

Abstract
The participation of non-heme dinuclear iron cluster-containing monooxygenases in natural product biosynthetic pathways has been recognized only recently. At present, two families have been discovered. The archetypal member of the first family, CmlA, catalyzes β-hydroxylation of L-p-aminophenylalanine (L-PAPA) covalently linked to the nonribosomal peptide synthetase (NRPS) CmlP, thereby effecting the first step in the biosynthesis of chloramphenicol by Streptomyces venezuelae. CmlA houses the diiron cluster in a metallo-β-lactamase protein fold instead of the 4-helix bundle fold of nearly every other diiron monooxygenase. CmlA couples O2 activation and substrate hydroxylation via a structural change caused by formation of the L-PAPA-loaded CmlP:CmlA complex. The other new diiron family is typified by two enzymes, AurF and CmlI, which catalyze conversion of aryl-amine substrates to aryl-nitro products with incorporation of oxygen from O2. AurF from Streptomyces thioluteus catalyzes the formation of p-nitrobenzoate from p-aminobenzoate as a precursor to the biostatic compound aureothin, whereas CmlI from S. venezuelae catalyzes the ultimate aryl-amine to aryl-nitro step in chloramphenicol biosynthesis. Both enzymes stabilize a novel type of peroxo-intermediate as the reactive species. The rare 6-electron N-oxygenation reactions of CmlI and AurF involve two progressively oxidized pathway intermediates. The enzymes optimize efficiency by utilizing one of the reaction pathway intermediates as an in situ reductant for the diiron cluster, while simultaneously generating the next pathway intermediate. For CmlI, this reduction allows mid-pathway regeneration of the peroxo intermediate required to complete the biosynthesis. CmlI ensures specificity by carrying out the multistep aryl-amine oxygenation without dissociating intermediate products.
Graphical Abstract
Two new families of diiron cluster-containing oxygenases serve as tailoring enzymes for NRPS and PKS biosynthetic systems.

1 Introduction
During the biosynthesis of natural products by nonribosomal peptide synthetases (NRPS) (and similar polyketide synthases (PKS)), a remarkably broad range of chemical modification (tailoring) reactions are employed to produce the final functional molecules.1, 2 These tailoring reactions are catalyzed either by domains of the NRPS or by independent enzymes that are often encoded in the same operon as the NRPS itself. One such tailoring reaction is the introduction of oxygen atoms into the natural product by oxygenases. These enzymes catalyze reactions in which molecular oxygen is activated to allow introduction of epoxides, hydroxyl groups, or other oxygenated functional groups into the mature natural product. In the past, oxygenases from several different families have been identified in these roles including: cytochrome P450s,3 2-oxoglutarate-linked nonheme iron-containing enzymes,4, 5 and flavoenzymes.6 Recent studies have revealed that another class of oxygenase that contains a nonheme dinuclear iron cluster in its active site can also fulfill this role.7 Diiron oxygenases and oxidases have been broadly studied in other contexts. Enzymes from this family are responsible for reactions as diverse as environmental methane oxidation (methane monooxygenase, MMO), deoxyribonucleotide production (Class 1A ribonucleotide reductase), desaturation of fatty acids (Δ9-stearoyl-ACP desaturase), and loading of the iron core of ferritin.8 The two types of reactions that these enzymes catalyze in natural product biosynthesis discovered thus far reflect this catalytic versatility. Both types of tailoring reactions are found in the biosynthetic pathway for chloramphenicol (CAM) by Streptomyces venezuelae shown in Figure 1. 7, 9 The diiron monooxygenase CmlA catalyzes the β-hydroxylation of L-p-aminophenylalanine (L-PAPA) covalently linked to the phosphopantetheine arm of the thiolation domain of the NRPS CmlP (CmlP~L-PAPA).10 A completely different type of oxygenation to form the aryl-nitro functional group of CAM is catalyzed by the diiron monooxygenase CmlI after the precursor aryl-amine has been released from the NRPS.9, 11 A homolog of CmlI, AurF from Streptomyces thioluteus, has been shown to catalyze aryl-amine oxygenation reaction in the biosynthesis of a precursor to the natural product biostatic aureothin.12, 13 Neither β-oxygenation nor aryl-amine oxygenation has been reported for other types of diiron oxygenases. Moreover, CmlA adopts a protein fold that has not been previously seen in this family.7, 14 Clearly, there is much to learn about both NRPS tailoring reactions and diiron oxygenase chemistry through the characterization of CmlA, CmlI, and AurF. The progress toward this goal is summarized and evaluated in this report. Recent reviews summarize the technical aspects of preparation, characterization, and structure determination of CmlI and CmlA.15, 16
Figure 1.

Biosynthetic pathway of chloramphenicol
2 CmlA – the β-hydroxylase in NRPS-linked chloramphenicol biosynthesis
2.1 CmlA reaction
CmlA catalyzes a regio- and stereo-specific monooxygenase reaction in which O2 is activated, resulting in fissure of the O-O bond, with one atom of oxygen inserted into a β-C–H bond of L-PAPA, while the second is reduced to water.7 The enzyme was purified from E. coli transformed with a plasmid containing the gene from S. venezuelae. It was shown to have an (α)2 subunit structure based on a monomer molecular weight of 62 kD, in accord with the 532 amino acids predicted by the coding sequence.17 Metal analysis revealed approximately 1.9 iron atoms present per monomer. As described below, the substrate for CmlA is CmlP~L-PAPA, and as a result, there is no reaction with free L-PAPA or similar small molecules. Like all monooxygenases, a source of two electrons per turnover is required for activity. The gene for the implied reductase was not found near those of the CAM biosynthetic suite and has not been identified. The reaction can be carried out in the single turnover mode by chemically reducing CmlA under anaerobic conditions with sodium dithionite using methyl viologen as a mediator.7 Upon mixing reduced CmlA (CmlAred) with CmlP~L-PAPA and O2, the hydroxylated product was produced in nearly stoichiometric yield. Use of 18O2 showed that the molecular oxygen is the source of all oxygen incorporated into L-PAPA during the reaction.
2.2 CmlA spectroscopic features
The UV-visible spectrum of CmlA consists of a broad feature between 300 and 400 nm with a shoulder at 340 nm (ε = 3.5 mM−1cm−1).7 The spectrum is largely bleached upon reduction of the enzyme. A reductive titration showed that 2 reducing equivalents per monomer are required to bleach the spectrum. Both the iron and reducing equivalent stoichiometries suggest a non-heme, carboxylate-bridged diiron cluster in the active site. This assignment was supported by observation of the characteristic optical spectrum of the complex with azide. Also, a characteristic EPR spectrum of the partially reduced, mixed-valent Fe(II)Fe(III) cluster was observed. In this case, antiferromagnetic coupling between the S = 2 and S = 5/2 irons gives an S = 1/2 system exhibiting resonances at g = 1.95, 1.80, and 1.75. The assignment of a diiron cluster was confirmed by the Mössbauer spectrum of the diferric enzyme, which consists of a single doublet with a splitting of 1.4 mm/s and an isomer shift of 0.51 mm/s. The observation of only one doublet showed that the two irons are in very similar environments, although slight broadening of the doublet over the theoretical value showed that the iron environments are distinguishable. The appearance as a quadrupole doublet rather than a multiline spectrum showed that the two inherently paramagnetic ferric ions are antiferromagnetically coupled to give a diamagnetic system. Changes in the spectrum in the presence of an applied magnetic field revealed an exchange coupling constant J of greater than 90 cm−1 for the antiferromagnetic coupling, consistent with the presence of an oxo-bridge. For this type of diiron cluster, charge transfer interactions between the oxo-bridge and the Fe(III) account for the relatively intense optical spectrum. Laser excitation into the optical band showed a feature at 481 cm−1 in its resonance Raman spectrum that downshifted 17 cm−1 when the enzyme was incubated in 18OH2 but was unaffected by incubation in 2H2O.18 These results are consistent with an unprotonated oxo-bridge derived from water in the resting enzyme. X-ray absorption spectra contained pre-edge features with an intensity that showed each of the iron centers to be 6-coordinate. 18, 19 Extended X-ray absorption fine structure spectra revealed an iron-iron distance of 3.32 Å. Each of these features is similar to those found in the past for other enzymes, proteins, and model complexes containing a diiron cluster.8, 20–22
2.3 CmlA structure
Analysis of the amino acid sequence of CmlA showed that the N-terminal portion does not resemble the sequence of any known protein, but the C-terminal half aligns reasonably well with the consensus sequence of a metallo-β-lactamase.7 Significantly, the His-x-His-x-Asp-His (HxHxDH) metal binding sequence motif is preserved. The metallo-β-lactamases often have di-zinc clusters in the active site. Accordingly, a conserved Cys or Ser metal ligand downstream of the consensus sequence is replaced by an Asp ligand in CmlA, as typically found in diiron clusters.
