

Abstract
Large mammalian herbivores greatly influence the functioning of grassland ecosystems. Through plant consumption, excreta, and trampling, they modify biodiversity, nutrient cycling, and soil properties. Grazing mammals can also alter soil and rhizosphere bacterial communities, but their effect on the microbiome of other animals in the habitat (i.e., insects) is unknown. Using an experimental field approach and Illumina MiSeq 16S rRNA gene sequencing, we analyzed the influence of cattle grazing on the microbial community of spring webworm caterpillars, Ocnogyna loewii. Our experimental setup included replicated grazed and non-grazed paddocks from which caterpillars were collected twice (first-second and fourth-fifth instar). The caterpillars’ microbiome is composed mostly of Proteobacteria and Firmicutes, and contains a potential symbiont from the genus Carnobacterium (55% of reads). We found that grazing significantly altered the microbiome composition of late instar caterpillars, probably through changes in diet (plant) composition and availability. Furthermore, the microbiome composition of early instar caterpillars significantly differed from late instar caterpillars in 221 OTUs (58 genera). Pseudomonas and Acinetobacter were dominant in early instars, while Carnobacterium and Acinetobacter were dominant in late instars. This study provides new ecological perspectives on the cascading effects mammalian herbivores may have on the microbiome of other animals in their shared habitat.
Introduction
By inducing changes in the environment, animals may consequently alter the composition and activity of microbiomes in their habitat (e.g., [1, 2]). The emergence of modern genomic approaches enable us to explore these cascading effects on microbial communities throughout the food chain [3]. Indeed, it has been shown recently that animals have important cascading effects on the microbiomes of other animals. The presence of salmon carcasses in streams, for example, altered the microbiome of mayflies [4]. In another example, antibiotic treatment of cattle altered the microbiota composition in dung feeding beetles [5].
Large mammalian herbivores are important habitat modifiers (ecosystem engineers) affecting the function, productivity, and diversity of grasslands [6, 7]. Through plant consumption, return of excreta, and trampling, they alter resource allocation, nutrient cycling, and soil physicochemical properties [8]. Hence, herbivore-mediated changes are likely to influence soil microbial diversity and activity, including that of the rhizosphere [9, 10]. While indirect effects of mammalian herbivores on soil and rhizosphere microbial communities are well recognized [8, 11–13], their effect on the bacterial communities of other animals in the habitat is practically unknown.
Mammalian herbivores have strong impacts on insect populations, directly or through plant-mediated mechanisms [14–17]. By modifying the environment, mammalian herbivores might also affect insect-associated microbiomes. Many insect species harbor microbial symbionts that affect their development, and contribute to their nutrition, reproduction, and survival [18]. Numerous factors may influence insect microbial communities, such as life stage, gut structure, or diet [18–21], and they may acquire beneficial bacteria from their environment [22].
The microbiome of different insect classes, including pests and beneficial insects, has been studied extensively (e.g., [23–28]). Yet, little is known about microbial associates of lepidopterans (butterflies and moths). Current knowledge on lepidopteran microbiomes is mostly limited to pest species (e.g., [21, 29–33]). These studies found that lepidopteran bacterial communities are composed mainly of Proteobacteria and Firmicutes, and the most common taxa in the larvae gut is Enterococcus. While no primary obligate symbiont was found in Lepidoptera, secondary intracellular symbionts, such as Wolbachia, have been reported [34–36]. Nonetheless, it is unclear whether disturbances in the natural environment, such as mammalian herbivore grazing, influence the microbial community structure in lepidopteran larvae, which are prevalent in grassland ecosystems.
We investigated the bacterial community of the spring webworm caterpillar (Ocnogyna loewii, Lepidoptera), a common herbivore in Mediterranean-type habitats (particularly grasslands) frequently grazed by cattle. The prevalence of this caterpillar species in grazed grasslands (Berman et al., submitted MS) makes it a convenient model system to study the impacts of mammalian herbivore grazing on insect microbiomes.
Using a replicated field experiment (grazed and non-grazed control sites), the objective of this study was to determine whether cattle grazing influences the microbial community composition of spring webworm caterpillars. In addition, we investigated how the microbial community varies between early- and late instar caterpillars. This study provides new ecological perspectives on how ecosystem engineers may alter the microbiome of other organisms by causing significant changes in their shared habitat.
Methods
Study organism
Caterpillars of the spring webworm feed on a wide variety of plants (polyphagous). They hatch toward the end of winter and the first three instars feed together as a colony inside a common web nest (the gregarious stage). About a month later, when they reach the fourth instar, the caterpillars disperse and feed solitarily (the solitary stage). The webworms pupate in the soil during summer and adults emerge in autumn. Winged adult males locate and mate with wingless females that later lay their eggs under stones or on the soil surface to complete their univoltine life cycle [37].
Experimental setup and sampling protocols
The study was carried out in “Karei Deshe” Experimental Farm, in the eastern Galilee, Israel (Table S1). The farm has a hilly topography, covered by 30% basaltic rocks. The Mediterranean grassland vegetation is dominated by Hordeum bulbosum L., Echinops spp. and Psoralea bituminosa L., and many annual species, such as Avena sterilis L., Bromus spp., Trifolium spp., and Medicago spp. [38]. Our experimental setup included three grazed paddocks (0.55–1.1 cows/ha, 27–35 ha) and three non-grazed paddocks (0.5–5 ha).
Samples of caterpillars and soil were collected twice from the grazed and non-grazed paddocks (Table S1): during the gregarious stage of the caterpillar (hereafter “early stage”); and over a month later, during the solitary stage (hereafter “late stage”, Fig. 1). All samples were collected under sterile conditions to avoid contamination.
Fig. 1.

Experimental and sampling setup. Caterpillars and soil were collected twice from three non-grazed (a–c) and grazed (d–f) paddocks, during the early stage (gregarious caterpillars) and over a month later, during the late stage (solitary caterpillars). Part of the solitary caterpillars were dissected to obtain their midgut (gregarious caterpillars were too small to dissect). Gregarious caterpillars: 5 caterpillars per nest, 5 nests per paddock, total of 30 samples; solitary caterpillars: 10 caterpillars per paddock, total of 60 samples, half were dissected; soil: 5 samples per paddock, total of 30 samples
Caterpillars
During the early stage, five first-second instar caterpillars were collected from five random nests in each paddock (excluding the margins). During the late stage, 10 fourth-fifth instar caterpillars were randomly collected from each paddock. The caterpillars were collected with tweezers, and placed individually in 50 ml sterile tubes (gregarious caterpillars from the same nest were placed together). On the day of collection (and within 6 h), the caterpillars were treated in a laminar flow hood under aseptic conditions. First, they were rinsed (vortexed for 10 s) twice with sterile saline (8.5 g sodium chloride [NaCl] with 1 l distilled water, autoclaved) to dislodge any particles from their surface (sterile blotting paper was used to remove fluids between rinses). Then, each gregarious caterpillar sample (composed of five caterpillars) and half of the solitary caterpillar samples were transferred into 2 ml sterile tubes containing 1.5 ml absolute ethanol. The other half of the solitary caterpillars were dissected to remove their midgut, on a sterilized wax dissection tray, under a stereo-microscope (in order to detect possible gut symbionts). We were unable to remove gregarious caterpillars’ midguts due to their small size. The midgut samples were rinsed in sterile saline 85% once before transferring them to 2 ml sterile tubes containing 1.5 ml absolute ethanol. The samples were kept at −20 °C until DNA extraction.
