

Abstract
Chronic neuroinflammation is a hallmark of the onset and progression of brain proteinopathies such as Alzheimer disease (AD) and it is suspected to participate in the neurodegenerative process. Transcription factor NRF2, a master regulator of redox homeostasis, controls acute inflammation but its relevance in low-grade chronic inflammation of AD is inconclusive due to lack of good mouse models. We have addressed this question in a transgenic mouse that combines amyloidopathy and tauopathy with either wild type (AT-NRF2-WT) or NRF2-deficiency (AT-NRF2-KO). AT-NRF2-WT mice died prematurely, at around 14 months of age, due to motor deficits and a terminal spinal deformity but AT-NRF2-KO mice died roughly 2 months earlier. NRF2-deficiency correlated with exacerbated astrogliosis and microgliosis, as determined by an increase in GFAP, IBA1 and CD11b levels. The immunomodulatory molecule dimethyl fumarate (DMF), a drug already used for the treatment of multiple sclerosis whose main target is accepted to be NRF2, was tested in this preclinical model. Daily oral gavage of DMF during six weeks reduced glial and inflammatory markers and improved cognition and motor complications in the AT-NRF2-WT mice compared with the vehicle-treated animals. This study demonstrates the relevance of the inflammatory response in experimental AD, tightly regulated by NRF2 activity, and provides a new strategy to fight AD.
Graphical abstract

1. Introduction
Low-grade chronic neuroinflammation is present at the onset and progression of all neurodegenerative diseases involving cognitive deficits, motor disturbance or both. The progressive deterioration of the brain parenchyma due to the loss of homeostatic capacity, either during ageing, trauma, or accumulation of toxic proteins or metabolites, leads to the release of damage-associated molecular patterns that elicit an inflammatory response. Far from solving the problem, this low-level but persistent activation of the immune system also participates in brain damage. Therefore, a new approach towards a brain protective therapy must consider the restoration of homeostatic functions including control of undesirable inflammation. Although numerous studies have addressed the use of anti-inflammatory therapy to alleviate neurodegeneration [1], [2], the fact is that standard non-steroidal anti-inflammatory drugs, generally designed to stop acute inflammation, have yielded mixed or inconclusive results [3], [4], [5], [6], [7], [8].
In recent years, transcription factor NRF2 (Nuclear factor-erythroid 2-related factor 2) has been identified as a regulator of the extent and duration of inflammatory responses [9], [10], [11]. While it is widely reported that NRF2 regulates oxidant metabolism and several cytoprotective responses, it is now being recognized that it also exerts immune regulatory functions by inducing the expression of anti-inflammatory genes such as CD36, MARCO or IL17D [12], [13], [14] and repressing the expression of the pro-inflammatory genes IL6 and IL1β [15]. Additional mechanisms involve the control of reactive oxygen species (ROS) levels, which regulate the NF-κB response [16], [17], [18], or inhibition of the infiltration of immune cells through the control of VCAM and MMP9 expression [19], [20], [21].
The transcriptional activity of NRF2 declines with aging [22], [23], in parallel to the dysregulation of the immune response and progression of neuropathological hallmarks [24], [25]. In fact, a direct link between NRF2 and neurodegeneration is now being recognized by the observation that functional haplotypes of the NRF2-coding gene (NFE2L2), that result in higher NRF2 levels are associated with protection against PD [26] while other haplotypes that result in a slight reduction in basal NRF2 protein levels are associated with increased risk of suffering AD, ALS or PD [26], [27], [28], [29]. Thus, in the case of AD, one haplotype allele of NFE2L2 was associated with 2 years earlier onset of the disease [27]. The relevance of NRF2 in neuroprotection has been analyzed mainly in transgenic mice developing amyloid beta plaques [30], [31] or tauopathy [10], [32] but not both, which is actually the case of the human pathology. These studies have only analyzed the cognitive effects and not the motor deficits, which are a general hallmark of tauopathies such as AD, Pick disease, progressive supranuclear palsy, corticobasal degeneration, frontotemporal dementia, parkinsonism linked to chromosome 17, etc.
The success of NRF2-targeted immune therapy has been demonstrated in multiple sclerosis (MS). The fumaric acid ester dimethyl fumarate (DMF) exhibits anti-inflammatory and cytoprotective actions in astrocytes by activating the NRF2-dependent production of glutathione and activation of heme oxygenase-1 [33], [34]. Although DMF has other targets besides NRF2 [35], at least part of its beneficial effects are channeled through activation of this transcription factor [36], [37].
In this study we assessed the relevance of NRF2 in the inflammatory response of a combined mouse model of amyloidopathy and tauopathy that recapitulates cognitive and motor deficits similar to those found in several neurodegenerative diseases including AD. We also used chronic administration of DMF as a potential anti-inflammatory drug. Our results indicate that loss of NRF2 activity critically worsens the inflammatory response to proteinopathy by anticipating onset and worsening progression of neuropathological cues.