The X-ray crystal structure of CmlA was subsequently solved at 2.17 Å resolution using phasing from a selenomethionine-substituted protein.14, 16 In accord with the sequence predictions, the enzyme was found to fold into two domains as shown in Figure 2A with a short linker region (residues 236-248) that connects these two domains. The N-terminal (residues 1-235) domain has no structural homologs. The C-terminal domain (residues 249-532) is clearly part of the metallo-β-lactamase superfamily with the characteristic αββα-motif and the HxHxDH metal binding sequence. A few other diiron cluster-containing enzymes have been shown to have this metallo-β-lactamase fold; however, none of these are oxygenases.23 In contrast, the typical fold for a diiron oxygenase such as the hydroxylase component of methane monooxygenase (MMOH) is the four helix bundle.24
Figure 2.

X-ray crystal structure of CmlA (A) Structure of diferric CmlA at 2.17 Å resolution. The N- and C-terminal domains are shown in gray and light blue respectively. (B) Space-filling surface representation showing the surface groove (yellow dashed line) formed at the interface of the N- and C-terminal domains. The active site cavity extends into the protein from the top end of the groove as shown. (C) Ligand environment of the diferric diiron cluster, PDB 4JO0. In the X-ray crystal structure of the diferrous enzyme, PDB 5KIK, the acetate ligand on Fe2 is replaced by two waters. Adapted with permission from T. M. Makris, C. J. Knoot, C. M. Wilmot and J. D. Lipscomb, Biochemistry, 2013, 52, 6662–6671. Copyright 2013 American Chemical Society and A. J. Jasniewski, C. J. Knoot, J. D. Lipscomb and L. Que, Jr., Biochemistry, 2016, 55, 5818–5831. Copyright 2016 American Chemical Society
The crystal structure suggests that the active enzyme is a homodimer, as predicted from the solution molecular weight determination. The interface between the protomers is established primarily by a long winding helix that forms one face of the roughly triangular shaped N-terminal domain. A second face of the N-terminal domain packs against the C-terminal domain, forming a long grove on the surface and terminating at the channel into the active site (Figure 2B). The C-terminal domain provides all of the ligands for the diiron cluster, which is located on one end of the characteristic central β-sandwich of the metallo-β-lactamase fold. The helical regions between the β-sheets provide the ligands for the diiron cluster as well as the residues that form one side of the surface groove in conjunction with the N-terminal region.
The diiron cluster is located at the end of a 10-Å-deep channel extending from the surface groove. The channel is lined with hydrophobic residues and has dimensions appropriate to accommodate the phosphopantetheine arm of CmlP and the bound L-PAPA substrate. The HxHxDH consensus sequence provides four of the cluster ligands. Fe1 is bound by His305 and His307 while Fe2 is bound by Asp 309 and His310 (Figure 2C). Two of the three conserved downstream residues provide an unusual μ-1,1-bridging carboxylate ligand (D403) and a bidentate carboxylate (E377) to Fe1. The other ligand sites are filled by a bridging oxo-moiety and an unusual chelated acetate derived from the crystallization buffer.
Chemical reduction of the CmlA in the crystal results in no changes in the overall structure and only small changes in the diiron cluster structure. All of the amino acid ligands to the cluster are retained. The bond lengths to the oxygen bridge increase, suggesting that it is converted to a hydroxo-bridge. The exogenous acetate ligand to Fe2 is replaced by 2 solvents. It is likely that one of the solvents is water while the second is hydroxide so that the overall charge on the cluster remains unchanged by reduction, a commonly encountered feature of diiron oxygenase reaction cycles.25 It is significant that reduction alone does not open a coordination site on either iron, so that no binding site for O2 is created. Thus, further structural changes are required to allow oxygen activation.
2.4 CmlA kinetics and regulation of O2 activation
Rapid mixing of oxygenated buffer with diferrous CmlA (CmlAred) resulted in very slow reoxidation of the diiron cluster in the presence or absence of CmlP.7, 14, 19 However, addition of CmlP~L-PAPA (or a truncated variant consisting of only the adenylation and thiolation domains, CmlPAT~L-PAPA) accelerated the diferrous cluster oxidation by more than 1000-fold (Figure 3) and resulted in the stereospecific hydroxylation of the bound L-PAPA at the β-carbon. The requirement for the presence of both CmlP and L-PAPA as a covalent entity for the commencement of the O2 reaction at the diiron cluster represents a novel twist on a familiar theme for diiron oxygenase enzymes.26, 27 The powerful oxygenating species generated by these enzymes and other types of oxygenases are prevented from carrying out non-specific reactions by a variety of strategies.28–30 In the case of CmlA, O2 reacts 1000-fold faster with the diiron cluster after the complex has formed between CmlA and L-PAPA-loaded CmlP. Similar type of regulation mediated by complex formation between tailoring enzymes and substrate-loaded NRPS enzymes have been reported for oxygenases with other types of metal centers. 31–33
Figure 3.

Time course of the reaction of CmlAred and variants with O2. (Green) Identical courses were observed for CmlAred alone and the E430A CmlAred in the presence or absence of CmlPAT with or without free L-PAPA. (Black) E430A CmlAred plus CmlPAT~L-PAPA. (blue) E377D CmlAred in the presence or absence of CmlPAT or CmlPAT~L-PAPA. (Orange) CmlAred plus CmlPAT~L-PAPA. Conditions: 4 °C, pH 7.5, CmlA = 75 μM (monomer), CmlPAT = 100 μM, O2 = 0.95 μM. Adapted with permission from T. M. Makris, C. J. Knoot, C. M. Wilmot and J. D. Lipscomb, Biochemistry, 2013, 52, 6662–6671. Copyright 2013 American Chemical Society, and A. J. Jasniewski, C. J. Knoot, J. D. Lipscomb and L. Que, Jr., Biochemistry, 2016, 55, 5818–5831. Copyright 2016 American Chemical Society
Two potential mechanisms by which formation of the CmlA:CmlP~L-PAPA complex initiates oxygen activation have been proposed. The first mechanism evolved from the unexpected observation of acetate in the ligand sphere of Fe2.14 Normally, exogenous anionic ligands occupy metal sites facing the substrate binding cavity, but this is not true in the case of CmlA. Instead, the acetate occupies the site where Glu430 (E430) is expected to bind based on sequence alignments with metallo-β-lactamases.7 E430 is in the vicinity of the binding site but slightly too distant to bind. However, it is on the structural loop where CmlP is expected to contact CmlA as it binds in the surface groove (Figure 4A). It is possible that, as the complex forms, E430 is shifted into a binding position, triggering a conformational change that opens a binding site for O2. The diferrous state of the variant E430A was found to react very slowly with O2 as observed for the wildtype CmlA (Figure 3).14 However, reaction with CmlP~L-PAPA was also slow, exhibiting a 25-fold decrease relative to the rate constant observed for unmodified CmlA, suggesting that the triggering mechanism for O2 activation had been compromised.