Soil
In order to determine whether cattle grazing in our study site had a significant impact on the bacterial populations of the habitat, soil samples were collected from both grazed and non-grazed paddocks (during the early and late stages) and the soil microbial population was examined. Soil samples were obtained from bare patches by scraping the upper part of the patch with a spatula. The samples were placed in 2 ml sterile tubes that contained 1.5 ml absolute ethanol and were kept in a cooler box with ice packs until returning to the lab (within 6 h of collection), where they were kept at −20 °C until DNA extraction.
DNA extraction
In order to remove ethanol residues from the caterpillar samples, each sample was rinsed (vortexed for 1 min) twice with sterile saline 85%. The caterpillars were then transferred into 2 ml sterile tubes containing 0.5 ml saline 85%, in which they were pulverized using a pestle. In the gregarious stage, DNA was extracted from five caterpillars per nest, and in the solitary stage, DNA was extracted from a single caterpillar. In order to remove ethanol residues from the midgut and soil samples, the tubes were centrifuged for 15 min at maximum speed, and the ethanol pipetted from the tube.
DNA was extracted from caterpillar, midgut and soil samples using a DNeasy Blood & Tissue kit (Qiagene, Germany) according to the manufacturer’s instructions, with minor modifications: prior to extraction, all samples were incubated while shaking for 10 min at 100 °C. Next, 180 µl enzymatic lysis buffer (20 mM Tris HCl (pH 8), 2 mM sodium EDTA, and 1.2% Triton-X-100) and 20 mg/ml lysozyme (SERVE, Germany) were added to all the samples and they were incubated while shaking for 60 min at 37 °C. DNA extraction then continued according to the manufacturer’s instructions.
Generation of the 16S rRNA gene libraries and illumina MiSeq sequencing
Genomic DNA was PCR-amplified using primers targeting the V4 variable region of the 16S rRNA ribosomal gene. The primers, CS1_515F (ACACTGACGACATGGTTCTACAGTGCCAGCMGCCGCGGTAA) and CS2_806R (TACGGTAGCAGAGACTTGGTCTGGACTACHVGGGTWTCTAAT) [39] were synthesized by Sigma Aldrich (Israel).
PCR amplification was performed in 25 µl reaction volume using the EmeraldAmp MAX HS PCR Master Mix (Takara Bio Inc., Otsu, Shiga, Japan). Primer concentrations were 0.5 ng/µl. A unit of 10–100 ng genomic DNA was added to each PCR reaction volume. PCR conditions were as follows: 95 °C for 5 min, followed by 28 cycles of 30 s at 95 °C; 45 s at 55 °C; and 30 s at 68 °C. A final elongation step of 7 min at 68 °C was included. The amplification was verified by agarose gel electrophoresis and the PCR products were stored at −20 °C. In order to improve the yield of soil samples, a second amplification was done: 1 µl of PCR product from the first stage was used as a template and all other reaction conditions were maintained.
MiSeq sequencing was performed at the DNA Services Facility, University of Illinois, Chicago. The sequencing protocol is described in ref. [40]. The procedure included a second PCR amplification where each sample received a separate primer set (obtained from the Access Array Barcode Library for Illumina), with a unique 10-base barcode (Fluidigm, South San Francisco, CA; Item# 100-4876). Pooled, diluted libraries were sequenced with Illumina MiSeq 600-cycle sequencing kit version 3, and analyzed with Casava 1.8 (pipeline 1.8). Reads were 200 nucleotides in length (paired end, 2 × 200). PhiX DNA was used as a spike-in control. The raw sequence data reported in this paper are available at the NCBI database under BioProject accession number PRJNA395145.
Sequence analysis
In total, 145 biological samples were analyzed (including replicates). Raw paired-end reads were first merged into contigs using Pear [41]. Only successfully merged reads were further processed. Contigs containing putative traces of PhiX genome (NC_001422), and NCBI Univec sequences, were removed using Fastq-screen. Sequences were further quality-trimmed using Trimmomatic [42], ensuring >27.0 phred33 quality scores on a sliding window of 15 bp. Primers were removed using cutaddapt. The merged and quality-filtered sequences were further cleaned from chimeras using the tool vsearch (https://github.com/torognes/vsearch), with the options “--uchime_ref --minh 0.2 --mindiv 1.5”. This chimera-filtering procedure was conducted using the “16S RDP trainset15 09-2015” database.
Following the quality control steps, sequences were clustered into operational taxonomic units (OTUs) at a sequence similarity level of 97% using the open reference clustering tool pick_open_reference_otus.py, in Qiime [43]. The green genes database “gg_13_8_otus” (http://greengenes.lbl.gov) was used for this procedure. Sequences that failed to match were de novo-clustered. The Qiime OTU table output was further cleaned from OTUs of low confidence (<0.01% of the reads). Next, sequences of bacterial genomic sources were selected (excluding sequences of Archaea, mitochondria, and chloroplasts). Rare OTUs (singletons and doubletons) were removed to reduce the potential data analysis “noise”. De novo-assembled OTUs were taxonomically classified using the assign_taxonomy.py tool in Qiime. Replicate samples were merged and an OTU abundance table was produced (Table S2). Representative sequences for each OTU were obtained using the pick_rep_set.py tool in Qiime.
Statistical analysis
Sample rarefaction analysis was performed using the iNEXT R package [44] (Fig. S1). The data were then subsampled to 9836 sequences per sample, in order to avoid sample-to-sample bias due to variable sequencing depth (different number of reads per sample), resulting in 115 final samples. Diversity analysis was conducted using the tool core_diversity_analyses.py. The entire Qiime analysis followed the microbiome-helper recommendations.