2. Material and methods
2.1. Transgenic mice
Colonies of NRF2-KO mice and NRF2-WT littermates were established from founders kindly provided by Dr. Masayuki Yamamoto (Tohoku University Graduate School of Medicine, Sendai, Japan) [38]. APPV717I mice (FVB/N), expressing in heterozygosis hAPP695 isoform with the V717I mutation under the control of the Thy1 promoter, were crossed with C57/BL6j-NRF2-WT (APP-NRF2-WT) or C57/BL6j-NRF2-KO (APP-NRF2-KO). Similarly, TAUP301L mice (FVB/N), expressing in homozygosis the longest isoform of protein TAU with the P301L mutation (TAU 4R/2N P301L) under control of the mouse Thy1 gene promoter, were crossed with C57/BL6j-NRF2-WT (TAU-NRF2-WT) or C57/BL6j-NRF2-KO (TAU-NRF2-KO). APP/TAU-NRF2-WT (AT-NRF2-WT) and APP/TAU-NRF2-KO (AT-NRF2-KO) in C57/BL6j background were obtained by crossing the proper mice during more than eight generations. Genotypic characterization of the APPV717I and TAUP301L transgenic mice was described previously [39], [40]. Animals were housed at room temperature under a 12 h light-dark cycle. Food and water was provided ad libitum. Animals were cared for according to a protocol approved by the Ethical Committee for Research of the Autonomous University of Madrid following institutional, Spanish and European guidelines (Boletín Oficial del Estado (BOE) of 18 March 1988; and 86/609/EEC, 2003/65/EC European Council Directives). Once the experimental schedule was completed, animals were anesthetized with 8 mg/kg ketamine and 1.2 mg/kg xylazine and perfused with PBS. The brains were divided sagittally and the right hemispheres were post-fixed in 4% paraformaldehyde for 16 h and cryoprotected by soaking in 30% sucrose solution in phosphate buffer until they sank. The left hemispheres were rapidly dissected and frozen for biochemical analysis. Spinal cord samples for immunohistochemistry were obtained from perfused animals with PBS followed by 4% paraformaldehyde.
2.2. Evaluation of motor alterations
A protocol for measurement of motor alterations was used to evaluate hind limb clasping, ledge, gait and kyphosis [41]. Each measure was recorded on a scale of 0–3, with a combined total of 0–12 for all four measures. This test was performed as double-blind.
2.3. Novel object recognition test (NOR)
During the exploration phase, animals were placed into an empty cage during 10 min. Following 24 h the animals were allowed to explore two identical objects during 8 min. During the test phase, 24 h later, one of the objects was exchanged by a new object (different shape and color) and memory was assessed for 6 min by comparing the time spent exploring the novel object as compared with the time spent exploring the familiar object. The objects were cleaned thoroughly with 40% ethanol followed by distilled water between trials to remove olfactory cues. Active exploration was defined as direct sniffing or whisking towards the objects or direct nose contact. Climbing over the objects was not counted as exploration. The relative exploration was quantified by normalizing the difference between the exploration time of the novel (Tn) and familiar object (Tf) by the total time of exploration (Ttot) to calculate the NOR discrimination index: NOR index = (Tn-Tf)/Ttot.
2.4. Analysis of mRNA levels
Total RNA extraction, reverse transcription and quantitative PCR were done as detailed elsewhere [9]. Primer sequences are shown in Suppl. Table S1. To ensure that equal amounts of cDNA were added to the PCR, the housekeeping genes ActB, Gapdh and Tbp were amplified. Data analysis was based on the ΔΔCT method with normalization of the average data of housekeeping genes. All PCRs were performed in triplicate.
2.5. Immunoblotting
Immunoblots were performed as described in [42]. Primary antibodies are reported in Suppl. Table S2. Membranes were analyzed using the appropriate peroxidase-conjugated secondary antibodies. Proteins were detected by enhanced chemiluminescence (GE Healthcare, Buckinghamshire, United Kingdom).
2.6. Immunohistochemistry and immunofluorescence
30 µm-thick sections from fixed brains were immunostained as indicated in [43]. For spinal cord immunohistochemistry, frozen 5 µm-thick spinal cord sections were stained with the adequate primary antibodies. For immunohistochemistry, the sections were subsequently incubated in 0.05% 30–30 diaminobenzidine tetrahydrochloride (Sigma-Aldrich) in Tris-HCl buffer, pH 8.0 for 25 min, and then developed in the same buffer containing 0.003% hydrogen peroxide (Sigma-Aldrich). The sections were mounted on gelatin-coated slides, air-dried, and finally dehydrated in graded alcohols, cleared in xylene and coverslipped. For immunofluorescence, the sections were incubated with secondary antibodies Alexa-Fluor546 or Alexa-Fluor488. Fluorescence images were captured using appropriate filters in a Leica DMIRE2TCS SP2 confocal microscope (Nussloch, Germany). The lasers used were Ar 488 nm for green fluorescence and Ar/HeNe 543 nm for red fluorescence.
2.7. Silver staining procedure
Sagittal series of 30-µm-thick sections were stained with FD Neurosilver TM kit II (FD Neurotecnologies, MD, USA) following manufacturer's instructions. The sections were dehydrated in ethanol and mounted in DePex (Thermo Fisher Scientific, MA, USA).
2.8. Image analysis and statistics
Calculation of p-values from ANOVA one-way followed by Newman-Keuls post-hoc test, ANOVA two-way followed by Bonferroni post-hoc test and Student's t-test was done with GraphPad Prism 5 software. A p value of ≤ 0.05 was considered significant. Unless otherwise indicated, all experiments were performed at least three times with similar results. Values presented in the graphs are means of at least three samples. Results are expressed as mean ± SEM.
3. Results
3.1. NRF2-deficiency accelerates mouse death induced by the expression of TAU P301L and APPV717I
The transgenic mice with neuronal expression of human hAPPV717I and hTAUP301L proteins (for simplicity from now on denoted as AT) were generated in C57/bl6 mice of wild type (AT-NRF2-WT) and Nrf2-knockout (AT-NRF2-KO) backgrounds. Memory performance was examined according to a double-blind novel object recognition test (NOR) in 6-months old animals. We observed an accelerated rate of cognitive decline in AT-NRF2-KO mice compared to AT-NRF2-WT animals (Fig. S1A). Moreover, AT-NRF2-KO mice exhibited a significant reduction in Nqo1, Osgin1, Gstm1, Aox1 and Sqstm1 compared to age-matched AT-NRF2-WT mice (Fig. S1B).
