

Abstract
The International Diabetes Federation predicts that by 2045 the number of individuals afflicted with diabetes will increase to 629 million. Furthermore, ~352 million individuals with impaired glucose tolerance are at increased risk for developing diabetes. Several mechanisms have been proposed for the onset of metabolic dysfunction and demise of the islet β-cell leading to the pathogenesis of diabetes. It is widely accepted that the onset of type 2 diabetes is due to an intricate interplay between genetic expression of the disease and a multitude of factors including increased oxidative and endoplasmic reticulum stress consequential to glucolipotoxicity and inflammation. Compelling experimental evidence from in vitro and in vivo studies implicates intracellular generation of ceramide (CER), a biologically-active sphingolipid, as a trigger in the onset of β-cell demise under above pathological conditions. Recent pharmacological and molecular biological evidence affirms regulatory roles for Ras-related C3 botulinum toxin substrate 1 (Rac1), a small G protein, in the islet β-cell function in health and diabetes. In this Commentary, we overviewed the emerging evidence implicating potential cross-talk between Rac1 and ceramide signaling pathways in the onset of metabolic dysregulation of the islet β-cell culminating in impaired physiological insulin secretion, loss of β-cell mass and the onset of diabetes. Further, we propose a model depicting contributory roles of defective protein lipidation (prenylation) pathway in the induction of metabolic defects in the β-cell under metabolic stress conditions. Potential avenues for the identification of novel therapeutic targets for the prevention/treatment of diabetes and its associated complications are highlighted.
Keywords: Ceramide, Rac1, Glucolipotoxicity, Pancreatic islet, Metabolic stress, Diabetes
Graphical Abstract

1. Introduction
Recent estimates by the International Diabetes Federation suggest that the incidence of diabetes has soared to an all-time high of 425 million in 2017. The federation predicts that by 2045 the number of individuals afflicted with this disease will increase to 629 million. Furthermore, an alarmingly high number of individuals (~352 million) with impaired glucose tolerance are at high risk for developing diabetes [1]. It is projected that the absolute global economic burden will escalate from U.S. $ 1.3 trillion in 2015 to $ 2.5 trillion in 2030, which represents a staggering increase in costs as a share of global GDP from 1.8% in 2015 to 2.2% in 2030 [2]. Therefore, efforts to understand the pathophysiology of diabetes are critical for moving toward the development of novel therapeutic strategies for this disease.
Insulin secretion from the pancreatic β-cell is regulated precisely by the ambient concentration of glucose in circulation. However, the molecular and cellular mechanisms underlying the stimulus-secretion coupling of glucose-stimulated insulin secretion (GSIS) remain only partially understood. It is well established that GSIS is mediated largely via the generation of soluble second messengers, such as cyclic nucleotides and hydrolytic products of phospholipids by phospholipases A2, C and D. The principal signaling cascade involves the glucose-transporter protein 2-mediated entry of glucose into the β-cell resulting in an increase in the intracellular ATP/ADP ratio as a consequence of glucose metabolism via the glycolytic and tricarboxylic acid pathways. Such an increase in intracellular ATP leads to the closure of membrane-associated ATP-sensitive potassium channels resulting in membrane depolarization followed by influx of the extracellular calcium through the voltage-gated calcium channels on the plasma membrane. A net increase in the intracellular calcium that occurs via the influx of extracellular calcium in addition to the mobilization of calcium from the intracellular storage compartments, has been shown to play critical roles in GSIS [3–5].
Several mechanisms have been proposed that underlie the onset of metabolic dysfunction and demise of the islet β-cell leading to the pathogenesis of diabetes [6–9]. In this context, Robertson proposed [10] that “the pathogenesis of T2D is akin to a double barrel shotgun. The first trigger causes an explosion that sets genetic expression of the disease in motion; the second trigger discharges a host of environmental factors that worsen its clinical course. Candidate shells include glucolipotoxicity, cytokines, oxidative and endoplasmic reticulum stress, and insulin resistance”. Based on emerging evidence in clonal β-cells, primary rodent islets and human islets, it may be concluded that ceramide (CER), a biologically active sphingolipid, plays a significant regulatory role in the onset of β-cell dysregulation under conditions of glucolipotoxicity, endoplasmic reticulum (ER) stress and inflammation [11–13]. These findings were confirmed in islets from animal models of impaired insulin secretion [14]. They are briefly discussed below.
2. CER biosynthetic pathway and roles of sphingolipids in islet (dys) function
A large number of studies have proposed a critical roles for CER and its metabolites in cellular function, specifically in the context of proliferation and apoptosis [15–17]. Intracellular generation of CER could result either from de novo synthesis from fatty acids (palmitate) or from the hydrolysis of sphingomyelin by sphingomyelinases (Figure 1) . In the de novo synthetic pathway, palmitoyl-CoA in the presence of serine is converted 3-ketosphingosine; a step catalyzed by the enzyme serine palmitoyl transferase. Consequential to several metabolic steps, 3-keto-sphingosine is converted to CER, which, in turn, is converted to sphingosine by ceramidase. Sphingosine is then phosphorylated to sphingosine-1-phosphate by sphingosine kinase. It should be noted that both CER and sphingosine are implicated in cell dysregulation, cell senescence, cell cycle arrest and cell apoptosis. Interestingly, however, sphinsone-1-phosphate has been shown to play key regulatory roles in cell proliferation, cell survival and cell motility [15–18; Figure 1].
Figure 1. Regulatory roles of CER signaling steps in cell survival and apoptosis.

CER can be generated intracellularly via the de novo and recycling pathways. In the de novo pathway, palmitoyl-CoA and serine are converted to CER, a step catalyzed by serine palmitoyl transferase. In the recycling pathway, CER is formed via degradation of sphingomyelins; these steps are mediated by a variety of sphingomyelinases. CER is converted sphingosine by ceramidase. Sphingosine is converted to sphingosine-1-phosphate by sphingosine kinase. It is noteworthy that both CER and sphingosine have been shown to promote cell cycle arrest and cell senescence and induce cell apoptosis. Interestingly, however, sphingosine-1-phosphate exerts positive modulatory effects on cell function including cell proliferation, motility and survival. Therefore, intermediates of these pathway play both positive and negative modulatory roles in cell function.
Seminal contributions from the laboratory of Unger have supported the concept of lipotoxicity in the onset of metabolic diseases including diabetes [11,14]. In this context, several recent reviews were dedicated to highlight regulatory roles of sphingolipids, particularly CER, in the onset of metabolic dysregulation and demise of the islet β-cell in type 1 and type 2 diabetes and its associated complications. Chaurasia and Summers [19] have reviewed existing evidence that strongly implicates CERs as “lipotoxic inducers of metabolic disorders” in multiple tissues including liver, eye, kidney, skeletal muscle and pancreas. Janikiewicz and associates [20] have suggested regulatory roles of CER as a “toxic lipid messenger” involved in the cross-talk between the ER and the mitochondria. Additional lines of investigations suggest that exposure of the islet β-cell to metabolic stress conditions including chronic hyperglycemia (glucotoxicity), hyperlipidemia (lipotoxicity) or both (glucolipotoxicity) leads to the pathogenesis of metabolic defects leading to aberrant functions of intracellular organelles, including ER (stress), mitochondria (loss in membrane permeability pore transition, cytochrome-C release and caspase-3 activation), and nucleus (lamin degradation and dissolution of nuclear envelop; [10,21,22]) . Similar metabolic defects have been reported in the islet β-cell following exposure to pro-inflammatory cytokines (IL-1β, TNFα and IFNγ; [23,24]) . Germane to this Commentary are data accrued in the studies suggesting regulatory roles for intracellularly generated CER in promoting islet β-cell dysregulation under the duress of metabolic stress (gluco-, lipo-, glucolipotoxicity) and exposure to cytokines [12,24,25]. As stated above, contributory roles of CER in the induction of metabolic defects in the islet β-cell have also been reported in animal models of diabetes [14,19]. Indeed, inhibition of CER biosynthesis significantly improved islet β-cell defects in animal models of diabetes [11,14]. More recent investigations by Campana and associates have demonstrated novel regulatory roles of de novo CER synthesis in hypothalamus on the onset of central insulin resistance and islet β-cell dysfunction in cultured hypothalamic neuronal GT1-7 cells and obese Zucker rats [26]. It is noteworthy that treatment of obese animals with myriocin, a known inhibitor de novo biosynthesis of CER, partially improved glucose tolerance by restoring GSIS and an increase in β-cell mass of obese Zucker rats. Together, based on the discussion above, it may be surmised that CER plays key regulatory roles in the pathophysiology of metabolic dysregulation of islet β-cell in vitro and in vivo.
