

Abstract
Background
Traumatic brain injury (TBI) induces cellular proliferation in the hippocampus, which generates new neurons and glial cells during recovery. This process is regulated by NMDA-type glutamate receptors, which are inhibited by ketamine. We hypothesized that ketamine treatment after TBI would reduce hippocampal cell proliferation, leading to worse behavioral outcomes in mice.
Methods
TBI was induced in mice using a controlled cortical impact injury, after which mice (N=118) received either ketamine or vehicle systemically for one week. We utilized immunohistochemical assays to evaluate neuronal, astroglial and microglial cell proliferation and survival 3 days, 2 weeks, and 6 weeks post-intervention. The Morris Water Maze reversal task was used to assess cognitive recovery.
Results
Ketamine dramatically increased microglial proliferation in the granule cell layer of the hippocampus 3 days after injury (Injury+Vehicle, 2800±2700 cells/mm3, N=4; Injury+Ketamine 11200±6600 cells/mm3, N=6; p=0.012). Ketamine treatment also prevented the production of astrocytes 2-weeks after injury (Sham+Vehicle, 2400±3200 cells/mm3, N=13; Injury+Vehicle, 10500±11300 cells/mm3, N=12; [p=0.013 vs Sham+Vehicle]; Sham+Ketamine, 3500±4900 cells/mm3, N=14; Injury+Ketamine, 4800±3000 cells/mm3, N=13 [p=0.955 vs Sham+Ketamine]). Independent of injury, ketamine temporarily reduced neurogenesis (Vehicle-exposed, 105100±66700, cells/mm3, N=25; Ketamine-exposed, 74300±29200 cells/mm3, N=27; p=0.031). Ketamine administration improved performance in the Morris Water Maze reversal test after injury, but had no effect on performance in Sham-treated mice.
Conclusions
Ketamine alters hippocampal cell proliferation after traumatic brain injury. Surprisingly, these changes were associated with improvement in a neurogenesis-related behavioral recall task, suggesting a possible benefit from ketamine administration after traumatic brain injury in mice. Future studies are needed to determine generalizability and mechanism.
Introduction
Traumatic brain injury (TBI) is a potentially devastating condition associated with significant long-term morbidity, including memory deficits, depression and seizure disorders. Though TBI involves neuronal loss and dysfunction, the post-injury response includes increased glial proliferation and activation as well as an increase in hippocampal neurogenesis1. Although adult-born hippocampal neurons have beneficial roles in healthy brains2, the contribution of post-traumatic neurogenesis to cognitive recovery after injury is less clear: it may be beneficial if new neurons compensate for circuits disrupted by injury, or it may be harmful, if neurons generated after TBI have maladaptive properties, and contribute to the formation of aberrant circuits3.
Hippocampal Radial Glial-like stem Cells (RGCs) reside in the subgranular zone of the dentate gyrus, and can differentiate into three types of cells: new RGCs, astrocytes or neurons4. The mitotic rate, fate specification, and long-term survival of cells generated from RGCs and their progeny are modulated by numerous endogenous and exogenous contingencies, including activity at ionotropic neurotransmitter receptors5, which might provide a link between activity-related signaling and hippocampal structure. Of particular importance is the N-methyl-D-aspartate-type inotropic glutamate receptor (NMDAR), which is involved in synaptic transmission and plasticity. Activation of NMDARs stimulates RGC mitosis6 and plays a vital role in the survival of adult-born neurons7 as well as in the formation of synapses8.
NMDARs are also known to potently modulate glial function and proliferation, either directly or indirectly via alterations in neuronal function and signaling9. Glial function likely has important roles in recovery from TBI, as two types of glia – microglia and astrocytes – are activated after injury10 and are known to modulate neuronal structure and function11. It is still not clear, however, whether various glial subtypes contribute positively or negatively to recovery after injury, as their activity likely has both beneficial and harmful consequences in terms of circuit function11.
NMDARs are targets of several anesthetic drugs, including ketamine, a noncompetitive NMDAR antagonist, which is frequently employed by medical providers in both ICUs and operating rooms. Although it has historically been contraindicated in the setting of head injury due to its perceived effects on intracranial pressure, it is increasingly recognized for its neuroprotective potential12. Based on positive outcomes from its use on battlefields and after trauma, it is no longer contraindicated in the setting of head injuries13. Thus, it is likely that increasing numbers of people will be exposed to ketamine early after head injury. In this study, we investigate the effects of ketamine administration after TBI on post-traumatic hippocampal cell proliferation in mice and their subsequent performance in hippocampus-dependent learning tasks.
Materials & Methods
Animals
All mouse housing, handling and procedures were performed in accordance with National Institutes of Health guidelines and were in compliance with Oregon Health & Science University (OHSU) Institutional Animal Care and Use Committee (IACUC) approved protocols. We utilized male and female adult (8-12 week old) C57BL/6J wildtype mice. Mice were euthanized at different time points after TBI for histologic analysis, and included 11 male and 12 female mice for the 3-day time point, 28 male and 26 female mice at the 2-week time point and 29 male and 18 female mice at the 6-week time point. All mice had access to food and water ad libitum and access to 24-hour/7-day veterinary consultation if needed. To minimize the number of mice needed for this study, the 6-week histology data was obtained on the same mice that underwent behavioral testing at 4-5 weeks.
Controlled Cortical Impact & Osmotic Pump Implantation
TBI was modeled using a controlled cortical impact (CCI) injury in mice as previously described14. Briefly, mice were anesthetized with isoflurane (1.8%) and placed on a stereotaxic frame. After 10% betadine sterilization and 2% lidocaine gel application, a scalp incision was made at midline, followed by a sterile 4 mm craniotomy to the right of the midline sagittal suture, between lambda and bregma, leaving dura intact. A 0.9 mm cortical deformation was applied using an electronic impactor (Leica Microsystems; speed 4.4 m/s; dwell time 800 msec) to the exposed area with a sterile 3.0 mm diameter stainless steel impact tip. After CCI, the scalp was sutured and bioadhesive glue was used to secure the incision site. Sham (non-injured) mice underwent similar anesthesia, frame mounting and the scalp incision/closure, but no craniotomy. Immediately after Sham or CCI, mice were implanted with an Alzet osmotic pump (Durect, Cupertino CA) delivering 1 μL/h of one of the following: vehicle (sterile 0.9% normal saline) or ketamine (± ketamine hydrochloride; Sigma Aldrich; 25 mg/ml in saline, for a dose of 30 mg/kg/day15) into the subcutaneous space on the back (dorsum) of the mouse. After pump placement, mice were marked using ear punches, and allowed to emerge from anesthesia in a heated, padded chamber, with access to acetaminophen-soaked food for 48 hrs. Mice typically resumed normal exploratory behaviors within 15 minutes after emergence. All subsequent experiments were performed by experimenters blinded to group assignment.
All mice survived Sham or CCI injury, apart from one mouse in the 3-day group that had been assigned to receive CCI injury/vehicle, which died within 1 hour of CCI and was excluded from analysis. In the 2-week and 6-week groups, osmotic pumps remained in place for 7 days, after which they were sterilely removed under anesthesia. In the 3-day group, pumps remained in place throughout the entire study period. Residual pump volumes were measured from all mice to confirm drug delivery, and in all cases, the volumes remaining were +/- 10% of that expected based on the calculated pump delivery rates.
Bromodeoxyuridine Injections
Bromodeoxyuridine (BrdU) was used to label cells undergoing mitosis after Sham or CCI injury. For mice undergoing delayed analysis of cell survival and fate specification at 2 and 6 weeks after CCI, BrdU (20 mg/mL in saline; Sigma-Aldrich, St. Louis MO) was injected intraperitoneally at a dose of 300mg/kg twice a day, 4 hours apart, on post-CCI days 2 and 3. Mice examined at 3 days after injury received 50 mg/kg BrdU three times a day, 2 hours apart, on post-CCI day 2 to assess proliferation rates.
Tissue Preparation and Immunohistochemistry
Mice were deeply anesthetized with isoflurane before receiving a lethal anesthetic dose of 2,2,2-tribromoethanol (Sigma-Aldrich) in accordance with IACUC-approved protocols. They were then transcardially perfused with chilled phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (in PBS) solution, and post-fixed overnight. A small, rostral-to-caudal nick was made to the ventral left hemisphere (contralateral to the CCI) to denote laterality. Free-floating coronal brain sections were prepared using a vibratome. For immunohistochemistry, four hippocampal sections were selected: two dorsal sections approximately 200 microns apart, and two ventral sections approximately 300 microns apart, and the rostrocaudal regions chosen were kept consistent between animals. In the rare event that damage to the dentate was apparent, samples were excluded and neighboring slices without dentate damage were chosen. Slices were permeabilized in 0.4% Triton in 0.5M potassium-PBS (KPBST) for 45 minutes, placed in 2N HCL KPBST at 37°C for 30 minutes, followed by a 10-minute wash in pH 8.5 KPBST, and 2 10-minute washes in 0.4% Triton KPBST (pH 7.4). Samples were blocked in 10% horse serum in 1% PBST and then incubated overnight at 4°C with primary antibodies and 1.5% horse serum in 0.4% KPBST. Primary antibodies and concentrations used for this study were: goat anti-doublecortin (1:500; SantaCruz), mouse anti-NeuN (1:500; EMD Millipore), rat anti-BrdU (1:500; Abcam), mouse anti-GFAP (cy3 conjugated; 1:500; Sigma-Aldrich), rabbit anti-Iba1 (1:500; Wako), rabbit anti-caspase-3 (1:500; Cell Signaling) and mouse anti-cfos (1:500; EMD Millipore). Following primary antibody incubation, samples were washed in 0.4% KPBST for 2 10-minute washes and then incubated for 4 hours at room temperature with secondary antibody. Secondary antibodies used in this study were Alexa-dye conjugated secondary antibodies at 1:500 concentrations (Invitrogen). Samples were then washed in KPBST for 2× 10-minutes, incubated in DAPI (1:20,000; Sigma-Aldrich) for 30 minutes, and subsequently mounted onto slides.