The tissue distribution of total hAPPV717I and hTAUP301L proteins (Suppl. Figs. S2B and S3B) was similar in both genetic backgrounds and affected neurons of the forebrain, including the isocortex (infragranular layers V/VI of the entorhinal cortex, M1 and M2 motor cortex) and the hippocampal formation (CA1, CA2, subiculum and hilum). In the brainstem, hAPPV717I and hTAUP301L were localized in thalamus and the vast majority of positive neurons and fibers were found in the reticular formation of the bulb and pons, including the pyramid of the pontine nuclei and spinal trigeminal nuclei. There were positive neurons and fibers in cerebellum (deep cerebellum nuclei and granular layer). The spinal cord showed positive neurons for hAPPV717I and hTAUP301L expression in the anterior horns. Despite the similar distribution of hAPPV717I and hTAUP301L in both genotypes (Suppl. Fig. S2C and S3C), we have previously reported that the AT-NRF2-KO mice exhibit increased levels of toxic Aβ*56 and insoluble phospho-TAU [43], [44].
Kaplan-Meier survival curves indicated that the mean survival time of AT-NRF2-WT mice was 14 months with some mice reaching 20 months. By contrast, 50% of AT-NRF2-KO mice died before the age of 12 months with hardly any survival at 14 months (Fig. 1A). These differences were similar for males and females (Suppl. Fig. S4). This premature death was associated with a rapid motor deterioration that in a time-lapse of 4 weeks presented with progressive alterations in ledge, clasping, gait and terminal kyphosis. Both mouse genotypes also displayed abnormal clasping, with an abnormal simultaneous retraction of both fore- and hind limbs (Fig. 1D). By the time that mice developed kyphosis, a significant weight loss was observed and death came in less than one week. Fig. 1B shows representative animals with spinal cord deformity and rigid thoracolumbar kyphosis that led to imminent death. X-ray analysis did not reveal any evidence of vertebral or limbs deformity that could be attributed to bone degeneration (Fig. 1C). To quantify the motor behavior of AT-NRF2-WT vs. AT-NRF2-KO mice, we used a standard double blind sensory-motor test [41]. This test measured the severity of ledge, clasping, gait and kyphosis on a scale of 0–3 for each parameter, where 0 value reflected non-affection and 3 value reflected high-affection. As shown in Fig. 1E, AT-NRF2-WT mice exhibited a progressive worsening of the motor disturbance at 11, 12 and 13 months while AT-NRF2-KO mice already had maximal plateau values from 11 months until spontaneous death or euthanized for ethical reasons. Therefore, while both genotypes revealed extensive parallelism in the expression pattern of hAPPV717I and hTAUP301L, AT-NRF2-KO mice presented accelerated appearance of the terminal motor phenotype.
Fig. 1.
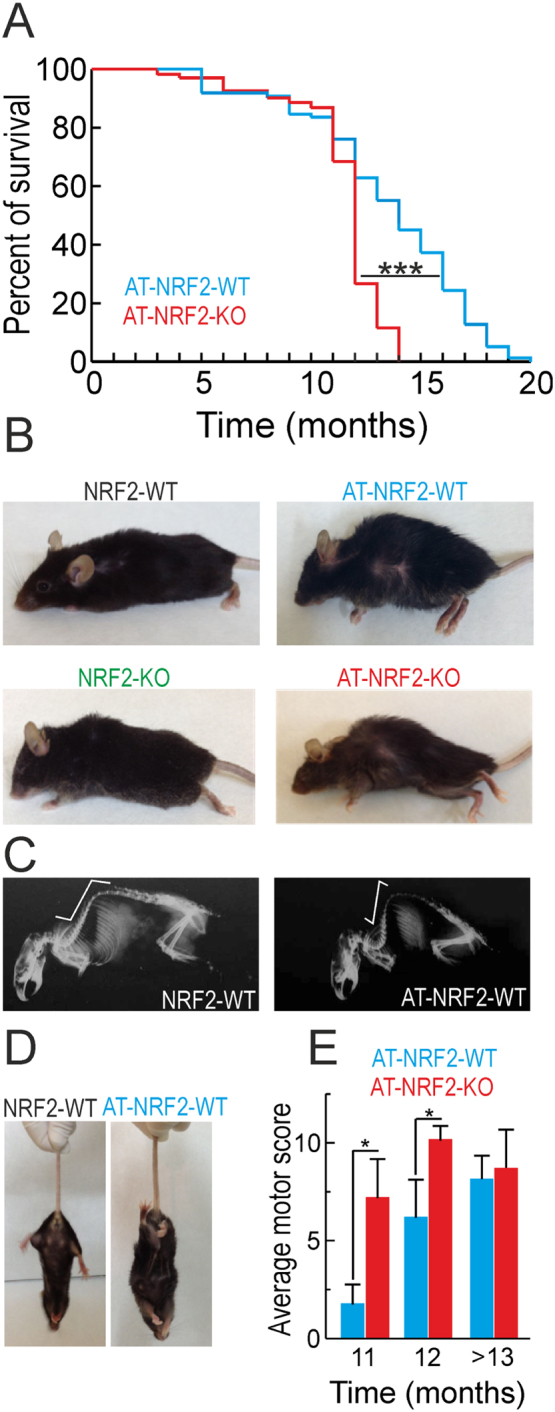
Death of the AT-mice occurs faster in the absence of NRF2. A, Kaplan-Meier curves of AT-NRF2-WT (n = 269) and AT-NRF2-KO (n = 209) over 3 years of observation. Statistical analysis was performed and a value of ***p < 0.001 was obtained comparing AT-NRF2-WT and AT-NRF2-KO curves. B, pictures from the indicated genotypes illustrating terminal postures. C, X-ray images of mice from the indicated genotypes showing the spinal deformity termed kyphosis. D, representative images of the clasping behavior of wild type and AT-mice when hanging from the tail. E, average score of the motor test performed in which we analyzed ledge, clasping, gait and kyphosis. We tested AT-NRF2-WT and AT-NRF2-KO animals at the indicated ages. Statistical analysis was performed with Student's t-test. *p < 0.5 comparing AT-NRF2-KO vs. AT-NRF2-WT groups.