Figure 4.
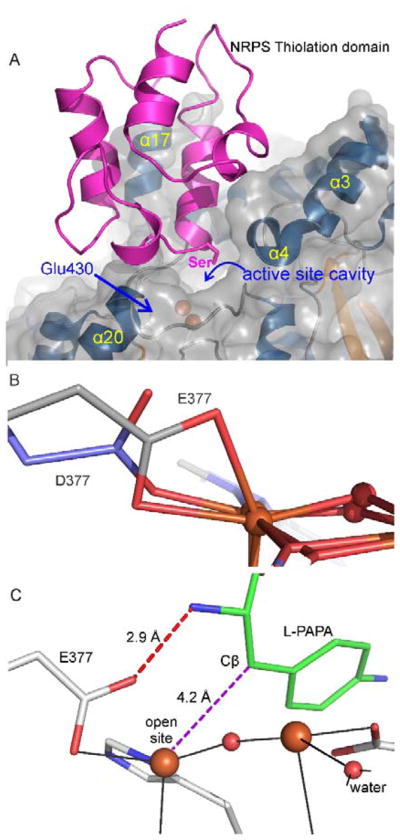
Interactions of CmlA and CmlP (A) Computational docking of CmlA and CmlP thiolation domain. The Ser residue shown is the attachment site for the phosphopantetheine moiety that binds the substrate. It is positioned over the channel into the active site. The location of E430 under the interface is shown. (B) Superimposition of the X-ray crystal structures of CmlA (blue, PDB 4JO0) and the E377D variant (gray, PDB 5KIL) showing the opening of a coordination site on the iron in the variant. (C) Computational docking of the substrate in the active site showing the alignment of the site of hydroxylation with the open coordination site on the diiron cluster proposed to be created through hydrogen bonding interactions with E377. Adapted with permission from T. M. Makris, C. J. Knoot, C. M. Wilmot and J. D. Lipscomb, Biochemistry, 2013, 52, 6662–6671. Copyright 2013 American Chemical Society and A. J. Jasniewski, C. J. Knoot, J. D. Lipscomb and L. Que, Jr., Biochemistry, 2016, 55, 5818–5831. Copyright 2016 American Chemical Society
Insight into the second potential regulatory mechanism stemmed from an X-ray absorption study of the CmlAred:CmlPAT complex, in which the CmlP was either loaded or unloaded with L-PAPA.19 It was found that only in the CmlAred:CmlP~ L-PAPA complex was at least one of the irons converted from 6 to 5 coordinate, thereby opening a site for O2 binding. This change was the result of an amino acid carboxylate shifting from bidentate to monodentate coordination as the complex formed. The crystal structure of CmlAred showed that the only bidentate carboxylate is E377. The diferrous E377D variant with a shorter side chain was structurally characterized and shown to be bound in a monodentate fashion (Figure 4B). Accordingly, E377D was found to react rapidly with O2 even in the absence of CmlP~L-PAPA, showing that regulation in the system had been disrupted (Figure 3). However, the reaction of diferrous E377D:CmlP~L-PAPA with O2 produced no product. As illustrated in Figure 4C, computational docking studies suggested that the binding orientation of L-PAPA in the active site would place the β-carbon immediately over the newly opened coordination site on Fe1 where O2 would bind and be activated.14, 16 This placement would also allow a hydrogen bond to form between one of the carboxylate oxygens of E377 and the amine of L-PAPA if E377 would rotate into a monodentate binding orientation. Thus, substrate binding both initiates O2 activation and fixes the substrate in the correct orientation to be hydroxylated. E377D ensures an open coordination site for O2 activation, but the side chain is too short to allow the orienting hydrogen bond to form with the substrate, thereby defeating effective oxygen transfer into the relevant C-H bond.
At present, the molecular mechanism of O–O bond cleavage and insertion is not known for CmlA. Given the strength of the C–H bond that must be broken (~85 kcal/mol), it is likely that the O–O bond is broken before attack on the substrate to yield a more potent high-valent iron-oxo intermediate of some sort. An intermediate similar to the bis-μ-oxo Fe(IV)Fe(IV) diiron cluster termed compound Q that is formed in the MMOH catalytic cycle is reasonable.34, 35 However, the CmlA diiron cluster and catalytic cycle differ in many respects from that reported for MMOH. The CmlA diiron cluster has one more His ligand, a different bridging carboxylate structure, and a different protonation state of its oxygen bridge (oxo rather than hydroxo) compared to MMOH.14, 24 Perhaps more significantly, the reactive intermediate Q in the MMOH cycle forms prior to substrate binding in the active site and employs a regulatory mechanism based on sequestration of the reactive intermediate until substrate becomes available.26, 34, 36 Both heterolytic and homolytic O–O bond cleavage mechanisms have been proposed for diiron oxygenases, although heterolytic cleavage is more likely in most cases (MMO being a notable exception).37 Heterolytic cleavage requires the donation of at least one proton to the bound O2. The source of such a proton in the case of CmlA is unknown, but there is an intriguing possibility that will be discussed following the introduction of the CmlI mechanism at the end of this report.
2.5 Other potential members of the CmlA family active in natural product biosynthesis
A BLAST search of the genome data base reveals at least fifty homologs of CmlA that are likely to contain dinuclear iron clusters and catalyze O2 activation and insertion reactions.7, 14, 16 These homologs are found in the genomes of many different types of bacteria, including some cyanobacteria. The CmlA homologues identified thus far are of similar size and have 30–45% sequence identity. Some of the homologs include a C-terminal thioester-reductase domain that may serve the role of the CmlP reductase domain in releasing the oxygenated natural product. The homologs are easily recognized by the characteristic HxHxHD metal binding motif that leads to complete conservation of the likely diiron cluster ligands. In general, the residues immediately surrounding the diiron cluster in CmlA are also highly conserved in the homologs. The composition of the probable entry channel residues varies widely, but substitutions are conservative, so that the channel remains highly hydrophobic.
In some cases, the biological roles are known for the CmlA homologs, but none of them has been fully characterized. Important examples are found in the biosynthetic pathways for the glycopeptide antibiotics such as A. teichomyceticus teicoplanin,38, 39 Streptomyces toyocaensis strain NRRL 15009 glycopeptide A47934, 40 and Nonomurea sp. glycopeptide A40926.41 Each of these enzymes utilizes an NRPS that binds L-Tyr to the thiolation domain. In contrast, bleomycin biosynthesis by Streptomyces verticillus utilizes an CmlA homolog that accepts an NRPS with a covalently linked L-His as the substrate.42 Similarly, an NRPS-linked L-His is likely to be the substrate for the CmlA homolog found in the biosynthetic pathway for Lysobacter sp. ATCC 53042 lysobactin. Indeed, this natural product contains β-hydroxylated Phe, Leu, and Asn residues, but there is only one CmlA homolog in the gene cluster. This observation suggests that the homolog may have a much broader substrate range than CmlA.43 Other bacteria that produce important biostatic molecules in pathways that are likely to involve CmlA homologs are: Streptomyces tsukubaensis,44Teredinibacter turnerae,45 and Nodularia spumigena.46
3 N-oxygenation reactions of CmlI and AurF
3.1 CmlI and AurF reactions
CmlI and AurF catalyze monooxygenase reactions in which an aryl-amine is converted to the aryl-nitro analog. As described below, characterization of the enzymes by methods similar to those described in the preceding sections for CmlA established the presence of a nonheme dinuclear iron cluster. CmlI catalyzes the ultimate step in the biosynthesis of CAM as illustrated in Figure 1,9, 11 while AurF catalyzes the conversion of p-aminobenzoate to p-nitrobenzoate.12, 13 The conversion of an amino- to a nitro-function is a six-electron oxidation, which cannot be carried out in one step by a diiron cluster, because there are not enough oxidizing equivalents available to allow this transformation. Consequently, a multistep reaction pathway must be envisioned. The most straightforward pathway consists of three successive two-electron oxidation steps passing through aryl-hydroxylamino and aryl-nitroso intermediates as illustrated for CmlI in Figure 5. Although amine oxygenation has not been previously reported for diiron cluster-containing enzymes, members of this class are known to carry out both oxygenation and oxidation reactions, so the formation of hydroxylamino- and nitroso-intermediates from a precursor amine is feasible. One way that the enzymes might carry out this reaction sequence is to repetitively activate O2 at each step as shown in Figure 5 with the formation of water as a byproduct. This process would require the input of a total of 6 exogenous electrons to re-reduce the diiron cluster three separate times. The studies described in the sections below indicate that Nature has evolved a more efficient pathway to achieve the same outcome.