Shannon H′ and Chao1 indices were calculated with PAST [45]. Comparison of alpha-diversity parameters was done with JMP. Due to unequal variance (Bartlett test), a non-parametric approach was used to statistically test differences between means and each sampling group was analyzed separately. The sampling groups were compared with the Kruskal–Wallis test (P < 0.05) and the Dunn method for joint ranking was used for post hoc multiple comparisons.
Similarity between bacterial communities was analyzed by nonmetric multidimensional scaling analysis (NMDS), based on Bray-Curtis similarity matrix using Primer v7 software (http://www.primer-e.com). In order to test whether bacterial community profiles differed between sampling groups, an analysis of similarity (ANOSIM) was done for all groups (P < 0.05), with post hoc pair-wise comparisons and Bonferroni corrections.
When significant differences were found between sampling groups, the Wilcoxon exact test was applied to check which of the OTUs within each sampling group contributed to the difference. This was done using vegan R package (version 2.4-3; [46]). The Benjamini–Hochberg procedure was applied for false discovery rate error correction (FDR < 0.05). Comparisons between the sampling groups at the phyla level were done using the Wilcoxon exact test for two inner group comparisons, since the data did not follow a normal distribution (using IBM SPSS software v.20).
Hierarchical clustering trees were prepared using the Ward’s method and plotted. The Fisher exact test was then applied to test whether sample composition in each branch was random (P < 0.05, using vegan R package).
Results
In all, 4 010 238 quality sequences were obtained. Subsampling according to the smallest sample (9836 sequences) resulted in 3 917 851 sequences (from 115 biological samples), which were classified into 7100 OTUs at 97% sequence similarity level across all samples, with an average of 1101 OTUs per sample.
Although each sample had at least 9836 sequences, the rarefaction curves did not reach an asymptotic level (Fig. S1), suggesting that our sampling effort was not sufficient to obtain a full estimate of OTU richness. Nevertheless, the rarefaction curves of caterpillars (gregarious, solitary, and midgut) seemed to be closer to an asymptotic level compared to soil.
The effects of grazing on the bacterial community composition of spring webworms
The similarity between the bacterial community composition of caterpillars (gregarious solitary and midgut, Fig. 2a) and soil (Fig. 2b) from grazed and non-grazed paddocks was evaluated using a NMDS clustering analysis, and tested for significance using an ANOSIM. The bacterial communities of gregarious and solitary caterpillars were clearly distinct from one another (ANOSIM: R = 0.623, P < 0.001, Fig. 2a). Within the solitary caterpillars group, no significant differences were found between the microbiota of whole caterpillars and their midguts.
Fig. 2.

Similarity between bacterial communities. a Caterpillars (gregarious, solitary and midgut). b Soil. A total of 3.9 million partial 16S rRNA gene sequences from 115 samples were clustered into OTUs at 97% sequence similarity threshold and resulting OTU abundance tables were analyzed. Plots show nonmetric multidimensional scaling (NMDS) analysis, based on Bray-Curtis dissimilarity matrix (stress = 0.16 for caterpillars and stress = 0.14 for soil)
While the bacterial community of soil significantly differed between grazed and non-grazed paddocks (ANOSIM: R = 0.048, P = 0.027), it was similar between soil samples collected during the early and late caterpillar instars (ANOSIM: R = 0.005, P = 0.319, Fig. 2b). Thus, cattle grazing significantly affected the soil microbiome.
The bacterial community of solitary caterpillars was significantly affected by grazing (ANOSIM: R = 0.384, P < 0.001). This effect was not evident in gregarious caterpillars or midgut samples (gregarious caterpillars: ANOSIM: R = 0.035, P = 0.223; midgut: ANOSIM: R = 0.024, P = 0.285). In order to verify these results, we performed a Fisher exact test to examine the homogeneity of clusters (hierarchal clustering) for gregarious (Fig. S2a) or solitary (Fig. S2b) caterpillar samples. This analysis also revealed that grazing significantly affected solitary but not gregarious caterpillars (gregarious caterpillars: Fisher’s exact test: P = 0.4; solitary caterpillars: Fisher’s exact test: P < 0.001). Hence, solitary caterpillars from non-grazed paddocks host a different bacterial community than those from grazed paddocks.
Thirty OTUs from eight genera (Table S3) contributed to the difference between the bacterial communities of solitary caterpillars from grazed and non-grazed paddocks (Fig. 3). Acinetobacter and Arthrobacter were the most abundant of these genera.
Fig. 3.

The relative abundance of the eight genera that contributed to the difference between solitary caterpillars from grazed and non-grazed paddocks. a Non-grazed paddocks. b Grazed paddocks. Only the OTUs that contributed to the difference (P < 0.05) were included within each genera. (Note that the scale of the X-axis is different for each graph). Bars indicate means ± SE
According to Chao1 and Shannon H′ indices (Table S4), richness and diversity values were similar between gregarious and solitary caterpillars (gregarious caterpillars from grazed paddocks had higher richness values compared to the remaining caterpillar groups). Midgut values were similar to those observed in whole-body caterpillars. Soil displayed similar diversity and richness values between grazed and non-grazed paddocks and early and late stages. Overall, the indices values were higher in soil than in caterpillars.
The bacterial community composition of early and late instar caterpillars
Collection of samples from the early and late caterpillar instars enabled us to analyze changes that might occur in the caterpillar microbiome between larval stages. Overall, 29 phyla were detected in the caterpillars. While Proteobacteria was the most abundant phylum in gregarious caterpillars, its relative abundance decreased in the later instar and Firmicutes became the most abundant phylum in the solitary caterpillars’ midgut (Fig. 4).
Fig. 4.
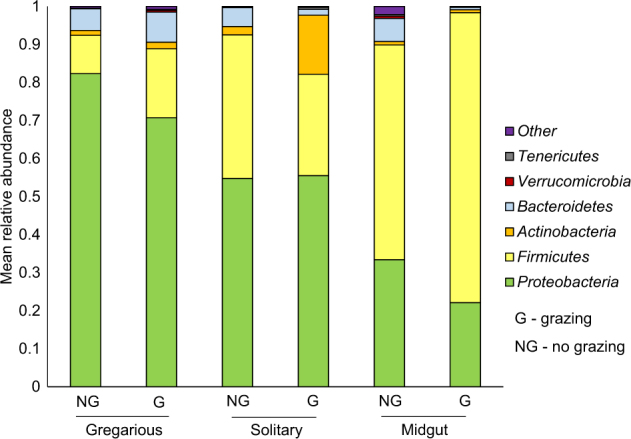
Mean phyla relative abundances among the different caterpillar groups. The caterpillars (gregarious, solitary and midgut) are divided according to grazed and non-grazed paddocks. Overall, 29 phyla were detected and the 6 most abundant phyla (over 10% of reads) across all groups are presented (less-abundant phyla were grouped as “other”)
In early instar gregarious caterpillars, the relative abundance of Tenericutes was significantly higher in grazed paddocks (0.2% compared to 0.02% in non-grazed paddocks, Wilcoxon signed-rank test: Z = −2.135, P = 0.033), whereas in the midgut, this phylum was higher in non-grazed paddocks (0.4% compared to 0.003% in grazed paddocks, Wilcoxon signed-rank test, Z = −2.226, P = 0.026). In solitary caterpillars, Bacteroidetes was significantly higher in non-grazed paddocks (5% compared to 1.5% in grazed paddocks, Wilcoxon signed-rank test, Z = −2.56, P = 0.01) and Actinobacteria was higher in grazed paddocks (15% compared to 2% in grazed paddocks, Wilcoxon signed-rank test: Z = −3.103, P = 0.002).