Confocal Microscopy & Cell Counting
Microscope slides were coded prior to microscopy so that blinding was maintained throughout imaging and analysis, until final results were tabulated. Four antibody-labeled hippocampal coronal sections from the hemisphere ipsilateral to Sham/CCI for each mouse were imaged using a Zeiss LSM780 confocal microscope with a 5x/0.16 numerical aperture (NA), 20x/0.8 NA or a 63x/1.4 NA lens. Contralateral sections were not analyzed, as prior studies demonstrated intermediate levels of proliferation in the contralateral hemisphere with high variability14,16. A z-stack of ~1.5 μm thick intervals was obtained for 20x images and 0.5-1.0 μm thick intervals for 63x images traversing the entire depth of the sample. Sections from each cohort were stained identically in parallel, and laser strength and detector gain settings were kept constant across all samples. Cell counting was performed using Imaris Image Analysis software (Bitplane, Concord MA) as follows: Three-dimensional images obtained from confocal microscopy were evaluated through the entire depth (z-stack) of the image. DAPI staining provided clear delineation of the granule cell layer and subgranular zone, and Imaris software provided a calculation of this volume as the region of interest through its “surfaces” function. BrdU+ cells within this defined region were identified by automated detection of antibody-stained objects with a diameter of 7 μm ± 2 μm. For all samples, cell counts were obtained from the dorsal blade of the hippocampal dentate gyrus granule cell layer (GCL) including the subgranular zone (SGZ), through the entire depth of the three-dimensional confocal images. Colocalization was determined blindly on coded slides and images; Dcx+/BrdU+, NeuN+/BrdU+ and Iba1+/BrdU+ colocalization was determined by presence of BrdU throughout the nuclear envelope of an antibody-outlined cell; GFAP+/BrdU+ colocalization was determined if GFAP surrounded the BrdU-labeled nucleus in 3 planes. For each image, cells that were selected for counting and which demonstrated co-localization were identified by the automated protocols (Imaris), and confirmed or rejected manually by an investigator blinded to group assignment prior to data tabulation. Cell densities were calculated by dividing cell count by GCL volume in cubic micrometers, and the 4 samples per subject were averaged.
Injury Size
Injury cavities were grossly visible in whole brains prior to slicing, and demonstrated no obvious asymmetries (cavities were round, based on gross visual inspection). Thus, we quantified injury magnitude using the cross-sectional area of the cavity in a single section through its mid-point using images from full coronal brain slices from both CCI groups (vehicle vs ketamine), at the same rostrocaudal level. The area of the dorsal quadrant ipsilateral to CCI was measured using FIJI, and subtracted from the area of the contralateral quadrant to provide a measurement of the area lost to injury.
Morris Water Maze and Reversal Testing
Four weeks after injury, mice underwent behavioral assessment by a blinded observer. Mice were singly housed each day for 1 hour prior to behavior in a holding room containing a white noise generator. The water maze consisted of a circular pool (122cm wide) filled with water (19-21°C) and located in the center of a room containing distal visual cues around the pool. A stationed platform was hidden from the mice by submerging it 1 cm below the water and by adding chalk to the water to make the water opaque. The drop location of the mice in the pool was pseudorandomized between 4 imaginary quadrants. Mice were first trained to locate the hidden platform using 2 sessions per day for 2 days (2-hour inter-session intervals). Each session included 3 trials (10-minute inter-trial intervals). Mice were given a maximum of 60 seconds to locate the platform and were guided to the platform if they did not locate it. Once on the platform, mice were allowed to remain on it for 3 seconds. A probe test, in which the hidden platform was removed in order to assess the preference for the platform location, was conducted 24-hours after the last training session (day 3). To avoid potential effects of the probe trial on the memory for the learned location of the first platform, a reinforcement session was conducted immediately after the probe trial in which the hidden platform was again placed in the original location.
The following day, the location of the platform was moved to a different quadrant and mice were trained to locate the hidden platform (reversal water maze), again using two sessions of three trials each with similar intervals as the first hidden platform training. A probe trial was then conducted on day 5. Three days later, all mice received 4 trials in which the platform was made visible using a clearly marked beacon. The location of the visible platform was moved to a different quadrant each trial. The time spent in the target quadrant, latency (time) to reach the platform, and swim speed were analyzed for potential group differences. Video recording and tracking software (Noldus, Ethovision) analyzed samples at a rate of 5 samples/sec.
Statistical Methods & Power Analysis
Mice from each litter were randomly allocated to treatment groups in balanced distribution. Data are presented as means and standard deviations (SD) in text and figures. Statistical analysis was conducted using RStudio version 1.0.136 (RStudio, Boston MA) with R version 3.3.2 (R CoreTeam 2016). All statistical tests were conducted using two-tailed hypothesis testing. Normality of data was assessed using the Shapiro-Wilk and Kolmogorov-Smirnov tests, and parametric tests were used in our analysis when data was normally distributed. Due to the variance observed in our data, unequal variance was assumed in all parametric comparisons. Immunohistochemistry results were analyzed using two-way ANOVA (drug × injury) followed by Tukey Honest Significant Difference post-hoc testing; results include weighted average main treatment effect comparisons of drug administration (all Vehicle-treated mice vs all Ketamine-treated mice) and injury state (all Sham-treated mice vs all CCI-treated mice), and post-hoc pairwise comparisons (Sham + Vehicle, CCI + Vehicle, Sham + Ketamine and CCI + Ketamine). For injury-size comparison between CCI + Vehicle and CCI + Ketamine groups, an independent t-test was used. Animal behavioral testing utilized paired t-tests, Mann-Whitney U tests, and repeated measures two-way ANOVA (drug × injury), with Newman-Keuls or Dunnett’s multiple comparison post hoc testing where stated. For all comparisons, p < 0.05 was considered statistically significant. Three mice from the 2-week time point (1 CCI + Vehicle, 2 CCI + Ketamine), two mice from the 6-week time point (1 CCI + Vehicle, 1 CCI + Ketamine), and one mouse from the 3-day time point (CCI + Vehicle) were excluded from histologic analysis due to predetermined criteria of extensive injury damage into the dentate itself. Power analysis of preliminary data indicated an N of 11 per group would be sufficient to detect significance with a power of 0.80 and an alpha of 0.05, based on pilot data, which demonstrated a 55% reduction in new neuron density, and variability of ± 35% in each injury group. Potential sex differences were evaluated and not found to be statistically significant across any experiments, so male and female results were grouped together for analysis. Figures were prepared using Prism Software version 7.0 (GraphPad, La Jolla CA).
Results
Ketamine Increases Hippocampal Cell Proliferation after CCI
To investigate the effect of ketamine on post-TBI hippocampal cell proliferation, we used a controlled cortical impact (CCI) model of TBI in mice (Figure 1A-B), and administered a continuous subanesthetic dose of ketamine for the first post-injury week via osmotic pump. This dose (30 mg/kg/day) demonstrated preclinical efficacy in a variety of other assays15,17, and was chosen to avoid sedation/immobility/anorexia that might be present at higher doses15, which could lead to potentially confounding effects. Ketamine did not cause gross differences in behavior or motor activity levels in either CCI or Sham-treated animals. Because weight loss or gain is another indicator of injury severity and can confound other results, we measured mouse weights at 1 week, 2 weeks and 6 weeks post-intervention. Mice in all groups gained weight throughout the evaluation period, as would be expected for healthy mice during this developmental window. There were no significant differences in weight gain induced by either CCI or ketamine administration (mean weight ± SD at 6 weeks post-CCI, in grams: Sham + Vehicle [1.8 ± 1.2, N = 12]; CCI + Vehicle [2.0 ± 1.4, N = 10]; Sham + Ketamine [1.6 ± 0.8, N = 12]; CCI + Ketamine [2.5 ± 1.5, N = 10]; two-way ANOVA: main effect (Injury vs Sham): F1,42 = 1.68, p = 0.203, main effect (Ketamine vs Vehicle): F1,42 = 0.10, p = 0.756).
Figure 1. Ketamine increases cell proliferation in the hippocampal granule cell layer and subgranular zone after CCI.
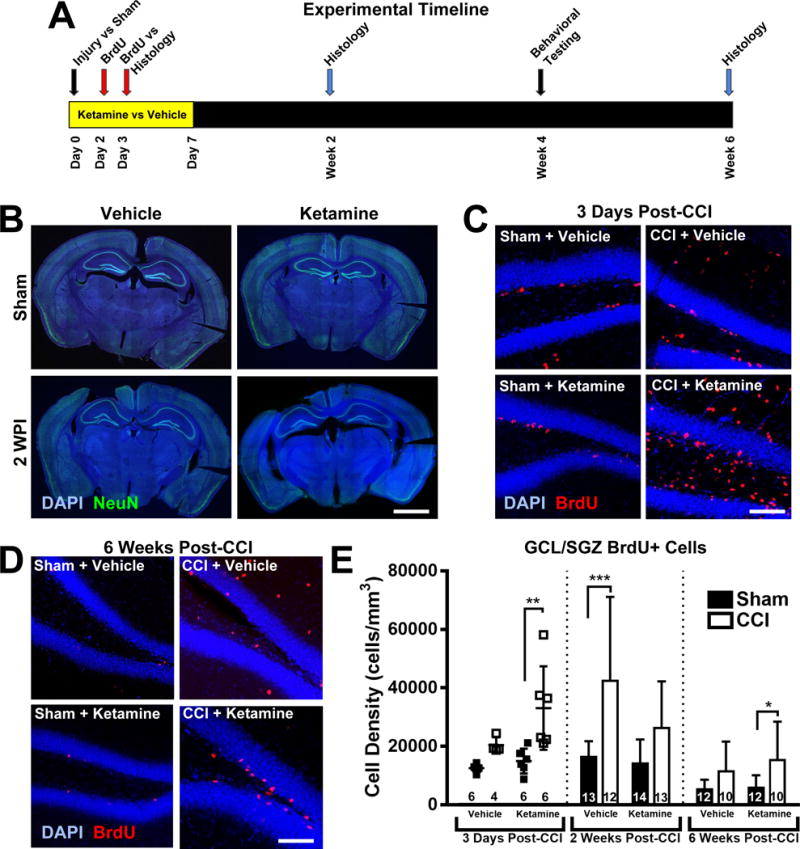
(A) Experimental timeline. Adult mice were randomized to undergo controlled cortical impact (CCI) or Sham injury, followed by continuous systemic administration of either Ketamine or Vehicle for one week, beginning on Day 0. Mice received the mitotic marker BrdU on days 2 and 3 after injury to permanently label actively dividing cells. Immunohistochemical analysis of the ipsilateral hippocampus was performed in separate cohorts of mice at three different time points: 3 days, 2 weeks and 6 weeks post-intervention, to distinguish patterns in cell proliferation and differentiation early and delayed stages. Mice in the 6-week arm underwent behavioral testing in weeks 4 and 5 post-intervention. (B) Representative images of coronal sections from Sham (non-injured) and CCI-injured mice at 2 weeks post-injury (WPI). Scale bar = 2mm. (C) Representative higher power images of the ipsilateral hippocampal dentate gyrus from injured and Sham-treated mice that received ketamine or vehicle after CCI. Ketamine administration after CCI induced a robust increase in newly born (BrdU+, red) cells in the granule cell layer and subgranular zone (GCL/SGZ, visualized with DAPI, blue) 3 days after injury. Scale bar = 150 μm. (D) Images of BrdU-stained dentate gyrus taken 6 weeks after CCI, with BrdU administered 2-3 days after CCI in the presence of ketamine or vehicle treatment. Scale bar = 150 μm. (E) Two-way ANOVA analysis of BrdU+ labeled cell density in the GCL/SGZ at the various time points evaluated in this study. Bars represent mean cell densities (cells/mm3) and error bars represent the standard deviation (SD); numbers inside bars represent the N for each group. For the three-day timepoint, individual values are displayed as well as the mean/SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Hippocampal cell proliferation was assessed using BrdU-mediated labeling of mitotic cells, as BrdU is incorporated into the DNA of dividing cells and can be detected long after cell maturation. BrdU was administered to mice 2-3 days after Sham or CCI injury, to coincide with the peak window of post-injury cell proliferation18. Mice were euthanized at various time points after CCI or Sham injury for histologic analysis of the hippocampal dentate gyrus using anti-BrdU immunohistochemistry.
