial of the microtubule-interacting peptide (NAP) in mice overexpressing α-synuclein shows improvement in motor function and reduction of α-synuclein inclusions Mol Cell Neurosci 2011. 46 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human α-synuclein J Neurosci 2004. 24 9434–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM et al. Olfactory deficits in mice overexpressing human wildtype α-synuclein Eur J Neurosci 2008. 28 247–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M et al. Widespread decrease of nicotinic acetylcholine receptors in Parkinson’s disease Ann Neurol 2006. 59 174–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee KW. et al. First in human trial of a type I positive allosteric modulator of α7-nicotinic acetylcholine receptors: Pharmacokinetics, safety, and evidence for neurocognitive effect of AVL-3288. J Psychopharmacol. 2017 doi: 10.1177/0269881117691590. 269881117691590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell JM et al. Smoking and Parkinson’s disease: a dose-response relationship Neurology 1999. 52 115–9 [DOI] [PubMed] [Google Scholar]
- Grace J et al. A double-blind comparison of galantamine hydrobromide ER and placebo in Parkinson disease J Neurol Neurosurg Psychiatry 2009. 80 18–23 [DOI] [PubMed] [Google Scholar]
- Hambsch B et al. Chronic nicotine improves short-term memory selectively in a G72 mouse model of schizophrenia Br J Pharmacol 2014. 171 1758–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanagasi HA et al. Dementia in Parkinson’s disease J Neurol Sci 2017. 374 26–31 [DOI] [PubMed] [Google Scholar]
- Harris JG et al. Effects of nicotine on cognitive deficits in schizophrenia Neuropsychopharmacology 2004. 29 1378–85 [DOI] [PubMed] [Google Scholar]
- Hatami A Chesselet MF Transgenic rodent models to study α-synuclein pathogenesis, with a focus on cognitive deficits Curr Top Behav Neurosci 2015. 22 303–30 [DOI] [PubMed] [Google Scholar]
- Henderson BJ Lester HA Inside-out neuropharmacology of nicotinic drugs Neuropharmacol 2015. 96 178–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BJ et al. Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors J Gen Physiol 2014. 143 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey LA Perlmutter JS Smoking and Parkinson disease: where there is smoke there may not be fire Neurology 2014. 83 1392–3 [DOI] [PubMed] [Google Scholar]
- Hong DP et al. Smoking and Parkinson’s disease: does nicotine affect α-synuclein fibrillation? Biochim Biophys Acta 2009. 1794 282–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja M et al. Nicotine partially protects against paraquat-induced nigrostriatal damage in mice; link to α6β2* nAChRs J Neurochem 2007. 100 180–90 [DOI] [PubMed] [Google Scholar]
- Kruger R et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease Nat Genet 1998. 18 106–8 [DOI] [PubMed] [Google Scholar]
- Lam HA et al. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein J Neurosci Res 2011. 89 1091–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW et al. Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model J Neurosci 2011. 31 6963–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelong-Boulouard V et al. Interactions of buprenorphine and dipotassium clorazepate on anxiety and memory functions in the mouse Drug Alcohol Depend 2006. 85 103–13 [DOI] [PubMed] [Google Scholar]
- Lester HA et al. Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery AAPS J 2009. 11 167–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I Chesselet MF Mouse models of cognitive deficits due to α-synuclein pathology J Parkinsons Dis 2011. 1 217–27 [DOI] [PubMed] [Google Scholar]
- Magen I et al. Cognitive deficits in a mouse model of pre-manifest Parkinson’s disease Eur J Neurosci 2012. 35 870–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I. et al. Intranasal NAP (davunetide) decreases tau hyperphosphorylation and moderately improves behavioral deficits in mice overexpressing α-synuclein. Pharmacol Res Perspect. 2014;2:e00065. doi: 10.1002/prp2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I. et al. Social Cognition Impairments in Mice Overexpressing α-Synuclein Under the Thy1 Promoter, a Model of Pre-manifest Parkinson’s Disease. J Parkinsons Dis. 2015 doi: 10.3233/JPD-140503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamikonyan E et al. Rivastigmine for mild cognitive impairment in Parkinson disease: a placebo-controlled study Mov Disord 2015. 30 912–8 [DOI] [PubMed] [Google Scholar]