Figure 5.

Simple three step pathway of aryl-amine oxygenation by CmlI
3.2 CmlI and AurF spectroscopic features
The spectroscopic features of CmlI and AurF are essentially indistinguishable. Consequently, those of CmlI are described here with references to the analogous studies for AurF. CmlI was cloned from S. venezuelae mycelia DNA and heterologously expressed in E. coli. The enzyme was found to have a homodimeric quaternary structure based on molecular weight, and metal analysis revealed 2.1 mol of Fe per monomer.9, 11, 13 This information, together with a relatively intense shoulder at 375 nm (ε ≈ 2700 M−1 cm−1) in the optical spectrum and a characteristic EPR spectrum with the principal g-values below 2 upon partial reduction, implied the presence of the dinuclear iron cluster.11, 13 The intensity of the optical spectrum suggested the presence of an oxo-bridge between the irons in the diferric state. This finding was confirmed by the resonance Raman spectrum, which showed a peak at 487 cm−1 at pH 9 characteristic of an Fe-O-Fe symmetric stretching mode. The feature downshifted the expected 18 cm−1 when the enzyme was exchanged into 18OH2.11, 47 The complexity of the EPR spectrum suggested the presence of more than one type of cluster. Accordingly, the Mössbauer spectrum of diferric CmlI (CmlIox) was found to be comprised of four nested quadrupole doublets, consistent with two types of clusters, each with antiferromagnetically coupled irons (Figure 6A).11 The individual irons in each cluster reside in distinguishable environments. The population of each type of cluster revealed by the Mössbauer spectrum of CmlIox and the distribution of species in the EPR spectrum of the mixed valent state were found to be pH dependent, suggesting that the structure of the cluster or its nearby surroundings is sensitive to protonation state in the pH 6 – 9 range. The Mössbauer spectrum of the CmlIred (Figure 6B) shows that the irons are both high spin ferrous and in similar but not identical environments.
Figure 6.

Mössbauer spectra of (A) diferric and (B) diferrous CmlI and (C) CmlI-P. Black hash marks show the data. Black lines are the fits composed of contributions from individual iron sites (blue and red lines). The gray line is a minor amount of CmlIox in the CmlI-P sample. Conditions: T = 4 K, 0 applied magnetic field, pH 9. Adapted with permission from T. M. Makris, V. V. Vu, K. K. Meier, A. J. Komor, B. S. Rivard, E. Münck, L. Que, Jr. and J. D. Lipscomb, J. Am. Chem. Soc., 2015, 137, 1608–1617. Copyright 2015 American Chemical Society.
3.3 CmlI and AurF X-ray crystal structures
AurF has been crystallized with either a diiron or dimanganese cluster in the active site.48–50 Comparison of the activities of the diiron and dimanganese forms indicates that the active form of the enzyme contains a diiron cluster.48 Both forms of AurF house the di-metal cluster in a 4-helix bundle motif, as found in most characterized diiron cluster-containing oxygenases such as MMOH.24, 51, 52 The only significant structural differences between the diiron and dimanganese forms of AurF occur in the di-metal cluster and its vicinity. Both enzymes share the same metal ligands from the protein: 3 His and 4 Glu residues. However, in the dimanganese form, the carboxylate moieties of Glu 227 and 136 (Glu 236 and 144, respectively, in CmlI) both bridge the metals, whereas the Glu 227 bridge is replaced by an oxo-bridge in the diiron form (illustrated in Figure 7A using CmlI, which has a structure like dimanganese AurF, see below). The bridging position of the Glu 227 carboxylate in the dimanganese form orients the peptide backbone so as to promote formation of a short alpha helix adjacent to the cluster (illustrated using CmlI in Figure 7C). The helix contains Ile 199 (Ile 208 in CmlI), which is forced into the substrate binding site (Figure 7D). The resulting active site cavity is too small to accommodate a substrate, thus accounting for the lack of activity of this form. The shift of the bridging Glu 227 carboxylate in the diiron form destabilizes the helix, allowing Ile 199 to rotate out of the active site, and thereby expanding it to the size required to bind substrate. It is notable that in each form of the enzyme, both metals are 5-coordinate (one iron may also have a weakly bound solvent). If this structure were to be maintained upon reduction, O2 could potentially bind by bridging the two irons and initiate catalysis.
Figure 7.

X-ray crystal structures of the CmlI and AurF active site regions. (A) Superimposition of the diiron form of AurFox (green, PDB 3CHH) and CmlIox (white, PDB 5HYG). The dimanganese form of AurF (PDB 2JCD) has a structure in this region almost identical to that of CmlI. (B) The inactive, spontaneously formed cis-μ-1,2-peroxo intermediate of CmlIox. (C and D) Superimposition of the diiron form of AurFox (blue) and CmlIox (dark gray) shows the conversion of helix (CmlIox) to a coil (AurF) in the vicinity of the diiron cluster. The helix forces CmlI I208 (AurF I199) into the space required to bind substrate defined by the position of the product (yellow) in a crystal structure of AurF (PDB 3CHT). Adapted with permission from C. J. Knoot, E. G. Kovaleva and J. D. Lipscomb, J. Biol. Inorg. Chem., 2016, 21, 589–603. Copyright 2016 Springer.
The X-ray crystal structure of CmlI was solved after truncation of the N-terminal 33 residues.53 Part of the comparable structural region is absent in AurF, and the truncation did not affect the activity, stability, or quaternary structure of CmlI. The overall structure of CmlI is nearly identical to that of AurF. Interestingly, while only the diiron form was observed for CmlI, the structure of the cluster and its vicinity were found to be nearly identical to those of dimanganese AurF. For the reasons stated above, this CmlI structure is unlikely to be representative of the active form of the enzyme (which is fully active in solution). Also, the optical spectrum of CmlI in solution shows definitively that an oxo-bridge is present in the diferric form of the active enzyme,11 while this bridge is missing in the crystal structure. One difference in conditions for these experiments is that the crystallization occurred at pH 6.8, whereas the optical and catalytic studies of CmlI were routinely carried out at pH 9. The (fully active) high pH form was studied because the Mössbauer spectra indicated the diferrous state of the metal center is most homogeneous and the reactive intermediate is most stable at pH 9 (see below).
Upon chemical reduction of the crystalline CmlI, very little change occurs in the structure. 53 Significantly, the active site remains blocked. The iron-iron distance expands by 0.3 Å, and both irons are 5-coordinate with adjacent vacant coordination sites. In most diiron cluster-containing enzymes and model complexes, this configuration would be expected to bind O2 between the irons to form a cis 1,2-μ-peroxo intermediate.