First, to evaluate cellular proliferation early after CCI, a non-saturating dose of BrdU (50 mg/kg x3 injections, 2h apart) was administered to mice 2 days after injury, and they were euthanized one day later for analysis. Consistent with previous observations14,18,19, CCI treatment increased hippocampal cell proliferation when compared with Sham-treated animals; however, the sub-hippocampal distribution of these cells differed between drug treatment groups. In the dentate molecular layer, CCI significantly increased proliferation in both injury groups compared to their respective shams (Figure 1C; Cells/mm3 means ± SD: Sham + Vehicle [400 ± 400, N = 6] vs CCI + Vehicle [11800 ± 5600, N = 4], two-way ANOVA, main effect (CCI vs Sham): F1,18 = 33.20, p < 0.001, Tukey HSD post-hoc test, p = 0.002; Sham + Ketamine [600 ± 600, N = 6] vs CCI + Ketamine [9500 ± 6200, N = 6], Tukey HSD post-hoc test, p = 0.006). However, when we focused exclusively on the dentate granule cell layer (GCL) including the subgranular zone, CCI only induced an early proliferative response in this subregion in the presence of ketamine (Figure 1C&E; two-way ANOVA, main effect (CCI vs Sham): F1,18 = 15.74, p = 0.001; main effect (Ketamine vs Vehicle): F1,18 = 6.02, p = 0.025; interaction effect (CCI:Ketamine): F1,18 = 2.21, p = 0.154; CCI + Ketamine vs Sham + Ketamine, Tukey HSD post-hoc test, p = 0.005). In contrast, vehicle-treated mice demonstrated a smaller and not statistically significant increase in GCL cell proliferation after CCI (Figure 1C+E, CCI + Vehicle vs Sham + Vehicle, Tukey HSD post-hoc test, p = 0.438). As the original goal of this study was to examine post-injury neurogenesis, from this point forwards we focus entirely on cell densities within the dentate GCL, as this area contains the radial glial stem cells with the capacity to differentiate into neurons.
Two weeks after CCI or sham surgery, a separate cohort of BrdU-labeled mice was examined to determine the effects of ketamine on continued proliferation and early survival of the cells born after injury. A saturating dose of BrdU20 was administered on the second and third post-injury day to label proliferating cells and their progeny (Figure 1A). At this time point, BrdU+ cell labeling was significantly increased in the GCL in vehicle-treated, CCI-exposed mice when compared with their vehicle-treated Sham littermates (Figure 1E; two-way ANOVA, main effect (CCI vs Sham): F1,48 = 17.35, p < 0.001; CCI + Vehicle vs Sham + Vehicle, Tukey HSD post-hoc test, p = 0.001). In contrast, however, although ketamine-treated mice demonstrated increased BrdU+ labeling in injured mice, this was not statistically significant (Figure 1E; two-way ANOVA, main effect (Ketamine vs Vehicle): F1,48 = 3.88, p = 0.055). Although the differences between Vehicle-treated mice at 3 days post-CCI and 2 weeks post-CCI could have resulted from differences in the exact timeframe of BrdU administration between the two time points examined (2nd post-injury day vs. 2nd/3rd post-injury days), the increase in cell labeling in the injured mice could also have resulted from ongoing proliferation of BrdU-labeled cells (whose daughters would also be labeled) or differences in cell survival between groups.
Thus, to examine longer-term survival of cells proliferating early after CCI, mice were allowed to recover for 6 weeks prior to histologic analysis. At 6 weeks after CCI, across both groups, CCI increased the number of BrdU+ cells compared to their respective sham-treated mice (two-way ANOVA, main effect (CCI vs Sham): F1,40 = 10.33, p = 0.003; main effect (Ketamine vs Vehicle): F1,40 = 0.76, p = 0.390). However, when analyzed via pairwise comparisons, BrdU labeling was significantly increased only in the CCI + Ketamine group compared to its respective sham (Figure 1D-E; Tukey HSD post-hoc test, CCI + Vehicle vs Sham + Vehicle: p = 0.288; CCI + Ketamine vs Sham + Ketamine: p = 0.043), indicating that ketamine exposure early after CCI increases the survival of cells that were between 2 and 6 weeks of age.
Overall, the patterns of post-TBI cell proliferation and longer-term cell survival indicate that ketamine has both an acute effect on cell proliferation during its administration, as well as longer-term effects on cell survival that outlast the window of ketamine exposure. These effects could result from early induction of long-term changes in gene expression that drive longer-term cell survival, or alternatively, by changes in cell fate specification, as different cell types would be expected to have differential proliferation and survival trajectories. Thus, we examined the phenotypes of BrdU-labeled cells after CCI using cell-type specific immunohistochemistry.
Ketamine Inhibits Early Post-Traumatic Neurogenesis
We analyzed the early phase of post-traumatic neurogenesis two weeks after TBI, using tissue from Sham and CCI-treated mice that had received either ketamine or vehicle during the first post-injury week. Cells that had assumed a neuronal fate were identified by staining for the immature neuronal marker Doublecortin (Dcx), and BrdU co-labeling was determined using confocal microscopy. As Dcx is expressed in neurons approximately 3-21 days after mitosis21, Dcx+/BrdU+ co-labeling can help define the fate of proliferating cells early after the injury, during the peak window for post-traumatic neurogenesis22. Additionally, the overall density of Dcx+ cells can serve to identify and quantify all of the recently born neurons generated over a wider (~2.5 week) timeframe.
We first examined whether ketamine administration altered the density of Dcx staining in injured brains. Ketamine administration significantly decreased neurogenesis in CCI-treated mice at this two-week time point, manifest as a decreased density of Dcx+ cells in ketamine-treated mice after CCI when compared with vehicle-treated, CCI-exposed mice (Figure 2A-B; two-way ANOVA, main effect (Ketamine vs Vehicle): F1,48 = 4.93, p = 0.031; Tukey HSD post-hoc test: CCI + Ketamine vs CCI + Vehicle, p = 0.048). CCI exposure itself did not significantly increase neurogenesis when analyzed at the two-week time point using Dcx immunohistochemistry (two-way ANOVA, main effect (CCI vs Sham): F1,48 = 0.80, p = 0.376).
Figure 2. Post-Injury Ketamine Inhibits Neurogenesis after CCI.
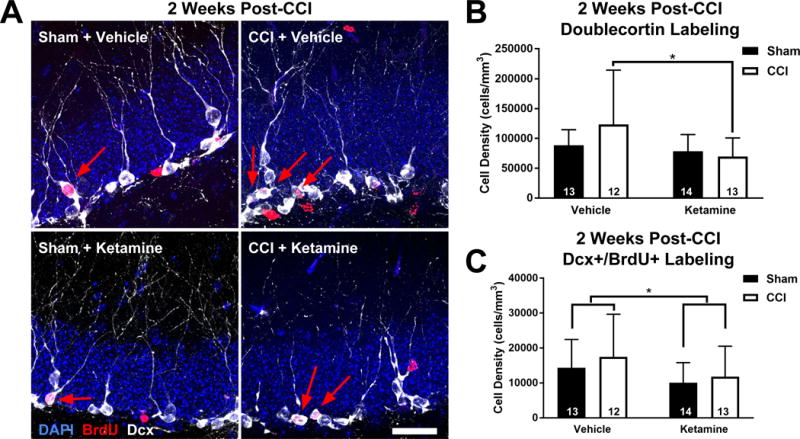
(A) Representative images of the dentate granule cell layer from each treatment group 2 weeks post-injury. Immature neurons are stained with for doublecortin (Dcx; white), along with BrdU-labeled cells (red) and DAPI (blue). Arrows indicate BrdU+/Dcx+ co-labeled cells. Scale bar = 25 μm. (B) Analysis of Dcx labeling at 2 WPI demonstrates that ketamine suppressed neurogenesis after CCI. (C) Ketamine administration suppressed the number of neurons born 2-3 days after injury, as identified using the density of BrdU+/Dcx+ co-labeled cells. Bars represent mean cell densities (cells/mm3) and error bars represent SD; numbers inside bars represent the N for each group; * p < 0.05.
To more precisely determine the fate of cells born early after TBI, we examined BrdU+/Dcx+ co-labeled cells in the dentate gyrus 2 weeks after CCI. Surprisingly, despite the fact that ketamine treatment alone did not affect overall levels of hippocampal cell proliferation (Figure 1), ketamine treatment decreased the number of neurons born 2-3 days after CCI across both sham and CCI conditions (Figure 2A&C; two-way ANOVA: F1,48 = 4.05; main effect (Ketamine vs Vehicle), p = 0.0499). However, CCI itself did not increase neurogenesis (two-way ANOVA, main effect (CCI vs Sham): F1,48 = 0.97, p = 0.329) at this time point.
