

Abstract
Dysregulation of the PI3K-AKT-mTOR signaling network is a prominent feature of breast cancers. However, clinical responses to drugs targeting this pathway have been modest, possibly due to dynamic changes in cellular signaling that drive resistance and limit drug efficacy. Using a quantitative chemoproteomics approach we mapped kinome dynamics in response to inhibitors of this pathway and identified signaling changes that correlate with drug sensitivity. Maintenance of AURKA after drug treatment was associated with resistance in breast cancer models. Incomplete inhibition of AURKA was a common source of therapy failure and combinations of PI3K, AKT or mTOR inhibitors with the AURKA inhibitor MLN8237 were highly synergistic and durably suppressed mTOR signaling resulting in apoptosis and tumor regression in vivo. This signaling map identifies survival factors whose presence limits the efficacy of targeted therapies and reveals a new drug combination to unlock the full potential of PI3K-AKT-mTOR pathway inhibitors in breast cancer.
INTRODUCTION
Mutations and aberrant signaling of the PI3K–AKT–mTOR pathway (PI3K-pathway) is a prominent feature of breast and many other cancers. Genomic alterations of PI3K-pathway components, including PTEN, PIK3CA, and AKT1 occur in over 60% of breast malignancies1. Despite this high prevalence, drugs targeting this pathway have demonstrated only modest responses across numerous clinical trials2,3. The clinical observation that most breast cancers fail to respond suggests that additional factors modulate cellular response and drive resistance. A prominent feature of this pathway is drug-induced signaling adaptation and feedback mechanisms resulting in suboptimal drug responses4–6. Therefore, it is likely that understanding and targeting these dynamic changes in signaling will be important in optimizing this class of agents.
In principle, the measurement of dynamic changes elicited by therapy can be used to develop novel drug combinations. While previous efforts have focused on acute signaling changes leading to pathway reactivation and drug resistance4,7, systematically contrasting global signaling changes with drug efficacy has not been performed. Such an analysis may reveal survival factors whose suppression is required for drug efficacy and hence could reveal new combinatorial strategies to enhance therapeutic responses. Previous identification of such factors have led to the understanding that drug-induced activation of apoptotic machinery8,9 and impairment of protein synthesis10 is required for sensitivity to a wide variety of drugs. In the context of breast cancer, multiple efforts in the field have identified mTORC1 as a survival factor whose suppression is necessary for PI3K-pathway inhibitor sensitivity11,12. This observation has led to clinical trials combining PI3K and mTOR inhibitors, yet reported clinical results have yielded suboptimal outcomes due to increased systemic toxicity and cytostatic tumor effects3. Hence, there remains a pressing need to uncover new combination targets in order to improve therapeutic efficiency of PI3K-pathway inhibitors. Identifying additional survival factors will require a comprehensive understanding of signaling dynamics in response to treatment and insight as to how these dynamics contribute to drug resistance.
Little is known about global kinome rewiring in response to drug treatment, which is due in part to limitations in available technologies. Recently, a kinase enrichment strategy has been developed using a chemoproteomics technique that combines kinase affinity capture with quantitative mass spectrometry (MS). This approach uses a multiplexed set of type I kinase inhibitors immobilized onto beads (MIBs), which are used to affinity purify a diverse set of active kinases through their increased avidity for ATP compared to inactive kinases. Enriched kinases are then identified and quantified by LC MS/MS (MIBs/MS), enabling simultaneous measurement of many endogenous kinases based on their activity state and abundance7. Because many drugs impinge on common pathways and cell lines often display unique behaviors, it is possible that a quantitative map of kinase dynamics spanning multiple cell lines and drug treatments may be used to identify more general responses to drug treatment that are linked to drug sensitivity.
Here we applied the MIBs/MS approach to identify signaling changes associated with drug efficacy by mapping the kinome following exposure to targeted therapies across a panel of breast cancer cell lines of various subtypes and genotypes. Comparing kinome activity profiles between drug-sensitive and resistant cells allowed us to generate a kinome-response signature associated with drug sensitivity. By performing a systematic analysis of signaling dynamics following drug treatment, we identified that failure to inhibit AURKA was associated with resistance to a diverse set of targeted therapies. Further analysis revealed that inhibition of AURKA was sufficient to engender strong synergistic responses when combined with inhibitors of PI3K, AKT, or mTOR. This provides an effective new framework for the unbiased identification of survival factors acting as molecular barriers to the efficacy of drugs, and we demonstrate the utility of this approach by developing rational combination strategies to enhance responses to PI3K-pathway inhibitors in breast cancer.
RESULTS
Generation and analysis of a dynamic kinome signaling map
We applied an unbiased proteomic strategy to measure kinome rewiring in response to drug treatment. Kinome profiling was performed via a chemoproteomics approach using Multiplexed Inhibitor Beads (MIBs) coupled with mass spectrometry (MIBs/MS). Our library of Multiplexed Inhibitor Beads (MIBs) consist of a mixture of sepharose beads covalently linked to 12 kinase inhibitors ranging from moderately selective (e.g. Lapatinib, Sorafenib) to pan-kinase inhibitors (e.g. Purvalanol B, Staurosporine) for broad kinome coverage (Fig. 1a and Supplementary Fig. 1). Because type I kinase inhibitors preferentially bind kinases in their active conformation, kinase capture by MIBs under the stringent binding conditions used here is a function of kinase expression, the affinity of kinases for the immobilized inhibitors, and the activation state of the kinase13. Vehicle or drug treated cell lysates were incubated with MIBs, and enriched kinases were eluted and quantified by LC MS/MS using label-free quantitation (see Methods)14. We estimate that our current approach is able to capture roughly 35% of highly expressed kinases in a given sample (Supplementary Fig. 2).
Figure 1. Measurement of kinome dynamics to identify correlates of drug sensitivity.
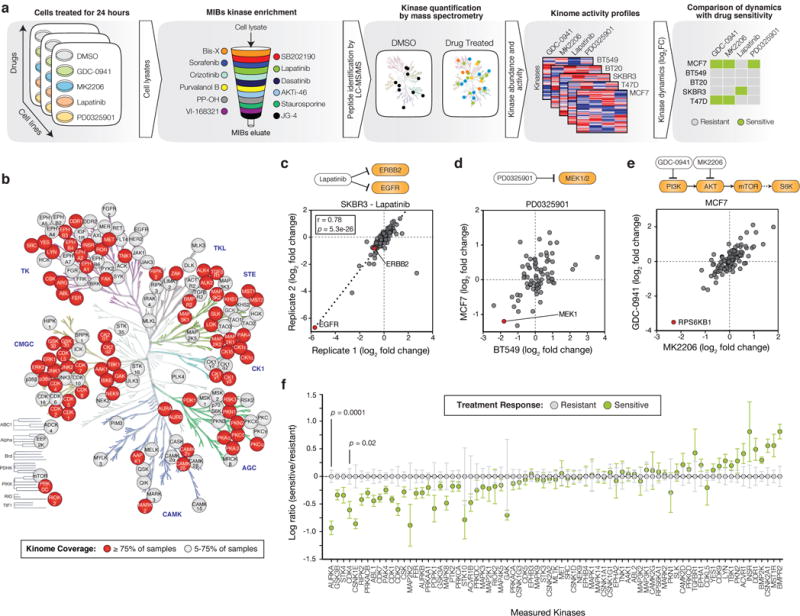
(a) Schematic of approach using multiplex inhibitor beads followed by mass spectrometry (MIBs/MS). Sample lysates are passed through a column containing the indicated kinase inhibitors covalently linked to beads. After washing, bound proteins are eluted, trypsin digested and quantified through label-free mass spectrometry. (b) Human kinome tree annotated with kinases identified in this study and colored based on the percentage of total samples where each particular kinase could be quantified. (c) Comparison of activity ratios between biological replicates for 122 kinases, expressed as a log ratio of measurements from SKBR3 cells treated with 200nM Lapatinib for 24 hours versus DMSO. Pearson correlation and p value shown. (d) Comparison of kinase activity ratios in BT549 and MCF7 cells treated with 100nM PD0325901 versus DMSO. Data represent 75 kinases with one outlier kinase (GAK, BT549 log2 fold change 8.3) removed. (e) Comparison of activity ratios for 70 kinases measured from MCF7 cells treated with either 250nM MK2206 or GDC-0941 versus DMSO. (f) Categorical analysis of kinome dynamics occurring in drug-sensitive treatment responses (n=6) versus resistant treatment responses (n=14) for all drugs pooled together. For visualization purposes, each kinase was centered on the mean of resistant samples. Data shown for 75 kinases which could be measured in >75% of samples. All drug treatments are 24 hours. Error bars are mean ± s.e.m. and p values calculated using a two-sided t-test.