A total of 221 OTUs, from 58 genera, contributed to the difference in the bacterial community between gregarious and solitary caterpillars (Table S5). The 22 most conspicuous genera (with P value of <0.001) are presented in Fig. 5.
Fig. 5.

The relative abundance of the 22 most conspicuous genera that contributed to the difference between gregarious and solitary caterpillars. a Gregarious caterpillars. b Solitary caterpillars. A total of 222 OTUs, from 58 genera, contributed to the difference in the bacterial community between gregarious and solitary caterpillars. Bars indicate means ± SE
In order to investigate which genera were dominant among the caterpillar groups (and detect possible symbionts), those with the highest relative abundances were examined. Twelve genera displayed a mean relative abundance of over 1% across all samples. The six most interesting genera (with potential biological significances) are presented in Fig. 6. Acinetobacter, Pseudomonas, Carnobacterium, and Erwinia contributed to the significant differences between gregarious and solitary caterpillars (Fig. 5). Acinetobacter and Arthrobacter contributed to the significant differences between solitary caterpillars from grazed and non-grazed paddocks (Fig. 3), yet these genera were hardly apparent in the midgut of solitary caterpillars (Fig. 6). Most of the reads belonged on average to Pseudomonas (18%) and Acinetobacter (14%) in gregarious caterpillars, to Carnobacterium (24%) and Acinetobacter (16%) in solitary caterpillars, and to Carnobacterium (55%) in midgut samples (Fig. 6).
Fig. 6.
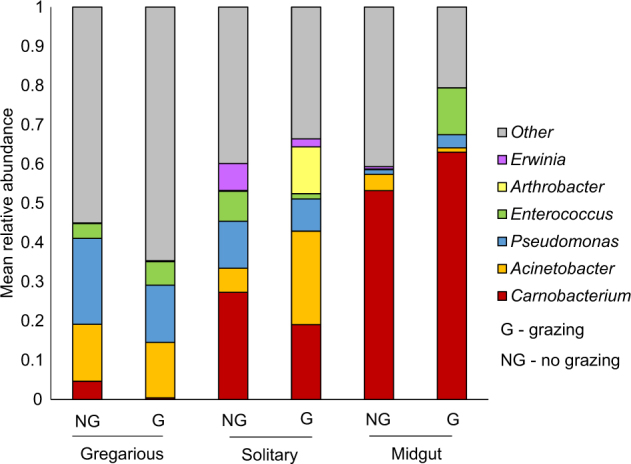
Mean relative abundance of six prominent genera among the different caterpillar groups. The caterpillars (gregarious, solitary and midgut) are divided according to grazed and non-grazed paddocks. The presented genera displayed a mean relative abundance of over 1% across all samples (less-abundant genera were grouped as “other”)
Interestingly, Buchnera (a primary endosymbiont of aphids) displayed a high relative abundance in the midgut of three solitary caterpillars (13, 56, and 70% of reads in these caterpillars, Table S6), collected from both grazed and non-grazed paddocks. The Buchnera OTUs shared 95–97% similarity with Buchnera species found in the aphid Myzus persica.
Carnobacterium
The most abundant genus in solitary caterpillars, especially in the midgut (over 55% of reads on average), was Carnobacterium (Fig. 6). This, together with the fact that Carnobacterium was recently detected as a gut symbiont in Plutella xylostella [29, 47], suggests it might also function as a gut symbiont in spring webworm caterpillars. Overall, 81 OTUs were identified as Carnobacterium in the caterpillars. No differences were found among the relative abundances of the dominant Carnobacterium OTUs in solitary caterpillars (and their midguts) from grazed and non-grazed paddocks (P > 0.05). Furthermore, the Carnobacterium OTU composition in solitary caterpillars (and their midguts) was similar between grazed and non-grazed paddocks.
In order to assess the phylogenetic position of these OTUs, a maximum likelihood dendrogram was calculated using the partial 16S rRNA gene sequence of nine dominant OTUs (over 50% of reads across all caterpillar samples), selected reference sequences of described Carnobacterium species (including the species identified in Lepidoptera) and reference sequences of other Lactobacillales (including Enterococcus found in Lepidoptera, Fig. 7). Based on the maximum likelihood tree, the partial 16S rRNA gene sequence data did not resolve between different Carnobacteirum at the species level. However, the analysis did suggest that Carnobacteiurm OTUs originating from the caterpillars, form a separate clade. The most abundant OTU (#949789, 390 970 reads) shared 96–98% similarity with other species of the genus Carnobacterium (Fig. 7).
Fig. 7.

A maximum likelihood phylogenetic tree, showing the nearest neighbors of the most abundant Carnobacterium OTU sequences. The nine most abundant OTUs (amounting to 55% of reads on average), representative closely related Carnobacterium and Enterococcus species, were analyzed. Lactobacillus acetotolerans was used as an outgroup. Strain and accessions follow the species names. Bootstrap values (as percent of 1000 replicates) are indicated at branching nodes. Bars represent substitution rate per nucleotide position. The most abundant OUTs found in caterpillars are marked in boldface. *Gut isolates of the diamondback moth (Plutella xylostella). **Midgut isolate of Choristoneura fumiferana
Discussion
It is well known that mammalian herbivores have great impacts on the diversity, distribution, and fitness of plant communities and other herbivores, including insects [15, 48]. Mammalian and insect herbivores often interact directly [49, 50]. Here we demonstrate for the first time that mammalian herbivores do not only affect the insects themselves but also alter the insect-associated microbiome.