CCI Suppresses Hippocampal Neurogenesis Late after Injury Independently of Ketamine
Altered cell proliferation in mice after CCI might have long-term effects on the ability of the hippocampus to sustain constitutive levels of adult neurogenesis, as has previously been observed in studies of post-seizure neurogenesis23. Thus, we examined the density of immature neurons 6 weeks after injury by staining for the immature neuronal marker Dcx, to focus on cells born several weeks after CCI.
6 weeks after injury, the density of immature neurons was significantly reduced when comparing all animals that received CCI to those that received Sham injury (Figure 3A&B; two-way ANOVA, main effect (CCI vs Sham): F1,40 = 4.25, p = 0.046), suggesting that in contrast to the burst of cell proliferation noted after CCI, the longer-term impact of CCI is to reduce neurogenesis at more remote time points. This effect was not significantly modified by ketamine (Figure 3B; two-way ANOVA, main effect (Ketamine vs Vehicle): F1.40 = 0.11, p = 0.744), indicating that ketamine treatment did not affect neurogenesis at remote time points after injury in either Sham or CCI-treated mice.
Figure 3. CCI Suppresses Hippocampal Neurogenesis Late After Injury Independently of Ketamine.
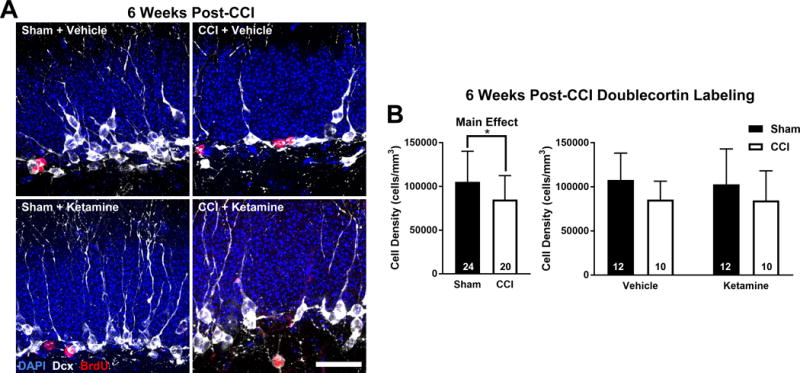
(A) High resolution images of the GCL of the 4 treatment groups 6 weeks after injury. Dcx-expressing immature adult-born neurons are shown in white, with BrdU (red) and DAPI (blue). Scale bar = 25 μm. (B) CCI decreased the density of Dcx+ cells across treatment groups with no difference between ketamine and vehicle exposed groups. Bars represent mean cell densities (cells/mm3) and error bars represent SD; numbers inside bars represent the N for each group; *p < 0.05.
Ketamine Does Not Affect the Long-Term Survival of Adult-Born Neurons after Injury
At 3-4 weeks after mitosis, granule cells begin expressing NeuN, a marker of mature neurons. During constitutive adult neurogenesis, most immature adult-born neurons do not survive to maturity24; similarly, during post-ischemic neurogenesis in the subventricular zone, many immature neurons born after stroke also do not survive to maturity25. As NMDAR activation is required for the long-term survival of constitutively generated adult-born neurons7, we hypothesized that survival of neurons born after TBI would be modulated by ketamine.
To evaluate ketamine’s impact on the long-term survival of newly born neurons, slices were evaluated 6 weeks after injury via antibody staining with the mature neuronal marker NeuN. Although neurogenesis (NeuN+/BrdU+ co-labeled cell density) increased in the CCI-treated groups versus the Sham-treated groups, this did not reach statistical significance (cells/mm3, mean ± SD: Sham + Vehicle = 3400 ± 3100; CCI + Vehicle = 4900 ± 5100; Sham + Ketamine = 3400 ± 1600; CCI + Ketamine = 5000 ± 3500; two-way ANOVA, main effect (CCI vs Sham): F1,40 = 2.44, p = 0.126). Importantly, there was no difference in the density of neurons born after CCI that survived until maturity when ketamine was administered for the week after CCI (main effect (Ketamine vs Vehicle): F1,40 = 0.00, p = 0.959; Tukey HSD post-hoc test: CCI + Ketamine vs CCI + Sham, p = 0.999), indicating that ketamine treatment did not negatively affect the ultimate survival of neurons born early after injury.
Ketamine Inhibits Astrogliogenesis after CCI
Because RGCs can differentiate into other cell types and only a minority of the BrdU+ cells observed co-labeled with neuronal markers, we stained tissues at various time points for glial markers to determine whether ketamine exposure altered cell fate and differentially affected the proliferation of glia. Astrocytes were identified via antibody staining against the astrocyte-selective cytoskeletal protein Glial Fibrillary Acidic Protein (GFAP), which also serves as a marker for astrocyte activation. Thus, increased GFAP staining could either result from increased astroglial reactivity, an increase in the number of astrocytes (or RGCs, which are also GFAP+), or both. Astrogliogenesis was quantified by co-staining tissue with GFAP and BrdU antibodies, to identify both overall GFAP staining, as well as to specifically identify astrocytes born after CCI or Sham injury.
Three days after the intervention, GFAP expression was significantly increased in the CCI-treated groups compared to their respective Sham groups (two-way ANOVA, main effect (CCI vs Sham): F1,18 = 18.46, p < 0.001; main effect (Ketamine vs Vehicle): F1,18 = 0.27, p = 0.609). This increase was similar between both drug treatment groups; however, when analyzed via pairwise comparison, GFAP expression was significantly increased only in the CCI + Ketamine group compared to its respective Sham (GFAP+ cells/mm3 ± SD, Tukey HSD post-hoc test: Sham + Vehicle [56200 ± 22200, N = 6] vs CCI + Vehicle [91100 ± 23000, N = 4], p = 0.095; Sham + Ketamine [52800 ± 14800, N = 6] vs CCI + Ketamine [97200 ± 26000, N = 6], p = 0.012). Examining astrocytes born specifically in the immediate post-injury period via GFAP+/BrdU+ colocalization, CCI-treated groups demonstrated an increase in astrocytosis when compared to Sham groups (two-way ANOVA, main effect (CCI vs Sham): F1,18 = 6.16, p = 0.023; main effect (Ketamine vs Vehicle): F1,18 = 1.30, p = 0.269), however no significant pairwise differences between any groups were observed (GFAP+/BrdU+ cells/mm3 ± SD, Tukey HSD post-hoc test: Sham + Vehicle [1400 ± 1300, N = 6] vs CCI + Vehicle [4500 ± 2000, N = 4], p = 0.210; Sham + Ketamine [2800 ± 1400, N = 6] vs CCI + Ketamine [4900 ± 3700, N = 6], p = 0.456). Taken together, these findings indicate that ketamine either facilitated the activation of glial cells or increased the density of CCI-induced astrocytes through proliferation prior to the 2nd post-CCI day.
To assess continued proliferation and survival of the astrocytes born after injury, we evaluated GFAP staining at 2 and 6 weeks post intervention. BrdU was administered 2 and 3 days after Sham or CCI treatment, to identify astrocytes generated during the early post-injury period. Despite no difference in astrocyte generation at 3 days after injury, at the 2 week time point we noted a dramatic increase in astrogliogenesis (GFAP+/BrdU+ co-labeled cell density) after CCI in vehicle-treated mice (Figure 4A&B; two-way ANOVA, main effect (CCI vs Sham): F1,48 = 6.65, p = 0.013; main effect (Ketamine vs Vehicle): F1,48 = 1.52, p = 0.224; interaction effect (CCI:Ketamine): F1,48 = 3.75, p = 0.059; Tukey HSD post-hoc test: Sham + Vehicle vs CCI + Vehicle, p = 0.013), which was almost completely abrogated by ketamine exposure (Figure 4A&B; Tukey HSD post-hoc test: Sham + Ketamine vs CCI + Ketamine, p = 0.955). By 6 weeks after intervention, the populations of GFAP+/BrdU+ cells equalized between all groups (data not shown).
Figure 4. Ketamine Inhibits Astrogliogenesis after CCI.
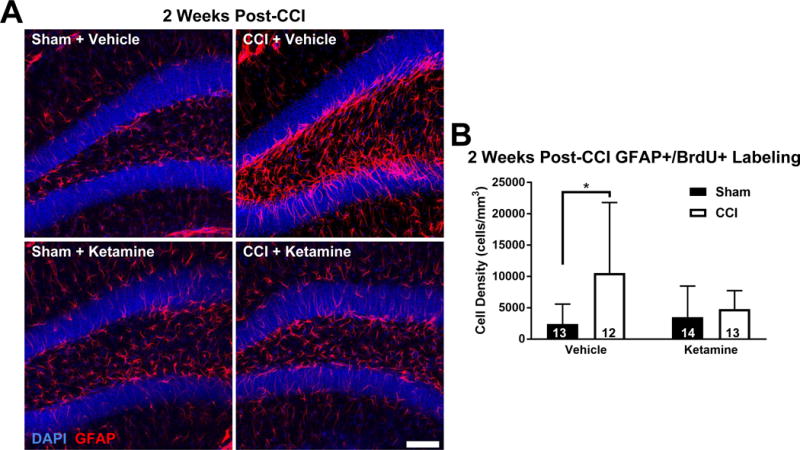
(A) Representative images of the four treatment groups 2 weeks after intervention. The CCI + Vehicle group demonstrated a robust astrocytic response (GFAP+, red; top right) not seen in the CCI + Ketamine group (bottom right). BrdU staining was omitted from the images for clarity. Scale bar = 150 μm. (B) Analysis of astrogliogenesis 2 weeks post-injury demonstrated a significant increase in new astroglia (GFAP+/BrdU+ cells) in the Vehicle + CCI group that was prevented by ketamine. Bars represent mean cell densities (cells/mm3) and error bars represent SD; numbers inside bars represent the N for each group; *p < 0.05.
Ketamine Treatment Facilitates Rapid Microgliogenesis after CCI
Microglia are the resident macrophages of the central nervous system and have a separate lineage than hippocampal radial glial-like stem cells and their progeny. In healthy tissue, microglia play important roles in sculpting neuronal circuits via synapse pruning and immune surveillance26. After TBI, microglia become activated and mount a dramatic response, including the release of pro- and anti-inflammatory cytokines and increased phagocytosis27. Moreover, activated microglia have complex effects on neurogenesis28. We evaluated microglial proliferation in the GCL after injury, using antibodies against the macrophage/microglia-specific marker Ionized calcium-binding adaptor molecule 1 (Iba1) and the marker for activated microglia, Galectin-3 (Mac2).