We applied this strategy to a panel of breast cancer cell lines of various subtype and genotype classifications, and measured kinome dynamics following treatment with a panel of targeted therapies. Cell lines were chosen to maximize transcriptional diversity and span the major subtypes of breast cancer (Supplementary Fig. 3). All lines harbored mutations in PI3K-pathway genes including PIK3CA-mutant MCF7 (ER+/PR+), BT20 (receptor negative) and T47D (ER+/PR+); PTEN-null BT549 (receptor negative); and HER2-amplified SKBR3 (HER2+) (Supplementary Fig. 4a). Cell lines were treated for 24 hours with DMSO or kinase inhibitors relevant to breast cancer signaling including the EGFR/HER2 inhibitor Lapatinib (200nM), the pan-Class I PI3K inhibitor GDC-0941 (250nM), the AKT inhibitor MK2206 (250nM), and the MEK inhibitor PD0325901 (100nM), and then profiled using MIBs/MS (Fig. 1a and Supplementary Fig. 4b). All together, we quantified changes across 151 kinases in total and 75 kinases which were present in over 75% (15/20) of samples (Fig. 1b, Supplementary Dataset 1). Significant drug-induced changes (defined based on the log2 fold change of drug versus DMSO treatment, logFC) were detected for 99 kinases at p<0.001 corresponding to 66% of kinases measured, indicating that the drugs had widespread and significant impacts on global kinome dynamics.
To assess quality and reproducibility of the MIBs/MS data, we initially compared biological replicates of SKBR3 (HER2+) cells treated with the dual EGFR/HER2 small-molecule inhibitor Lapatinib. We observed a high correlation of 0.78 between replicates for identified kinases (p=5e-26) (Fig. 1c). The MIBs/MS screening strategy also accurately captured activity inhibition of direct drug targets by Lapatinib indicated by the significant decrease in levels for EGFR (logFC=−5.8, p=6e-5) and HER2 (−0.7, p=1e-4) (Fig. 1c). We observed a decrease in MEK1 activity upon treatment with the MEK inhibitor PD0325901 in BT549 and MCF7 cells (logFC=−1.8 and −1.2 respectively, Fig. 1d). We also observed indirect pathway-specific events, such as a decrease in the activity of the mTOR effector kinase RPS6KB1 when treated with either the PI3K inhibitor GDC-0941 or AKT inhibitor MK2206 in MCF7 cells (logFC=−3.5 and −2.3 respectively, Fig. 1e). Comparison of observed kinome changes with previous MIBs/MS data revealed a high degree of concordance (Supplementary Fig. 5)15. These results highlight the reproducibility of the MIBs/MS approach as well as its ability to identify direct and indirect drug targets based on reductions in both activity and abundance.
We hypothesized that the identification of shared responses across lines and drugs may lead to a more robust understanding of signaling dynamics, as opposed to changes specific to a particular drug or cell type. We therefore sought to identify changes that were generally associated with treatment sensitivity or resistance in a drug-agnostic fashion. First, cell lines were classified as sensitive or resistant to each of the drugs in our panel based on dose-response analysis (Supplementary Fig. 4b, Supplementary Fig. 6a-d). Next, fold changes for each kinase were compared between these sensitive and resistant classifications for all drugs pooled together to identify candidate kinases whose inhibition was associated with drug sensitivity (Fig. 1f). This analysis revealed that suppression of 12 kinases was significantly associated with drug sensitivity (p<0.05). Among the identified candidates were kinases involved in cell cycle processes including mitotic kinases AURKA (p=0.0001) and CDK1 (p=0.04), and kinases involved in interphase, CDK4 (p=0.02) and CDK2 (p=0.05). Other kinases identified were involved in YAP signaling (STK4, p=0.01) and WNT signaling (GSK3B, p=0.005 and CSNK1E, p=0.02). These results were not linked to general impairment of the cell cycle per se. We observed no correlation with sensitivity for other cyclin-dependent kinases (CDKs) measured in our screen such as CDK6, a closely related CDK to CDK4. In addition, the AURKA paralog AURKB was not significantly associated with sensitivity even though it is regulated during mitosis in a similar manner (Fig. 1f)16. We performed a similar analysis using a three-response categorization (i.e., sensitive, moderately sensitive, and resistant) and found that these results were largely independent to how sensitivity was classified (Supplementary Fig. 6e-g). We postulate that this drug-agnostic approach identifies changes that are general to drug sensitivity and reveals factors that may be missed by studies limited to a single drug analyses. For example, the top candidate from our analysis, AURKA, was trending towards but not found to be significantly associated with resistance or even among the top several candidates with any single drug. However, by pooling responses across all drugs it emerged as the most associated with resistance in terms of both magnitude and significance (Supplementary Fig. 7). Therefore, by performing a systematic screen of signaling dynamics following drug exposure, we identified a set of specific kinases whose maintenance was associated with resistance to targeted therapies in breast cancer.
AURKA associates with PI3K and AKT inhibitor resistance
We focused our validation of molecular correlates of drug sensitivity on the PI3K-pathway given its central importance to breast cancer. We observed a significant association between maintenance of AURKA after treatment and drug resistance (Fig. 2a). To confirm this result, we measured molecular responses to treatment with the pan-PI3K inhibitor GDC-0941 in two sensitive (T47D and MCF7, IC50 < 200nM) and two new cell lines that were robustly resistant (HCC38 and MDAMB453, IC50 > 40μM). A critical output of the PI3K-pathway is the activation of the mTORC1 complex, whose inhibition is necessary for sensitivity to PI3K inhibitors11. After treatment we observed suppression of mTORC1 activity only in sensitive cells, as evidenced by decreased phosphorylation of its effector protein S6 (Fig. 2b). Confirming our MIBs/MS data, in response to treatment we observed decreases in the abundance and auto-phosphorylation of AURKA in sensitive cells, whereas resistant cells maintained these levels throughout (Fig. 2b, Supplementary Fig. 9a,b). Similar results were observed using the AKT inhibitor MK2206, representing the next step in the PI3K-pathway (Supplementary Fig. 9c-e). These results confirm that failure to suppress AURKA activity is associated with resistance to PI3K and AKT inhibition in breast cancer cells.
Figure 2. Maintenance of AURKA is associated with resistance to PI3K inhibition.