The effects of grazing on the bacterial community composition of spring webworms
Gregarious caterpillars (early instar) began their life with a similar microbiome composition, regardless of their location (grazed or non-grazed paddocks). Yet, solitary caterpillars (late instar) from non-grazed paddocks hosted a different microbiome to those from grazed paddocks (Fig. S2), suggesting that the microbiome composition is modified by time and location. During the gregarious stage, the caterpillars feed together in a web nest (in a small area), with limited exposure to the external environment. During the solitary stage, however, the caterpillars exit the common web shelter and walk freely across the habitat while feeding on a variety of plant species [51]. Consequently, solitary caterpillars are exposed to more heterogeneous vegetation bacteria (and soil bacteria) in the environment than gregarious caterpillars. Although gregarious caterpillars were collected shortly after they hatched, differences between their microbiomes in grazed and non-grazed paddocks, if existent, were not yet detectable. Once the caterpillars reached the late instar, the accumulated differences between the microbiomes became eminent.
While whole-body solitary caterpillars from grazed and non-grazed paddocks hosted distinct microbiomes, the bacterial community within their midguts was quite similar, implying that the difference in the microbiomes stems from other body parts, such as the hindgut or foregut, as observed in other insects [52]. This was further supported by the fact that Acinetobacter and Arthrobacter, which were responsible for the difference between the microbiomes of solitary caterpillars from grazed and non-grazed paddocks, were hardly apparent in their midgut (Fig. 6).
The distinct bacterial communities between caterpillars from grazed and non-grazed paddocks is likely a result of the different food plants available to them. Mammalian herbivores greatly influence the composition of vegetation [53]. Similarly, a long-term study carried out in the “Karei Deshe” farm showed that cattle grazing altered the vegetation composition by reducing the relative cover of tall annual grasses, leading to an increased cover of other plant species, such as annual and perennial thistles, forbs, legumes, and short grasses [54]. Cattle grazing also maintained younger plants and induced re-growth of existing plants [55]. These effects were most prominent at the peak of the growing season (spring), when spring webworm caterpillars thrive.
Diet is known to affect the composition of insect microbiomes by selecting for bacteria that can metabolize the ingested food [20, 56]. For example, different diet contents caused a shift in the gut microbial communities of omnivorous cockroaches and of two lepidopteran larvae, Spodoptera littoralis and Helicoverpa armigera [19, 57]. Furthermore, bacteria ingested with the food (such as those present on the phyllosphere) may later alter the gut bacteria composition [58]. We were unable to examine whether the plant microbiome affected the bacterial community composition of the caterpillars due to the highly polyphagous nature of the spring webworm and the large variety of plant species in our study site (over 150 species during spring [38]). Although different diets are likely accountable for the distinct microbiomes between solitary caterpillars from grazed and non-grazed areas, other factors caused by cattle grazing, such as changes in vegetation heterogeneity, soil compaction, and predator assemblages (e.g., [59–61]), might also contribute to the shift in the caterpillar microbiome composition.
Our study showed that the soil microbiome varied significantly between grazed and non-grazed paddocks (Fig. 2b), hence, cattle grazing in our study site had a significant impact on the bacterial communities within it. By physically contacting soil and cattle feces while foraging in grazed paddocks (TS Berman, personal observations), late instar caterpillars may possibly ingest fecal and soil bacteria through contaminated food. Lepidopteran larvae have an extremely alkaline gut, which mostly eliminates bacteria ingested from the environment [62], yet if certain bacterial species are able to survive the extreme conditions of the gut, they may colonize it [63].
While studies regarding how ecosystem engineers may affect the microbial communities of other species are scarce, a few reports indicate that they do have such an impact [3–5]. Here we demonstrate that cattle have cascading effects on the microbiome of spring webworm caterpillars that share their habitat.
The bacterial community composition of spring webworms
Overall, the caterpillars displayed a low phylum-level diversity compared to termites, ants, and beetles [20, 64–66]. Proteobacteria and Firmicutes were the most dominant phyla (Fig. 4), as observed in other lepidopterans [29, 30, 32, 67]. Firmicutes became more abundant than Proteobacteria in the later instar, and was most prevalent in the solitary caterpillars’ midgut. A similar shift in microbiome composition was also observed in the moth Spodoptera littoralis [30, 68], and was associated with the ability to efficiently utilize food during development.
The different microbiome composition between gregarious and solitary caterpillars may be attributed to the general changes that occur during insect development, such as molting and growth. During molting, bacteria associated with the foregut and hindgut are removed as the cuticle lining is shed, leading to changes in the bacterial community [69]. As the caterpillars grow, less oxygen can penetrate the gut wall and it becomes anoxic [57]. Anoxic conditions favor the development of facultative anaerobic bacteria, like Carnobacterium and Enteroccocus (Fig. 6). Furthermore, bacteria ingested together with the plant (phyllosphere) can affect the gut bacteria composition [58]. As described above, solitary caterpillars disperse and feed on more plant species (and plant parts) than gregarious caterpillars, which may explain part of the difference in their microbiome composition.
Most of the dominant genera identified in the caterpillars in this study (Fig. 6) are known from other insects, including Lepidoptera (e.g., [30, 70, 71]). Enterococcus, which was abundant in all our caterpillar samples (Fig. 6), is a common gut bacteria in lepidopteran larvae [21, 31, 57]. Enterococcus is involved in the metabolism of nutrients and prevention of pathogenesis in lepidopterans [30, 72]. Interestingly, Buchnera, a primary endosymbiont of aphids [73], was quite abundant in the midgut of a few solitary caterpillars. It is most likely that these caterpillars incidentally consumed aphids while feeding on the plants. Our Buchnera sequences were highly similar to Buchnera found in Myzus persica (see also ref. [74]).
The most conspicuous genus in solitary caterpillars, especially in the midgut, was Carnobacterium (Fig. 6), which is a facultative anaerobic bacteria similar to Enterococcus [75]. C. maltaromaticum was recently identified as a gut symbiont of the moth P. xylostella [29, 47]. This bacteria encoded a series of enzymes involved in digestion, production of amino acids, and detoxification of plant defense compounds [29]. Comparisons of the dominant Carnobacterium OTUs to other reference sequences (Fig. 7) suggest that spring webworm caterpillars harbor novel species within the Carnobacterium genus and possibly even a novel genus within the order Lactobacillales. Lactobacillales (phylum Firmicutes) are widespread in the environment and many species within this order are associated with insects (e.g., [76–78]), including Lepidoptera (e.g., [21, 31, 57]). The prevalence of Carnobacterium in the caterpillars’ midgut, its role as a gut symbiont in other lepidopteran species, and the association of Lactobacillales species with insect symbionts indicate that Carnobacterium may function as a gut symbiont in spring webworm caterpillars.