Three days after the CCI, ketamine treatment dramatically increased the density of newly generated (BrdU+) Iba1-labeled microglia within the GCL (Figure 5A&B; two-way ANOVA, main effect (CCI vs Sham): F1,18 = 16.45, p = 0.001; main effect (Ketamine vs Vehicle): F1,18 = 6.70, p = 0.019; Tukey HSD post-hoc tests: Sham + Ketamine vs CCI + Ketamine, p = 0.001; CCI + Ketamine vs CCI + Vehicle, p = 0.012; CCI + Ketamine vs Sham + Vehicle, p = 0.001). This increase was statistically significant compared with all other groups and included a significant interaction effect with injury, indicating a vastly different effect of ketamine on microglial proliferation after CCI that was not present in Sham-treated mice (Figure 5B; interaction effect (CCI:Ketamine): F1,18 = 7.46, p = 0.014). Surprisingly, there was no CCI-associated increase in Iba1+/BrdU+ microglia in the GCL of the vehicle-treated mice group at this early time point (Figure 5B; Tukey HSD post-hoc test: CCI + Vehicle vs Sham + Vehicle, p = 0.907).
Figure 5. Ketamine Treatment Facilitates Rapid Microgliogenesis after CCI.
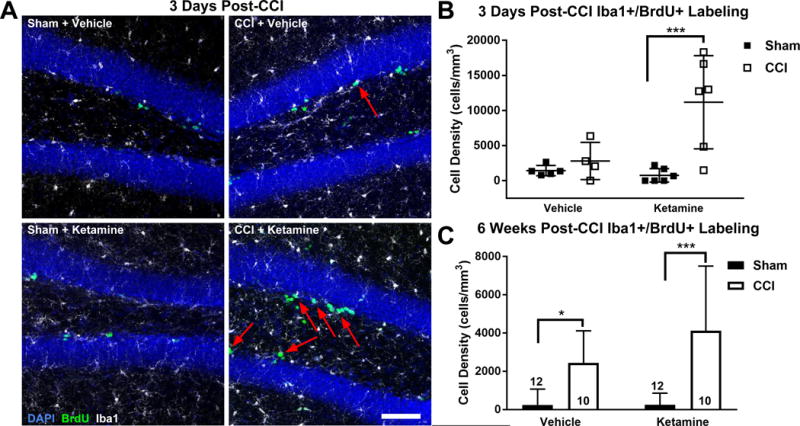
(A) Representative images of the GCL (DAPI, blue), microglia (Iba1+, white) and newly generated BrdU-labeled cells (green) 3 days after CCI or sham treatment. Newly generated microglia (Iba1+/BrdU+) are designated by arrows. Scale bar = 150 μm. (B) Quantification of microgliogenesis 3 days after injury demonstrated a significant increase in newly born microglia within the GCL/SGZ only in the Ketamine + CCI group. (C) At 6 weeks post-intervention, microglia born after CCI/Sham were significantly increased in both CCI groups versus their respective Sham groups. Bars represent mean cell densities (cells/mm3) and error bars represent SD; numbers inside bars represent the N for each group; * p < 0.05; ***p < 0.001.
Mac2 staining of these samples demonstrated no significant differences between any groups, indicating the microglia present in the GCL were not more activated with exposure to CCI or ketamine at this time point (Mac2+ cells/mm3, mean ± SD: Sham + Vehicle 55700 ± 11100; CCI + Vehicle 61400 ± 7500; Sham + Ketamine 42500 ± 11200; CCI + Ketamine 44800 ± 14200; two-way ANOVA, main effect (CCI vs Sham): F1,18 = 0.10, p = 0.753; main effect (Ketamine vs Vehicle): F1,18 = 1.42, p = 0.248).
Two weeks after injury, there was an overall increase in the density of Iba1+/BrdU+ microglia in all CCI-treated mice relative to Sham mice, but no significant difference between groups induced by ketamine treatment (two-way ANOVA: F1,48 = 6.24; main effect (CCI vs Sham), p = 0.016; main effect (Ketamine vs Vehicle): F1,48 = 0.73, p = 0.398). This result indicates that either delayed microglial maturation or ongoing microglial proliferation in the vehicle-treated group resulted in similar densities of new microglia at this more delayed time point. By 6 weeks post-injury, the co-labeled (BrdU+/Iba1+) population of microglia in the CCI-injured groups remained significantly increased versus their Sham counterparts, suggesting increased persistence of injury-induced microglia in the GCL, with no significant difference between vehicle and ketamine-treated groups (Figure 5C; two-way ANOVA, main effect (CCI vs Sham): F1,40 = 28.69, p < 0.001; main effect (Ketamine vs Vehicle): F1,40 = 1.87, p = 0.179; Tukey HSD post-hoc test: Sham + Vehicle vs. CCI + Vehicle, p = 0.045; Sham + Ketamine vs. CCI + Ketamine, p < 0.001).
Ketamine Does Not Affect Other Indicators of Injury Severity
If ketamine has neuroprotective properties, ketamine-induced modification of injury severity could indirectly alter the nature of post-TBI cell proliferation between groups. For example, if ketamine treatment modified the timing or degree of cell death, it could alter the proliferative response in a quantitative or qualitative manner. Moreover, as ketamine could also alter neuronal activity after TBI, including potential modulation of subclinical seizure frequency/occurrence29, this could also alter neurogenesis and gliogenesis via activity-dependent mechanisms. We therefore attempted to identify possible indirect consequences of ketamine treatment that could lead to secondary changes in cell proliferation.
To grossly assess the impact of ketamine on neuronal injury, we first measured the degree of cortical cavitation using low-power images of coronal brain sections. We compared the amount of tissue lost after injury at both 2 and 6 weeks post-CCI. The cross-sectional area of cortical cavity size was not affected by post-injury ketamine treatment at either time point (Figure 6A&C; independent t-tests: CCI + Vehicle vs CCI + Ketamine, 2 week: p = 0.105; 6 week: p = 0.517). Although all cavities appeared round on gross inspection prior to sectioning, it remained possible that our coronal cross-sectional analysis may have missed subtle differences in three-dimensional cavity shape or profile. Thus, we performed additional analyses of injury severity.
Figure 6. Ketamine Does Not Modulate Other Indicators of Injury Severity.
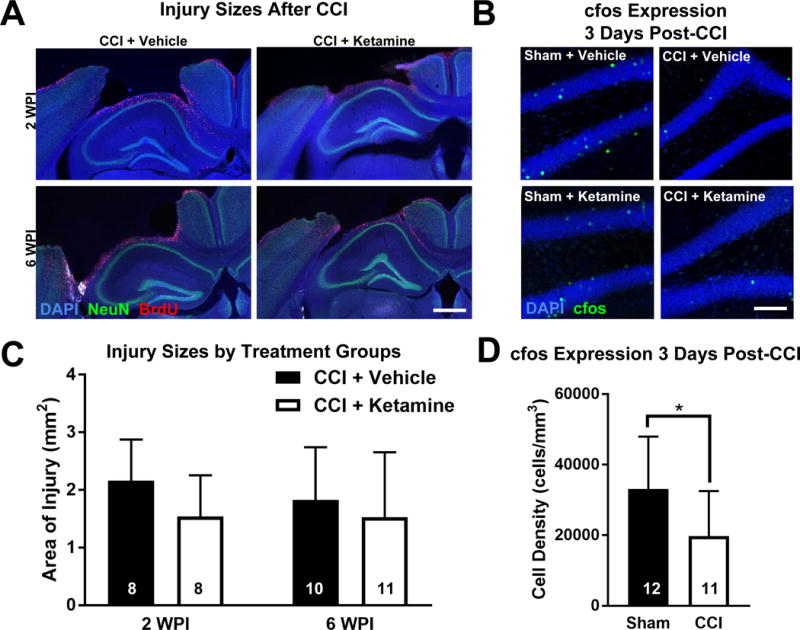
(A) Low-power images demonstrate typical CCI-induced cortical cavities above the hippocampus at 2 and 6 weeks post-injury (WPI). Scale bar = 500 μm. (B) CCI decreased cfos activation (cfos+, green) 3 days after injury, which was not affected by ketamine treatment. Scale bar = 150 μm. (C) Ketamine administration did not alter cortical injury size at either 2 or 6 weeks post-injury. Independent t-test: CCI + Vehicle vs CCI + Ketamine 2 weeks: p = 0.105; 6 weeks: p = 0.517. (D) Quantification of cfos expression demonstrated a significant decrease in neuronal activity in CCI groups compared to Sham groups. Bars represent means and error bars represent SD; numbers inside bars represent the N for each group; *p < 0.05.
The amount of cortical loss would likely not be sensitive to differences in hippocampal cell apoptosis early after CCI, which might be more directly related to cell proliferation in the hippocampus. To evaluate hippocampal cell death, we performed immunohistochemical staining for activated caspase-3, a marker for the induction of apoptosis, at the same time point at which mitosis was measured (3 days post-injury or sham), and quantified caspase-3 labeling in the dentate gyrus of the ipsilateral hippocampus. There were no significant differences in caspase-3 expression between any of the groups at 3 days post-intervention (caspase-3+ cells/mm3, mean ± SD: Sham + Vehicle [1100 ± 1100, N = 6]; CCI + Vehicle [500 ± 400, N = 4]; Sham + Ketamine [1000 ± 700, N = 6]; CCI + Ketamine [800 ± 600, N = 6]; two-way ANOVA, main effect (CCI vs Sham): F1,18 = 1.50, p = 0.236; main effect (Ketamine vs Vehicle): F1,18 = 0.03, p = 0.875). This lack of caspase activation suggests a lack of ongoing apoptosis at this time point, but might also have resulted from a rapid clearance of any apoptotic cells such that ongoing caspase-3 expression was not detected.
Subclinical seizures or changes in neuronal activity can influence post-injury histopathology and recovery30. To evaluate this, we performed antibody staining for cfos, a metabolic marker upregulated by neurons after recent activity. Though paradoxical, it has previously been shown that TBI attenuates neuronal activity for up to a month after injury31. Consistent with this prior finding, we observed a significant decrease in neuronal activity in all CCI-treated mice versus all Sham-treated mice three days after injury, with no difference in cfos activity as a result of ketamine administration (Figure 6B&D; two-way ANOVA, main effect (CCI vs Sham): F1,19 = 4.68, p = 0.044; main effect (Ketamine vs Vehicle): F1,19 = 0.12, p = 0.735).