3. G proteins in islet function in health, metabolic stress and diabetes
Emerging evidence suggests important roles for a wide variety of GTP-binding proteins (G proteins) in GSIS from the islet β-cell [27–29]. At least three classes of G proteins have been described to date in β-cells. The first group is heterotrimeric in nature, and comprised of α, β and γ subunits. The trimeric G proteins are involved in coupling membrane-associated receptors (GPCRs) to their intracellular effectors (e.g., ion channels, phospholipases, adenylyl cyclases, and phosphodiesterases) . Examples of this class of G proteins include the inhibitory (Gi) and the stimulatory (Gs) class of G proteins. The second group of G proteins (focus of this Commentary) is monomeric in nature, which, to a large extent, regulate protein sorting and trafficking of secretory vesicles in many cell types. Some examples of these include Cdc42, Rho, Rac1, and ARF6. The third (less studied and understood) group of G proteins is represented by elongation factors and Tau proteins, which are involved in a variety of cellular functions, including protein synthesis and stabilization of microtubular networks.
From a mechanistic standpoint, G proteins cycle between their GDP-bound (inactive) and GTP-bound (active) conformations; this is often referred to as the GTP-hydrolytic cycle, which is tightly controlled by specific regulatory proteins/factors [28–30]. At least three major types of such regulatory proteins/factors have been described for small G proteins. The first group is comprised of the guanine nucleotide exchange factors (GEFs), which promote the conversion of the GDP-bound form of G proteins to their GTP-bound forms. The second group consists of the GDP-dissociation inhibitors (GDIs), which prevent the dissociation of GDP from G proteins thereby retaining them in their inactive confirmation. Therefore, GDIs are considered negative modulators of the G protein activation cascade. The third group represents the GTPase-activating proteins (GAPs) ; these proteins promote conversion of the GTP-bound G proteins to their GDP-bound conformation to complete the GTP hydrolytic cycle. Interestingly, however, emerging evidence suggests additional, and somewhat non-canonical, regulatory roles for these factors/proteins to regulate G protein function. For example, certain GDIs have been shown to complex with GTP-bound G proteins and prevent their degradation [31]. This remains to be verified in the islet β-cell. It should be also noted that specific post-translational modifications of G proteins (e.g., isoprenylation; see below) appear to be requisite for optimal regulation of the G protein function by GEFs, GDIs and GAPs [32].
4. Regulatory roles of Rac1 in β-cell (dys) function
Evidence from multiple laboratories suggests novel roles for Ras-related C3 botulinum toxin substrate 1 (Rac1), a small G protein belonging to the Rho subfamily of G proteins, in cellular function in health and diabetes [27–30]. Rac1 undergoes activation-deactivation cycles [30]. The GDP-bound inactive Rac1 is converted to its active, GTP-bound conformation by GEFs, including T-lymphocyte invasive and metastasis protein 1 (Tiam1) and the guanine nucleotide exchange factor Vav2 (Vav2) . Using pharmacological and gene depletion (siRNA) approaches, we have demonstrated novel roles for Tiam1 and Vav2 in islet function in health and metabolic stress (glucolipotoxicity and ER stress) in clonal β-cells, rat islets and human islets [30]. It should be noted that while this review is largely focused on the islet β-cell, investigations from numerous laboratories have suggested roles for chronic activation of Rac1 in multiple pathologies including cancer, neurodegenerative diseases (Parkinson and Alzheimer’s), cardiac disease as well as metabolic disease, including diabetes [33–39].
What then are potential target (effector) proteins, which are regulated by Rac1? In this context, studies from several laboratories, including our own, have demonstrated that activation of phagocyte-like NADPH oxidase (Nox2) as one of the early signaling events in the induction of cellular defects seen under glucolipotoxicity, ER stress and exposure to pro-inflammatory cytokines and cell-permeable analogues of CER [23,29,40–43]. Rac1 is an integral part (member of the cytosolic core) of the Nox2 holoenzyme, and activation of Rac1 is critical for its translocation to the membrane to facilitate Nox2 holoenzyme assembly, activation and associated generation of reactive oxygen species (ROS) . Indeed, suppression of Rac1 activation protects β-cells against noxious effects of glucolipotoxicity and cytokines [23,29,40–43].
The main objective of this Commentary is to highlight the available evidence implicating roles of Rac1 in metabolic dysregulation of various cells including the islet β-cell as it relates to the biological effects of CER. Several lines of experimental evidence in human leukemic Jurkat cells [44], Rat2 fibroblasts [45], NIH3T3 cells [46], human umbelical endothelial cells [47], retinal pericytes [47], and retinal endothelial cells [48] have suggested potential interplay between CER and Rac1 signaling pathways culminating in cell dysregulation and demise. Following is our current understanding of regulatory roles of these two signaling pathways in the onset of islet dysfunction under conditions of metabolic stress and inflammation.
5. Evidence suggesting regulatory roles of Rac1-CER signaling steps in islet β-cell dysfunction
Recent investigations from our laboratory have implicated sustained activation of Rac1 in metabolic dysregulation in clonal β-cells, normal rodent islets and human islets exposed to glucolipotoxic conditions [29,30,41–43,50–52]. Further, we also identified Nox2 as one of the target proteins, which is regulated by Rac1. It should be noted that Rac1 represents one of the cytosolic core proteins of the Nox2 holoenzyme, which upon activation (Rac1.GTP) translocates, alongside other cytosolic core proteins (p47phox and p67phox) to the membrane fraction to associate with the membranous core (p22phox, gp91phox) for completion of holoenzyme assembly leading to the activation of Nox2 and subsequent generation of superoxides [29,41,43]. In the following section, we will highlight findings from some studies that investigated potential roles of Rac1-CER signaling pathway in promoting β-cell dysfunction under the duress of metabolic stress and exposure to pro-inflammatory cytokines.
5.1. Regulation of islet dysfunction by PA and CER
Using normal rat islets and clonal INS 832/13 cells, we tested the hypothesis that activation of Rac1, which is a member of the Nox2 holoenzyme, is necessary for PA-induced (lipotoxicity) generation of superoxides in pancreatic β-cells [42]. We observed that Incubation of isolated β-cells with PA markedly augmented Nox2 activity culminating in a significant increase in the generation of superoxides and lipid peroxides in these cells. Such effects of PA were attenuated by diphenyleneiodonium, a known inhibitor of Nox2. In addition, fumonisin B-1 (FB1), which inhibits de novo synthesis of CER from PA, suppressed PA-induced superoxide and lipid peroxide generation and Nox2 activity implicating intracellularly generated CER in the metabolic effects of PA. In further support of this postulation, the above effects were also demonstrable in the presence of the cell-permeable C2-CER. Based on these observations, we concluded that sustained activation of Rac1 signaling pathway may underlie PA-induced, CER-dependent superoxide generation and mitochondrial dysfunction in pancreatic β-cells.
5.2. Regulation of islet dysfunction by deoxysphingolipids
Zuellig and associates [53] recently provided the first direct evidence implicating deoxy-sphingolipids in induction of metabolic dysfunction of insulin-secreting INS-1 cells and primary rodent islets. Preincubation of these cells with 1-deoxysphinganine resulted in significant alterations in GSIS and eventually leading to cell demise. Notably, 1-deoxysphinganine also increased the expression and activation of Rac1 (not RhoA) and caused alterations in cytoskeletal makeup, including intracellular accumulation of F-actin, and activation of stress kinases. A significant upregulation of CER synthase 5 activity in 1-deoxysphinganine-treated cells resulted in its conversion to 1-deoxydihydroCER. More importantly, pharmacological inhibition of intracellular trafficking of sphingolipids partially restored the cytotoxic effects 1-deoxysphinganine. Based on these observations, these researchers concluded that 1-deoxysphinganine and its metabolic intermediate 1-deoxydihydroCER exert deleterious effects on islet β-cell function, including inhibition of GSIS and induction of loss in metabolic cell viability [53].
5.3. Regulation of islet dysfunction by water-soluble CERs
In 2006, Szulc and associates [54] have developed a new class of cationic CER mimics (dubbed ceramidoids) to determine their organelle-targeted effects. This was achieved by conjugating CERs with pyridinium salts, and due to their cationic nature, these CER analogs showed a significantly improved water solubility, fast cellular uptake and higher anti-cancer activity in MCF-7 breast carcinoma cells. We recently investigated [55] the effects of L-threo-C6-pyridinium-CER bromide, a water soluble cationic CER (Ws-CER), on mitochondrial function and cell viability in insulin-secreting INS 832/13 cells. We observed a marked increase in the Nox2 activation (i.e., Nox2 subunit expression, its functional activation and associated ROS generation) in these cells exposed to Ws-CER. A significant reduction in MMP, increase in caspase-3 activity and associated loss in cell viability were demonstrable in Ws-CER-treated cells. These findings confirmed our earlier investigations demonstrating inhibitory effects of C2- CER, but not C2-dihydroCER, mitochondrial membrane potential leading to cytochrome C release and loss in metabolic cell viability [56]. Furthermore, in earlier studies, we identified a novel okadaic acid-sensitive serine, threonine phosphatase activity (PP2A), which was stimulated by C2-CER, but not its dihydro derivative. Based on these data we conclude that CER-induced Nox2-mediated oxidative stress couples mitochondrial dysfunction (presumably via stimulation of a CER-activated PP2A) to loss in cell viability in the pancreatic β-cell [56].