Conclusion
Using an experimental field approach, we show for the first time that cattle grazing influences the microbial community composition of spring webworm caterpillars in an age-dependent manner. Furthermore, we identified a potential gut symbiont in spring webworm caterpillars from the genera Carnobacterium. It seems that by changing the composition and availability of plants, the cattle altered the caterpillars’ diet, ultimately influencing their microbiome. Overall, the shift detected in the bacterial community of caterpillars from grazed paddocks offers insights into how the modification of plant communities (insect diet) by mammalian herbivores may indirectly influence bacterial community dynamics. It will be highly interesting to further investigate the underlying mechanisms by which mammalian herbivores (and other ecosystem engineers) affect the microbiomes of animals throughout the food chain.
Electronic supplementary material
Acknowledgements
We thank the staff of ‘Karei Deshe’ for their valuable support. We thank Amir Viner, Daniel Schlesinger, Natalie Amar and Achiad Sade for their technical assistance. We also thank Dr. Assaf Malik from the Bioinformatics Unit at the University of Haifa for his help. We thank the two anonymous reviewers for their helpful comments. Finally, we would like to thank Dr. Elah Pick and Dr. Einat Zchori-Fein for fruitful discussions.
Funding:
This research was supported by The Israel Science Foundation (ISF, grant No. 248/17) and by the German Research Foundation (DFG, the Deutsche Forschungsgemeinschaft) grant No. GZ: HO 930/5-2.
Author contributions:
Conceived and designed the experiments: TSB, MH, and MI. Performed the experiments: TSB with technical advice from SL-S. Analyzed the data and prepared the figures: TSB, SL-S, and ML. Wrote the paper: TSB, MH, and MI. Reviewed and commented on the paper: ML. Contributed reagents/materials: MH and MI. All authors read and approved the chapter.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0102-4) contains supplementary material, which is available to authorized users.
References
- 1.Eisenhauer N, Schlaghamerský J, Reich PB, Frelich LE. The wave towards a new steady state: effects of earthworm invasion on soil microbial functions. Biol Invasions. 2011;13:2191–6. doi: 10.1007/s10530-011-0053-4. [DOI] [Google Scholar]
- 2.Fukami T, Wardle DA, Bellingham PJ, Mulder CPH, Towns DR, Yeates GW, et al. Above- and below-ground impacts of introduced predators in seabird-dominated island ecosystems. Ecol Lett. 2006;9:1299–307. doi: 10.1111/j.1461-0248.2006.00983.x. [DOI] [PubMed] [Google Scholar]
- 3.McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, et al. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci. 2013;110:3229–36. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pechal JL, Benbow ME. Microbial ecology of the salmon necrobiome: evidence salmon carrion decomposition influences aquatic and terrestrial insect microbiomes. Environ Microbiol. 2016;18:1511–22. doi: 10.1111/1462-2920.13187. [DOI] [PubMed] [Google Scholar]
- 5.Hammer TJ, Fierer N, Hardwick B, Simojoki A, Slade E, Taponen J, et al. Treating cattle with antibiotics affects greenhouse gas emissions, and microbiota in dung and dung beetles. Proc R Soc B. 2016;283:20160150. doi: 10.1098/rspb.2016.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawley MJ. Herbivory: the dynamics of animal-plant interactions. Oxford: Blackwell Scientific Publications; 1983. [Google Scholar]
- 7.McNaughton SJ, Oesterheld M, Frank DA, Williams KJ. Ecosystem-level patterns of primary productivity and herbivory in terrestrial habitats. Nature. 1989;341:142–4. doi: 10.1038/341142a0. [DOI] [PubMed] [Google Scholar]
- 8.Harrison KA, Bardgett R. Impacts of grazing and browsing by large herbivores on soils and soil biological properties. In: Gordon IJ, Prins HHT, editors. The ecology of browsing and grazing. Springer; Berlin Heidelberg, 2008; p. 201–16.
- 9.Hamilton EW, Frank DA, Hinchey PM, Murray TR. Defoliation induces root exudation and triggers positive rhizospheric feedbacks in a temperate grassland. Soil Biol Biochem. 2008;40:2865–73. doi: 10.1016/j.soilbio.2008.08.007. [DOI] [Google Scholar]
- 10.Olsen YS, Dausse A, Garbutt A, Ford H, Thomas DN, Jones DL. Cattle grazing drives nitrogen and carbon cycling in a temperate salt marsh. Soil Biol Biochem. 2011;43:531–41. doi: 10.1016/j.soilbio.2010.11.018. [DOI] [Google Scholar]
- 11.Liu T, Nan Z, Hou F. Grazing intensity effects on soil nitrogen mineralization in semi-arid grassland on the Loess Plateau of northern China. Nutr Cycl Agroecosystems. 2011;91:67–75. doi: 10.1007/s10705-011-9445-1. [DOI] [Google Scholar]
- 12.Shan Y, Chen D, Guan X, Zheng S, Chen H, Wang M, et al. Seasonally dependent impacts of grazing on soil nitrogen mineralization and linkages to ecosystem functioning in inner Mongolia grassland. Soil Biol Biochem. 2011;43:1943–54. doi: 10.1016/j.soilbio.2011.06.002. [DOI] [Google Scholar]
- 13.Zhou X, Wang J, Hao Y, Wang Y. Intermediate grazing intensities by sheep increase soil bacterial diversities in an inner Mongolian steppe. Biol Fertil Soils. 2010;46:817–24. doi: 10.1007/s00374-010-0487-3. [DOI] [Google Scholar]
- 14.Gish M, Ben-Ari M, Inbar M. Direct consumptive interactions between mammalian herbivores and plant-dwelling invertebrates: prevalence, significance, and prospectus. Oecologia. 2017;183:1–6. doi: 10.1007/s00442-016-3775-2. [DOI] [PubMed] [Google Scholar]
- 15.van Klink R, van der Plas F, van Noordwijk CGE, Wallisdevries MF, Olff H. Effects of large herbivores on grassland arthropod diversity. Biol Rev. 2015;90:347–66. doi: 10.1111/brv.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohgushi T. Indirect interaction webs: herbivore-induced effects through trait change in plants. Annu Rev Ecol Evol Syst. 2005;36:81–105. doi: 10.1146/annurev.ecolsys.36.091704.175523. [DOI] [Google Scholar]