At the cerebellum, Purkinje and GABAergic neurons as well as astroglia and microglia were not affected by the transgenes expression (Suppl. Fig. S5). However, at the pontine reticular formation, FD Neurosilver staining evidenced the presence of degenerating fibers (Fig. 2A) in both AT-NRF2-WT and AT-NRF2-KO mice but the number of neurons with arginophilic inclusions was higher in the AT-NRF2-KO mice (Fig. 2B). Taken together, these results suggest that the brainstem neuronal circuits that control motor activity are the most vulnerable to APP/TAU overexpression and that this toxic effect is exacerbated in the absence of NRF2.
Fig. 2.

Neurodegeneration in the brainstem was exacerbated in the absence of NRF2. A and B, silver staining of sagittal brain sections from 11-months old mice of the indicated genotypes. Pictures show dystrophic neurites (A) and neurons with arginophilic inclusions (B) in the pontine reticular formation. C, quantification of the number of positive neurons with arginophilic inclusions from B. Data are mean ± SEM (n = 3). Statistical analysis was performed with Student's t-test. *p < 0.05, comparing AT-NRF2-KO vs. AT-NRF2-WT groups.
3.2. NRF2 modulates brain glial and inflammatory responses
Based on the Kaplan-Meier survival curves and the motor alterations described above, we took the age of 11 months as the most discriminating time window to compare pathophenotypes of AT-NRF2-WT and AT-NRF2-KO mice. We examined astrogliosis in hippocampi with antibodies against glial fibrillar acidic protein (GFAP). Immunofluorescence staining of CA1 layer and subiculum demonstrated activated astrocytes in both genotypes, as determined by morphology of thick cell bodies and small cytoplasmic branches. Abolishment of NRF2 expression did not change the levels of GFAP immunostaining or mRNA in hippocampus (Fig. 3A and C). These results are in accordance with no significant loss of neurons at this region in either AT-NRF2-KO or AT-NRF2-WT mice. Next, we analyzed microglial activation in hippocampi of these mice with antibodies against CD11b. We found a mild increase in the number of CD11b positive microglial cells in AT-NRF2-KO compared with AT-NRF2-WT mice (Fig. 3B and D). This change was quantified by qRT-PCR evidencing a modest but significant increase in CD11b mRNA levels (Fig. 3B). We also found higher mRNA levels of the pro-inflammatory markers, IL6 and NOS2 in AT-NRF2-KO vs. age-matched AT-NRF2-WT mice (Fig. 3E).
Fig. 3.

Gliosis is exacerbated by NRF2 deficiency in hippocampi of AT-NRF2-KO mice. A and B, brain sections from 11-months old AT-NRF2-WT and AT-NRF2-KO mice were stained with GFAP or CD11b, respectively. Pictures show CA1 and subiculum regions of the hippocampus. C and E, mRNA levels determined by qRT-PCR of the indicated genes normalized to the expression of ActB. Data are mean ± SEM (n = 4). Statistical analysis was performed with Student's t-test. *p < 0.05, comparing AT-NRF2-KO vs. AT-NRF2-WT mice. D, number of CD11b-positive microglial cells in CA1 and subiculum of the indicated genotypes. Data are mean ± SEM (n = 3) represented as % of the total number of cells. Statistical analysis was performed with Student's t-test. *p < 0.05, comparing AT-NRF2-KO vs. AT-NRF2-WT mice.
Glial activation was also evaluated in the brainstem (Fig. 4) with anti-GFAP or anti-IBA1 antibodies. GFAP and IBA1 staining was increased in the AT-NRF2-KO mice in both locations (Fig. 4A). These results were further confirmed in immunoblots from AT-NRF2-WT and AT-NRF2-KO brainstem lysates (Fig. 4B). GFAP and IBA1 levels were about two-fold higher in the AT-NRF2-KO mice compared with age-matched AT-NRF2-WT mice in brainstem samples (Fig. 4C). Similar experiments were conducted to analyze astroglial and microglial activation in spinal cord (Fig. 5). In accordance with the results showed in Fig. 1, macroscopic analysis of the spinal cord dissected from AT-NRF2-KO mice evidenced higher torsion angle compared with AT-NRF2-WT. In line with the results obtained in brainstem, NRF2-deficiency led to increased staining of GFAP and IBA1 in white and grey matter of the spinal cord (Fig. 5A). Then, we confirmed these observations in immunoblots from AT-NRF2-WT and AT-NRF2-KO spinal cords samples. The protein levels of GFAP and IBA1 were slightly increased in the AT-NRF2-KO compared with AT-NRF2-WT mice (Fig. 5B and C). These results suggest that NRF2-deficiency exacerbates glial activation in these locations.
Fig. 4.

Increased glial activation in the brainstem of AT-NRF2-KO mice. A, immunostaining with anti-GFAP and anti-IBA1 in sagittal sections of the pontine reticular formation of the brainstem in the indicated genotypes. B, immunoblot analysis of GFAP and IBA1 levels in brainstem and spinal cord homogenates from 11-month old AT-NRF2-WT and AT-NRF2-KO mice. Protein levels of GAPDH were analyzed to ensure similar load per lane. C, densitometric quantification of representative blots from B. Data are mean ± SEM (n = 3). Statistical analysis was performed with Student's t-test. *p < 0.05, comparing AT-NRF2-KO vs. AT-NRF2-WT mice.
Fig. 5.

Glial activation in the spinal cord of AT-NRF2-WT and AT-NRF2-KO mice. A, immunostaining with anti-GFAP or anti-IBA1 in sagittal sections of the spinal cord in the indicated genotypes. Pictures show white (a) and grey (b) matter of the anterior horn. Spinal cord sections were counterstained with hematoxylin. B, immunoblot analysis of GFAP and IBA1 levels in spinal cord homogenates from 11-month old AT-NRF2-WT and AT-NRF2-KO mice. Protein levels of GAPDH were analyzed to ensure similar load per lane. C, densitometric quantification of representative blots from B. Data are mean ± SEM (n = 3). Statistical analysis was performed with t-Student test.