5.4. Regulation of islet dysfunction by pro-inflammatory cytokines
Published evidence in the literature suggests that cytokine-induced cell dysfunction is mediated via generation of intracellular CERs [24–26,57]. Chronic exposure of islet β-cells to pro-inflammatory cytokines activates Nox2 leading to increased generation of ROS and intracellular oxidative stress culminating in the metabolic dysregulation of pancreatic β-cells [23,58]. Although the mitochondrial electron transport chain is felt to be the primary source of ROS, several lines of recent evidence suggest that Nox2 plays a central role in cytokine-mediated ROS generation and apoptosis of β-cells. In this context, we recently investigated potential mechanisms underlying the regulation of Nox2 in INS-1 832/13 cells exposed to proinflammatory cytokines. A significant, time-dependent increase in Nox2 activation/ intracellular ROS production, p47phox subunit, but not p67phox subunit, expression of the phagocyte-like NADPH oxidase were demonstrable under these conditions. Cytomix-mediated mitochondrial dysfunction in INS 832/13 cells was evidenced by a significant loss of MMP and upregulated caspase 3 activity. Cytomix treatment also caused activation of Rac1, a component of the Nox2. Furthermore, GGTI-2147 and NSC23766, known Rac1 inhibitors, not only attenuated the cytomix-induced Rac1 activation but also significantly prevented loss of MMP. Together, these findings suggested that Rac1-mediated regulation of Nox2 contributes to cytokine-mediated mitochondrial dysfunction in the β-cell [23,58]. Based on the above discussion, we conclude that Rac1-CER signaling pathway could potentially contribute to cell dysfunction in multiple cell types including the islet β-cell.
6. Is CER-induced Rac1 activation mediated by specific regulatory proteins/factors?
Several lines of evidence implicate the intermediary of additional regulatory factors in CER-induced activation of Rac1. These include Tiam1 and Vav2, which are known GEFs for Rac1. For example, using rat mesangial cells, Yi and associates [59] have proposed novel roles for Rac1-NADPH oxidase signaling axis in homocysteine-induced glomerular injury and end-stage renal disease. Genetic silencing (siRNA) of Rac1 expression significantly reduced homocysteine-induced superoxide production. Furthermore, siRNA-mediated depletion and functional inactivation of Vav2, a known GEF for Rac1, markedly suppressed homocysteine-induced superoxide generation suggesting that Vav2-Rac1 module might regulate homocysteine-induced NADPH-oxidase activation. In addition, siRNA-Vav2 attenuated C16-CER-induced Rac1 activation. Finally, homocysteine-treatment significantly increased tyrosine phosphorylation (and activation) of Vav2, which, in turn, was inhibited by FB1. Based on these observations, the authors concluded that homocysteine-mediated generation of intracellular oxidative stress may, in part, be due to increased intracellular generation of CER. This, in turn, facilitates Rac1 activation via tyrosine phosphorylation and activation of Vav2 leading to Nox2 activation and superoxide generation. These findings, thus, provide evidence in support of a key regulatory role for Vav2 in CER-induced activation of Rac1.
Along the above lines, Syed et al [42] reported novel roles for Tiam1, another GEF for Rac1, in PA and CER-induced metabolic defects in pancreatic islet β-cells. These studies utilized NSC23766, a selective inhibitor of Rac1, but not Cdc42 or Rho [29,30], to assess the regulatory roles for Tiam1 in PA and CER induced metabolic dysregulation of islet β-cells. NSC23766 significantly reduced PA-induced Rac1 activation and associated generation of superoxides and lipid peroxides. It also prevented C2-CER-induced Rac1 activation and production of superoxides and lipid peroxides. Lastly, C2-CER, but not its inactive analogue, markedly suppressed the mitochondrial membrane potential, which was prevented to a large degree by NSC23766. Taken together, these findings indicate novel roles for Tiam1-Rac1 signaling module in PA and CER induced Rac1 activation in β-cells.
Studies by Subasinghe et al [23] identified Rac1 as one of the targets involved in metabolic dysregulation induced by pro-inflammatory cytokines (IL-1β, TNFα and IFNγ; cytomix) in insulin-secreting INS-1 832/13 cells. NSC23766 not only attenuated the cytomix-induced Rac1 activation but also prevented loss of MMP seen following cytokine exposure. Interestingly, however, NSC23766 had no effect on cytomix-induced nitric oxide generation or caspase 3 activation, suggesting additional regulatory mechanisms might underlie these signaling steps. Lastly, the aforementioned in vitro findings were confirmed by Veluthakal and associates in the NOD mouse, a well-studied model for type 1 diabetes [60]. Administration of NSC23766 to NOD mice significantly suppressed Rac1 expression, activation and endoplasmic reticulum stress in isolated islets from these mice. Furthermore, NSC23766 treatment prevented the development of spontaneous diabetes in the NOD mice [60]. Altogether, data from these studies highlight the requisite nature of Tiam1-Rac1 signaling pathway in pro-inflammatory cytokine-induced metabolic dysfunction of the islet. Despite the evidence suggesting regulatory roles of CER in the genesis of islet dysfunction under the duress of inflammation [24,25], potential contributory roles of this signaling pathway in cytokine-induced and CER-Rac1 mediated islet β-cell dysregulation remains to be validated further.
More recent investigations by Kumar et al in retinal endothelial cells provided evidence to implicate Tiam1 as the GEF for Rac1 activation under the duress of lipotoxicity, glucolipotoxicity and exposure to cell permeable CER [49]. They noted a marked suppression by NSC23766 of activation of Rac1 and Nox2 and associated increase in ROS generation under these conditions. NSC23766 also prevented mitochondrial damage induced by glucolipotoxicity and CER exposure. In summary, based on the findings reviewed above, it can be concluded that metabolic stress induced cell dysregulation involves significant cross-talk between CER and Rac1 signaling pathways, and such a regulatory mechanism also involves additional regulatory factors, including Tiam1 and Vav2.
7. Other G proteins, besides Rac1, are also targets for CER-mediated cellular regulation
In addition to Rac1 (above), published evidence suggests modulation of other G protein-mediated signaling pathways by sphingolipids, including CER. Select studies are highlighted in this section. Using mouse embryonic stem cells, Park et al [61] have demonstrated a dose-and time-dependent increase in cell migration by C16-CER. From a mechanistic standpoint, these studies implicated phosphorylation of protein kinase C, focal adhesion kinase and paxillin as intermediary steps involved in CER-induced cell migration. In addition, exposure of these cells to CER markedly promoted activation of Cdc42 thereby promoting the formation of a complex between Cdc42, the Neural Wiscott-Aldrich Syndrome Protein (N-WASP) and the Actin-Related Protein 2/3 (Arp2/3) . Based on data accrued in gene knock-out studies, these investigators concluded novel roles for N-WASP/Cdc42/Arp2/3 in CER-induced protein kinase C, focal adhesion kinase and paxillin-mediated cytoskeletal organization and cell migration.
Gupta et al [62] have implicated RhoA, another small G protein, in angiotensin-induced actin stress fiber reorganization, cell detachment and demise in human endothelial cells. Preexposure of these cells to angiotensin resulted in transient increase in CER production and RhoA activation leading to cytotoxicity. Interestingly, N-acetylcysteine, a known antioxidant, prevented structural and metabolic defects in these cell under the duress of angiotensin treatment. Based on these findings the researchers postulated critical regulatory roles for CER-RhoA signaling axis in angiotensin-induced free radical production (oxidative stress) and cellular dysregulation in human endothelial cells.
Brindley and coworkers [63] have tested the hypothesis that TNFα-mediated effects on the onset of insulin resistance are mediated via CER. They reported a significant increase in 2-deoxyglucose uptake by differentiated 3T3-L1 adipocytes following exposure to cell permeable CER analogues under basal conditions (i.e., absence of insulin) . Such effects of CER were significantly attenuated following inhibition of mitogen-activated protein kinase, PI 3-kinase and S6 kinases, thus suggesting key roles for these kinases in CER-mediated effects. These findings were further confirmed in rat2 fibroblasts in that exposure of these cells to TNFα, bacterial sphingomyelinase, or cell-permeable CERs significantly increased tyrosine kinase activity, Ras G protein activation leading to nearly five-fold increase in PI 3-kinase activity. These findings implicated novel roles for CER-Ras signaling pathway in TNFα-induced uptake of glucose under basal conditions [63]. Follow-up studies [64] by these investigators in rat2 fibroblasts implicated additional regulatory roles and involvement of Cdc42, Rac1 and RhoA in TNFα-mediated CER-induced effects. For example, treatment of these cells to TNFα, sphingomyelinase or CER promoted translocation of these G proteins to the membrane fraction, a signaling step necessary for their biological activation and effector interaction. Roles of RhoA in this signaling cascade was further confirmed by transfection of a dominant negative mutant of RhoA, which lead to inhibition of TNFα and CER-induced stress fiber formation. Based on these findings these investigator proposed novel roles of CER-G protein signaling axis in TNFα-induced stress fiber formation and cytoskeletal reorganization.