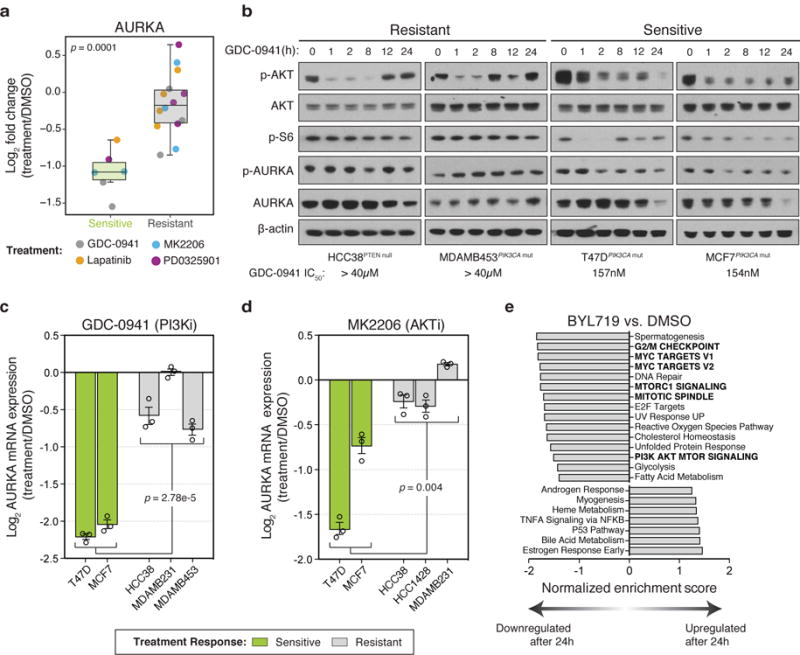
(a) Changes in activity of AURKA as measured by MIBs in drug-sensitive versus drug-resistant treatment responses after 24 hours of exposure to the indicated compounds. Each point reflects a single cell line and drug treatment (n=20 biologically independent samples). Box-and-whisker plots show median (centerline), upper/lower quartiles (box limits), and whiskers spanning the interquartile range from 25-75 percentiles. P value calculated using a two-sided t-test. (b) Western blot showing PI3K and AURKA signaling in GDC-0941-resistant and GDC-0941-sensitive cell lines. Protein lysates from cells treated with 1μM GDC-0941 were extracted at different points time, separated by SDS-PAGE, and analyzed by immunoblot with the indicated antibodies. Representative image of n=3 independent experiments (full blots shown in Supplementary Fig. 8). (c,d) Log ratio expression values of AURKA mRNA measured by RT-PCR from the indicated cell lines treated with (c) 1μM of GDC-0941 or (d) 1μM MK2206 for 24 hours and compared to DMSO treatment. Data represents n=3 biological replicates. Error bars are mean ± s.e.m. and p values calculated using one-way ANOVA. (e) Gene Set Enrichment Analysis (GSEA) of top gene sets significantly upregulated or downregulated after 24 hours in response to 1μM BYL719 treatment in MCF7 and T47D cells compared to DMSO. Data in panel (e) based on transcriptomic data from Bosch, et al.17
We next asked how AURKA is regulated in response to PI3K-pathway inhibition in drug-sensitive cells. AURKA regulates centrosome alignment, mitotic spindle formation and chromosome segregation during mitosis and its activity and abundance is tightly regulated16. We observed a robust and significant change in AURKA protein levels after 24 hours in drug-sensitive cells leading us to hypothesize that changes in transcription of AURKA might account for its loss after treatment. AURKA mRNA levels were decreased in response to GDC-0941 and MK2206 when comparing drug-sensitive and resistant cell lines (p=2.8e-5 and p=0.004 respectively, Fig. 2c,d). In addition, transcriptomes of MCF7 and T47D cells treated with the PI3Kα-specific inhibitor BYL719 for 24 hours17 reflected a significant reduction of AURKA after drug treatment in both of these BYL719-sensitive cell lines (IC50 ≤ 250nM, Supplementary Fig. 10a)11,18. Interestingly, Gene Set Enrichment Analysis (GSEA)19 of these transcriptomes revealed that a prominent component of response to PI3K inhibition was the suppression of genes involved in the G2/M checkpoint, including AURKA, suggesting that transcriptional control of this aspect of the cell cycle is a major output of the PI3K-pathway (Fig. 2e, Supplementary Fig. 10b, Supplementary Dataset 2).
AURKA mediates survival during PI3K-pathway inhibition
We next asked if the downregulation of AURKA was functionally relevant and whether the presence of AURKA limits efficacy of PI3K-pathway directed therapies. We tested whether AURKA inhibition was sufficient to confer sensitivity to PI3K-pathway inhibitors using a combination profiling approach to measure drug synergy across an extended panel of 13 breast cancer cell lines. We applied a dose matrix of increasing concentrations of the AURKA-specific inhibitor MLN8237 alone and in combination with a PI3K (GDC-0941), AKT (MK2206), or mTOR (RAD001) inhibitor and measured effects on cell proliferation. To evaluate drug synergy we: (1) visualized Loewe excess values, (2) scored combination index values measuring shifts in drug potency, (3) calculated synergy scores based on Loewe excess values, and (4) visualized and scored combinations using a Bliss independence model20 (see Methods). Our results in MCF7 cells indicated that MLN8237 in combination with GDC-0941, MK2206 or RAD001 was synergistic using all four approaches (Fig. 3a, Supplementary Fig. 11-13, Supplementary Dataset 3). Testing the combination with GDC-0941 across the extended panel of cell lines we found significant synergy based on the Loewe excess model in 38% of models (5/13) based on a synergy score > 1, which we determined through simulation to represent a less than 5% chance of non-synergy (i.e. FDR<5%) (Fig. 3b, Supplementary Fig. 11). We extended this analysis to drug combinations of MLN8237 with either MK2206 or RAD001 and found significant synergy in 54% and 85% of models, respectively (Fig. 3b, Supplementary Fig. 12,13). Overall we found no significant trend towards synergy based on PIK3CA or PTEN mutational status, but did observe slightly increased synergy in receptor positive cell lines (ER+ or HER2+, p=0.04 for GDC-0941 and p=0.035 for MK2206, based on a two-tailed t-test) (Supplementary Dataset 3).
Figure 3. AURKA suppression enhances sensitivity and drives cell death in response to PI3K-pathway inhibitors in breast cancer cell lines.
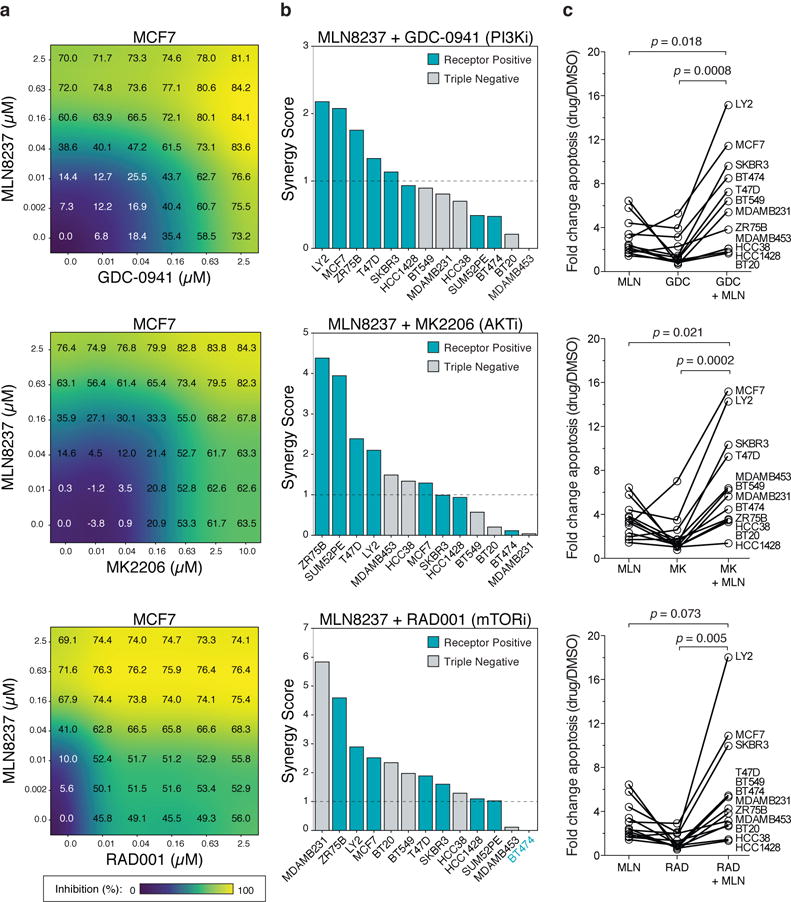
(a) A dose matrix of GDC-0941 (PI3K), MK2206 (AKT), or RAD001 (mTOR) in combination with the AURKA inhibitor MLN8237 in MCF7 cells. Cell proliferation was assessed after 72 hours. Percent growth inhibition at each dose shown. (b) Synergy scores based on a Loewe excess inhibition model across 13 breast cancer cell lines that were treated with the indicated combination using a escalating dose matrix for 72 hours. Dashed line indicates a 5% FDR cutoff to define synergistic combinations (see Methods). (c) Cell lines were treated with 625nM of the indicated single agents or combined together for 72 hours and apoptosis measured by YO-PRO1 positivity. Data represents n=4 biologically independent samples. Error bars are mean ± s.d. and p values calculated using a two-sided t-test.