- 17.Stewart AJA. The impact of deer on lowland woodland invertebrates: a review of the evidence and priorities for future research. Forestry. 2001;74:259–70. doi: 10.1093/forestry/74.3.259. [DOI] [Google Scholar]
- 18.Dillon RJ, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol. 2004;49:71–92. doi: 10.1146/annurev.ento.49.061802.123416. [DOI] [PubMed] [Google Scholar]
- 19.Pérez-Cobas AE, Maiques E, Angelova A, Carrasco P, Moya A, Latorre A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol Ecol. 2015;91:1–14. doi: 10.1093/femsec/fiv022. [DOI] [PubMed] [Google Scholar]
- 20.Yun JH, Roh SW, Whon TW, Jung MJ, Kim MS, Park DS, et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microbiol. 2014;80:5254–64. doi: 10.1128/AEM.01226-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broderick NA, Raffa KF, Goodman RM, Handelsman J. Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl Environ Microbiol. 2004;70:293–300. doi: 10.1128/AEM.70.1.293-300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kikuchi Y, Hosokawa T, Fukatsu T. Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl Environ Microbiol. 2007;73:4308–16. doi: 10.1128/AEM.00067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boucias DG, Cai Y, Sun Y, Lietze VU, Sen R, Raychoudhury R, et al. The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol Ecol. 2013;22:1836–53. doi: 10.1111/mec.12230. [DOI] [PubMed] [Google Scholar]
- 24.Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors. 2013;6:146. doi: 10.1186/1756-3305-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engel P, Martinson VG, Moran NA. Functional diversity within the simple gut microbiota of the honey bee. Proc Natl Acad Sci USA. 2012;109:11002–7. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Köhler T, Dietrich C, Scheffrahn RH, Brune A. High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.) Appl Environ Microbiol. 2012;78:4691–701. doi: 10.1128/AEM.00683-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulsen M, Sapountzis P. Behind every great ant, there is a great gut. Mol Ecol. 2012;21:2054–7. doi: 10.1111/j.1365-294X.2012.05510.x. [DOI] [PubMed] [Google Scholar]
- 28.Mohr KI, Tebbe CC. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field. Environ Microbiol. 2006;8:258–72. doi: 10.1111/j.1462-2920.2005.00893.x. [DOI] [PubMed] [Google Scholar]
- 29.Xia X, Gurr GM, Vasseur L, Zheng D, Zhong H, Qin B, et al. Metagenomic sequencing of Diamondback moth gut microbiome unveils key holobiont adaptations for herbivory. Front Microbiol. 2017;8:1–12. doi: 10.3389/fmicb.2017.00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B, Teh BS, Sun C, Hu S, Lu X, Boland W, et al. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci Rep. 2016;6:29505. doi: 10.1038/srep29505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston PR, Rolff J. Host and symbiont jointly control gut microbiota during complete metamorphosis. PLoS Pathog. 2015;11:1–11. doi: 10.1371/journal.ppat.1005246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammer TJ, McMillan WO, Fierer N. Metamorphosis of a butterfly-associated bacterial community. PLoS ONE. 2014;9:e86995. doi: 10.1371/journal.pone.0086995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson CJ, Schloss P, Ramos Y, Raffa K, Handelsman J. Robustness of the bacterial community in the cabbage white butterfly larval midgut. Microb Ecol. 2010;59:199–211. doi: 10.1007/s00248-009-9595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dyson EA, Kamath MK, Hurst GDD. Wolbachia infection associated with all-female broods in Hypolimnas bolina (Lepidoptera: Nymphalidae): evidence for horizontal transmission of a butterfly male killer. Heredity (Edinb) 2002;88:166–71. doi: 10.1038/sj.hdy.6800021. [DOI] [PubMed] [Google Scholar]
- 35.Hiroki M, Kato Y, Kamito T, Miura K. Feminization of genetic males by a symbiotic bacterium in a butterfly, Eurema hecabe (Lepidoptera: Pieridae) Naturwissenschaften. 2002;89:167–70. doi: 10.1007/s00114-002-0303-5. [DOI] [PubMed] [Google Scholar]
- 36.Tagami Y, Miura K. Distribution and prevalence of Wolbachiain Japanese populations of Lepidoptera. Insect Mol Biol. 2004;13:359–64. doi: 10.1111/j.0962-1075.2004.00492.x. [DOI] [PubMed] [Google Scholar]
- 37.Swailem SM, Amin AH. On the biology of the spring webworm Ocnogyna loewii Z. (Arctiidae: Lepidoptera) Mesop J Agric. 1979;14:183–95. [Google Scholar]
- 38.Sternberg M, Gutman M, Perevolotsky A, Ungar ED, Kigel J. Vegetation response to grazing management in a Mediterranean herbaceous community: a functional group approach. J Appl Ecol. 2000;37:224–37. doi: 10.1046/j.1365-2664.2000.00491.x. [DOI] [Google Scholar]
- 39.Caporaso JG, Lauber CL, Walters WA, Berg-lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aizenberg-Gershtein Y, Izhaki I, Santhanam R, Kumar P, Baldwin IT, Halpern M. Pyridine-type alkaloid composition affects bacterial community composition of floral nectar. Sci Rep. 2015;5:11536. doi: 10.1038/srep11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–20. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high- throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh TC, Ma KH, Chao A. iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers) Methods Ecol Evol. 2016;7:1451–6. doi: 10.1111/2041-210X.12613. [DOI] [Google Scholar]
- 45.Hammer Oslash, Harper DAT, Ryan PD. Paleontological statistics software: Package for education and data analysis. Palaeontol Electron. 2001;4:1–9. [Google Scholar]
- 46.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. vegan: community ecology package. R package version 2.0-10; 2015. R Packag ver 24–3; 2013. Retrieved from: http://vegan.r-forge.r-project.org.