Lastly, published evidence also implicates novel roles for CER in modulation of enzymes involved phospholipid metabolism. For example, phospholipase D (PLD), which hydrolyzes phosphatidylcholine to yield phosphatidic acid, has been shown to be regulated by intracellularly generated CER. Further, if CERs are converted to sphingosine and its phosphorylated derivative, sphingosine-1-phosphate, it would lead to functional activation of PLD and further propagation of mitogenic effects. Interestingly, however, CERs, directly influence PLD activity by preventing interaction of PLD with small G proteins including ADP ribosylation factor-6 (ARF6) and Rho, a step which is necessary for the functional activation of PLD. Along these lines Venable and Obeid have proposed [65] that CER could directly affect PLD activation by inhibiting RhoA activation, its translocation to the membrane and gene expression. Therefore, it is likely that CER-induced metabolic dysfunction in cells (and increased senescence) could involve its direct effects on G proteins. Potential implication of these findings in CER-induced islet β-cell dysfunction need to be validate further. Based on the above discussion, it can be concluded that CER-induced effects of cellular function involves a variety of small G proteins including Arf6, Cdc42, Rac1, Ras and RhoA. Indeed, studies from many laboratories, including our own have clearly implicated these G proteins in islet β-cell function, including GSIS [27–30]. They represent the positive modulatory roles of these G proteins in islet function. However, as highlighted in the above section, it appears that some of these G-proteins (Rac1) are constitutively activated in the islet β-cell under conditions of metabolic stress, including exposure to glucolipotoxic, ER stress conditions as well as exposure to proinflammatory cytokines [29,41]. The two important questions that need to be answered are: what are putative mechanisms that mediate constitutive activation of these G proteins, and where does CER signaling fit into this scheme? These questions are addressed in the following section.
8. Proposed model for CER-induced defects in the G protein prenylation signaling pathway
At least 300 prenylated proteins are identified in the human genome; the majority of which partake in a variety of cellular processes including growth, differentiation, cytoskeletal organization/dynamics and vesicle trafficking [66,67]. Aberrant prenylation of proteins is implicated in human pathologies including cancer; neurodegenerative diseases, retinitis pigmentosa, and premature ageing syndromes [66–69]. Available evidence in multiple cell types, including the islet β-cell, suggests that chronic inhibition (pharmacological, dominant negative mutants and conditional deletion of prenylating enzymes) of prenylation the C-terminal cysteine of small G proteins leads to functional activation [29,70,71] and mis-localization (nuclear accumulation) of these G proteins, such as Rac1 [29]. Therefore, it is likely that metabolic stress conditions (glucolipotoxicity, ER stress) and exposure to pro-inflammatory cytokines, and potentially, CER could promote aberrant prenylation of these G proteins leading to their activation constitutively (Figure 2) . To test such a formulation, we recently assessed the impact of metabolic stress on the functional status of protein prenylation pathway in pancreatic β-cells. We observed that metabolic stress activates caspase-3 and promotes degradation of the common α-subunit of farnesyl transferase (FTase) and geranylgeranyl transferase (GGTase) in INS-1 832/13 cells, normal rodent islets and human islets leading to inhibition of FTase and GGTase activities [72]. Furthermore, we observed significant reduction in the catalytic activities of FTase/GGTase under these conditions. It is noteworthy that caspase-3 activation and FTase/GGTase-α degradation were also seen in islets from the ZDF rat, a model for Type 2 diabetes. Consequential to defects in FTase/GGTase-α signaling, a significant accumulation of unprenylated proteins (Rap1) was also observed in β-cells exposed to glucotoxic conditions [72]. Furthermore, transfection of INS-1 832/13 cells with a caspase 3-resistant mutant of FTase/GGTaseα prevented loss in GSIS and cell viability under glucotoxic conditions (Kowluru, unpublished) . These findings implicate that caspase-3 induced degradation of prenylating enzymes may, in part, be responsible for aberrant prenylation of G proteins (Rac1) in insulin-secreting cells.
Figure 2. A proposed model for CER-induced defects in protein prenylation, aberrant activation of G proteins culminating in cell dysfunction and demise.

We propose that intracellularly generated CER promotes defects in protein prenylation via two mechanisms. In the first (left), it induces degradation of the common α-subunit of FTase and GGTase via caspase 3 activation. Degradation of FTase/GGTase-α leads to its functional inactivation [72]. While the later was reported in clonal β-cells, rodent and human islets exposed to metabolic stress conditions [72], this has not been verified in β-cells under the duress of CER exposure. The second mechanism (right) involves direct effects of CER on the expression and catalytic function of HMG CoA reductase [75,76]. This, in turn, leads to depletion of mevalonate-derived farnesyl pyrophosphate (Fpp) and geranylgeranyl pyrophosphate (GGpp), which are substrates for FTase and GGTase, respectively. This postulation has not been tested in the islet β-cell. We propose that inhibition of prenylation leads to sustained activation of candidate G proteins (Rac1) leading to their mislocalization in inappropriate localization (nuclear association) and accelerated apoptosis [74].
Recent evidence also suggests similar caspase 3-mediated degradation of FTase/GGTase activities in pancreatic β-cells under apoptotic conditions promoted by a different stimulus. Exposure of insulin-secreting INS 832/13 cells or normal rat islets to etoposide leads to significant activation of caspase-3 and subsequent degradation of the common α-subunit of FTase/GGTase [73]. Furthermore, etoposide-induced caspase 3 activation and FTase/GGTase degradation were prevented by Z-DEVD-FMK, a known inhibitor of caspase-3. In addition, treatment of cell lysates with recombinant caspase-3 also caused FTase/GGTase α-subunit degradation. Nifedipine, a calcium channel blocker, markedly attenuated etoposide-induced caspase-3 activation, FTase/GGTase α-subunit degradation in INS 832/13 cells and normal rat islets. Further, nifedipine significantly restored etoposide-induced loss in metabolic cell viability in INS 832/13 cells. Based on these findings, we concluded that etoposide induces loss in cell viability by inducing mitochondrial dysfunction, caspase-3 activation and degradation of FTase/GGTase α-subunit. Together, as we originally proposed, protein prenylation plays a significant regulatory role in β-cell survival and function including insulin secretion [66]. Inhibition of these signaling steps leads to mistargeting of unprenylated and constitutively active G proteins into “improper” cellular compartments leading to cell dysfunction [51,52,66,74; Figure 2]. Therefore, it is likely that exposure or generation of intracellular CER (glucolipotoxicity, ER stress and cytokine exposure) leads to aberrant prenylation and constitutive activation of G-proteins, including Rac1 to favor cell dysfunction, i.e., non-friendly roles of Rac1 as we proposed earlier [41]. Even though it seems likely, potential roles of intracellularly generated CER in the functional inactivation of prenylating enzymes and the downstream signaling events need to be verified in the islet β-cell.
9. Proposed model for CER induced defects in the G protein prenylation via inhibition of HMG CoA-reductase pathway
In addition to the direct effects of CER on FTase/GGTase α degradation and inhibition, there might be other signaling steps that is potentially regulated by intracellularly generated CER leading to inhibition of protein prenylation pathway leading to constitutive activation of G proteins (Rac1; Figure 2) . For example, studies by Gallardo and associates [75] have demonstrated direct effects of TNFα, and its second messenger CER on HMGCoA reductase, the rate-limiting enzyme in the mevalonate pathway. Mevalonate is the precursor for biosynthesis of isoprenyl-pyrophosphates (farnesyl pyrophosphate and geranylgeranyl pyrophosphate), which are substrates for FTase and GGTase, respectively. Treatment of human U-937 and HL-60 cells with TNFα or C2-CER significantly attenuated the expression (mRNA levels) and catalytic activity of HMGCoA reductase in a time-dependent fashion. Consequentially, maturation of Ras was also inhibited under these conditions culminating in significant accumulation of unprenylated Ras. Provision of exogenous mevalonate prevented defects in TNFα and CER-induced processing of Ras and apoptosis. These findings afford further strength to the hypothesis that alterations in prenylation of proteins could contribute to cytokine- and CER-induced cell apoptosis in these cells [75]. Along these lines, Subbaiah and associates investigated effects of CER on cholesterol trafficking and HMG CoA reductase activity in fibroblasts [76]. They observed that incubation of these cells with sphingomyelinase C caused a significant reduction (90%) in HMG CoA reductase activity. Much lower levels on inhibition of this activity was seen in cells incubated with sphingomyelinase D. Furthermore, treatment of these cells to exogenous CER significantly attenuated (60–80%) HMG CoA reductase activity. A significant increase in the phosphorylation of HMG CoA reductase enzyme (a trigger for its functional inactivation) was also noted in cells incubated with exogenous CER. These data raise an interesting possibility that intracellularly generated CER could inhibit protein prenylation via depleting prenylpyrophosphates (Figure 2) . These possibilities will need to be assessed in the islet β-cell.