Since PI3K-pathway inhibitors are primarily cytostatic5,21 and AURKA is known to regulate apoptosis22, we next asked whether AURKA inhibition could enhance responses to PI3K-pathway inhibitors by inducing cytotoxic responses. Across 12 cell lines, we found that the addition of MLN8237 caused an increase in apoptotic cell death (Fig. 3c), which was independent of the particular dose used (Supplementary Fig. 14a,b). This enhancement in cell death generally occurred in conditions where synergy was also observed (Supplementary Fig. 14b). We compared this response with the combination of CDK4/6 and PI3K inhibitors which are known to be synergistic12. While we observed synergy between PI3K, AKT and mTOR inhibitors and the CDK4/6 inhibitor LEE011, the response was primarily cytostatic indicating that CDK4 is only necessary for proliferation rather than tumor cell survival in the presence of PI3K-pathway inhibitors (Supplementary Fig. 14c-e, Supplementary Dataset 3). Therefore, AURKA mediates cellular survival in the context of PI3K-pathway inhibition, and since the drug combinations are synergistic in inducing apoptosis in breast cancer cells, we propose that it may be a promising companion target in order to enhance the efficacy of PI3K-pathway inhibitors.
MLN8237 and Everolimus (RAD001) induce cell death in vivo
We next evaluated the efficacy of this combination in vivo and focused on the combination of MLN8237 with the only FDA-approved inhibitor targeting this pathway in breast cancer, the mTOR inhibitor RAD001 (Everolimus). Clinically, RAD001 overwhelmingly results in disease stabilization rather than regression23. This is reflected in vitro where all lines have a high RAD001 Emax indicating cytostatic effects. In particular, MCF7 cells have a high Emax of 0.54 and do not display evidence of PARP cleavage at high doses (Supplementary Fig. 15). To investigate whether AURKA suppression enhances response to RAD001 treatment, we tested the combination in MCF7 orthotopic transplants. While RAD001 or MLN8237 monotherapy only partially impaired tumor growth, the combination showed significantly greater tumor growth inhibition than either single agent alone (Fig. 4a). Furthermore, all animals receiving the combination (9/9) showed marked tumor regression, while no regressions were observed with monotherapy (0/13 in total, p=2e-6 by Fisher’s exact test, Fig. 4b). Post-treatment tumor specimens displayed an induction of apoptosis specific to the combination as demonstrated by an increase in the number of TUNEL-positive cells (Fig. 4c,d). During the course of study we did not observe any significant weight loss in animals receiving the combination as compared to the RAD001 single-agent group (Supplementary Fig. 16), suggesting tolerability and no added toxicity from co-inhibiting Aurora kinase A. Therefore, the addition of MLN8237 to RAD001 treatment results in tumor regression and a strong cytotoxic response in vivo.
Figure 4. The Aurora kinase inhibitor MLN8237 enhances sensitivity to Everolimus (RAD001) and induces cell death in vivo.
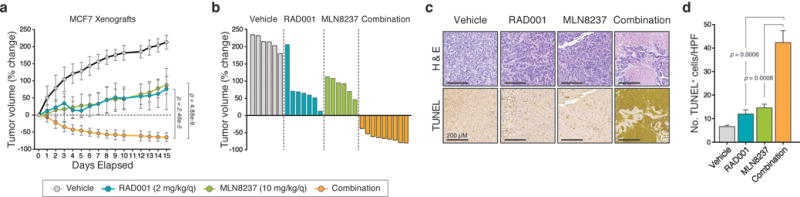
(a) MCF7 orthotopic xenograft tumors were treated with vehicle (n=6 biologically independent mice), RAD001 (2 mg/kg/day, n=7 biologically independent mice), MLN8237 (10 mg/kg/day, n=6 biologically independent mice) or the combination of the two single-agents (n=9 biologically independent mice) via oral gavage, daily, over 15 days. The percentage change in tumor volume was calculated for each animal from baseline. (b) Individual tumor profiles compared to baseline for each tumor treated with vehicle (n=6 biologically independent mice), RAD001 (n=7 biologically independent mice), MLN8237 (n=6 biologically independent mice) or the combination (n=9 biologically independent mice) over a 15-day period. (c) Representative images of tumor tissue extracted from mice after 15 days treatment with the indicated agents and stained for H&E and TUNEL. Images shown using a 10× objective. Scale bars represent 200 μm. (d) Quantification of the number of TUNEL+ cells/field from TUNEL staining of MCF7 tumors following 15 days of treatment. Data is an average of five high-powered (20×) fields analyzed per tumor and are representative of n=3 biologically independent animals. In all graphs, error bars are mean ± s.d. and p values calculated using a two-sided t-test.
Co-inhibition durably suppresses mTORC1 signaling via AKT
We next turned to identify mechanisms driving the increased efficacy of the drug combination. Since most PI3K-pathway inhibitors (including rapamycin or RAD001) elicit feedback signals resulting in incomplete suppression of mTOR and drug resistance11,24, we first asked if the combination of MLN8237 enhanced the activity of RAD001 on mTOR signaling to effectors, RPS6 (S6) and 4E-BP1, in vivo. While we observed an incomplete and partial suppression of S6 in RAD001-treated MCF7 xenografts, the addition of MLN8237 resulted in a durable and complete loss of S6 in all 9 tumors (Fig. 5a). Though RAD001 is a relatively potent inhibitor of S6, it is a weak inhibitor of 4E-BP1 and therefore only partially impairs cap-dependent protein synthesis24. We therefore investigated the activity of phospho-4E-BP1, which can be stimulated by rapamycin treatment24. While phospho-4E-BP1 levels were enhanced with RAD001 single-agent treatment, co-treatment with MLN8237 suppressed these levels back to nearly baseline (Fig. 5a). This surprising finding led us to ask how Aurora kinase inhibition might alter this key signaling output of mTOR. We investigated AKT activity via phosphorylation of serine 473, which activates mTOR and is catalyzed by a variety of kinases25. Single-agent MLN8237 reduced phospho-AKT levels both in monotherapy and combination treatment, indicating that Aurora kinases sustain mTOR levels by promoting AKT activity (Fig. 5a). We next examined whether Aurora kinase driven maintenance of mTOR was a general feature of PI3K-pathway inhibitors. Using MCF7 cells in vitro, we observed that MLN8237 treatment impaired phospho-AKT and that the combination of MLN8237 with either GDC-0941 (targeting PI3K) or MK2206 (targeting AKT) led to robust ablation of phospho-S6 and phospho-4E-BP1 levels (Fig. 5b). Therefore, Aurora kinases contribute to resistance to PI3K-pathway inhibitors through the maintenance of AKT and residual mTORC1 activity. Hence targeting this survival mechanism results in a more durable and complete repression of the PI3K-pathway.
Figure 5. Aurora kinase co-inhibition durably suppresses mTORC1 signaling and alters the BAX/BCL2 ratio.