- McDowell KA et al. Sleep dysfunction and EEG alterations in mice overexpressing α-synuclein J Parkinsons Dis 2014. 4 531–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer PM et al. Reduced α4β2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease Arch Gen Psychiatry 2009. 66 866–77 [DOI] [PubMed] [Google Scholar]
- Moy SS et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice Genes Brain Behav 2004. 3 287–302 [DOI] [PubMed] [Google Scholar]
- Nashmi R et al. Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path J Neurosci 2007. 27 8202–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi N et al. Quantification of nicotinic acetylcholine receptors in Parkinson’s disease with (123)I-5IA SPECT J Neurol Sci 2007. 256 52–60 [DOI] [PubMed] [Google Scholar]
- Ono K et al. Anti-fibrillogenic and fibril-destabilizing activity of nicotine in vitro: implications for the prevention and therapeutics of Lewy body diseases Exp Neurol 2007. 205 414–24 [DOI] [PubMed] [Google Scholar]
- Perry EK et al. Autoradiographic distribution of [3H]nicotine binding in human cortex: relative abundance in subicular complex J Chem Neuroanat 1992. 5 399–405 [DOI] [PubMed] [Google Scholar]
- Pfeiffer HC et al. Cognitive impairment in early-stage non-demented Parkinson’s disease patients Acta Neurol Scand 2014. 129 307–18 [DOI] [PubMed] [Google Scholar]
- Picciotto MR Zoli M Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease Front Biosci 2008. 13 492–504 [DOI] [PubMed] [Google Scholar]
- Pimlott SL et al. Nicotinic acetylcholine receptor distribution in Alzheimer’s disease, dementia with Lewy bodies, Parkinson’s disease, and vascular dementia: in vitro binding study using 5-[(125)i]-a-85380 Neuropsychopharmacology 2004. 29 108–16 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease Science 1997. 276 2045–7 [DOI] [PubMed] [Google Scholar]
- Poorthuis RB et al. Nicotinic actions on neuronal networks for cognition: general principles and long-term consequences Biochem Pharmacol 2009. 78 668–76 [DOI] [PubMed] [Google Scholar]
- Postuma RB et al. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease Mov Disord 2012. 27 617–26 [DOI] [PubMed] [Google Scholar]
- Preskorn SH et al. Normalizing effects of EVP-6124, an α-7 nicotinic partial agonist, on event-related potentials and cognition: a proof of concept, randomized trial in patients with schizophrenia J Psychiatr Pract 2014. 20 12–24 [DOI] [PubMed] [Google Scholar]
- Quik M et al. Nicotine and Nicotinic Receptor Drugs: Potential for Parkinson’s Disease and Drug-Induced Movement Disorders Int Rev Neurobiol 2015. 124 247–71 [DOI] [PubMed] [Google Scholar]
- Quik M Jeyarasasingam G Nicotinic receptors and Parkinson’s disease Eur J Pharmacol 2000. 393 223–30 [DOI] [PubMed] [Google Scholar]
- Quik M et al. Nicotine reduces established levodopa-induced dyskinesias in a monkey model of Parkinson’s disease Mov Disord 2013. 28 1398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M et al. Nicotine as a potential neuroprotective agent for Parkinson’s disease Mov Disord 2012. 27 947–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy Neurotherapeutics 2014a. 11 840–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F et al. Chronic administration of cholesterol oximes in mice increases transcription of cytoprotective genes and improves transcriptome alterations induced by α-synuclein overexpression in nigrostriatal dopaminergic neurons Neurobiol Dis 2014b. 69 263–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne JO et al. A postmortem study of brain nicotinic receptors in Parkinson’s and Alzheimer’s disease Brain Res 1991. 547 167–70 [DOI] [PubMed] [Google Scholar]
- Ritz B et al. Parkinson disease and smoking revisited: ease of quitting is an early sign of the disease Neurology 2014. 83 1396–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E et al. Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters J Neurosci Res 2002. 68 568–78 [DOI] [PubMed] [Google Scholar]
- Ross GW et al. Association of olfactory dysfunction with incidental Lewy bodies Mov Disord 2006. 21 2062–7 [DOI] [PubMed] [Google Scholar]