10. Additional possibilities that need to be considered in validation of the above model
Based on the experimental evidence available thus far it is reasonable to conclude that metabolic stress induces alterations in protein prenylation leading to constitutive activation of unprenylated G proteins (Rac1) . Aberrant prenylation might result from direct inhibition of HMG CoA reductase thereby depleting intracellular pools of farnesyl pyrophosphates (Fpp) or geranylgeranyl pyrophosphates (GGpp), which are needed for prenylation of G-proteins. This has not been verified in the islet β-cell yet. Alternatively, defects in protein prenylation could be due to degradation of prenylating enzymes leading to functional activation of these enzymes. This has been verified in clonal β-cells, normal rat islets and human islets. Lastly, based on data from multiple cell types, inhibition of Tiam1-Rac1 signaling pathway (NSC23766) prevents activation of Rac1 under metabolic stress [29]. However, it has been shown in multiple cell types, albeit not in islet β-cell, that Tiam1 is also degraded and inactivated by caspase 3. Some of these studies are highlighted below.
Using three cell model systems (Jurkat, PC12 and HMN1), Qi et al [77] reported cleavage of Tiam1 by caspase 3 under conditions of exposure to CER or Fas or serum deprivation, which lead to functional inactivation of Tiam1 as evidenced by its inability to activate Rac1, and activation of downstream effectors of Rac1, including c-jun NH2 terminal kinase and serum deprivation factor. It is noteworthy that cleavage of Tiam1 coincided with inactivation of Rac1 in CER-treated cells. These data suggest that inactivation of Rac1 might represent a critical signaling step in cellular apoptosis induced by three different stimuli including CER. Along these lines, studies by Tsai and coworkers reported [78] alterations in mitochondrial function, caspase 3 activation, Tiam1 cleavage and inactivation of Rac1 in hippocampal neurons following siRNA-mediated knockdown of sigma-1 receptors, which lead to dendritic spine deficits. A caspase 3 resistant mutant of Tiam1 (C1199DN) or constitutively active Rac1 reversed the dendritic spine defects. These two studies provide evidence for potential regulation of Tiam1-Rac1 signaling in cells under conditions of caspase 3 activation and cell apoptosis. Although caspase 3 mediated degradation of Tiam1 has not been addressed in islet β-cell, it is more than likely that might occur, and needs to be verified experimentally. This raises an interestingly possibility that activation of Tiam1-Rac1 signaling pathway during the early stages of cellular dysfunction prior to activation of mitochondrial pathway and caspase 3 mediated degradation would be sufficient to trigger metabolic defects leading to loss in islet β-cell function including loss of GSIS. Time course studies, therefore, are necessary to assess the magnitude of effects of Tiam1-Rac1 activation on metabolic indices in cells under metabolic stress conditions, including generation of intracellular CER. In this context, it may be germane to highlight observations of O’Brien et al who examine roles of Tiam1-Rac-ERK signaling axis in protective effects of cytokines against apoptosis induced by serum deprivation in rabbit corneal stromal fibroblasts [79]. Using a culture model system they demonstrated significant increase steady state levels of Tiam1 (mRNA and protein), a GEF for Rac1, during the initial 6 hrs. of post-serum deprivation. A significant increase in membrane-associated Rac1 was also demonstrable within this time period. After 6 hour post-serum deprivation the levels of native Tiam1 decreased as evidenced by a 75 kDa caspase 3 degradation product. Based on these observations the investigators concluded that Tiam1-Rac1-ERK pathway may function in promoting cell dysfunction in corneal stromal fibroblasts under conditions of inflammation.
11. Conclusions and future directions
It is evident from the above discussion that sustained activation of Rac1 may represent one of the underlying steps in metabolic dysregulation of the islet β-cell. This appears to be the case in cells following exposure to noxious conditions including glucotoxicity, lipotoxicity, glucolipotoxicity, exposure to pro-inflammatory cytokines, and ER stress. Potential intermediary roles of sphingolipids, such as CER, in the onset of Rac1-mediated metabolic dysfunction of the cells appear plausible. It should be noted that aberrant activation of Rac1 and CER signaling pathways leads to functional impairments of numerous target proteins/transcriptional factors (e.g., Akt, ERK1/2, FoXo1, NFkb, p38 MAPK, p53, JNK1/2, and PP2A) . Based on the available evidence in multiple cell types, including the islet β-cell, we propose a working model which states that Rac1-CER signaling plays novel roles in the onset of metabolic dysfunction of the islet, including loss of GSIS, loss of functional β-cell mass, and pathogenesis of diabetes (Figure 3) . Exposure of islet β-cell to glucolipotoxic conditions or pro-inflammatory cytokines results in intracellular generation of CER via the de novo (sensitive to FB1) or recycling (sensitive to sphingomyelinase inhibitors) pathways. Increased levels of intracellular CER, in turn, promote sustained activation of Rac1, which is mediated via activation of two GEFs for Rac1, namely Tiam1 and Vav2. Tiam1/Rac1 and Vav2/Rac1 signaling steps are inhibited by NSC23766 and Ehop-016, respectively [30]. It is also proposed that sustained activation of Rac1 leads to increased generation of ROS mediated via Nox2 activation, which, in turn, promotes stress kinase activation and mitochondrial dysfunction. These defects manifest into impaired GSIS and loss in metabolic cell viability eventually resulting in cell demise [80].
Figure 3. A proposed model for Rac1-CER signaling cascade in the genesis of islet dysfunction under conditions of metabolic stress and exposure to pro-inflammatory cytokines.
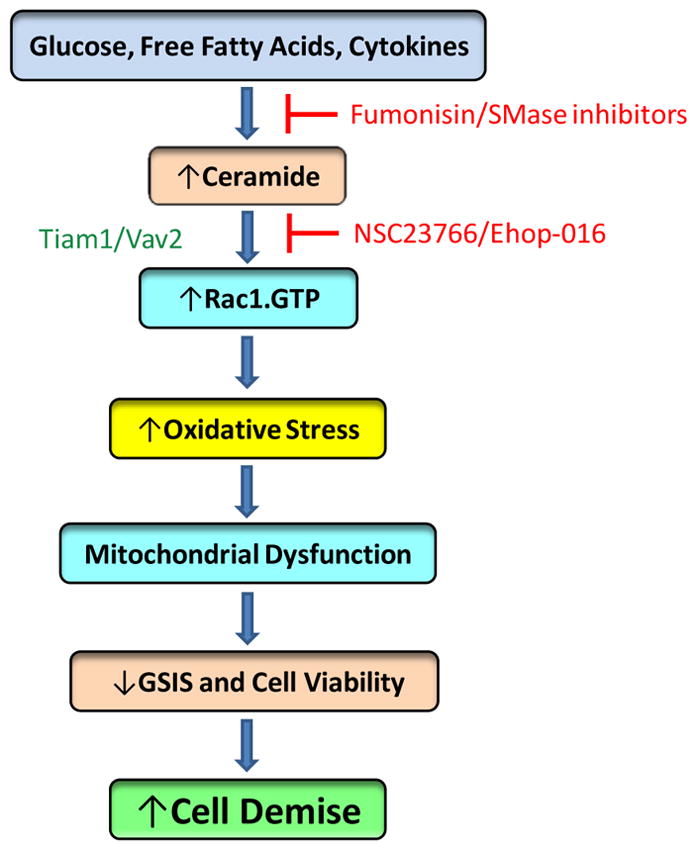
Exposure of islet β-cells to high glucose, saturated fatty acids and cytokines leads to generation of intracellular CER, which, in turn, promotes Rac1 activation by its GEFS, namely Tiam1/Vav2. It should be noted that, at least, in the context of islet β-cell, roles of Vav2 in CER-induced activation of Rac1 have not been demonstrated. Constitutively activated Rac1 promotes activation of Nox2 resulting in increased oxidative stress, mitochondrial dysregulation, impaired GSIS, loss in metabolic cell viability resulting in cell apoptosis (see text for additional details) .