Ketamine Ameliorates the CCI-Induced Impairment in Water Maze Reversal Learning
To determine how ketamine administration after TBI might affect cognitive recovery, mice underwent behavioral testing 4 weeks after injury using a modified Morris Water Maze (MWM) task. Although the standard reference memory water maze paradigm is sensitive to changes in spatial learning, there is considerable discrepancy in its ability to capture performance differences when hippocampal neurogenesis is altered32. Because the function of new neurons is believed to involve context separation, we focused our water maze test on a MWM reversal paradigm, which requires distinction between very similar contexts (i.e. new versus old target location)33. Details of the water maze test are described in the methods section, but in general, once all groups of mice were sufficiently trained such that they showed equal levels of preference for the first target location (platform), we changed the target location and assessed whether mice could distinguish between the two learned target locations. Performance in this MWM reversal task requires intact and properly functioning hippocampal neurogenesis, which is thought to allow mice to distinguish between different contextual location cues between the two different platform locations that are learned in the task33.
In the first phase of the water maze test, mice attempted to locate and then recall the location of a submerged platform. Across the average of the training sessions, CCI-treated mice required more time to locate the hidden platform (mean time in seconds ± SD: Sham = 28.7 ± 6.2, N = 24; CCI = 36.2 ± 7.3, N = 23; two-way ANOVA, main effect (CCI vs Sham): F1,43 = 13.75, p = 0.001). Despite this difference, all groups of mice improved their ability to locate the hidden platform during repeated sessions (repeated measures ANOVA, effect of session: F4,172 = 34.82, p = 0.001) and had no group differences in their learning rates (Figure 7A; repeated measures ANOVA, CCI × session: F4,172 = 1.45; p = 0.220; Drug × session: F4.172 = 0.52; p = 0.725). Furthermore, there were no group differences between sham and CCI-treated mice on session 4, the last session before the first probe trial (Figure 7A; two-way ANOVA, main effect (CCI vs Sham): F1,43 = 2.55, p = 0.117). In the first probe trial, all groups of mice showed a preference for the learned location of the hidden platform as all groups of mice spent significantly more time in the target quadrant versus any of the other three non-target quadrants (Figure 7A&B; Dunnett’s multiple comparison post-hoc test: p < 0.050 for each quadrant vs the target quadrant, for all groups of mice).
Figure 7. Ketamine Ameliorates the CCI-Induced Impairment in Water Maze Reversal Learning.
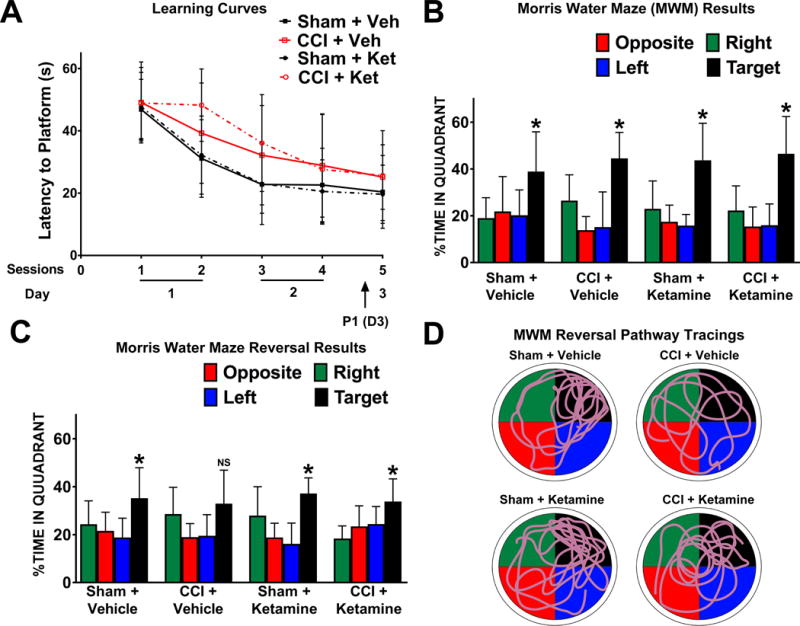
(A) Learning curves for the Morris Water Maze (MWM) illustrate the time required for mice in each group to locate a submerged platform (latency) during each subsequent session. CCI-treated mice were initially slower to learn the platform location (Session 2), but by session 4 there were no group differences between Sham and CCI treated mice in locating the platform. P1 represents the probe trial during which the platform is removed and mice are evaluated for their ability to recall the platform in the target location. Markers represent means and error bars represent SD. (B) All groups successfully demonstrated preference for the target location during the first probe trial after training (P1). (C) In the reversal task of the MWM, the platform was moved to a different quadrant, requiring subjects to learn the new location and engage in pattern separation. In this trial, the CCI + Vehicle group failed to successfully adapt to the new location, whereas the CCI + Ketamine group was able to achieve the task. (D) Characteristic path tracings from each group during the reversal probe trials, in which the appropriate target quadrant (black) is in the upper right. Bars represent means and error bars represent SD; *p < 0.05.
After all mice had learned the first platform location, mice were subjected to a reversal MWM test, in which the platform location was changed to another quadrant of the pool, a task more specific for the assessment of neurogenesis-dependent hippocampal function33. An effect of CCI was again observed across the average of the hidden platform sessions, as CCI-treated mice took longer to locate the hidden platform (mean time in seconds ± SD: Sham = 29.9 ± 10.0, N = 24; CCI = 37.3 ± 13.4, N = 20; two-way ANOVA, main effect (CCI vs Sham): F1,43 = 4.43, p = 0.041). Similar to the first training, all groups of mice improved in their ability to locate the hidden platform during training (repeated measures ANOVA, effect of session: F1,43 = 22.77, p = 0.001) without any group differences in the learning rates (repeated measures ANOVA, CCI × session: F1,43 = 0.02; p = 0.895; drug × session: F1,43 = 1.58; p = 0.216). Although across the average of the sessions CCI treated mice took longer to locate the hidden platform, there were no significant group differences in their acquisition of the new target location during the last session before the second probe trial (two-way ANOVA, main effect (CCI vs Sham): F1,43 = 2.92; p = 0.095).
During the reversal task probe trial the following day, all Sham-treated mice (both Vehicle and Ketamine-exposed) demonstrated a clear bias towards the new location of the platform (target quadrant versus any other non-target quadrant: Dunnett’s multiple comparison post-hoc test, p < 0.050 for each quadrant vs the target quadrant). This correct preference was deficient in CCI-injured mice treated with vehicle (target quadrant versus other quadrants: Dunnett’s multiple comparison post-hoc test, p = 0.637) indicating this group had a deficit in water maze reversal memory (Figure 7C&D). Surprisingly, ketamine treatment after CCI rescued the ability of injured mice to retain the new platform location during the probe trial, as these mice demonstrated preservation of spatial memory retention during reversal water maze testing (Figure 7C&D; Dunnett’s multiple comparison post-hoc test, p < 0.050 for each quadrant vs the target quadrant). These differences were not attributable to altered motor coordination, vision or motivation to perform the task, as there was no effect of CCI, drug treatment or interaction on swim speed or latency of mice to reach the “visible” platform when it was later clearly marked with a beacon and there were no group differences across the average of these visible platform trials (two-way ANOVA, main effect (CCI vs Sham): F1,42 = 2.48; p = 0.123; main effect (Ketamine vs Vehicle): F1,42 = 2.36; p = 0.132).
Although vehicle-treated CCI mice did not spend statistically more time in the target quadrant during the reversal probe trial, we found that the general pattern between groups appeared grossly similar (all spent the most time in the target quadrant) and thus we performed additional analyses of our data. First, we performed a different version of the quadrant preference analysis, in which all non-target quadrants are averaged together in an attempt to minimize the effect of a single spurious quadrant, and then compared this average with the time spent in the target quadrant. This analysis again demonstrated significant target preference in the two Sham groups and the CCI + Ketamine group (Sham + Vehicle, p = 0.0174, N = 12; Sham + Ketamine, p < 0.001, N =12; CCI + Ketamine, p = 0.008, N = 12; paired t-tests) but not the vehicle-treated CCI group (p = 0.088, N = 11, paired t-test). Additionally, we re-analyzed the trajectory data during the probe trial, to determine the amount of time spent above the target platform location during the reversal probe trial, and also found that this data had the same pattern. Compared to Sham + Vehicle mice, CCI + Vehicle treated mice had significantly fewer crossings (Sham + Vehicle = 4.0 ± 2.9; CCI + Vehicle = 2.0 ± 1.3, p = 0.046, Mann-Whitney test), whereas the number of crossings between Sham + Ketamine and CCI + Ketamine treated mice were not different from each other (Sham + Ketamine = 2.9 ± 2.1; CCI + Ketamine = 2.5 ± 1.6, p = 0.610, Mann-Whitney test). As all groups of mice successfully learned the location of the new target location during training (see above), but only the CCI + Vehicle mice were selectively deficient in recalling during the probe trial 24 hours later, our data suggest that their deficit was more likely related to recall rather than the ability to acquire the new location per se.
Discussion
TBI induces robust cellular proliferation in the hippocampus, which includes the generation of immature neurons and astrocytes from Radial Glial-like Cells18,19. In this study, when ketamine was administered after CCI, hippocampal cell proliferation in the dentate granule cell layer was markedly accelerated, as was evident by an increase in BrdU labeling of mitotic cells at the 3-day time point, which was mostly a product of early increases in microgliogenesis – a surprising finding given ketamine’s anti-inflammatory properties34. Interestingly, these changes in proliferation were accompanied by improvement in behavioral outcomes, suggesting there may be a benefit to ketamine administration in the setting of head injury.
Mechanisms Driving Post-Traumatic Neurogenesis
Neurologic insults such strokes, seizures and traumatic brain injuries are associated with increased hippocampal cell proliferation, including neurogenesis4. These pathologies often involve pathologic neuronal excitation, leading to the release of glutamate and other neurotransmitters, which can increase metabolic demands and cause excitotoxic neuronal death. Injury-associated glutamate release can also activate NMDARs on neuronal stem cells and immature neurons. As NMDAR activation modulates both the proliferation5,6 and the survival of adult-born neurons7, these receptors could mediate injury-induced changes in adult neurogenesis. Additionally, glutamatergic signaling via NMDARs can also influence glial cell signaling both directly and indirectly9, which could mediate additional quantitative and qualitative changes in both gliogenesis and neurogenesis11.