- Ryan RE et al. Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in α4 nicotinic receptor subunit knockout mice Br J Pharmacol 2001. 132 1650–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R et al. Abnormal social behavior in nicotinic acetylcholine receptor β4 subunit-null mice Nicotine Tob Res 2013. 15 983–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M et al. nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms Biochem Pharmacol 2009. 78 658–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB. et al. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Soleimani MA et al. Disrupted social connectedness in people with Parkinson’s disease Br J Community Nurs 2014. 19 136–41 [DOI] [PubMed] [Google Scholar]
- Sossi V et al. Dopamine turnover increases in asymptomatic LRRK2 mutations carriers Mov Disord 2010. 25 2717–23 [DOI] [PubMed] [Google Scholar]
- Spillantini MG Goedert M The α-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy Ann N Y Acad Sci 2000. 920 16–27 [DOI] [PubMed] [Google Scholar]
- Spillantini MG et al. α-synuclein in Lewy bodies Nature 1997. 388 839–40 [DOI] [PubMed] [Google Scholar]
- Srinivasan R et al. Pharmacological chaperoning of nAChRs: A therapeutic target for Parkinson’s disease Pharmacol Res 2014. 83C 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R et al. Smoking-Relevant Nicotine Concentration Attenuates the Unfolded Protein Response in Dopaminergic Neurons J Neurosci 2016. 36 65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam SR et al. Region specific mitochondrial impairment in mice with widespread overexpression of α-synuclein Neurobiol Dis 2014. 70 204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita M et al. Meta-analysis for epidemiologic studies on the relationship between smoking and Parkinson’s disease J Epidemiol 2001. 11 87–94 [DOI] [PubMed] [Google Scholar]
- Tanner CM et al. Smoking and Parkinson’s disease in twins Neurology 2002. 58 581–8 [DOI] [PubMed] [Google Scholar]
- Timmermann DB et al. Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of α2- and α4-containing nicotinic acetylcholine receptors Br J Pharmacol 2012. 167 164–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulorge D et al. Neuroprotection of midbrain dopamine neurons by nicotine is gated by cytoplasmic Ca2+ FASEB J 2011. 25 2563–73 [DOI] [PubMed] [Google Scholar]
- Vieregge A et al. Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study Neurology 2001. 57 1032–5 [DOI] [PubMed] [Google Scholar]
- Villafane G et al. Chronic high dose transdermal nicotine in Parkinson’s disease: an open trial Eur J Neurol 2007. 14 1313–6 [DOI] [PubMed] [Google Scholar]
- Wall PM Messier C Infralimbic κ opioid and muscarinic M1 receptor interactions in the concurrent modulation of anxiety and memory Psychopharmacology (Berl) 2002. 160 233–44 [DOI] [PubMed] [Google Scholar]
- Wallace TL Porter RH Targeting the nicotinic α7 acetylcholine receptor to enhance cognition in disease Biochem Pharmacol 2011. 82 891–903 [DOI] [PubMed] [Google Scholar]
- Wang HF et al. Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: systematic review with meta-analysis and trial sequential analysis J Neurol Neurosurg Psychiatry 2015. 86 135–43 [DOI] [PubMed] [Google Scholar]
- Wang L et al. Mice overexpressing wild-type human α-synuclein display alterations in colonic myenteric ganglia and defecation Neurogastroenterol Motil 2012. 24 e425–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JB et al. Alterations in corticostriatal synaptic plasticity in mice overexpressing human α-synuclein Neuroscience 2009. 159 501–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MB et al. Regionally-specific microglial activation in young mice over-expressing human wildtype α-synuclein Exp Neurol 2012. 237 318–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters E Non-motor extranigral signs and symptoms in Parkinson’s disease Parkinsonism Relat Disord 2009. 15 Suppl 3 S6–12 [DOI] [PubMed] [Google Scholar]
- Wrasidlo W et al. A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease Brain 2016. 139 3217–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N et al. α-synuclein overexpression in mice alters synaptic communication in the corticostriatal pathway J Neurosci Res 2010. 88 1764–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. et al. Parkinson’s Disease and Cognitive Impairment. Parkinsons Dis. 2016 doi: 10.1155/2016/6734678. 2016, 6734678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C et al. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases Arch Neurol 2003. 60 337–41 [DOI] [PubMed] [Google Scholar]