It may be germane to point out that while a significant degree of discussion was devoted to Rac1 as an effector molecule (or target) for CER-mediated effects on islet β-cell, potential roles of Rac1 in the generation of intracellular CER should not be ignored, and should be studied methodically. This can be achieved via use of siRNA-mediated depletion of endogenous Rac1, dominant negative mutant of Rac1 (N17Rac1) or pharmacological inhibition of Rac1 (e.g., EHT-1864), and quantifying generation of CER under various pathological conditions in cells in which Rac1 functions are negated. Furthermore, use of specific inhibitors of GEFs for Rac1 (i.e., NSC23677 for Tiam1 and Ehop-016 for Vav2) might shed light into putative roles of these GEFs in promoting Rac1 activation and CER generation.
While the experimental evidence (reviewed above) affirm support to our proposed model, some aspects remain speculative and needed to be confirmed experimentally. For example, experimental evidence is still lacking to substantiate putative cross-talk and/or interdependence, if any, between Tiam1 and Vav2 in mediating their effects on Rac1 activation under conditions of metabolic stress, including regulation by intracellularly generated CER under these conditions. Furthermore, putative signaling mechanisms which are upstream to activation of these two GEFs need to be identified. It is also important to understand the subcellular localization/distribution of Tiam1/Vav2 as well as Rac1 under acute stimulatory conditions leading to GSIS and chronic exposure conditions culminating in cellular dysfunction and demise. In this context, we recently demonstrated inappropriate movement (mis-targeting) of Rac1 into distinct cellular compartments (e.g., nucleus) under conditions of metabolic stress. Therefore, it will be interesting to assess the effects of CER (and other biologically active sphingolipids) on the subcellular distribution and activation of Rac1 in the islet β-cell. These insights might help us better understand the nature and potential mediators of activation of Rac1, which, as discussed above, plays damaging roles in the induction of metabolic defects.
Another question that needs to be addressed relates to regulatory effects of CER (and other sphingolipids) on prenylation of G proteins, including Rac1 (Figure 2) . If CER induces defects in this signaling pathway, it is plausible that defective prenylation of Rac1 could lead to alterations in its interaction with its regulatory and scaffolding proteins (e.g., GDI) resulting in its dissociation from GDI, a condition which is conducive to its activation by GEFs. Furthermore, data from these studies will further affirm the postulation that Rac1 translocates to the nuclear fraction in a prenylation independent pathway leading to activation of pro-apoptotic proteins (e.g., p53) and induction of cell dysfunction.
Significant knowledge gaps exist on potential defects in prenylation [farnesylation/ geranyl geranylation] of Gγ subunits under conditions of metabolic stress/diabetic conditions. At least 12 Gγ genes are encoded in the human genome, and all are either farnesylated or geranylgeranylated. As reviewed by Hildebrandt prenylation of Gγ is critical for optimal cell function, including membrane attachment of prenylated proteins, intracellular membrane trafficking of prenylated proteins, reversible trafficking between plasma membrane and endomembranes and protein-protein interactions [81]. Therefore, it is conceivable that defects in FTase/GGTase activities that we reported in the islet β-cell under the duress of metabolic stress (and intracellularly generated CER) could result in defects in Gγ prenylation status in the islet β-cell. Potential defects in this signaling pathway could lead to defects in the activation-deactivation cycle of trimeric G-proteins leading to alterations in coupling of GPCR signaling pathways under these pathological conditions. This remains to be verified in in vitro and in vivo model systems of metabolic stress and diabetes.
Lastly, published evidence in multiple cell types, including the pancreatic islet β-cell have demonstrated stimulation of protein phosphatase 2A (PP2A) activity by cell permeable analogues of CER [82–86]. In this context, we also reported hyper-activation of PP2A in islets under the duress of metabolic stress [87,88]. However, the mechanisms underlying CER- induced activation remain unknown. It is noteworthy that recent evidence appears to indicate that CER binds directly to Inhibitor 2 of PP2A (SET/I2PP2A), an endogenous inhibitor of PP2A, thereby relieving PP2A from the inhibitory effects of PP2A [89]. Along these lines, observations from studies of Bharath and coworkers have further confirmed such a postulation in that they demonstrated association of CER with I2PP2A in endothelial cells leading to translocation of catalytically-active PP2A to the plasma membrane fraction and perturbing the downstream signaling steps culminating in arterial dysfunction in animal models of diet-induced obesity [86]. Unpublished evidence from our laboratory suggests that SET/I2PP2A is expressed in INS-1 832/13 cells and primary rodent islets. Putative roles of small G proteins (Rac1) in this signaling cascade will also need to be assessed experimentally. Studies are underway in our laboratory to address these important questions to gain much needed insights to mechanisms of CER-Rac1 signaling module in the onset of islet dysfunction under the duress of glucolipotoxicity.
Acknowledgments
Our research findings highlighted in this review were accrued through the support from a MERIT Review Award from the US Department of Veterans Affairs [BX002801] and the National Institutes of Health [DK-74921 and EY-022230]. AK also received a Senior Research Career Scientist Award [13S-RCS-006] from the Department of Veterans Affairs.
Abbreviations used
- Arf 6
ADP ribosylation factor 6
- CER
CER
- C-fos-SRE
c-fos serum response element
- ER stress
Endoplasmic reticulum stress
- FTase
Farnesyltransferase
- Fpp
Farnesylpyrophosphate
- GAP
GTPase activating protein
- GDI
GDP dissociation inhibitor
- GEF
Guanine nucleotide exchange factor
- GGpp
Geranylgeranylpyrophosphate
- GGTase
Geranylgeranyltransferase
- GGTI-2147
Geranylgeranyltransferase inhibitor-2147
- GPCR
G protein-coupled receptor
- GSIS
Glucose-stimulated insulin secretion
- HMG CoA reducatse
Hydroxylmethylglutaryl coA reductase
- IL-1β
Interleukin-1β
- IFNγ
Interferon γ
- MMP
Mitochondrial membrane potential
- Nox2
Phogocyte-like NADPH oxidase
- NSC23766
N6-[2-[4-(Diethylamino) -1-methylbu-tyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-qu-inolinediamine trihydrochloride
- PA
Palmitate
- PI-3-kinase
Phosphatidyl inositol-3-kinase
- PLD
Phospholipase D
- PP2A
Protein phosphatase 2A
- Rac1
Ras-related C3 botulinum toxin substrate 1
- ROS
Reactive oxygen species
- Tiam1
T-lymphoma invasion and metastasis-inducing protein 1
- TNFα
Tumor necrosis factor α
- Vav2
Guanine nucleotide exchange factor VAV2
- Ws-CER
Water soluble cationic CER
Footnotes
Conflict of interest
There is no conflict of interest among the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.IDF Diabetes Atlas. 8. International Diabetes Federation; Brussels, Belgium: 2017. pp. 1–146. [Google Scholar]
- 2.Bommer C, Sagalova V, Heesemann E, Manne-Goehler J, Atun R, Barnighausen T, et al. Global Economic Burden of Diabetes in Adults: Projections From 2015 to 2030. Diabetes Care. 2018:dc171962. doi: 10.2337/dc17-1962. [DOI] [PubMed] [Google Scholar]
- 3.Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signaling. Diabetologia. 2010;53:1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Komatsu M, Takei M, Ishii H, Sato Y. Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Investig. 2013;4:511–516. doi: 10.1111/jdi.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasnain SZ, Prins JB, McGuckin MA. Oxidative and endoplasmic reticulum stress in beta-cell dysfunction in diabetes. J Mol Endocrinol. 2016;56:R33–54. doi: 10.1530/JME-15-0232. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee A, Morales-Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med. 2015;21:439–49. doi: 10.1016/j.molmed.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eguchi K, Nagai R. Islet inflammation in type 2 diabetes and physiology. J Clin Invest. 2017;127:14–23. doi: 10.1172/JCI88877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberston RP. Beta-cell deterioration during diabetes: what’s in the gun? Trends Endocrinol Metab. 2009;20:388–393. doi: 10.1016/j.tem.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA. 1998;95:2498–2502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lang F, Ullrich S, Gulbins E. Ceramide formation as a target in beta-cell survival and function. Expert Opinion on Therapeutic Targets. 2011;15:1061–1107. doi: 10.1517/14728222.2011.588209. [DOI] [PubMed] [Google Scholar]