Our data demonstrating that ketamine alters cell proliferation after TBI are consistent with a role for NMDARs in the control of cell proliferation after injury. Several unexpected findings, however, suggest potentially novel mechanisms at play in mediating these responses. Ketamine administration notably reduced neurogenesis in both injured and non-injured mice 2 weeks post-injury. Ketamine has proven antidepressant efficacy in human subjects35, yet classic antidepressants have been shown to increase neurogenesis36; this finding suggests a potentially different mechanism of action for ketamine’s antidepressant action. Prior studies of short-term ketamine administration did not demonstrate changes in adult neural cell proliferation in healthy rodents37,38. One study did report that intermittent ketamine administration could increase neurogenesis in some portions of the hippocampus, but not others39, and a separate study demonstrated that repeated ketamine administration could decrease survival of adult-born neurons40. However, no studies have previously examined a longer term continuous administration of ketamine after TBI, which might be a better model for continuous human exposure via infusion, particularly in light of the extremely short serum half-life of ketamine in mice (13 minutes).
Although one week of ketamine administration reduced post-injury neurogenesis, its most dramatic effects were on glial proliferation (both microglia and astrocytes), suggesting that ketamine might not be working exclusively via the control of neuronal function. In this regard, microglial activation directly inhibits neurogenesis26 and astrocytes release factors that have positive influences on neurogenesis11, raising the possibility that either the early increase in microglial proliferation or ketamine-associated decrease in astrocytosis could have contributed to the reduction in Dcx+ cell counts at 2 weeks after injury in the ketamine-treated group. Moreover, a lack of a ketamine-induced change in cfos activity after injury argue against alterations in neuronal circuit activity as the primary mediator of ketamine’s effects. Future studies will hopefully delineate the specific receptor mechanisms mediating ketamine’s actions on post-injury neurogenesis.
Although ketamine reduced the density of Dcx-stained immature neurons 2 weeks after injury, we did not find that it substantially altered the long-term survival of neurons born after TBI. One limitation of our study resulted from an unexpectedly high variability in post-traumatic neurogenesis within our CCI-treated mice, such that our data displayed enhanced injury-induced neurogenesis, which was not statistically significant. It has been well established in the literature that CCI induces neurogenesis14,18, however experimental variability has led to negative results in some studies41. This higher-than-expected variability may have left our study underpowered to determine statistically significant increases in neurogenesis, and thus left us unable to find group differences between drug conditions. Our sample sizes were based on power analyses using data obtained in our own lab using the same model, and thus we did not feel it appropriate to continue to add additional mice in the quest for statistical significance in regards to neurogenesis, particularly in light of the multiple findings in regards to glial proliferation. However, it does have implications for sample sizes necessary for future studies.
Ketamine’s Effects on Gliogenesis
One of our most surprising findings involved the ketamine-associated changes in post-injury gliogenesis. To our knowledge, no prior studies have reported an effect of ketamine on the generation of new glial cells after traumatic brain injury. There are, however, prior studies evaluating the effects of ketamine on glial function, which suggest possible anti-inflammatory effects. Ketamine suppressed astrocyte activation in a mouse model of neuropathic pain42, and ketamine can directly alter astrocyte function in vitro43. Ketamine also reduces cytokine production by both astrocytes and microglia in vitro44, and pro-inflammatory cytokines have been shown to favor astrocytogenesis45, suggesting that ketamine could be inhibiting astrocytosis after injury by decreasing injury-induced cytokine signaling.
Furthermore, although ketamine is an NMDAR antagonist, it interacts with numerous other receptors17,46, and its metabolites have therapeutic effects that are not mediated by actions at NMDARs47. Thus, the effects of ketamine we noted in this study may have been due to non NMDAR-mediated mechanisms. Interestingly, activation of the pro-inflammatory fibroblast growth factor (FGF) receptor promotes astrocytosis, and as ketamine downregulates FGF signaling48, this could provide one potential mechanism through which ketamine suppresses astrocytosis after TBI.
Improved Learning after Ketamine
Fascinatingly, despite no differences in the long-term survival of neurons generated during early post-traumatic neurogenesis, mice treated with ketamine after injury had improved learning in the water maze reversal task, suggesting a benefit to receiving ketamine after injury. The MWM reversal test relies on the presumed neurogenesis-dependent ability to distinguish between similar environments and to supplant previously learned preferences given new, subtle environmental cues. Although performance in this task has been interpreted as related to rodents’ ability to successfully engage in pattern separation between the environmental contexts between the two different platform locations33, this task also involves other cognitive processes such as behavioral adaptability and we expect that future work will help elucidate the specific features that might be improved by ketamine treatment.
Increased neurogenesis after TBI has previously been associated with improved MWM performance, and Blaiss et al demonstrated that ablation of neurogenesis after TBI specifically impairs reversal MWM49. Conversely, Gradari et al detailed a very specific association between decreased immature neuron populations and improved performance in the reversal MWM50, which may best explain the improvement seen in this study, as ketamine exposure reduced neurogenesis at the 2 week time point. It is not immediately clear, however, how an early decrease in neurogenesis after injury might explain such an association, given that we did not find any ketamine-induced change in neurogenesis at the 6-week time point. One potential explanation would be that the improved performance after ketamine exposure related to qualitative, and not quantitative, differences in post-injury neurogenesis.
Another explanation for the improved behavioral outcome after ketamine exposure might relate to the changes in glial function after injury. Intense astrocyte activation and scar formation has been associated with behavioral deficits, raising the possibility that muting this reactivity (as was noted two weeks after CCI in the ketamine-treated group) was beneficial. Similarly, the accelerated proliferation and sustained survival of microglia generated after TBI might also altered the functional properties of hippocampal neurons, as it has recently become clear that microglia play important roles in sculpting neuronal circuit function26. As the behavioral improvements and histologic findings in our study are purely associative, and not causal, further evaluation of either of these possibilities through the specific targeting of glial function using genetic or pharmacologic means would hopefully shed light on these potential mechanisms.
Alternatively, hippocampal neurons born after various forms of neuronal injury have altered structural and functional properties14, and ketamine could serve to improve the functional capacity of neurons born after TBI. Ketamine rapidly induces synaptogenesis36, although it is not clear how long these changes would persist or translate to cells which had not yet received excitatory inputs during the period of ketamine administration. Although we attempted to morphologically characterize immature Dcx+ neurons in this study, neither dendritic morphology nor dendritic spine density (a surrogate for excitatory synaptic innervation) could be accurately discerned using this method (data not shown), thus the potential effect of ketamine on structural maturation was not assessed. Further studies using genetic or retrovirus-based cell labeling techniques, in conjunction with electrophysiologic analyses, could help define the morphologic and functional properties of neurons born after CCI in the presence of ketamine to identify potential differences. Additionally, a broader array of behavioral assessments will allow us to more precisely delineate the scope of ketamine’s effects on outcomes after TBI, as changes in hippocampal cell proliferation after injury could also have deleterious consequences that were not detected in our current study.
Conclusion
Ketamine administration after TBI alters hippocampal cell proliferation. These changes are associated with an improvement in learning weeks after ketamine cessation, as determined by the MWM reversal task. Additional studies focusing on the cellular mechanisms underlying these responses to ketamine after TBI are needed, so that we may help refine strategies to improve neurologic outcomes after brain injury and to determine whether ketamine might also improve outcomes in humans after TBI.
Acknowledgments
We would like to thank the following: Gary Westbrook, MD, senior scientist at the Vollum Institute, Portland, OR USA for project guidance; members of the Westbrook and Schnell labs for helpful discussions; David Yanez, PhD, associate professor of biostatistics and co-director of the Biostatistics & Design Program, OHSU, Portland, OR USA, for statistical guidance and interpretation; and Sarah Mader, research assistant at OHSU Department of Anesthesiology, Portland, OR USA, for outstanding technical support. The contents of this manuscript do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Sources of Funding:
Primary funding for this project was provided by a Foundation for Anesthesia Education & Research (FAER) Research Fellowship Grant (AJP). Additional funding was provided by a BIRCWH K12 award made possible through the Eunice Kennedy Shriver National Institute of Child Health & Human Development and the Office of Research on Women’s Health (NIH K12 HD 043488) (LEV), a Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development CDA-2 Award 005-10S (ES), a Department of Veterans Affairs Merit Review Award I01-BX002949 (ES) and Oregon Health & Science University (OHSU) Anesthesiology & Perioperative Medicine departmental funds. Supplementary equipment and developmental funds were provided by the Oregon Clinical and Translational Research Institute (OCTRI) grant TL1TR000129 and National Institute for Neurological Disorders and Stroke (NINDS) P30 NS061800 (Aicher, PI).
Footnotes
Prior Presentations: Peters A, Villasana L and Schnell E. Ketamine Alters Hippocampal Cell Proliferation after Traumatic Brain Injury and Improves Learning in Mice. Society for Neuroscience and Critical Care. Boston, MA. October 2017. American Society of Anesthesiologists. Boston, MA. October 2017.
Peters A, Villasana L and Schnell E. Ketamine Reduces Post-Traumatic Brain Injury Neurogenesis and Improves Outcomes in Mice.
Western Anesthesia Residents’ Conference. Portland, OR. April 2017.‡
Association of University Anesthesiologists. Washington D.C. May 2017.
International Anesthesia Research Society. Washington D.C. May 2017.*,†
‡First Place: Best Overall Oral Presentation
*Kosaka Best of Meeting Abstract Award Finalist
†Top Finalist in Basic Science Research
Conflicts of Interest: The authors have no conflicts of interest to disclose.