- 47.Lin XL, Kang ZW, Pan QJ, Liu TX. Evaluation of five antibiotics on larval gut bacterial diversity of Plutella xylostella (Lepidoptera: Plutellidae) Insect Sci. 2015;22:619–28. doi: 10.1111/1744-7917.12168. [DOI] [PubMed] [Google Scholar]
- 48.Milchunas DG, Lauenroth WK. Quantitative effects of grazing on vegetation and soils over a global range of environmnents. Ecol Monogr. 1993;63:327–66. doi: 10.2307/2937150. [DOI] [Google Scholar]
- 49.Gish M, Dafni A, Inbar M. Mammalian herbivore breath alerts aphids to flee host plant. Curr Biol. 2010;20:628–9. doi: 10.1016/j.cub.2010.06.065. [DOI] [PubMed] [Google Scholar]
- 50.Berman TS, Ben-Ari M, Glasser TA, Gish M, Inbar M. How goats avoid ingesting noxious insects while feeding. Sci Rep. 2017;7:1–10. doi: 10.1038/s41598-017-14940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yathom S. Distribution and flight period of two Ocnogyna species in Israel (Lepidoptera: Arctiidae) Isr J Entomol. 1984;18:63–6. [Google Scholar]
- 52.Engel P, Moran NA. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol Rev. 2013;37:699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- 53.Skarpe C, Hester AJ. Plant traits, browsing and grazing herbivores, and vegetation dynamics. In: Gordon I, Prins HHT, editors. The ecology of browsing and grazing. Berlin: Springer; 2008. pp. 217–47. [Google Scholar]
- 54.Sternberg M, Golodets C, Gutman M, Perevolotsky A, Ungar ED, Kigel J, et al. Testing the limits of resistance: a 19-year study of Mediterranean grassland response to grazing regimes. Glob Chang Biol. 2015;21:1939–50. doi: 10.1111/gcb.12866. [DOI] [PubMed] [Google Scholar]
- 55.Henkin Z, Ungar ED, Dvash L, Perevolotsky A, Yehuda Y, Sternberg M, et al. Effects of cattle grazing on herbage quality in a herbaceous Mediterranean rangeland. Grass Forage Sci. 2011;66:516–25. doi: 10.1111/j.1365-2494.2011.00808.x. [DOI] [Google Scholar]
- 56.Colman DR, Toolson EC, Takacs-Vesbach CD. Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol. 2012;21:5124–37. doi: 10.1111/j.1365-294X.2012.05752.x. [DOI] [PubMed] [Google Scholar]
- 57.Tang X, Freitak D, Vogel H, Ping L, Shao Y, Cordero EA, et al. Complexity and variability of gut commensal microbiota in polyphagous lepidopteran larvae. PLoS ONE. 2012;7:e36978. doi: 10.1371/journal.pone.0036978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Priya NG, Ojha A, Kajla MK, Raj A, Rajagopal R. Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS ONE. 2012;7:e30768. doi: 10.1371/journal.pone.0030768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beylich A, Oberholzer HR, Schrader S, Höper H, Wilke BM. Evaluation of soil compaction effects on soil biota and soil biological processes in soils. Soil Tillage Res. 2010;109:133–43. doi: 10.1016/j.still.2010.05.010. [DOI] [Google Scholar]
- 60.Adler P, Raff D, Lauenroth W. The effect of grazing on the spatial heterogeneity of vegetation. Oecologia. 2001;128:465–79. doi: 10.1007/s004420100737. [DOI] [PubMed] [Google Scholar]
- 61.Belovsky GE, Slade JB, Stockhoff BA. Susceptibility to predation for different grasshoppers: an experimental study. Ecology. 1990;71:624–34. doi: 10.2307/1940316. [DOI] [Google Scholar]
- 62.Brinkmann N, Martens R, Tebbe CC. Origin and diversity of metabolically active gut bacteria from laboratory-bred larvae of Manduca sexta (Sphingidae, Lepidoptera, Insecta) Appl Environ Microbiol. 2008;74:7189–96. doi: 10.1128/AEM.01464-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vallet-Gely I, Lemaitre B, Boccard F. Bacterial strategies to overcome insect defences. Nat Rev Microbiol. 2008;6:302–13. doi: 10.1038/nrmicro1870. [DOI] [PubMed] [Google Scholar]
- 64.Ceja-Navarro JA, Nguyen NH, Karaoz U, Gross SR, Herman DJ, Andersen GL, et al. Compartmentalized microbial composition, oxygen gradients and nitrogen fixation in the gut of Odontotaenius disjunctus. ISME J. 2014;8:6–18. doi: 10.1038/ismej.2013.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones RT, Sanchez LG, Fierer N. A cross-taxon analysis of insect-associated bacterial diversity. PLoS ONE. 2013;8:e61218. doi: 10.1371/journal.pone.0061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–5. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 67.Xia X, Zheng D, Zhong H, Qin B, Gurr GM, Vasseur L, et al. DNA sequencing reveals the midgut microbiota of Diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS ONE. 2013;8:e68852. doi: 10.1371/journal.pone.0068852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shao Y, Arias-Cordero E, Guo H, Bartram S, Boland W. In vivo Pyro-SIP assessing active gut microbiota of the cotton leafworm, Spodoptera littoralis. PLoS ONE. 2014;9:e85948. doi: 10.1371/journal.pone.0085948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Douglas AE. Lessons from studying insect symbioses. Cell Host Microbe. 2011;10:359–67. doi: 10.1016/j.chom.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.WenHong L, Jin D, Li F, Jin J, Ying C. Phenotypic fingerprints of bacterium Erwinia persicina from larval gut of the diamondback moth, Plutella xylostella (Lepidoptera Plutellidae) Acta Entomol Sin. 2016;59:456–63. [Google Scholar]
- 71.Hernández-Flores L, Llanderal-Cázares C, Guzmán-Franco AW, Aranda-Ocampo S. Bacteria present in Comadia redtenbacheri larvae (Lepidoptera: Cossidae) J Med Entomol. 2015;52:1150–8. doi: 10.1093/jme/tjv099. [DOI] [PubMed] [Google Scholar]
- 72.Frankenhuyzen K, van, Liu Y, Tonon A. Interactions between Bacillus thuringiensis subsp. kurstaki HD-1 and midgut bacteria in larvae of gypsy moth and spruce budworm. J Invertebr Pathol. 2010;103:124–31. doi: 10.1016/j.jip.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 73.Douglas AE. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu Rev Entomol. 1998;43:17–37. doi: 10.1146/annurev.ento.43.1.17. [DOI] [PubMed] [Google Scholar]
- 74.Inbar M, Doostdar H, Mayer RT. Effects of sessile whitefly nymphs (Homoptera: Aleyrodidae) on leaf-chewing larvae (Lepidoptera: Noctuidae) Environ Entomol. 1999;28:353–7. doi: 10.1093/ee/28.3.353. [DOI] [Google Scholar]
- 75.Hammes WP, Hertel C. The genera Lactobacillus and Carnobacterium. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E, editors. The prokaryotes. New York: Springer; 2006. pp. 320–403. [Google Scholar]
- 76.Schmid RB, Lehman RM, Brözel VS, Lundgren JG. An indigenous gut bacterium, Enterococcus faecalis (Lactobacillales: Enterococcaceae), increases seed consumption by Harpalus pensylvanicus (Coleoptera: Carabidae) Fla Entomol. 2014;97:575–84. doi: 10.1653/024.097.0232. [DOI] [Google Scholar]
- 77.Vásquez A, Forsgren E, Fries I, Paxton RJ, Flaberg E, Szekely L, et al. Symbionts as major modulators of insect health: lactic acid bacteria and honeybees. PLoS ONE. 2012;7:e33188. doi: 10.1371/journal.pone.0033188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Storelli G, Defaye A, Erkosar B, Hols P, Royet J, Leulier F. Lactobacillus plantarum promotes drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011;14:403–14. doi: 10.1016/j.cmet.2011.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.