3.4 Peroxo-intermediates of CmlI and AurF
A significant difference between the X-ray crystal structures of CmlIox and those of both forms of AurF is that a cis-μ-1,2-peroxo intermediate forms spontaneously in the crystal of resting CmlIox (Figure 7B),53 making both irons 6-coordinate. The source of the oxygen to form this intermediate is unlikely to be O2 because the metals would have to be reduced to allow such an intermediate to form. Rather, the peroxo intermediate is likely to have formed by binding the H2O2 released as polyethylene glycol in the crystallization solution breaks down.54 The availability of adjacent open metal coordination sites in the di-metal cluster would allow facile formation of this type of peroxo-intermediate, which has been trapped and studied in a few other diiron cluster containing enzymes.55–58 Often, this type of peroxo intermediate is formed as a step in the process of cleaving the O–O bond to form a more powerful oxidizing intermediate.35, 52, 59 Addition of H2O2 to CmlIox in solution resulted in a long-lived intermediate with a broad spectrum in the 600–700 nm region typical of cis-μ-1,2-peroxo species.60–62 This intermediate did not appear to react with aryl-amine substrates, suggesting that it forms adventitiously and is not the reactive intermediate of CmlI. Direct observation of the optical spectrum of the intermediate in the crystal showed a similar long wavelength absorbance and no evidence for the intense shoulder at 375 nm characteristic of the oxo-bridge seen in the solution optical spectrum.53
Another type of peroxo-intermediate that is much more likely to be the reactive species was reported to occur in AurF and later in CmlI.11, 63 This species, termed P, formed upon addition of O2 to the diferrous enzymes. The orange species is characterized by a broad absorbance centered at 500 nm (ε = 500 M−1cm−1), which differs substantially from the blue color of the peroxo species seen in the crystal of CmlI as well as the cis-μ-1,2-peroxo species found in other enzyme systems. The species in CmlI is remarkably stable, exhibiting a half-life of 3 h at 4 °C (Figure 8A). Nevertheless, upon addition of an aryl-amine substrate, the species reacted in less than 1 s to yield the oxygenated product (Figure 8B). A range of alternative substrates has been tested for both CmlI and AurF.11, 64 As shown by the plots in Figure 8C, the rate constant for decay of CmlI-P increases linearly with substrate concentration. Also, the slopes of these plots increase as the aromatic ring substituents become more electron supplying. Both of these characteristics support P as the reactive species for aryl-amine oxygenases.
Figure 8.

Dependence of the CmlI-P decay rate constant on the type and concentration of substrate. Time courses and exponential fits of: (A) CmlI-P decay in the absence of substrate and (B) CmlI-P decay in the presence of p-aminobenzoate (pABA) or NH2-CAM. (C) Rate constants for CmlI-P decay plotted versus the type and concentration of substrate. The slopes of the plots yield the apparent second order rate constants shown in the inset. (pAP = p-aminophenol) Adapted with permission from T. M. Makris, V. V. Vu, K. K. Meier, A. J. Komor, B. S. Rivard, E. Münck, L. Que, Jr. and J. D. Lipscomb, J. Am. Chem. Soc., 2015, 137, 1608–1617. Copyright 2015 American Chemical Society.
The long lifetime of CmlI-P in the absence of substrate at pH 9 allowed it to be easily trapped for further spectroscopic studies, corroborating and expanding upon what had been established for AurF. 63 Again, the properties of CmlI-P will be described here, but those of AurF-P are very similar and are cited. Mössbauer spectra showed that the iron centers in CmlI-P are antiferromagnetically coupled with distinguishable isomer shifts indicative of diferric species (Figure 6C). 11, 63 The spectra are markedly different from those of resting (diferric) CmlIox (Figure 6A), consistent with formation of a peroxo-species. However, the Mössbauer spectra are quite distinct from those observed for the typical cis-μ-1,2- peroxo species characterized in other enzymes and model complexes.55, 62, 65, 66 Resonance Raman spectra revealed a vibration at 789 cm−1 that downshifted 43 cm−1 when 18O2 was used.11, 47 The use of mixed labeled O2 gave an intermediate shift, demonstrating that this is a peroxo- rather than an oxo-intermediate. This resonance Raman vibration occurs at a much lower frequency than that of any known biological or synthetic diiron peroxo species, showing that a new type of peroxo species was formed. The vibrational frequency is similar to those found for mixed di-metal complexes in which O2 binds side-on to iron and then forms one or two bonds to the second metal such as Cu, Sc, or Y (μ-η2:η1 peroxo binding mode).67 Thus, CmlI-P was originally hypothesized to have a μ-η2:η1 peroxo bridging structure. Subsequent resonance Raman studies revealed a νs(Fe–O–Fe) at 485 cm−1that downshifted 18 cm−1 in 18OH2 buffer characteristic of an oxo-bridge (Figure 9).47 In order to assume the μ-η2:η1 bridging structure, the oxo-bridge would have to be lost, thus the originally proposed type of peroxo structure is not supported. The current proposal for the CmlI-P structure is based on EXAFS analysis, which revealed two diagnostic Fe–O distances at 1.83 and 2.82 Å within the cluster structure.47 The 1.83-Å distance is characteristic of the oxo-bridge, but the 2.82-Å distance could not be associated with any known structure. The intensity of the feature showed that the oxygen is equidistant from the irons, suggesting a novel μ-1,1-peroxo ligand (Figure 10). This type of peroxo species has often been postulated but not observed thus far in biological or synthetic systems. Significantly, it would place the terminal oxygen of the peroxo in a position where it could reasonably add to the amino- or nitroso-functional groups of pathway intermediates based on the position of the aryl-nitro-functional group in a crystal structure of the product complex of AurF (Figure 7D).48 This type of peroxo species would have quite different chemical properties than the rather inert cis-μ–1,2-peroxo, which would presumably enhance the reactivity such that it could transfer oxygen to an aryl-amine. The resonance Raman spectrum for a CmlI-P sample in 2H2O buffer showed no shift in the vibrational frequency for the new peroxo ligand, suggesting that it is not protonated.47 However, in a computational study of the peroxo intermediate of AurF calibrated by MCD and NRVS data, it was proposed that a protonated cis-μ-1,2-peroxo is the reactive species.68 This study used a structure similar to that of the dimanganese enzyme as a starting point for computation, which as detailed above, is unlikely to be catalytically relevant. A subsequent computational study concurred with the spectroscopic studies predicting a reactive μ-1,1-peroxo ligand structure of AurF/CmlI-P.69 However, it also predicted that the terminal oxygen of the peroxo would be protonated. This aspect of the structure remains unresolved, but it is clear that Nature has evolved a new type of reactive intermediate to carry out this specialized chemistry.
Figure 9.

Resonance Raman spectra of CmlI-P in buffers made with the isotope of water shown. The vibration at 485 cm−1 originates from the symmetric Fe-O-Fe stretch. The asymmetric stretch at 780 cm−1 is obscured by spectral overlap, but detectable by spectral fitting. The vibration at 789 cm−1 originates from the novel O–O stretch of the μ-1,1-peroxo bridge. Conditions: excitation = 561 nm, pH/D = 9, T = 4 °C. Protein concentration for all panels is ~ 1 mM. Adapted with permission from A. J. Jasniewski, A. J. Komor, J. D. Lipscomb and L. Que, Jr., J. Am. Chem. Soc., 2017, 139, 10472–10485 Copyright 2017 American Chemical Society.
Figure 10.

Proposed structure of CmlI-P. Fe to ligand distances are shown in blue (angstroms). The Fe-Fe distance is 3.35 Å. Adapted with permission from A. J. Jasniewski, A. J. Komor, J. D. Lipscomb and L. Que, Jr., J. Am. Chem. Soc., 2017, 139, 10472–10485 Copyright 2017 American Chemical Society.