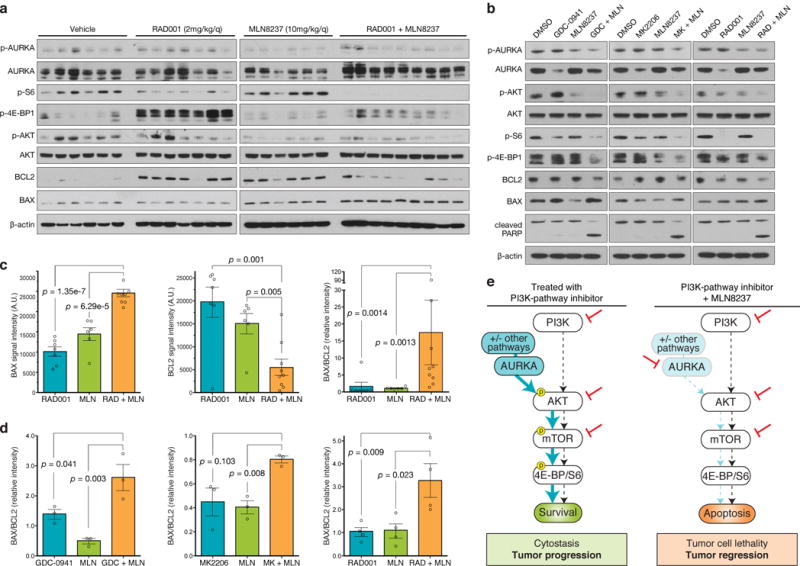
(a) MCF7 orthotopic xenografts were treated with vehicle (n=6 biologically independent mice), RAD001 (2 mg/kg/day, n=7 biologically independent mice), MLN8237 (10 mg/kg/day, n=6 biologically independent mice) or the combination of the two single-agents (n=9 biologically independent mice) for 15 days, at which point tumors were harvested and snap frozen. Western blot of protein lysates from individual tumors were probed with the indicated antibodies (full blots shown in Supplementary Fig. 17a). (b) MCF7 cells were treated with either 250nM GDC-0941, 250nM MK2206, 5nM RAD001, 100nM MLN8237 or the indicated combinations for 24 hours and protein lysates subjected to immunoblot using the indicated antibodies. Representative image from n=3 independent experiments (full blots shown in Supplementary Fig. 17b). (c) BAX, BCL2 and BAX/BCL2 ratio in MCF7 orthotopic xenografts treated for 15 days with the indicated drugs based on quantification of western blot images (RAD001, n=7; MLN8237, n=6; combination, n=9 biologically independent mice analyzed). (d) BAX/BCL2 ratio in MCF7 cells treated for 24 hours with the indicated drugs based on quantification of western blot images from n=3 independent experiments. (e) Proposed model of mechanism of Aurora kinase inhibitor synergy. De novo resistance to single agent inhibition of PI3K, AKT or mTOR is due to incomplete suppression of the pathway due to Aurora kinase signaling which activates AKT. Drug combinations that simultaneously inhibit the PI3K-pathway and block Aurora kinase signaling completely suppress mTOR signaling to 4E-BP1 and S6 resulting in tumor cell death. In all graphs, error bars are mean ± s.e.m. and p values calculated using a two-sided t-test.
Co-inhibition unbalances pro- and anti-apoptotic factors
Since we observed cell death in response to these drug combinations (Fig. 5b, Fig. 4d), we next sought to elucidate how Aurora kinase mediates cell survival in response to PI3K-pathway suppression. Both Aurora kinases and mTOR regulate a number of components of the intrinsic apoptosis pathway22,26, and we hypothesized that deregulation of the balance of pro- and anti-apoptotic factors may cause cell death in response to drug combinations containing MLN8237. BAX promotes apoptosis while BCL2 prevents apoptosis by inhibiting the activity of BAX and together the balance of these two proteins forms a molecular rheostat for apoptosis27. In MCF7 xenografts, combination treatment resulted in increased BAX levels and a reduction in BCL2 levels leading to an increase in the ratio of BAX/BCL2 compared to either MLN8237 or RAD001 treatment alone (Fig. 5c). Furthermore, the BAX/BCL2 ratio was also increased by the addition of MLN8237 to GDC-0941, MK2206 or RAD001 in MCF7 cells in vitro where it was associated with the presence of cleaved PARP (Fig. 5b,d). Taken together, we propose a model whereby Aurora kinase inhibitors potentiate the activity of PI3K-pathway inhibitors through enabling a durable and complete suppression of AKT/mTOR signaling, and drive cell death by altering the balance of pro and anti-apoptotic factors (Fig. 5e).
MYC regulates AURKA downstream of the PI3K-pathway
We next sought to identify factors that regulate AURKA in response to treatment. We noted that a MYC target gene signature was among the most suppressed gene sets after treatment with BYL719 suggesting MYC may play a significant role in regulating the transcriptional response to PI3K inhibition and therefore potentially AURKA (Fig. 2e). To directly define if MYC activity is suppressed by PI3K-pathway inhibition, we transcriptionally profiled an isogenic pair of MCF10A breast epithelial cells over-expressing MYC to derive a gene signature of the top 150 most up-regulated genes by MYC (Supplementary Dataset 4). Comparison of this signature with transcriptional changes induced by BYL719 treatment in MCF7 and T47D cells revealed that most MYC signature genes were strongly repressed during PI3K inhibition (Fig. 6a,b). Therefore MYC is regulated by the PI3K-pathway in these cells, likely via mTORC1-mediated translation and AKT-mediated stabilization of MYC28–30. AURKA was among the signature genes and we found that MYC over-expressing cells had an 8-fold increase in AURKA transcript levels as well as higher levels of total and phosphorylated AURKA protein (Fig. 6c,d). These data provide direct evidence that MYC regulates AURKA abundance and activity and suggest that both are controlled by the PI3K-pathway in breast cancer.
Figure 6. AURKA transcription is regulated by MYC downstream of the PI3K-pathway.
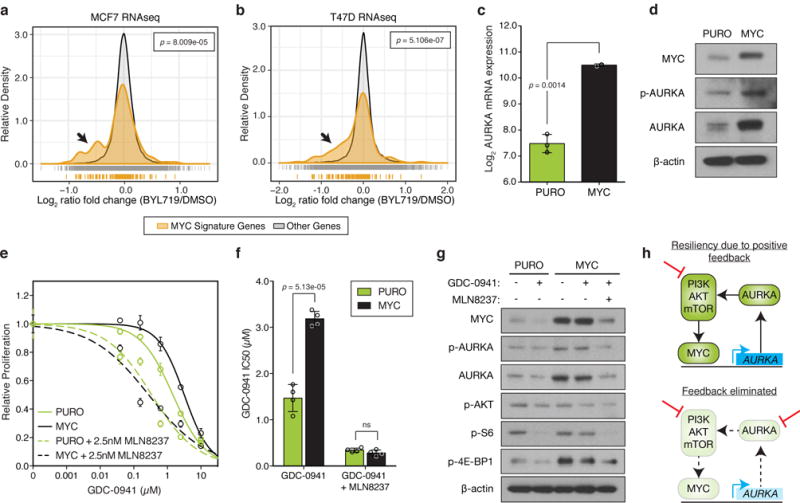
(a,b) Histogram of normalized gene expression of all 150 genes in the MYC gene signature compared to genes not in this signature for (a) MCF7 or (b) T47D cells treated with 1μM BYL719 or DMSO for 24 hours. BYL719 treatment data obtained from Bosch, et al.17 and p values determined by Kolmogorov–Smirnov test. (c) Relative levels of AURKA mRNA in an isogenic pair of control (PURO, n=3 independent samples) or MYC expressing MCF10A cells (n=2 independent samples) measured by RT-PCR. (d) Immunoblot of protein lysates from PURO or MYC cells representative of n=3 independent experiments with similar results (full blots shown in Supplementary Fig. 18a). (e) Proliferation of control or MYC cells in response to GDC-0941 or treated with the combination of 2.5nM MLN8237. Combinations were normalized to MLN8237 alone. Data represents n=4 biologically independent samples. (f) IC50 analysis of dose-response curves shown in (e) from n=4 independent samples. (g) Immunoblot of lysates from control and MYC MCF10A cells treated with 1μM of GDC-0941 or 100nM of MLN8237 for 24 hours. Representative image of n=3 independent experiments with similar results (full blots shown in Supplementary Fig. 18b). (h) Proposed model of positive feedback loop between the PI3K-pathway, MYC and AURKA. In all graphs, error bars are mean ± s.d. and p values calculated using a two-sided t-test, unless otherwise indicated.