- 13.Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem. 2003;278:30015–30021. doi: 10.1074/jbc.M302548200. [DOI] [PubMed] [Google Scholar]
- 14.Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–32490. doi: 10.1074/jbc.273.49.32487. [DOI] [PubMed] [Google Scholar]
- 15.Szpigel A, Hainault I, Carlier A, Venteclef N, Batto AF, Hajduch E, Bernard C, Ktorza A, Gautier JF, Ferre P, Bourron O, Foufelle F. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia. 2015;61:399–412. doi: 10.1007/s00125-017-4462-5. [DOI] [PubMed] [Google Scholar]
- 16.Zuzmenko DI, Klimentyeva TK. Role of cermaide in apoptosis and development of insulin resistance. Biochemistry. 2016;81:913–927. doi: 10.1134/S0006297916090017. [DOI] [PubMed] [Google Scholar]
- 17.Aburasayn H, Al Batran R, Ussher JR. Targeting ceramide metabolism in obesity. Am J Physiol Endocrinol Metab. 2016;311:E423–435. doi: 10.1152/ajpendo.00133.2016. [DOI] [PubMed] [Google Scholar]
- 18.Adada M, Luberto C, Canals D. Inhibitors of the sphingomyelin cycle: Sphingomyelin synthases and sphingomyelinases. Chem Phys Lipids. 2016;197:45–59. doi: 10.1016/j.chemphyslip.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Chaurasia B, Summers S. Ceramides- Lipotoxic Inducers of Metabolic Disorders. Endocrinology & Metabolism. 2015;26:538–550. doi: 10.1016/j.tem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Janikiewicz J, Hanzelka K, Kozinski K, Kolcynska K, Dobrzyn A. Islet β-cell failure in type 2 diabetes within the network of toxic lipids. Biochem and Biophys Res Commun. 2015;460:401–96. doi: 10.1016/j.bbrc.2015.03.153. [DOI] [PubMed] [Google Scholar]
- 21.Sidarala V, Kowluru A. The regulatory roles of mitogen-activated protein kinase (MAPK) pathways in health and diabetes: Lessons learned from the pancreatic beta cell. Recent Pat Endocri Metab Immune Drug Discov. 2017;10:76–84. doi: 10.2174/1872214810666161020154905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khadija SG, Chen F, Hadden T, Commissaris RL, Kowluru A. Biology and regulatory roles of nuclear lamins in cellular function and dysfunction. Recent Pat Endocr Metab Immune Drug Discov. 2015;9:111–120. doi: 10.2174/1872214809666151009120402. [DOI] [PubMed] [Google Scholar]
- 23.Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine- induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol. 2011;300:R12–20. doi: 10.1152/ajpregu.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei X, Bone RN, Ali T, Zhang S, Bohrer A, Tse HM, Bidasee KR, Ramanadham S. Evidence of contribution of iPLA2-mediated events during islet beta-cell apoptosis due to proinflammatory cytokines suggests a role for iPLA2 in T1D development. Endocrinology. 2014;155:3352–3364. doi: 10.1210/en.2013-2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boslem E, Meikle P, Biden T. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets. 2012;4:177–187. doi: 10.4161/isl.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campana M, Bellini L, Rouch C, Rachdi L, Butin NCL, et al. Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances β-cell function of obsese Zucker rats. Mol Metab. 2018;8:23–36. doi: 10.1016/j.molmet.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis- roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:896–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowluru A. Small G proteins in islet beta-cell function. Enocr Rev. 2010;31:52–78. doi: 10.1210/er.2009-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kowluru A. Role of G-proteins in islet function in health and diabetes. Diabetes Obes Metab. 2017;19:63–75. doi: 10.1111/dom.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kowluru A. Tiam1/Vav2-Rac1 axis: A tug-of-war between islet function and dusfunction. Biochem Pharmacol. 2017;132:9–17. doi: 10.1016/j.bcp.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signaling. Cell Signal. 1999;11:545–554. doi: 10.1016/s0898-6568(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 32.Philips MR. The perplexing case of the geranylgeranyltransferase-deficient mouse. J Clin Invest. 2011;121:510–513. doi: 10.1172/JCI45952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen E, Li Y, Shan L, Zhu H, Feng Q, Arnold JM, Peng T. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009;58:2386–2395. doi: 10.2337/db08-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marei H, Malliri A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases. 2017;8:139–163. doi: 10.1080/21541248.2016.1211398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haga RB, Ridley AJ. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases. 2016;7:207–221. doi: 10.1080/21541248.2016.1232583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aguilar BJ, Zhu Y, Lu Q. Rho STPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res Ther. 2017;9:97. doi: 10.1186/s13195-017-0320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marinokovic G, Heemskerk N, Van Buul JD, De Waard V. The Ins and Outs of Small GTPase Rac1 in the Vasculture. J Parmacol Exp Ther. 2015;354:91–102. doi: 10.1124/jpet.115.223610. [DOI] [PubMed] [Google Scholar]
- 38.Zou T, Mao X, Yin J, Li X, Chen J, Zhu T, et al. Emerging roles of RAC1 in treating lung cancer patients. Clin Genet. 2017;91:520–528. doi: 10.1111/cge.12908. [DOI] [PubMed] [Google Scholar]
- 39.Kazanietz MG, Caloca MJ. The Rac GTPase in Cancer: From Old Concepts to New Paradigms. Cancer Res. 2017;77:5445–5451. doi: 10.1158/0008-5472.CAN-17-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newsholme P, Morgan D, Rebelato E, Oliveira-Emillo HC, Procopio J, Curi R, et al. Insights into the critical role of NADPH oxidases(s) in the normal and dysregulated pancreatic beta cell. Diabetologia. 2009;52:2489–2498. doi: 10.1007/s00125-009-1536-z. [DOI] [PubMed] [Google Scholar]
- 41.Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lesions learnt from pharmacological and molecular biological approaches. Biochem Parmacol. 2011;81:965–975. doi: 10.1016/j.bcp.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Syed I, Jayaram B, Subsinghe W, Kowluru A. Tiam1/ Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic β-cells. Biochem Pharmacol. 2010;80:874–883. doi: 10.1016/j.bcp.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, et al. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60:2843–2852. doi: 10.2337/db11-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brenner B, Koppenhoefer U, Weinstock C, Linderkamp O, Langs F, Gulbins E. Fas- or cermide-induced Apoptosis Is Mediated by a Rac1-regulated Activation of Jun N-terminal Kinase/p38 Kinases and GADD153. J Biol Chem. 1997;272:22173–22181. doi: 10.1074/jbc.272.35.22173. [DOI] [PubMed] [Google Scholar]
- 45.Kim B, Kim J. Exogenous C2-ceramide activates c-fos serum response element via Rac- dependent signaling pathway. Biochem. 1998;330:1009–1014. doi: 10.1042/bj3301009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Embade N, Valeron PF, Aznar S, Lopez-Collazo E, Lacal JC. Apoptosis induced by Rac GTPase correlates with induction of FasL and ceramide production. Mol Biol Cell. 2000;11:4347–4358. doi: 10.1091/mbc.11.12.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deshpande S, Qi B, Park Y, Irani K. Constitutive activation of rac1 results in mitochondiral oxidative stress and induces premature entothesial cell senscence. Arterioscler Thromb Vase Biol. 2003;23:e1–e6. doi: 10.1161/01.atv.0000047869.13737.53. [DOI] [PubMed] [Google Scholar]
- 48.Cacicedo J, Benjacharowong S, Chou E, Ruderman N, Ido Y. Palmitate induced apoptosis in cultured bovine retinal pericytes: Roles of NADPH oxidase, oxidant stress, and ceramide. Diabetes. 2005;54:1938–1845. doi: 10.2337/diabetes.54.6.1838. [DOI] [PubMed] [Google Scholar]
- 49.Kumar B, Kowluru A, Kowluru R. Lipotoxicity augments glucotoxicity-induced mitochondrial damage in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015;56:2985–2992. doi: 10.1167/iovs.15-16466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sidarala V, Veluthakal R, Syeda K, Vlaar C, Newsholme P, Kowluru A. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1) Biochem Pharmacol. 2015;95:301–310. doi: 10.1016/j.bcp.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sidarala V, Kowluru A. Exposure to chronic hyperglycemic conditions results in Ras-related C3 botulinum toxin substrate 1 (Rac1) -mediated activation of p53 and ATM kinase in pancreatic β-cells. Apoptosis. 2017;22:597–607. doi: 10.1007/s10495-017-1354-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baidwan S, Chekuri A, Hynds DL, Kowluru A. Glucotoxicity promotes aberrant activation and mislocalization of Ras-realted C3 botulinum toxin substrate 1 [Rac1] and metabolic dysfunction in pancreatic islet β-cells: reversal of such metabolic defects by metformin. Apoptosis. 2017;22:1380–1393. doi: 10.1007/s10495-017-1409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuellig R, Hornemann T, Othman A, Hehl A, Bode H, Guntert T, et al. Deoxysphingolipids: novel biomarkers for Type 2 diabetes are cytotoxic for insulin- producing cells. Diabetes. 2014;63:1326–1339. doi: 10.2337/db13-1042. [DOI] [PubMed] [Google Scholar]