3.5 Steps in the biosynthetic pathways of CmlI and AurF
The exceptional stability of the P intermediates of CmlI and AurF has allowed detailed studies of the biosynthetic pathways. Two different proposals have emerged for these enzymes, both of which differ from the original notion shown in Figure 5 that three P intermediates react progressively with the aryl-amine substrate and the subsequent 2-electron-oxidized intermediates to form the final aryl-nitro product.13 The respective P intermediates of both enzymes were shown to react with the aryl-amine substrate to first form the aryl-hydroxylamino intermediate.11, 63, 70–72 In a remarkable observation, Bollinger, Krebs and coworkers used optical stopped-flow and rapid freeze quench (RFQ) Mössbauer studies to show that AurF-P can react with the synthetically prepared p-hydroxylamino-benzoate to yield the p-nitrobenzoate product while regenerating the diferrous form of the enzyme.72 If O2 was present, AurF-P could then reform and react again, allowing the reaction to become catalytic. Because no external reducing equivalents were added in the experiment, this observation suggested that one of the pathway intermediates was capable of reducing the diferric cluster that results from the reaction of AurF-P with a substrate. The authors proposed that the initial product of the reaction of AurF-P with the p-hydroxylamino-benzoate was p-dihydroxylamino-benzoate rather than p-nitroso-benzoate (Figure 11, left). The former intermediate could re-reduce the cluster while being itself oxidized to the p-nitrobenzoate final product. Other groups had reported the formation of p-nitroso-benzoate intermediate,48, 73 but the most recent work with AurF showed that synthetically prepared p-nitroso-benzoate did not react with AurF-P and no accumulation of any four-electron oxidized intermediate was detected. 72,72
Figure 11.

Two new pathways proposed for aryl-amine oxygenases. (Left) Pathway proposed for AurF in which an aryl-dihydroxylamino intermediate is formed and acts as the mid-pathway reducing agent for the diiron cluster. (Right) Pathway proposed for CmlI in which an aryl-hydroxylamino intermediate acts as the internal reducing agent. Adapted with permission from A. J. Komor, B. S. Rivard, R. Fan, Y. Guo, L. Que, Jr. and J. D. Lipscomb, J. Am. Chem. Soc., 2016, 138, 7411–7421. Copyright 2016 American Chemical Society.
The second proposed pathway emerged from studies showing that the aryl-nitroso precursor of CAM (NO-CAM) could be directly observed during the reaction of the aryl-amine precursor of CAM (NH2-CAM) with CmlI-P.70, 71 Moreover, synthetically prepared NO-CAM was converted to CAM by reaction with diferrous CmlI (CmlIred) and O2, showing that it is a likely reaction pathway intermediate. The exceptional stability of CmlI-P allowed it to be prepared with 18O2 and then used in experiments over the next hour or more. During this time, incubation of the 18O-CmlI-P in an atmosphere of 16O2 did not result in oxygen exchange. When the 18O-CmlI-P was reacted with a stoichiometric amount of NH2-CAM in an 18O2 atmosphere, the oxygen of the nitroso-group of NO-CAM and both of the oxygens of the nitro group of CAM became fully labeled with 18O. Carrying out the same reaction in an 16O2 atmosphere resulted in complete 18O labeling of the NO-CAM, but the nitro group of CAM had substantial incorporation of 16O. Because CmlI-P itself does not exchange with the headspace oxygen, these findings indicated that that CmlI-P must be regenerated at some point in the pathway to allow incorporation of O2 from the headspace into CAM. In contrast to the results for AurF described above, the diiron cluster of CmlI was found to be fully oxidized after this single turnover. The most direct interpretation of the CmlI results is that the aryl-hydroxylamine adduct of CAM (NH(OH)-CAM) functions as the internal reducing species in the pathway. The 18O-CmlI-P reaction with NH2-CAM would yield NH(18OH)-CAM), which could then act to reduce the diiron cluster while becoming oxidized itself to N18O-CAM (Figure 11, right). Subsequent reformation of CmlI-P from the diferrous cluster in the 16O2 atmosphere and reaction with N18O-CAM would yield mixed labeled CAM and the diferric cluster, as observed. The ability of NH(OH)-CAM to reduce CmlI was directly demonstrated using Mössbauer spectroscopy of 57Fe-labeled enzyme. In this experiment, an aerobic mixture of NH(OH)-CAM with CmlIox yielded CmlI-P and then CAM without requiring additional external reducing equivalents. Notably, Bollinger, Krebs and co-workers found that this is not a significant pathway in AurF by showing that the reaction of AurFox with the hydroxylamine substrate resulted in reduction of less than 10% of the diiron cluster.72
Differentiating between these two pathway proposals has been difficult given the instability of some of the proposed intermediates. It is possible that both pathways are valid depending on the specific enzyme being used and the reaction conditions. This possibility was evaluated by studying the kinetics of the reactions in the pathway of CmlI as described below.
3.6 Reaction pathway kinetics of CmlI
The orange chromophore of CmlI-P and the large differences in the chromophores of CmlIox and CmlIred allowed the single turnover transient kinetics of the individual steps in the reaction pathway to be observed.71 The reaction of CmlIred with O2 to form CmlI-P is a fast process (~58 s−1 mM−1 at 4 °C, pH 9 or 58 s−1 at 1 mM O2) compared with the overall rate constant of turnover (a rate constant of 5.2 s−1is rate-limiting during the single turnover cycle, see Figure 12). CmlIred was found to react with stoichiometric O2 and excess NH2-CAM to yield a CmlI-P:NH2-CAM complex. This complex then transformed to a CmlIox:NH(OH)-CAM complex, which rapidly converted to the CmlIred:NO-CAM complex. The rate constant for each of these steps exceeds that of the overall rate-limiting step by at least a factor of eight. Similarly, mixing of Cmlred with a mixture of excess NO-CAM and O2 yielded a CmlI-P:NO-CAM complex that rapidly converted to CmlIox-CAM from which CAM was released in the overall rate-limiting step.
Figure 12.

Single turnover kinetics for CmlI. Rate constants were determined for the individual steps in a single turnover of CmlIred mixed with NH2-CAM and O2. The version shown is simplified to illustrate the main pathway. In fact, NH2-CAM and O2 can add in either order with somewhat different kinetics. The dissociation and reassociation of intermediates are actually multi-step processes. It is postulated that all of the reactions inside the box occur in a single active site of CmlI without kinetically significant intermediate dissociation. See reference 71 for details.
An important regulatory aspect of catalysis by CmlI emerged from these studies. It was found that the order of addition of stoichiometric O2 and NH2-CAM to CmlIred had little effect on the kinetics and rapidly led to the same intermediate CmlIred:NO-CAM complex. In contrast, mixing CmlIox with NH(OH)-CAM resulted in a very slow reduction of the diiron cluster and formation of the CmlIred:NO-CAM complex. The acceleration of diiron cluster reduction accruing from formation of NH(OH)-CAM in situ in the active site versus addition of NH(OH)-CAM from solution was approximately 400 fold. Indeed, if NH(OH)-CAM dissociated and had to rebind to CmlIox to continue the biosynthetic process (there is no excess CmlI-P in the experiment), the reaction step would be rate-limiting in the overall pathway. In actuality, the rate-limiting step is product release when the complete biosynthetic reaction is observed by mixing CmlIred with excess O2 and NH2-CAM. The most direct explanation for this observation is that the enzyme actively retains the intermediates in the active site at each step of the pathway. Additional evidence for this phenomenon is seen in the NO-CAM oxygenation step. This step is fast when NO-CAM is formed in situ in the CmlI active site, but it does not occur at all if CmlI-P is mixed with NO-CAM. Trapping the intermediates in the active site provides for a very efficient and specific biosynthetic pathway that avoids the release of species that could potentially react with other cellular components. This pathway chemistry illustrated in Figure 12 is made possible by the use of an internal reductant that generates the next intermediate in the pathway in the active site while priming the cluster to reform the reactive peroxo intermediate. The complete process requires only two external electrons to prime the pathway by formation of the initial CmlI-P.
The direct observation of the steps in the CmlI reaction shows that a pathway proceeding through NO-CAM is viable, but it does not rule out the pathway through an aryl-dihydroxylamino intermediate analogous to that proposed for AurF. Indeed, when CmlI-P is mixed with NH(OH)-CAM, the end product is CmlIred and CAM, an outcome completely analogous to the observations reported for AurF-P mixed with p-hydroxylamino-benzoate.71 It seems likely that in the case of CmlI, the retention of intermediates in the active site prevents the reaction of NH(OH)-CAM with CmlI-P in vivo. However, it may be that the smaller intermediates in the AurF pathway can leave the active site more rapidly, thereby allowing access to the alternative pathway under conditions where excess AurF-P is present.