Considering AURKA activates AKT (Fig. 5b)31,32, our results suggest a model whereby the PI3K-pathway regulates the abundance of its upstream activator AURKA through the control of MYC. Hence, MYC-driven AURKA signaling may constitute a positive feedback loop that helps to continuously activate the PI3K-pathway, even in the context of single agent drug treatment. In support, we observed that MCF10A-MYC cells were more resistant to GDC-0941 and MK2206 compared to parental cells consistent with previous reports of MYC driving resistance to inhibitors of this pathway (Fig. 6e,f, Supplementary Fig. 19a,b)33–36. Although MYC expressing cells were drug-resistant, they could be re-sensitized to GDC-0941 or MK2206 by the addition of MLN8237 back to approximately the same relative IC50 as parental cells given the combination (Fig. 6e,f, Supplementary Fig. 19a,b), indicating that AURKA is principally responsible for causing the resistance to PI3K inhibition seen as a result of MYC activation in this model.
To test this model, we asked if MYC-driven resistance to PI3K inhibitors is through the maintenance of PI3K-pathway activity and if this is dependent on AURKA. GDC-0941 treatment in MCF10A cells led to a reduction in MYC and AURKA signaling as well as phospho-S6 and phospho-4E-BP1, indicating that MYC and AURKA are regulated by the PI3K-pathway (Fig. 6g). However, constitutive expression of MYC resulted in the maintenance of all of these factors after PI3K inhibition suggesting that MYC also acts upstream of the PI3K-pathway and can maintain its activity. Furthermore, maintenance of mTORC1 signaling by MYC over-expression was reversed by co-inhibition of AURKA thus designating AURKA as the critical link between MYC and activation of the PI3K-pathway in these cells (Fig. 6g). Similar results were observed using the AKT inhibitor MK2206 (Supplementary Fig. 19c). Taken together, our data define a novel circuit whereby the PI3K-pathway regulates the abundance of its own activator through MYC-mediated transcription of AURKA (Fig. 6h).
DISCUSSION
Through an unbiased proteomics approach to assay kinase activity, we measured dynamic changes elicited by therapy as a means to develop novel drug combinations. The systematic measurement of kinome dynamics across a diverse set of cell lines allowed us to map molecular changes associated with resistance to a variety of inhibitors, which is unique from previous approaches limited to a single drug or cell line7,15,37. We found a number of cases where failure to inhibit a particular kinase was associated with drug resistance. Since our proteomic screen included multiple drugs that impinge on distinct oncogenic pathways, we found it surprising that a set of common survival factors were identified. This may be due to the convergence of both the PI3K and MAPK pathways on protein synthesis38,39. Beyond AURKA, we identified that CDK4 suppression was associated with drug sensitivity and that the combination of CDK4 and PI3K-pathway inhibitors was synergistic, consistent with previous work12. Future work may determine if other candidates we identified also act as survival factors and how they might do so.
We show that the expression of AURKA limits the efficacy of PI3K-pathway targeted therapy and thus represents a new vulnerability to enhance therapeutic responses to this class of drugs. Investigating AURKA regulation we found that the reduction in AURKA abundance in drug-sensitive cells appears to be the result of transcriptional control by MYC, which is in turn regulated by the PI3K-pathway. MYC has been shown to regulate AURKA transcription in multiple tumor types40-42. MYC has been associated with resistance to PI3K inhibitors which may be clinically relevant but remains mechanistically ambiguous33–36. Here we show that one potential mechanism of resistance is through MYC-driven AURKA activation resulting in maintenance of the PI3K-pathway in response to PI3K inhibition. Future work may gauge the relative importance of AURKA versus other outputs of MYC in driving resistance to PI3K inhibitors.
Maintenance of AURKA was sufficient to confer drug resistance in a variety of cell lines as evident by the widespread drug synergy we observed. We show that in response to treatment with PI3K-pathway inhibitors Aurora kinase maintains the activation of AKT and drives residual mTOR activity. Co-inhibition of the PI3K-pathway and AURKA with MLN8237 fully blocks this residual mTOR activity resulting in cell death. These findings also highlight the importance of AKT activation through serine 473 as a route of drug resistance. Since a number of kinases have been shown to operate at this site including mTORC225, it remains unclear whether Aurora kinases act on this site directly or indirectly. These studies elaborate a positive feedback loop whereby the PI3K-pathway promotes the expression of AURKA, which in turn activates the pathway via AKT. One feature of such a positive feedback loop is the creation of switch-like outputs resulting in heightened stability and resistance to perturbation43. We postulate that such loops are common and may lead to the resiliency and adaptation that is a hallmark of the PI3K-pathway and a major cause of the challenges in targeting it therapeutically. Delineating such loops may be an important strategy in identifying effective drug combinations. As a case in point, we show that eliminating this positive feedback loop by blocking AURKA renders cells more sensitive to PI3K inhibitors.
Our findings reveal that the combination of Aurora kinase inhibitors and PI3K-pathway inhibitors is synergistic and could be a promising clinical strategy to enhance treatment response in breast cancer. These data are consistent with observations made in other settings44–46. Clinical data of PI3K and mTOR inhibitors have shown only modest benefit in breast cancers, at best resulting in short term disease stabilization in patients23,47. Consistent with these clinical observations, most inhibitors in this class cause only a proliferative arrest in vitro5,21 and it has been proposed that combinations that induce apoptosis may be used to enhance responses48. In contrast to cytostatic combinations with the CDK4/6 inhibitor (i.e. synthetic sickness), we found that combinations with Aurora kinase inhibitors were synergistic and potently induced cell death. As clinical trials testing CDK4/6 inhibitor combinations are ongoing, it remains to be seen the impact this distinction will play on patient responses. These results warrant an expanded analysis of combinations with AURKA inhibitors in additional patient-derived models of breast and other cancer types. Tested as monotherapy, Aurora kinase inhibitors have reached phase 3 clinical trials for lymphoma with manageable toxicities but limited efficacy49. Given that the most common adverse events of PI3K-pathway inhibition are hyperglycemia, rash, and gastrointestinal toxicity, and those of Aurora kinase inhibition are primarily neutropenia, we are encouraged that the non-overlapping toxicity profile between the two agents may be tolerated in patients as they were in our in vivo studies. As single-agent responses to both PI3K-pathway and Aurora kinase inhibitors have been modest, these findings may unlock the full potential of these agents in realizing a clinical benefit.
ONLINE METHODS
Breast cancer cell lines and reagents
BT549 and SKBR3 cells were obtained from the UCSF Cell Culture Facility. BT20, BT474, HCC1428, HCC38, LY2, MCF7, MDAMB231, MDAMB453, T47D, SUM52PE, and ZR75B cell lines were obtained from the American Type Culture Collection (ATCC). Cell lines used for proteomic profiling and molecular analyses were authenticated by STR analysis. Lines were grown according to published protocols50 except for SKBR3 which was cultured using RPMI media supplemented with 10% fetal bovine serum (FBS) and 1% pen/strep. All cell lines tested negative for mycoplasma contamination. Drugs used for cell culture experiments in this study were purchased from Selleck Chemicals (GDC-0941, MK2206, PD0325901, Lapatinib, MLN8237, and LEE011) and LC Laboratories (RAD001).
Multiplex inhibitor bead (MIB) analysis
Multiplexed Inhibitor Bead enrichment and MS analysis (MIBs/MS) were performed as described previously14. In summary, a selection of bait compounds were purchased or synthesized and immobilized on sepharose using standard peptide coupling chemistry. The following compounds were purchased commercially: Bisindolylmaleimide X (Enzo Life Sciences); SB202190, Staurosporine (LC Labs); Purvalanol B (Tocris); Lapatinib, Crizotinib, Dasatinib (Selleckchem). When not commercially available without modification, linkable versions of previously described compounds were synthesized based on prior methods: VI-1683251,52, Akti-4653, PP-hydroxyl54, sorafenib55, and JG-456 with minor adjustments made for synthetic tractability. After initial pilot syntheses and validation, compounds were synthesized by Pharmaron, Inc. Louisville KY. Couplings were performed overnight at room temperature on a rotator. Beads and compounds were mixed in 1:1 Dimethyl formamide: Ethanol with 0.1 M 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide.