- 54.Szulc ZM, Bielawski J, Gracz H, Gustilo M, Mayroo N, Hannun YA, et al. Tailoring structure-function and targeting properties of ceramides by site-specific cationization. Bioorg Med Chem. 2006;14:7083–7104. doi: 10.1016/j.bmc.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 55.Syed I, Szulc ZM, Ogretmen B, Kowluru A. L-threo-C6-pyridinium-ceramide bromide, a novel cationic ceramide, induces NADPH oxidase activation, mitochondrial dysfunction and loss in cell viability in INS 832/13 β-cells. Cell Physiol Biochem. 2012;30:1051–1058. doi: 10.1159/000341481. [DOI] [PubMed] [Google Scholar]
- 56.Veluthakal R, Palanivel R, Zhao Y, McDonald P, Gruber S, Kowluru A. Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: Potential mechanisms underlying ceramide-mediated metabolic dysfunction of the β-cell. Apoptosis. 2005;10:841–850. doi: 10.1007/s10495-005-0431-4. [DOI] [PubMed] [Google Scholar]
- 57.Welsh N. IL-1beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription of ATF-2 in the insulin producing cell line RINm5F. J Biol Chem. 1996;271:8307–8312. doi: 10.1074/jbc.271.14.8307. [DOI] [PubMed] [Google Scholar]
- 58.Mohammed AM, Syeda K, Hadden T, Kowluru A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: attenuation of oxidative stress and nitrosative stress by 2-bromopalmitate. Biochem Pharmacol. 2013;85:109–114. doi: 10.1016/j.bcp.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yi F, Chen Q, Jin S, Li P. Mechanism of Homocysteine-Induced Rac1/NADPH Oxidase Activation in Mesangial Cells: Role of Guanine Nucleotide Exchange Factor Vav2. Cell Physiol Biochem. 2007;20:909–918. doi: 10.1159/000110451. [DOI] [PubMed] [Google Scholar]
- 60.Veluthakal R, Sidarala V, Kowluru A. NSC23766, a known inhibitor of Tiam1-Rac1 signaling module, prevents the onset of type 1 diabetes in the NOD mouse model. Cell Physiol Biochem. 2016;39:760–767. doi: 10.1159/000445666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park SS, Kim MO, Yun SP, Ryu JM, Park JH, Seo BN, et al. C(16) -Ceramide-induced F-actin regulation stimulates mouse embryonic stem cell migration: involvement of N-WASP/Cdc42/ARP2/3 complex and cofilin-1/α-actinin. Biochim Biophys Acta. 2013;1831:350–360. doi: 10.1016/j.bbalip.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 62.Gupta N, Nodzenski E, Khodarev NN, Yu J, Khorasani L, Beckett MA, et al. Angiostatin effects on endothelial cells mediated by ceramide and RhoA. EMBO Rep. 2001;2:536–540. doi: 10.1093/embo-reports/kve115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brindley DN, Wang CN, Mei J, Xu J, Hanna AN. Tumer necrosis factor-alpha and ceramides in insulin resistance. Lipids. 1999;34:S85–8. doi: 10.1007/BF02562240. [DOI] [PubMed] [Google Scholar]
- 64.Hanna AN, Berthiamume LG, Kikuchi Y, Begg D, Bourgoin S, Brindley DN. Tumor necrosis factor-alpha induces stress fiber formation through ceramide production: role of sphingosine kinase. Mol Biol Cell. 2001;12:3618–3630. doi: 10.1091/mbc.12.11.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venable ME, Obeid LM. Phospholipase D in cellular senescence. Biochim Biophys Acta. 1999;1439:291–298. doi: 10.1016/s1388-1981(99)00101-8. [DOI] [PubMed] [Google Scholar]
- 66.Kowluru A, Kowluru RA. Protein prenylation in islet beta-cell function in health and diabetes: Putting the pieces of the puzzle together. Biochem Pharmacol. 2015;98:363–370. doi: 10.1016/j.bcp.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63:255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Novelli G, D'Apice MR. Protein farnesylation and disease. J Inherit Metab Dis. 2012;35:917–926. doi: 10.1007/s10545-011-9445-y. [DOI] [PubMed] [Google Scholar]
- 69.Resh MD. Targeting protein lipidation in disease. Trends Mol Med. 2012;18:206–214. doi: 10.1016/j.molmed.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khan OM, Ibrahim MX, Jonsson IM, Karlsson C, Liu M, Sjogren AK, et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J Clin Invest. 2011;121:628–639. doi: 10.1172/JCI43758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dunford JE, Rogers MJ, Ebetino FH, Phipps RJ, Coxon FP. Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J Bone Miner Res. 2006;21:684–694. doi: 10.1359/jbmr.060118. [DOI] [PubMed] [Google Scholar]
- 72.Veluthakal R, Arora DK, Goalstone ML, Kowluru RA, Kowluru A. Metabolic stress induces caspase-3 mediated degradation and inactivation of farnesyl and geranylgeranyl transferase activities in pancreatic beta-cells. Cell Physiol Biochem. 2016;39:2110–2120. doi: 10.1159/000447907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arora DK, Mohammed AM, Kowluru A. Nifedipine prevents etoposide-induced caspase-3 activation, prenyl transferase degradation and loss in cell viability in pancreatic beta-cells. Apoptosis. 2013;18:1–8. doi: 10.1007/s10495-012-0763-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kowluru A. Inappropriate movement of Rac1 contributes to glucotoxicity of the islet beta-cell. Cell Cycle. 2017;16:1387–1388. doi: 10.1080/15384101.2017.1345229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gallardo G, Lopez-Blanco F, Galarreta C, Fanjul L. HMGCoA reductase inhibition partially mediates tumor necrosis factor α-induced apoptosis in human U-937 and HL-60 cells. Biochem Biophys Res Commun. 2003;300:397–402. doi: 10.1016/s0006-291x(02)02846-2. [DOI] [PubMed] [Google Scholar]
- 76.Subbaiah PV, Sowa JM, Singh DK. Sphingolipids and cellular cholesterol homeostasis. Effect of ceramide on cholesterol trafficking and HMG CoA reductase activity. Arch Biochem Biophys. 2008;474:32–38. doi: 10.1016/j.abb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qi H, Juo P, Masuda-Robens J, Caloca M, Zhou H, Stone N, et al. Capase-mediated Cleavage of the TIAM1 Guanine Nucleotide Exchange Factor during Apoptosis. Cell Growth and Differentiation. 2001;12:603–611. [PubMed] [Google Scholar]
- 78.Tsai SY, Hayashi T, Harvey BK, Wang Y, Wu WW, Shen RF, et al. Sigma-1 receptors regulate hippocamapal dedritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc Natl Acad Sci USA. 2009;106:22468– 22473. doi: 10.1073/pnas.0909089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’Brien WJ, Heimann T, Rizvi F, Conklyn D. TIAM1/Rac/ERK Signal Transduction in Cell Survival of Serum Deprived/Cytokine Treated Rabbit Corneal Stromal Fibroblasts. Investigative Ophthalmology. 2010;51:6207. [Google Scholar]
- 80.Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucotoxicity and diabetes. Biochem Pharmacol. 2014;88:275–283. doi: 10.1016/j.bcp.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hildebrandt JD. Heterogeneous prenyl processing of the heterotrimeric G protein gamma subunits. In: Tamanoi F, Hrycyna CA, Bergo MO, editors. The Enzymes. Vol. 39. Academic Press; 2011. pp. 97–124. [Google Scholar]
- 82.Kowluru A, Metz S. Ceramide-activated protein phophatase-2A activity in insulin- secreting cells. FEBS. 1997;418:179–182. doi: 10.1016/s0014-5793(97)01379-3. [DOI] [PubMed] [Google Scholar]
- 83.Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268:15523–15530. [PubMed] [Google Scholar]
- 84.Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. j Biol Chem. 1999;274:20296–20300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- 85.Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inatctve dephosphorylated state. Diabetes. 2001;50:2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- 86.Bharath PL, Ruan T, Li Y, Ravindran A, Wan X, Nhan JK, Walker ML, Deeter L, Goodrich R, Johnson E, Munday D, Mueller R, Kunz D, Jones D, Reese V, Summers S, Babu PVA, Holland WL, Zhang Q-J, Abel ED, Symons Jd. Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes. 2015;64:3914–3926. doi: 10.2337/db15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kowluru A, Matti A. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: Potential mechanisms and functional consequences. Biochem Pharmacol. 2012;84:591–597. doi: 10.1016/j.bcp.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 88.Arora DK, Machhadieh B, Matti A, Wadzinski BE, Ramanadham S, Kowluru A. High glucose exposure promotes activation of protein phosphatase 2A in rodent islets and INS- 832/13 beta-cells by increasing the posttranslational carboxylmethylation of its catalytic subunit. Endocrinology. 2014;155:380–391. doi: 10.1210/en.2013-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oaks J, Ogretmen B. Regulation of PP2A by sphingolipid metabolism and signaling. Front Oncol. 2015;4:1–7. doi: 10.3389/fonc.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]