3.3. DMF reduces motor alteration and improves memory
Our next goal was to evaluate whether NRF2 activation with dimethyl fumarate (DMF) could modulate the pathological outcomes triggered by APP and TAU expression. In a previous study we already established the conditions of DMF administration by oral gavage and demonstrated that it reaches the brain and activates the NRF2 transcriptional signature in this organ [45]. As shown in Suppl. Fig. S6A, 9 months-old AT-NRF2-WT mice were treated by oral gavage with vehicle or DMF (100 mg/kg) once every two days for six weeks. This age was chosen because motor alterations and cognitive deficits were still absent. During the whole time of treatment, we did not detect changes in weight that would be attributed to feeding problems after oral gavage (Suppl. Fig. S6B). Five days after the first administration, animals were weighted and submitted to double-blind motor and novel object recognition (NOR) tests to obtain baseline values of memory. This protocol was repeated once every two weeks. As shown in Fig. 6A, mRNA levels of NRF2-target genes were analyzed by qRT-PCR. We found a statistically significant increase in the expression of Nrf2, Nqo1, Osgin1, and Gstm1 in the brain, in agreement with NRF2-activation by DMF. Next, we evaluated astrocytosis and microgliosis employing GFAP and IBA1 antibodies, respectively. As shown in Fig. 6B, DMF ameliorated the levels of GFAP positive cells in hippocampus and in brainstem indicating preservation of the neural tissue. Similarly, microgliosis was also reduced in both regions of the DMF-treated mice (Fig. 6C). Moreover, in response to DMF we found a downward trend in the protein levels of the pro-inflammatory mediators COX2 and NOS2, as well as the gliosis markers GFAP, IBA1 and MHCII (Fig. 6D and E). The average motor score slightly increased over the duration of the experiment in the vehicle treated mice but remained unchanged in the DMF-treated group, suggesting that NRF2 activation attenuates the progression of the motor alterations (Fig. 6F). In parallel, we analyzed the mice ability to discriminate a novel object in the environment. The discrimination index of the DMF-treated group raised 2.5–3-fold compared with the value of the vehicle-treated group, indicating a tendency to better memory function in parallel to NRF2 activation (Fig. 6G). Although, the differences achieved in both tests were modest, the tendency was statistically significant at 2-weeks. Taken together, these results indicate that NRF2 activation by DMF alleviates brain inflammation as well as motor and cognitive disability triggered by hAPPV717I and hTAUP301L expression.
Fig. 6.

DMF improves memory and motor deficits in AT-NRF2-WT mice. 9-month old AT-NRF2-WT mice received intragastric doses of vehicle (n = 10) or DMF (100 mg/kg, n = 10) once every two days during six weeks. A, qRT-PCR determination of brain mRNA levels of the NRF2-regulated genes Nrf2, Nqo1, Osgin1, and Gstm1 normalized by the average of Actb, Tbp and Gapdh. Data are mean ± SEM (n = 4). Statistical analysis was performed with Student's t-test. **p < 0.01 and *p < 0.05, comparing DMF vs. vehicle treated mice. B and C, immunostaining with anti-GFAP (B) and anti-IBA1 (C) in sagittal brain sections of the indicated groups. Pictures show CA1 and subiculum of the hippocampus as well as the pontine reticular formation of the brainstem. D, immunoblot analysis of the indicated protein levels in brain homogenates from vehicle- or DMF-treated mice. Protein levels of ACTB were analyzed to ensure similar load per lane. E, densitometric quantification of representative blots from D. Data are mean ± SEM (n = 3). Statistical analysis was performed with Student's t-test. **< 0.01 and *p < 0.05, comparing DMF vs. vehicle treated mice. F, average score of the motor test performed in which we analyzed ledge, clasping, gait and kyphosis at the indicated times. Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-hoc test. *p < 0.05, comparing DMF vs. vehicle treated mice. G, discrimination index obtained in the novel object recognition task (total time spent with new object/total time of object exploration) by the two experimental groups. Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-hoc test. *p < 0.05, comparing DMF vs. vehicle treated mice.
4. Discussion
In the present study, we used a new mouse model of combined amyloidopathy and tauopathy to analyze the effect of NRF2 on neuroinflammation, which is a crucial pathological hallmark of neurodegenerative diseases such as AD. In a previous study, we employed these mice to characterize the effect of NRF2-deficiency in autophagy and several pathophenotypes associated with AD [43], [44]. Here we describe the effect of NRF2-deficiency on motor and cognitive deficits, as well as gliosis and neuroinflammation.
Motor alterations were produced mainly in response to tauopathy, since they were detected even in transgenic mice expressing only the hTAUP301L protein (data not shown and [46], [47]). Motor deficits are common in human tauopathies such as AD, Pick disease, progressive supranuclear palsy, corticobasal degeneration, frontotemporal dementia and parkinsonism linked to chromosome 17, spinocerebellar ataxia type 11, etc. Regarding amyloidopathy, an analysis of five Han Chinese EOFAD families with the APPV717I mutation exhibited clinical symptoms of cerebellar ataxia. Mutations in presenilin 1 have also been associated with cerebellum alterations and movement disorders [48], [49]. However, although we detected a modest expression of APP and TAU protein in cerebellar regions, we did not appreciate any alteration in Purkinje, GABAergic or glial cells that could explain motor disturbance (Supl. Fig. S5). Most likely, motor alterations were related to the damage to somatomotor neurons located at the nuclei of the brainstem and the anterior horn of the spinal cord. In these regions, we detected exacerbated microgliosis and astrogliosis and spheroid axons [44]. Previous studies with hTAUP301L expressing mice in a Swiss/3T3 genetic background under the control of either the prion or the Thy1 promoter demonstrated a role of progressive tauopathy in the alteration of brainstem nuclei, leading to motor alterations similar to those described here [50], [51], [52]. However, our C57/Bl6 mice lived several months longer than the Swiss/3T3 mice, thus suggesting strain specific properties. Independent studies have correlated the motor deficits observed in AT-mice with TAUP301L overexpression [46], [47] as well as with redox imbalance [53]. Our observations extended the impact, because we directly connected TAU pathology with redox, inflammatory and proteostasis stress through NRF2-deficiency.