After 24-hour treatment with drug or DMSO, cell lysates were diluted in binding buffer with 1 mol/L NaCl and kinase enrichment was performed using gravity chromatography following pre-clearing. After washing, the bound kinases were eluted with SDS followed by extraction/precipitation, tryptic digest and desalting. Liquid chromatography-tandem mass spectrometry (LC/MS-MS) was performed on a Velos Orbitrap (Thermo Scientific) with in-line high-performance liquid chromatography (HPLC) using an EASY-spray column (Thermo Scientific). Peptide identifications were made using ProteinProspector (v5.10.10) and input into Skyline for label-free quantification57.
Peptide quantification data were pre-processed before analysis with MSstats v2.3.358. First, library peptides and peptides that map to non-kinase proteins were removed. Kinase peptide peak area values were log2-transformed and quantile-normalized to correct for variation between replicates. Finally, peptides that mapped to multiple kinases were removed, as well as peptides that were entirely missing in one or more conditions. For each kinase, the log2 ratio of each drug-treated condition to the DMSO control was estimated using the mixed-effects regression model in MSstats.
Drug combination studies
Cell lines were seeded in 384-well assay plates at a density of 1,000 cells/well in a total volume of 40 μL/well, and incubated at 37°C, 5% CO2 overnight. Dose matrices were assembled containing 6-point, 4-fold serial dilutions from the top concentration for each agent on the x- and y-axes. Following 72 hours of drug exposure, proliferation and cell death was measured by staining with Hoescht (Life Technologies) nuclear dye and YO-PRO-1 (Life Technologies), respectively, and analyzed using a Thermo CellInsight High Content microscope. Raw phenotype measurements from each treated well were normalized to the median of vehicle-treated control wells and examined for synergistic effects between both compounds.
To evaluate drug combinations we used a Loewe model of drug additivity and calculated a synergy score. First, we fit a sigmoidal function to each of the single agent responses. Next we calculated the expected inhibition for each combination using the Loewe additivity model20. The synergy score S was calculated as previously defined59 as a positive-gated inhibition-weighted volume over of Loewe additivity:
Where fX and fY are the dilution factors used for compounds X and Y respectively, Idata is the matrix of inhibition data at this dilution factor, and ILoewe is the expected inhibition according to Loewe additivity. Synergy score calculations were also derived using Bliss independence20, based on a model where drugs act independently of each other. CI50 values for equal-dose combinations were calculated as previously defined20:
Where (D)1 and (D)2 are the given doses of the two drugs, and (D50)1 and (D50)2 are the IC50 values for each drug as a single agent.
To determine a cutoff for the synergy score we simulated the distribution of scores generated by an additive drug combination. We generated two hypothetical compounds by sampling random shape parameters for their dose-response functions, and calculated the expected Loewe model of the combination. We then added normally distributed noise to the model with variance estimated from our experimental data and calculated the resulting synergy score. This process was repeated 100,000 times to simulate the distribution of synergy scores for different additive combinations. The 95th percentile of this distribution was 0.91 and so we conservatively identified combinations with S ≥ 1as synergistic.
Western blotting and antibodies
Proteins were extracted using RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM EDTA pH 8.0, 1% NP-40) containing proteinase (Roche) and phosphatase (Roche) inhibitor cocktails. Samples were resolved using 4–12% SDS-PAGE gels (Life Technologies) and transferred to PVDF membranes (Millipore). Membranes were probed overnight on a 4°C shaker with primary antibodies (1:1,000 dilution unless indicated) recognizing the following proteins: p-AKT (Ser473) (9271, Cell Signaling), AKT (4691, Cell Signaling), p-S6 (Ser240/244) (5364, Cell Signaling, 1:20,000), p-4E-BP1 (Thr37/46) (2855, Cell Signaling), p-AURKA (Thr288) (3079, Cell Signaling), AURKA (4718, Cell Signaling), Cleaved PARP (Asp214) (9541, Cell Signaling), BCL2 (2870, Cell Signaling), BAX (2772, Cell Signaling), MYC (ab32072, Abcam), and β-actin (3700, Cell Signaling).
Mouse xenograft studies
All animal studies were conducted in compliance with all relevant ethical regulations set forth by the UCSF Institutional Animal Care and Use Committee (IACUC). 4-week old immunocompromised NOD/SCID female mice were purchased from Taconic Biosciences, and MCF7 cells used for in vivo transplant were obtained from the UCSF Preclinical Therapeutics Core. Xenograft tumors were initiated in the cleared mammary fat pads of mice bearing slow release estrogen pellets (Innovative Research of America) by orthotopic injection of 1e6 MCF7 cells in a 1:1 mixture of serum-free medium and Matrigel (BD Biosciences). When tumors reached ≥ 1 cm in any direction via electronic caliper measurements, mice were randomized into cohort groups and treatment was initiated.
Treatment arms received either vehicle (1:1 mixture of single-agent diluents), RAD001 formulated as a microemulsion (2mg/kg/q; 30% Propylene glycol, 5% Tween 80), MLN8237 (10mg/kg/q; 10% 2-hydroxypropyl-β-cyclodextrin, 1% sodium bicarbonate), or the combination daily, via oral gavage. Animals were monitored daily for evidence of toxicity including weight and skin effects, and changes in tumor size (mm3) through bidirectional measurements of perpendicular diameters using electronic calipers, and calculated as V = 1/2 (length ×width2). Mice were sacrificed after 15 days of treatment, following which tumors were excised and a portion of the tissue fixed in 4% paraformaldehyde. The remaining tumor tissue was flash-frozen in liquid nitrogen.
Immunohistochemical analysis
PFA-fixed tumor samples were paraffin-embedded, and immunohistochemical staining of tissue sections was performed. TUNEL staining was carried out using the ApopTag Peroxidase In situ Apoptosis Detection Kit (Millipore), according to the manufacturer’s instructions (n=15 data points per group; five high-powered (20×) fields analyzed from separate areas of each tumor from 3 mice per experimental group). Stained slides were digitized using the Leica DMi1 Microscope (Leica Microsystems) with a 20× objective. Images were scored as the number of TUNEL-positive cells per captured field, and quantification was performed in a manner that was blinded to treatment group.
Real-time PCR
RNA was isolated according to the manufacturer’s instructions (TRIzol, Life Technologies). One microgram of total RNA from each sample was subjected to first-strand cDNA synthesis according to the manufacturer’s recommendations (Promega). Quantitative PCR was performed on a CFX96 Real-Time PCR detection system with a PrimeTime Gene Expression Master Mix (IDT technology) according to the manufacturer’s protocol. AURKA was amplified with the following primers: 5′-AGTTGGCAAACGCTCTGTCT-3′ (forward primer) and 5′-GTGCCACACATTGTGGTTCT-3′ (reverse primer). RPL13A was used as an endogenous control with the following primers: 5′-CGGATTTGGTCGTATTGG-3′ (forward primer) and 5′-TCCTGGAAGATGGTGATG-3′ (reverse primer). The cycling conditions for AURKA and RPL13A were as follows: one cycle at 95°C for 3 min; 40 cycles of 95°C for 15 s, and 60°C for 60 s. The specificity of the PCR amplification was validated by the presence of a single peak in the melting curve analyses.
Gene Set Enrichment Analyses (GSEA)
Gene set enrichment analysis (GSEA) of hallmark cancer gene signatures in the Molecular Signatures Database (MSigDB v6.0) was performed using GSEA v3.0 software (http://www.broadinstitute.org/gsea/)19 under the following parameters: permutation, phenotype; metric, Signal2Noise; scoring scheme, weighted; and number of permutations, 1,000. Gene sets were considered significantly enriched following a nominal P < 0.05 and FDR < 0.25 cutoff.
Statistical analysis
Data are expressed as means ± s.d., unless otherwise indicated. Statistical analyses were performed using GraphPad Prism 6 (v6.0g) and R (v3.32). Two-tailed Student t tests (with unequal variance) were used in all comparisons unless otherwise noted. P < 0.05 was considered statistically significant throughout the study.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The raw mass spectrometry data is accessible via http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer under the search key: lixlgarvea.