3.7 Mechanism of reaction of peroxo-intermediates
A comparison of the very distinct reactions catalyzed by the relatively similar diiron clusters of CmlA and CmlI is informative. Both CmlAred and CmlIred are likely to initially form peroxo-intermediates of some sort upon reaction with O2. CmlI and AurF appear to utilize their peroxo intermediate directly to carry out the amine oxygenation reaction. In contrast, the strong C–H bond that must be broken during the CmlA reaction will probably require a more potent intermediate than a peroxo (or hydroperoxo). By analogy with other diiron oxygenases, it is reasonable to postulate that the peroxo O–O bond breaks in the CmlA reaction to form an Fe(IV)-oxo or MMO compound Q-like intermediate before reaction with the substrate. What aspect of structure dictates whether the O–O bond breaks before or after reaction with substrate? The mechanism for O–O bond cleavage from an initially formed diiron-peroxo intermediate is thought to involve protonation of either the peroxo moiety itself or a nearby amino acid residue.37 An intriguing possibility is suggested by the fact that one of the irons in the diiron cluster of CmlAred has two water ligands (Figure 2, legend). Displacement of one of these waters during cis-μ-1,2-peroxo intermediate formation (Figure 4C) would still leave one water to provide the proton needed to break the O–O bond and form a high-valent species. In contrast, the μ-1,1-peroxo species in CmlI, would not have a metal site for a water ligand (Figure 10), thereby stabilizing the peroxo species.
The rationale for the amine oxygenases to utilize a peroxo rather than a high-valent oxo intermediate is a current topic of experimental and computational investigation. As neither CmlI nor AurF has been shown to oxygenate unactivated C–H bonds, the reliance on a peroxo intermediate to carry out aryl-amine hydroxylation may prevent adventitious reactions that could lead to enzyme inactivation. Also, unlike CmlA and oxygenases such as MMO, the amine oxygenases must promote reactions with the chemically quite distinct substrates encountered in the biosynthetic pathway. For example, one could imagine that the oxygenation of electron rich NH2-CAM is very different than the oxygenation of electron poor NO-CAM, but both reactions are carried out by the same CmlI-P intermediate. Peroxo species differ from high-valent oxo species in that they can react as both electrophilic and nucleophilic reagents. Thus, the ambiphilic peroxo intermediate may be ideal for application in a pathway where the extreme oxidizing potential of a high-valent species is not required, but versatility in dealing with a broad range of chemically distinct substrates is a necessity. Detailed studies of the mechanism of amine oxygenation by peroxo intermediates of amine oxygenases have only recently commenced, but it has been shown that the reaction with aryl-amines accelerates markedly as the electron donating capacity of other aromatic ring substituents is increased (Figure 8C).11 Thus, the ability of the peroxo moiety to act as an electrophile at the outset of the pathway is strongly supported. One proposal for the ambiphilic reactions of the peroxo species with the aryl-amino and aryl-nitroso pathway intermediates is illustrated in Figure 13.
Figure 13.

Possible mechanisms for the (A) electrophilic and (B) nucleophilic oxygenation by CmlI-P along the reaction pathway. Adapted with permission from A. J. Jasniewski, A. J. Komor, J. D. Lipscomb and L. Que, Jr., J. Am. Chem. Soc., 2017, 139, 10472–10485. Copyright 2017, American Chemical Society.
3.8 Other amine oxygenases in natural product biosynthesis
BLAST searches show that there are likely to be hundreds of additional AurF and CmlI homologs. Those that have been identified so far are present in soil bacteria and/or plant pathogenic bacteria, and, like CmlI and AurF, are associated with PKS or NRPS containing operons.12, 74, 75 As in the case of CAM, the nitro-group conveys valuable properties to the natural product, so the investigation of this new diiron family is likely to yield new bioactive molecules as well as new insight into this largely unexplored chemistry.76
4 Conclusions
The investigation of the role of diiron cluster containing oxygenases in natural product biosynthesis is just beginning. The observations related here showing that the first two diiron enzymes found to be associated with an NRPS pathway have completely different structures, chemical mechanisms, and strategies of regulation suggests that this field has great potential. These enzymes show that Nature has the capability to produce oxygen-activating cofactors that can be tuned up or down in potency with great precision by the protein environment to enforce specificity. In the case of CmlI, a new type of reactive species was developed to allow exquisite control of the type of functional group that can serve as the target of oxygenation. Every oxygenase in our experience has a means to protect the reactive oxygen intermediate from carrying out adventitious chemistry. The exclusive ability of CmlP~L-PAPA to convert nearly inert CmlA into a powerful oxygen-activating enzyme is a superb example. Given the need to produce exceptionally complex natural products with exact three dimensional structures, it seems likely that more insight into how Nature can control the temporal and spatial generation of these potent species will evolve from the study of this new diiron family.
Acknowledgments
The authors acknowledge the long-term support of the National Institutes of Health (GM40466, GM100943 and GM118030 to J.D.L and GM38767 to L.Q and graduate traineeship GM08700 to A.J.K.)
References
- 1.Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM. Curr Opin Chem Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Walsh CT. Chem Biol. 2001;8:301–312. doi: 10.1016/s1074-5521(01)00009-6. [DOI] [PubMed] [Google Scholar]
- 4.Neary JM, Powell A, Gordon L, Milne C, Flett F, Wilkinson B, Smith CP, Micklefield J. Microbiology. 2007;153:768–776. doi: 10.1099/mic.0.2006/002725-0. [DOI] [PubMed] [Google Scholar]
- 5.Hollenhorst MA, Bumpus SB, Matthews ML, Bollinger JM, Jr, Kelleher NL, Walsh CT. J Am Chem Soc. 2011;133:1609. doi: 10.1021/ja1072367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh CT, Wencewicz TA. Nat Prod Rep. 2013;30:175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makris TM, Chakrabarti M, Münck E, Lipscomb JD. Proc Natl Acad Sci USA. 2010;107:15391–15396. doi: 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 9.Lu HG, Chanco E, Zhao HM. Tetrahedron. 2012;68:7651–7654. doi: 10.1016/j.tet.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pacholec M, Sello JK, Walsh CT, Thomas MG. Org Biomol Chem. 2007;5:1692–1694. doi: 10.1039/b703356g. [DOI] [PubMed] [Google Scholar]
- 11.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Jr, Lipscomb JD. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He J, Hertweck C. J Am Chem Soc. 2004;126:3694–3695. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- 13.Simurdiak M, Lee J, Zhao H. ChemBioChem. 2006;7:1169–1172. doi: 10.1002/cbic.200600136. [DOI] [PubMed] [Google Scholar]
- 14.Makris TM, Knoot CJ, Wilmot CM, Lipscomb JD. Biochemistry. 2013;52:6662–6671. doi: 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerjee R, Komor AJ, Lipscomb JD. Meth Enzymol. 2017;596:239–290. doi: 10.1016/bs.mie.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 16.Knoot CJ, Makris TM, Lipscomb JD. In: Encyclopedia of Inorganic and Bioinorganic Chemistry. Scott RA, editor. John Wiley & Sons, Ltd; Chichester, UK: 2015. pp. 1–10. [DOI] [Google Scholar]