The anatomopathological and clinical phenotype of terminal AT-NRF2-KO mice could not be distinguished from terminal AT-NRF2-WT mice. However, the pathology spread over a narrower age-window when NRF2 was absent: 12 months in AT-NRF2-KO mice as opposed to 14 months in AT-NRF2-WT mice. The AT-NRF2-KO mice recapitulate the main AD hallmarks including loss of redoxtasis and proteostasis [31], [32], [43], [44] and, as described here, the presence of neuroinflammation. Our previous results strongly support that NRF2-deficiency mimics transcriptomic features found in AD brains [44]. It is therefore reasonable to speculate that neurons should be more susceptible to APP and TAU toxicity in the NRF2-deficient than in the wild type background. Indeed, AT-NRF2-KO mice exhibited more damaged neurons in brainstem, exacerbated cognitive [44] and motor impairments, and premature death.
Experimental and clinical evidence suggests that DMF plays a prominent role in targeting brain inflammation mainly through NRF2 activation [35], [54], [55], [56]. Accordingly, we detected induction of NRF2, a reduction of inflammatory markers and improvement of motor and memory cues in the brain of 9-month old AT-NRF2-WT mice treated with DMF. These results are in line with protective functions reported in diverse models of neurodegenerative diseases and its effects in prevention of spatial memory impairment and hippocampal neurodegeneration mediated by intracerebroventricular injection of streptozotocin in rats [57]. From the immunomodulatory point of view, additional mechanisms independent of NRF2 have been proposed to explain the therapeutic value of DMF [35], [58]. We did not test NRF2-independent effects of DMF in the AT-NRF2-KO mice since as indicated in Suppl. Fig. S4 they exhibit high mortality during the time of experimentation. However, the fact that the DMF targeted the NRF2 transcriptional signature in the brain together with the established evidence of the immunomodulatory signals, points towards NRF2-dependent beneficial effects of DMF. Future clinical work will be needed to definitely determine if pharmacological activation of NRF2 may be a valid strategy to ameliorate neurodegeneration but surely DMF will be a suitable candidate because it is the only FDA- and EAE-approved NRF2 activator.
5. Conclusion
We have developed a new mouse model that combines amyloidopathy and tauopathy with deficiency in the master regulator of homeostatic responses, the transcription factor NRF2. The study demonstrates that low-grade chronic neuroinflammation is a driver of neurodegeneration, and not just a consequence, since an antioxidant modulator of inflammation, DMF, which targets NRF2, protected from disease progression. The fact that DMF is already in clinical use for multiple sclerosis provides a rationale for repurposing this drug in AD.
Acknowledgements
This work was funded by SAF2016-76520-R of the Spanish Ministry of Economy and Competitiveness, a Pathfinder grant of the Centres of Excellence in Neurodegeneration (COEN) of the EU-Joint Program for Neurodegenerative diseases, and European Regional Development Fund, Competitiveness Operational Program 2014–2020, through the grant P_37_732/2016 REDBRAIN and a collaborative CIBERNED project PI2017/04-3. M.P. is recipient of a FPU fellowship of Autonomous University of Madrid.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.07.006.
Contributor Information
Ana I. Rojo, Email: airojo@iib.uam.es.
Antonio Cuadrado, Email: antonio.cuadrado@uam.es.
Appendix A. Supplementary material
Supplementary material
References
- 1.Gasparini L., Ongini E., Wenk G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer's disease: old and new mechanisms of action. J. Neurochem. 2004;91:521–536. doi: 10.1111/j.1471-4159.2004.02743.x. [DOI] [PubMed] [Google Scholar]
- 2.Vlad S.C., Miller D.R., Kowall N.W., Felson D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.in t′ Veld B.A., Ruitenberg A., Hofman A., Launer L.J., van Duijn C.M., Stijnen T., Breteler M.M., Stricker B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 4.de Craen A.J., Gussekloo J., Vrijsen B., Westendorp R.G. Meta-analysis of nonsteroidal antiinflammatory drug use and risk of dementia. Am. J. Epidemiol. 2005;161:114–120. doi: 10.1093/aje/kwi029. [DOI] [PubMed] [Google Scholar]
- 5.Aisen P.S., Schmeidler J., Pasinetti G.M. Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology. 2002;58:1050–1054. doi: 10.1212/wnl.58.7.1050. [DOI] [PubMed] [Google Scholar]
- 6.Rogers J., Kirby L.C., Hempelman S.R., Berry D.L., McGeer P.L., Kaszniak A.W., Zalinski J., Cofield M., Mansukhani L., Willson P. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 7.Reines S.A., Block G.A., Morris J.C., Liu G., Nessly M.L., Lines C.R., Norman B.A., Baranak C.C., Rofecoxib G. Protocol 091 Study, Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 8.Scharf S., Mander A., Ugoni A., Vajda F., Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999;53:197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- 9.Rojo A.I., Innamorato N.G., Martin-Moreno A.M., De Ceballos M.L., Yamamoto M., Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 10.Lastres-Becker I., Innamorato N.G., Jaworski T., Rabano A., Kugler S., Van Leuven F., Cuadrado A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain: J. Neurol. 2014;137:78–91. doi: 10.1093/brain/awt323. [DOI] [PubMed] [Google Scholar]