Supplementary Material
Acknowledgments
The authors would like to thank members of the Bandyopadhyay laboratory for helpful discussions and technical assistance. We also thank A. Beardsley, E. Markegard, D. Ruggero and W. Weiss for helpful discussions and reagents. This work was supported in part by NCI U01CA168370 (S.B.), NIGMS R01GM107671 (S.B.), NCI R01CA170447 (A.G.), Prospect Creek Foundation (S.B., A.G.), OHSU Pilot Project Funding (S.B., J.K.), American Cancer Society Postdoctoral Fellowship (J.D.G.), the Gazarian Foundation (A.G.), DOD W81XWH-12-1-0272 and DOD W81XWH-16-1-0603 (A.G.).
Footnotes
AUTHOR CONTRIBUTIONS
H.J.D., J.T.W., K.M.S, J.K., J.D.G., and S.B. contributed towards study conceptualization. H.J.D., J.T.W., and J.D.G. performed data analyses supporting the study. H.J.D. designed and performed the majority of experiments. N.B. assisted with samples for initial MIBs/MS profiling. R.S.L. J.T.W., and J.D.G. provided technical advice and guided the interpretation of mass spectrometry data from MIB/MS profiling. R.C. and O.M. assisted with animal studies, and K.N.S. helped with additional experiments. H.J.D. and S.B. composed the original draft and all authors contributed towards manuscript finalization. S.B. and A.G. supervised the study.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janku F, et al. Assessing PIK3CA and PTEN in Early-Phase Trials with PI3K/AKT/mTOR Inhibitors. Cell Rep. 2014;6:377–387. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol Ther. 2017;175:91–106. doi: 10.1016/j.pharmthera.2017.02.037. [DOI] [PubMed] [Google Scholar]
- 4.O’Reilly KE, et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3:1345–1354. doi: 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah PD, Chandarlapaty S. Resistance to PI3K Pathway Inhibition. In: Dey N, De P, Leyland-Jones B, editors. PI3K-mTOR in Cancer and Cancer Therapy. Springer International Publishing; 2016. pp. 125–147. [DOI] [Google Scholar]
- 7.Duncan JS, et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corcoran RB, et al. TORC1 Suppression Predicts Responsiveness to RAF and MEK Inhibition in BRAF-Mutant Melanoma. Sci Transl Med. 2013;5:196ra98–196ra98. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson GR, et al. PIK3C A mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci Transl Med. 2016;8:369ra175–369ra175. doi: 10.1126/scitranslmed.aae0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boussemart L, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 11.Elkabets M, et al. mTORC1 Inhibition Is Required for Sensitivity to PI3Kp110α Inhibitors in PIK3CA-Mutant Breast Cancer. Sci Transl Med. 2013;5:196ra99–196ra99. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vora SR, et al. CDK 4/6 Inhibitors Sensitize PIK3CA Mutant Breast Cancer to PI3K Inhibitors. Cancer Cell. 2014;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bantscheff M, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 14.Sos ML, et al. Oncogene Mimicry as a Mechanism of Primary Resistance to BRAF Inhibitors. Cell Rep. 2014;8:1037–1048. doi: 10.1016/j.celrep.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stuhlmiller TJ, et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015;11:390–404. doi: 10.1016/j.celrep.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lens SMA, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 17.Bosch A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci Transl Med. 2015;7:283ra51–283ra51. doi: 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leroy C, et al. Activation of IGF1R/p110β/AKT/mTOR confers resistance to α-specific PI3K inhibition. Breast Cancer Res BCR. 2016;18:41. doi: 10.1186/s13058-016-0697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3:e00149. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Golemis EA. Aurora-A kinase (AURKA) in normal and pathological cell growth. Cell Mol Life Sci CMLS. 2013;70:661–687. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piccart M, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2†. Ann Oncol Off J Eur Soc Med Oncol. 2014;25:2357–2362. doi: 10.1093/annonc/mdu456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vadlakonda L, Dash A, Pasupuleti M, Anil Kumar K, Reddanna P. The Paradox of Akt-mTOR Interactions. Front Oncol. 2013;3:165. doi: 10.3389/fonc.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laplante M, Sabatini D. M mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volkmann N, Marassi FM, Newmeyer DD, Hanein D. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 2014;21:206–215. doi: 10.1038/cdd.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene. 1998;17:769. doi: 10.1038/sj.onc.1201990. [DOI] [PubMed] [Google Scholar]
- 29.Sears R, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119:2304–2312. doi: 10.1002/ijc.22154. [DOI] [PubMed] [Google Scholar]
- 32.Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28:866–875. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martins MM, et al. Linking Tumor Mutations to Drug Responses via a Quantitative Chemical–Genetic Interaction Map. Cancer Discov. 2015;5:154–167. doi: 10.1158/2159-8290.CD-14-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muellner MK, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway–dependent and PI3K pathway–independent mechanisms. Nat Med. 2011;17:1116. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nomanbhoy TK, et al. Chemoproteomic Evaluation of Target Engagement by the Cyclin-Dependent Kinase 4 and 6 Inhibitor Palbociclib Correlates with Cancer Cell Response. Biochemistry (Mosc) 2016;55:5434–5441. doi: 10.1021/acs.biochem.6b00629. [DOI] [PubMed] [Google Scholar]
- 38.Roux PP, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.She QB, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.den Hollander J, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu L, et al. Aurora kinase A mediates c-Myc’s oncogenic effects in hepatocellular carcinoma. Mol Carcinog. 2014;54:1467–1479. doi: 10.1002/mc.22223. [DOI] [PubMed] [Google Scholar]
- 42.Zheng F, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. 2016;7:10180. doi: 10.1038/ncomms10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrell JE. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol. 2002;14:140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- 44.Katsha A, et al. Activation of EIF4E by Aurora Kinase A Depicts a Novel Druggable Axis in Everolimus-Resistant Cancer Cells. Clin Cancer Res. 2017;23:3756–3768. doi: 10.1158/1078-0432.CCR-16-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savannah KJB, et al. Dual Targeting of mTOR and Aurora-A Kinase for the Treatment of Uterine Leiomyosarcoma. Clin Cancer Res. 2012;18:4633–4645. doi: 10.1158/1078-0432.CCR-12-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu LL, et al. Inhibition of mTOR Pathway Sensitizes Acute Myeloid Leukemia Cells to Aurora Inhibitors by Suppression of Glycolytic Metabolism. Mol Cancer Res. 2013;11:1326–1336. doi: 10.1158/1541-7786.MCR-13-0172. [DOI] [PubMed] [Google Scholar]
- 47.Baselga J, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017 doi: 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zwang Y, et al. Synergistic interactions with PI3K inhibition that induce apoptosis. eLife. 2017;6:e24523. doi: 10.7554/eLife.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barr PM, et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:2399–2404. doi: 10.1200/JCO.2014.60.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental References
- 50.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barvian M, et al. Pyrido[2,3-d]pyrimidin-7-one Inhibitors of Cyclin-Dependent Kinases. J Med Chem. 2000;43:4606–4616. doi: 10.1021/jm000271k. [DOI] [PubMed] [Google Scholar]
- 52.Daub H, et al. Kinase-Selective Enrichment Enables Quantitative Phosphoproteomics of the Kinome across the Cell Cycle. Mol Cell. 2008;31:438–448. doi: 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Blake JF, et al. Discovery of pyrrolopyrimidine inhibitors of Akt. Bioorg Med Chem Lett. 2010;20:5607–5612. doi: 10.1016/j.bmcl.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka M, et al. An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol. 2005;3:e128. doi: 10.1371/journal.pbio.0030128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bankston D, et al. A Scaleable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer. Org Process Res Dev. 2002;6:777–781. [Google Scholar]
- 56.Statsuk AV, et al. Tuning a Three-Component Reaction For Trapping Kinase Substrate Complexes. J Am Chem Soc. 2008;130:17568–17574. doi: 10.1021/ja807066f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinforma Oxf Engl. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi M, et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 2014;30:2524–2526. doi: 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- 59.Lehár J, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The raw mass spectrometry data is accessible via http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer under the search key: lixlgarvea.