- 17.He J, Magarvey N, Piraee M, Vining LC. Microbiology. 2001;147:2817–2829. doi: 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- 18.Vu VV, Makris TM, Lipscomb JD, Que L., Jr J Am Chem Soc. 2011;133:6938–6941. doi: 10.1021/ja201822v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jasniewski AJ, Knoot CJ, Lipscomb JD, Que L., Jr Biochemistry. 2016;55:5818–5831. doi: 10.1021/acs.biochem.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]
- 21.Sjöberg BM, Loehr TM, Sanders-Loehr J. Biochemistry. 1982;21:96–102. doi: 10.1021/bi00530a017. [DOI] [PubMed] [Google Scholar]
- 22.Zheng H, Zang Y, Dong Y, Young VG, Jr, Que L., Jr J Am Chem Soc. 1999;121:2226–2235. [Google Scholar]
- 23.Silaghi-Dumitrescu R, Ng KY, Viswanathan R, Kurtz DM., Jr Biochemistry. 2005;44:3572–3579. doi: 10.1021/bi0477337. [DOI] [PubMed] [Google Scholar]
- 24.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 25.Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 26.Zheng H, Lipscomb JD. Biochemistry. 2006;45:1685–1692. doi: 10.1021/bi051605g. [DOI] [PubMed] [Google Scholar]
- 27.Moe LA, McMartin LA, Fox BG. Biochemistry. 2006;45:5478–5485. doi: 10.1021/bi0601611. [DOI] [PubMed] [Google Scholar]
- 28.Fielding AJ, Lipscomb JD, Que LJ. J Biol Inorg Chem. 2014;19:491–504. doi: 10.1007/s00775-014-1122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 30.Bollinger JM, Jr, Diao Y, Matthews ML, Xing G, Krebs C. Dalton Trans. 2009:905–914. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci USA. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 34.Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 35.Lee SK, Nesheim JC, Lipscomb JD. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 36.Wallar BJ, Lipscomb JD. Biochemistry. 2001;40:2220–2233. doi: 10.1021/bi002298b. [DOI] [PubMed] [Google Scholar]
- 37.Banerjee R, Proshlyakov Y, Lipscomb JD, Proshlyakov DA. Nature. 2015;518:431–434. doi: 10.1038/nature14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li TL, Huang F, Haydock SF, Mironenko T, Leadlay PF, Spencer JB. Chem Biol. 2004;11:107–119. doi: 10.1016/j.chembiol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 39.McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Li W. Biopolymers. 2002;66:261–284. doi: 10.1002/bip.10296. [DOI] [PubMed] [Google Scholar]
- 40.Pootoolal J, Thomas MG, Marshall CG, Neu JM, Hubbard BK, Walsh CT, Wright GD. Proc Natl Acad Sci USA. 2002;99:8962–8967. doi: 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sosio M, Stinchi S, Beltrametti F, Lazzarini A, Donadio S. Chem Biol. 2003;10:541–549. doi: 10.1016/s1074-5521(03)00120-0. [DOI] [PubMed] [Google Scholar]
- 42.Du L, Sanchez C, Chen M, Edwards DJ, Shen B. Chem Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 43.Hou J, Robbel L, Marahiel MA. Chem Biol. 2011;18:6555–6664. doi: 10.1016/j.chembiol.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Blazic M, Starcevic A, Lisfi M, Baranasic D, Goranovic D, Fujs S, Kuscer E, Kosec G, Petkovic H, Cullum J, Hranueli D, Zucko J. Appl Environ Microbiol. 2012;78:8183–8190. doi: 10.1128/AEM.01891-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trindade-Silva AE, Machado-Ferreira E, Senra MVX, Vizzoni VF, Yparraguirre LA, Leoncini O, Soares CAG. Genet Mol Biol. 2009;32:572–581. doi: 10.1590/S1415-47572009005000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu L, Budnjo A, Jokela J, Haug BE, Fewer DP, Wahlsten M, Rouhiainen L, Permi P, Fossen T, Sivonen K. ACS Chem Biol. 2014;10:725–733. doi: 10.1021/cb5004306. [DOI] [PubMed] [Google Scholar]
- 47.Jasniewski AJ, Komor AJ, Lipscomb JD, Que L., Jr J Am Chem Soc. 2017;139:10472–10485. doi: 10.1021/jacs.7b05389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci USA. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zocher G, Winkler R, Hertweck C, Schulz GE. J Mol Biol. 2007;373:65–74. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 50.Winkler R, Zocher G, Richter I, Friedrich T, Schulz GE, Hertweck C. Angew Chem Int Ed. 2007;46:8605–8608. doi: 10.1002/anie.200703089. [DOI] [PubMed] [Google Scholar]
- 51.Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tinberg CE, Lippard SJ. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knoot CJ, Kovaleva EG, Lipscomb JD. J Biol Inorg Chem. 2016;21:589–603. doi: 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray WJ, Puvathingal JM. Anal Biochem. 1985;146:307–312. doi: 10.1016/0003-2697(85)90544-5. [DOI] [PubMed] [Google Scholar]
- 55.Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que L., Jr Proc Natl Acad Sci USA. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han Z, Sakai N, Boettger LH, Klinke S, Hauber J, Trautwein AX, Hilgenfeld R. Structure. 2015;23:882–892. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 57.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 58.Bailey LJ, Fox BG. Biochemistry. 2009;48:8932–8939. doi: 10.1021/bi901150a. [DOI] [PubMed] [Google Scholar]
- 59.Banerjee R, Meier KK, Münck E, Lipscomb JD. Biochemistry. 2013;52:4331–4342. doi: 10.1021/bi400182y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Solomon EI, Park K. JBIC, J Biol Inorg Chem. 2016;21:575–588. doi: 10.1007/s00775-016-1372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srnec M, Rokob TA, Schwartz JK, Kwak Y, Rulisek L, Solomon EI. Inorg Chem. 2012;51:2806–2820. doi: 10.1021/ic2018067. [DOI] [PubMed] [Google Scholar]
- 62.Trehoux A, Mahy JP, Avenier F. Coord Chem Rev. 2016;322:142–158. [Google Scholar]
- 63.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winkler R, Richter MEA, Knuepfer U, Merten D, Hertweck C. Angew Chem Int Ed. 2006;45:8016–8018. doi: 10.1002/anie.200603060. [DOI] [PubMed] [Google Scholar]
- 65.Frisch JR, Vu VV, Martinho M, Münck E, Que L., Jr Inorg Chem. 2009;48:8325–8336. doi: 10.1021/ic900961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cranswick MA, Meier KK, Shan X, Stubna A, Kaizer J, Mehn MP, Münck E, Que L., Jr Inorg Chem. 2012;51:10417–10426. doi: 10.1021/ic301642w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li F, Van HKM, Meier KK, Münck E, Que L., Jr J Am Chem Soc. 2013;135:10198–101201. doi: 10.1021/ja402645y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park K, Li N, Kwak Y, Srnec M, Bell CB, Liu LV, Wong SD, Yoda Y, Kitao S, Seto M, Hu M, Zhao J, Krebs C, Bollinger JM, Solomon EI. J Am Chem Soc. 2017;139:7062–7070. doi: 10.1021/jacs.7b02997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang C, Chen H. J Am Chem Soc. 2017;139:13038–13046. doi: 10.1021/jacs.7b06343. [DOI] [PubMed] [Google Scholar]
- 70.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. J Am Chem Soc. 2016;138:7411–7421. doi: 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. Biochemistry. 2017;56:4940–4950. doi: 10.1021/acs.biochem.7b00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li N, Korboukh VK, Krebs C, Bollinger JM., Jr Proc Natl Acad Sci USA. 2010;107:15722–15727. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winkler R, Hertweck C. Angew Chem Int Ed. 2005;44:4083–4087. doi: 10.1002/anie.200500365. [DOI] [PubMed] [Google Scholar]
- 74.Platter E, Lawson M, Marsh C, Sazinsky MH. Arch Biochem Biophys. 2011;508:39–45. doi: 10.1016/j.abb.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 75.Indest KJ, Eberly JO, Hancock DE. J Gen Appl Microbiol. 2015;61:217–223. doi: 10.2323/jgam.61.217. [DOI] [PubMed] [Google Scholar]
- 76.Fries A, Winkler R, Hertweck C. Chem Commun (Cambridge, U K) 2010;46:7760–7762. doi: 10.1039/c0cc02811h. [DOI] [PubMed] [Google Scholar]