- 11.Rojo A.I., McBean G., Cindric M., Egea J., Lopez M.G., Rada P., Zarkovic N., Cuadrado A. Redox control of microglial function: molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014;21:1766–1801. doi: 10.1089/ars.2013.5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey C.J., Thimmulappa R.K., Sethi S., Kong X., Yarmus L., Brown R.H., Feller-Kopman D., Wise R., Biswal S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011;3:78ra32. doi: 10.1126/scitranslmed.3002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishii T., Mann G.E. Redox status in mammalian cells and stem cells during culture in vitro: critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox Biol. 2014;2:786–794. doi: 10.1016/j.redox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saddawi-Konefka R., Seelige R., Gross E.T., Levy E., Searles S.C., Washington A., Jr., Santosa E.K., Liu B., O'Sullivan T.E., Harismendy O., Bui J.D. Nrf2 induces IL-17D to mediate tumor and virus surveillance. Cell Rep. 2016;16:2348–2358. doi: 10.1016/j.celrep.2016.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi E.H., Suzuki T., Funayama R., Nagashima T., Hayashi M., Sekine H., Tanaka N., Moriguchi T., Motohashi H., Nakayama K., Yamamoto M. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toledano M.B., Leonard W.J. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA. 1991;88:4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuadrado A., Martin-Moldes Z., Ye J., Lastres-Becker I. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014;289:15244–15258. doi: 10.1074/jbc.M113.540633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banning A., Brigelius-Flohe R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 2005;7:889–899. doi: 10.1089/ars.2005.7.889. [DOI] [PubMed] [Google Scholar]
- 20.Wenzel P., Rossmann H., Muller C., Kossmann S., Oelze M., Schulz A., Arnold N., Simsek C., Lagrange J., Klemz R., Schonfelder T., Brandt M., Karbach S.H., Knorr M., Finger S., Neukirch C., Hauser F., Beutel M.E., Kroller-Schon S., Schulz E., Schnabel R.B., Lackner K., Wild P.S., Zeller T., Daiber A., Blankenberg S., Munzel T. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur. Heart J. 2015;36:3437–3446. doi: 10.1093/eurheartj/ehv544. [DOI] [PubMed] [Google Scholar]
- 21.Bourdonnay E., Morzadec C., Fardel O., Vernhet L. Redox-sensitive regulation of gene expression in human primary macrophages exposed to inorganic arsenic. J. Cell Biochem. 2009;107:537–547. doi: 10.1002/jcb.22155. [DOI] [PubMed] [Google Scholar]
- 22.Rahman M.M., Sykiotis G.P., Nishimura M., Bodmer R., Bohmann D. Declining signal dependence of Nrf2-MafS-regulated gene expression correlates with aging phenotypes. Aging Cell. 2013;12:554–562. doi: 10.1111/acel.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suh J.H., Shenvi S.V., Dixon B.M., Liu H., Jaiswal A.K., Liu R.M., Hagen T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montecino-Rodriguez E., Berent-Maoz B., Dorshkind K. Causes, consequences, and reversal of immune system aging. J. Clin. Investig. 2013;123:958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539:180–186. doi: 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Otter M., Landgren S., Nilsson S., Celojevic D., Bergstrom P., Hakansson A., Nissbrandt H., Drozdzik M., Bialecka M., Kurzawski M., Blennow K., Nilsson M., Hammarsten O., Zetterberg H. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson's disease. BMC Med. Genet. 2010;11:36. doi: 10.1186/1471-2350-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Otter M., Landgren S., Nilsson S., Zetterberg M., Celojevic D., Bergstrom P., Minthon L., Bogdanovic N., Andreasen N., Gustafson D.R., Skoog I., Wallin A., Tasa G., Blennow K., Nilsson M., Hammarsten O., Zetterberg H. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer's disease and age-related cataract. Mech. Ageing Dev. 2010;131:105–110. doi: 10.1016/j.mad.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Bergstrom P., von Otter M., Nilsson S., Nilsson A.C., Nilsson M., Andersen P.M., Hammarsten O., Zetterberg H. Association of NFE2L2 and KEAP1 haplotypes with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemp. Degener. 2014;15:130–137. doi: 10.3109/21678421.2013.839708. [DOI] [PubMed] [Google Scholar]
- 29.von Otter M., Bergstrom P., Quattrone A., De Marco E.V., Annesi G., Soderkvist P., Wettinger S.B., Drozdzik M., Bialecka M., Nissbrandt H., Klein C., Nilsson M., Hammarsten O., Nilsson S., Zetterberg H. Genetic associations of Nrf2-encoding NFE2L2 variants with Parkinson's disease - a multicenter study. BMC Med. Genet. 2014;15:131. doi: 10.1186/s12881-014-0131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanninen K., Heikkinen R., Malm T., Rolova T., Kuhmonen S., Leinonen H., Yla-Herttuala S., Tanila H., Levonen A.L., Koistinaho M., Koistinaho J. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2009;106:16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joshi G., Gan K.A., Johnson D.A., Johnson J.A. Increased Alzheimer's disease-like pathology in the APP/ PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging. 2015;36:664–679. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jo C., Gundemir S., Pritchard S., Jin Y.N., Rahman I., Johnson G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014;5:3496. doi: 10.1038/ncomms4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin S.X., Lisi L., Dello Russo C., Polak P.E., Sharp A., Weinberg G., Kalinin S., Feinstein D.L. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro. 2011;3 doi: 10.1042/AN20100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scannevin R.H., Chollate S., Jung M.Y., Shackett M., Patel H., Bista P., Zeng W., Ryan S., Yamamoto M., Lukashev M., Rhodes K.J. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 35.Schulze-Topphoff U., Varrin-Doyer M., Pekarek K., Spencer C.M., Shetty A., Sagan S.A., Cree B.A., Sobel R.A., Wipke B.T., Steinman L., Scannevin R.H., Zamvil S.S. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc. Natl. Acad. Sci. USA. 2016;113:4777–4782. doi: 10.1073/pnas.1603907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linker R.A., Lee D.H., Ryan S., van Dam A.M., Conrad R., Bista P., Zeng W., Hronowsky X., Buko A., Chollate S., Ellrichmann G., Bruck W., Dawson K., Goelz S., Wiese S., Scannevin R.H., Lukashev M., Gold R. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain: J. Neurol. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 37.Mills E.A., Ogrodnik M.A., Plave A., Mao-Draayer Y. Emerging Understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front. Neurol. 