References
- 1.Morganti-Kossmann M, Rancan M, Stahel P, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–5. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Arruda-Carvalho M, Sakaguchi M, Akers K, Josselyn S, Frankland P. Posttraining Ablation of Adult-Generated Neurons Degrades Previously Acquired Memories. J Neurosci. 2011;31:15113–27. doi: 10.1523/JNEUROSCI.3432-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parent J, Jessberger S, Gage F, Gong C. Is neurogenesis reparative after status epilepticus? Epilepsia. 2007;48:69–71. doi: 10.1111/j.1528-1167.2007.01355.x. [DOI] [PubMed] [Google Scholar]
- 4.Bond A, Ming G, Song H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Stem Cell. 2015;116:1477–90. doi: 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron H, McEwen B, Gould E. Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J Neurosci. 1995;15:4687–92. doi: 10.1523/JNEUROSCI.15-06-04687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrus D, Fabel K, Kronenberg G, Winter C, Steiner B, Kempermann G. NMDA and benzodiazepine receptors have synergistic and antagonistic effects on precursor cells in adult hippocampal neurogenesis. Eur J Neurosci. 2009;29:244–52. doi: 10.1111/j.1460-9568.2008.06579.x. [DOI] [PubMed] [Google Scholar]
- 7.Tashiro A, Sandler V, Toni N, Zhao C, Gage F. NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature. 2006;442:929–33. doi: 10.1038/nature05028. [DOI] [PubMed] [Google Scholar]
- 8.Gambrill A, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011;108:5855–60. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verkhratsky A, Kirchhoff F. NMDA Receptors in glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- 10.Kou Z, VandeVord P. Traumatic white matter injury and glial activation: From basic science to clinics. Glia. 2014;62:1831–55. doi: 10.1002/glia.22690. [DOI] [PubMed] [Google Scholar]
- 11.Morrens J, Broeck W van den, Kempermann G. Glial cells in adult neurogenesis. Glia. 2012;60:159–74. doi: 10.1002/glia.21247. [DOI] [PubMed] [Google Scholar]
- 12.Bell J. In Vogue: Ketamine for Neuroprotection in Acute Neurologic Injury. Anesth Analg. 2017;124:1237–43. doi: 10.1213/ANE.0000000000001856. [DOI] [PubMed] [Google Scholar]
- 13.Chang L, Raty S, Ortiz J, Bailard N, Mathew S. The emerging use of ketamine for anesthesia and sedation in traumatic brain injuries. CNS Neurosci Ther. 2013;19:390–5. doi: 10.1111/cns.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villasana L, Kim K, Westbrook G, Schnell E. Functional Integration of Adult-Born Hippocampal Neurons after Traumatic Brain Injury. eNeuro. 2015;2 doi: 10.1523/ENEURO.0056-15.2015. ENEURO.0056-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeung L, Rudd J, Lam W, Mak Y, Yew D. Mice are prone to kidney pathology after prolonged ketamine addiction. Toxicol Lett. 2009;191:275–8. doi: 10.1016/j.toxlet.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Villasana L, Westbrook G, Schnell E. Neurologic impairment following closed head injury predicts post-traumatic neurogenesis. Exp Neurol. 2014;261:156–62. doi: 10.1016/j.expneurol.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan S, Rudd J, Yew D. Gene Expression Changes in GABAA Receptors and Cognition Following Chronic Ketamine Administration in Mice. PLoS One Edited by McAlonan GM. 2011;6:e21328. doi: 10.1371/journal.pone.0021328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dash P, Mach S, Moore A. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. 2001;63:313–9. doi: 10.1002/1097-4547(20010215)63:4<313::AID-JNR1025>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Chirumamilla S, Sun D, Bullock M, Colello R. Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. JNeurotrauma. 2002;19:693–703. doi: 10.1089/08977150260139084. [DOI] [PubMed] [Google Scholar]
- 20.Cameron H, Mckay R. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol. 2001;435:406–17. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Mu Y, Gage F. Development of Neural Circuits in the Adult Hippocampus. Curr Top Dev Biol. 2009;87:149–74. doi: 10.1016/S0070-2153(09)01205-8. [DOI] [PubMed] [Google Scholar]
- 22.Rice A, Khaldi A, Harvey H, Salman N, White F, Fillmore H, Bullock M. Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol. 2003;183:406–17. doi: 10.1016/s0014-4886(03)00241-3. [DOI] [PubMed] [Google Scholar]
- 23.Hattiangady B, Shetty A. Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia. 2008;49:26–41. doi: 10.1111/j.1528-1167.2008.01635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dayer A, Ford A, Cleaver K, Yassaee M, Cameron H. Short-term and long-term survival of new neurons in the rat dentate gyrus. J Comp Neurol. 2003;460:563–72. doi: 10.1002/cne.10675. [DOI] [PubMed] [Google Scholar]
- 25.Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–70. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 26.Tremblay M-È, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–9. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loane D, Byrnes K. Role of Microglia in Neurotrauma. Neurotherapeutics. 2010;7:366–77. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekdahl C, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: The dual role of microglia. Neuroscience. 2009;158:1021–9. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 29.Arndt D, Lerner J, Matsumoto J, Madikians A, Yudovin S, Valino H, McArthur D, Wu J, Leung M, Buxey F, Szeliga C, Hirtum-Das M Van, Sankar R, Brooks-Kayal A, Giza C. Subclinical early posttraumatic seizures detected by continuous EEG monitoring in a consecutive pediatric cohort. Epilepsia. 2013;54:1780–8. doi: 10.1111/epi.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao Y, Bramlett H, Atkins C, Truettner J, Lotocki G, Alonso O, Dietrich W. Post-Traumatic Seizures Exacerbate Histopathological Damage after Fluid-Percussion Brain Injury. J Neurotrauma. 2011;28:35–42. doi: 10.1089/neu.2010.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall K, Lifshitz J. Diffuse traumatic brain injury initially attenuates and later expands activation of the rat somatosensory whisker circuit concomitant with neuroplastic responses. Brain Res. 2010;1323:161–73. doi: 10.1016/j.brainres.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shors T, Townsend D, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–84. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garthe A, Kempermann G. An old test for new neurons: refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis. Front Neurosci. 2013;7:63. doi: 10.3389/fnins.2013.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loix S, DeKock M, Henin P. The anti-inflammatory effects of ketamine : State of the art. Acta Anaesthesiolologica Belgica. 2011;62:47–58. [PubMed] [Google Scholar]
- 35.Berman R, Cappiello A, Anand A, Oren D, Heninger G, Charney D, Krystal J. Antidepressant effects of ketamine in depressed patients. Soc Biol Psychiatry. 2000;47:351–4. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 36.Duman R, Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond B Biol Sci. 2012;367:2475–84. doi: 10.1098/rstb.2011.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tung A, Herrera S, Fornal C, Jacobs B. The effect of prolonged anesthesia with isoflurane, propofol, dexmedetomidine, or ketamine on neural cell proliferation in the adult rat. Anesth Analg. 2008;106:1772–7. doi: 10.1213/ane.0b013e31816f2004. [DOI] [PubMed] [Google Scholar]
- 38.Winkelheide U, Lasarzik I, Kaeppel B, Winkler J, Werner C, Kochs E, Engelhard K. Dose-dependent effect of S(+) ketamine on post-ischemic endogenous neurogenesis in rats. Acta Anaesthesiol Scand. 2009;53:528–33. doi: 10.1111/j.1399-6576.2009.01905.x. [DOI] [PubMed] [Google Scholar]
- 39.Clarke M, Razmjou S, Prowse N, Dwyer Z, Litteljohn D, Pentz R, Anisman H, Hayley S. Ketamine modulates hippocampal neurogenesis and pro-inflammatory cytokines but not stressor induced neurochemical changes. Neuropharmacology. 2017;112:210–20. doi: 10.1016/j.neuropharm.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 40.Soumier A, Carter R, Schoenfeld T, Cameron H. New Hippocampal Neurons Mature Rapidly in Response to Ketamine But Are Not Required for Its Acute Antidepressant Effects on Neophagia in Rats. eNeuro. 2016;3:1–13. doi: 10.1523/ENEURO.0116-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bye N, Carron S, Han X, Agyapomaa D, Ng S, Yan E, Rosenfeld J, Morganti-Kossmann C. Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats. J Neurosci Res. 2011;89:986–1000. doi: 10.1002/jnr.22635. [DOI] [PubMed] [Google Scholar]
- 42.Mei X, Wang W, Wang W, Li Y, Zhang H, Wu S, Li Y, Xu L. Inhibiting astrocytic activation: A novel analgesic mechanism of ketamine at the spinal level? J Neurochem. 2009;109:1691–700. doi: 10.1111/j.1471-4159.2009.06087.x. [DOI] [PubMed] [Google Scholar]
- 43.Yuhas Y, Ashkenazi S, Berent E, Weizman A. Ketamine upregulates eNOS expression in human astroglial A172 cells: Possible role in its antidepressive properties. J Neuroimmunol. 2017;305:75–81. doi: 10.1016/j.jneuroim.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 44.Shibakawa Y, Sasaki Y, Goshima Y, Echigo N, Kamiya Y, Kurahashi K, Yamada Y, Andoh T. Effects of ketamine and propofol on inflammatory responses of primary glial cell cultures stimulated with lipopolysaccharide. Br J Anaesth. 2005;95:803–10. doi: 10.1093/bja/aei256. [DOI] [PubMed] [Google Scholar]
- 45.Monje M, Toda H, Palmer T. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–5. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 46.Sleigh J, Harvey M, Voss L, Denny B. Ketamine – more mechanisms of action than just NMDA blockade. Trends Anaesth Crit Care. 2014;4:76–81. [Google Scholar]
- 47.Zanos P, Moaddel R, Morris P, Georgiou P, Fischell J, Elmer G, Alkondon M, Yuan P, Pribut H, Singh N, Dossou K, Fang Y, Huang X-P, Mayo CL, Wainer I, Albuquerque E, Thompson S, Thomas C, Zarate C, Gould T. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:1–18. doi: 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wesseling H, Rahmoune H, Tricklebank M, Guest P, Bahn S. A targeted multiplexed proteomic investigation identifies ketamine-induced changes in immune markers in rat serum and expression changes in protein kinases/phosphatases in rat brain. J Proteome Res. 2015;14:411–21. doi: 10.1021/pr5009493. [DOI] [PubMed] [Google Scholar]
- 49.Blaiss C, Yu T-S, Gui Z, Powell C, Kernie S. Temporally specified genetic ablation of neurogenesis impairs cognitive recovery following traumatic brain injury. J Neurosci. 2011;454:42–54. doi: 10.1523/JNEUROSCI.5265-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gradari S, Pérez-Dómper P, Butler R, Martínez-Cué C, Polavieja G de, Trejo J. The relationship between behavior acquisition and persistence abilities: Involvement of adult hippocampal neurogenesis. Hippocampus. 2016;26:857–74. doi: 10.1002/hipo.22568. [DOI] [PubMed] [Google Scholar]