2018;9:5. doi: 10.3389/fneur.2018.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 39.Moechars D., Dewachter I., Lorent K., Reverse D., Baekelandt V., Naidu A., Tesseur I., Spittaels K., Haute C.V., Checler F., Godaux E., Cordell B., Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 40.Terwel D., Lasrado R., Snauwaert J., Vandeweert E., Van Haesendonck C., Borghgraef P., Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J. Biol. Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- 41.Guyenet S.J., Furrer S.A., Damian V.M., Baughan T.D., La Spada A.R., Garden G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010 doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rojo A.I., Medina-Campos O.N., Rada P., Zuniga-Toala A., Lopez-Gazcon A., Espada S., Pedraza-Chaverri J., Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic. Biol. Med. 2012;52:473–487. doi: 10.1016/j.freeradbiomed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Pajares M., Jimenez-Moreno N., Garcia-Yague A.J., Escoll M., de Ceballos M.L., Van Leuven F., Rabano A., Yamamoto M., Rojo A.I., Cuadrado A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016;12:1902–1916. doi: 10.1080/15548627.2016.1208889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rojo A.I., Pajares M., Rada P., Nunez A., Nevado-Holgado A.J., Killik R., Van Leuven F., Ribe E., Lovestone S., Yamamoto M., Cuadrado A. NRF2 deficiency replicates transcriptomic changes in Alzheimer's patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–451. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lastres-Becker I., Garcia-Yague A.J., Scannevin R.H., Casarejos M.J., Kugler S., Rabano A., Cuadrado A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson's disease. Antioxid. Redox Signal. 2016;25:61–77. doi: 10.1089/ars.2015.6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X., Duff K., Hardy J., Corral A., Lin W.L., Yen S.H., Dickson D.W., Davies P., Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 47.Crespo-Biel N., Theunis C., Borghgraef P., Lechat B., Devijver H., Maurin H., Van Leuven F. Phosphorylation of protein Tau by GSK3beta prolongs survival of bigenic Tau.P301LxGSK3beta mice by delaying brainstem tauopathy. Neurobiol. Dis. 2014;67:119–132. doi: 10.1016/j.nbd.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Appel-Cresswell S., Guella I., Lehman A., Foti D., Farrer M.J. PSEN1 p.Met233Val in a complex neurodegenerative movement and neuropsychiatric disorder. J. Mov. Disord. 2018;11:45–48. doi: 10.14802/jmd.17066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lemere C.A., Lopera F., Kosik K.S., Lendon C.L., Ossa J., Saido T.C., Yamaguchi H., Ruiz A., Martinez A., Madrigal L., Hincapie L., Arango J.C., Anthony D.C., Koo E.H., Goate A.M., Selkoe D.J., Arango J.C. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat. Med. 1996;2:1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 50.Dutschmann M., Menuet C., Stettner G.M., Gestreau C., Borghgraef P., Devijver H., Gielis L., Hilaire G., Van Leuven F. Upper airway dysfunction of Tau-P301L mice correlates with tauopathy in midbrain and ponto-medullary brainstem nuclei. J. Neurosci. 2010;30:1810–1821. doi: 10.1523/JNEUROSCI.5261-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Menuet C., Borghgraef P., Matarazzo V., Gielis L., Lajard A.M., Voituron N., Gestreau C., Dutschmann M., Van Leuven F., Hilaire G. Raphe tauopathy alters serotonin metabolism and breathing activity in terminal Tau.P301L mice: possible implications for tauopathies and Alzheimer's disease. Respir. Physiol. Neurobiol. 2011;178:290–303. doi: 10.1016/j.resp.2011.06.030. [DOI] [PubMed] [Google Scholar]
- 52.Menuet C., Borghgraef P., Voituron N., Gestreau C., Gielis L., Devijver H., Dutschmann M., Van Leuven F., Hilaire G. Isoflurane anesthesia precipitates tauopathy and upper airways dysfunction in pre-symptomatic Tau.P301L mice: possible implication for neurodegenerative diseases. Neurobiol. Dis. 2012;46:234–243. doi: 10.1016/j.nbd.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 53.Treiber N., Maity P., Singh K., Kohn M., Keist A.F., Ferchiu F., Sante L., Frese S., Bloch W., Kreppel F., Kochanek S., Sindrilaru A., Iben S., Hogel J., Ohnmacht M., Claes L.E., Ignatius A., Chung J.H., Lee M.J., Kamenisch Y., Berneburg M., Nikolaus T., Braunstein K., Sperfeld A.D., Ludolph A.C., Briviba K., Wlaschek M., Florin L., Angel P., Scharffetter-Kochanek K. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell. 2011;10:239–254. doi: 10.1111/j.1474-9726.2010.00658.x. [DOI] [PubMed] [Google Scholar]
- 54.Wilms H., Sievers J., Rickert U., Rostami-Yazdi M., Mrowietz U., Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflamm. 2010;7:30. doi: 10.1186/1742-2094-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Albrecht P., Bouchachia I., Goebels N., Henke N., Hofstetter H.H., Issberner A., Kovacs Z., Lewerenz J., Lisak D., Maher P., Mausberg A.K., Quasthoff K., Zimmermann C., Hartung H.P., Methner A. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm. 2012;9:163. doi: 10.1186/1742-2094-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellrichmann G., Petrasch-Parwez E., Lee D.H., Reick C., Arning L., Saft C., Gold R., Linker R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington's disease. PLoS One. 2011;6:e16172. doi: 10.1371/journal.pone.0016172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Majkutewicz I., Kurowska E., Podlacha M., Myslinska D., Grembecka B., Rucinski J., Plucinska K., Jerzemowska G., Wrona D. Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav. Brain Res. 2016;308:24–37. doi: 10.1016/j.bbr.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 58.Kornberg M.D., Bhargava P., Kim P.M., Putluri V., Snowman A.M., Putluri N., Calabresi P.A., Snyder S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360:449–453. doi: 10.1126/science.aan4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material