

Abstract
Protein arginine methyltransferase 5 (PRMT5) regulates gene expression either transcriptionally by symmetric dimethylation of arginine residues on histones H4R3, H3R8, and H2AR3 or at the posttranslational level by methylation of nonhistone target proteins. Although emerging evidence suggests that PRMT5 functions as an oncogene, its role in metabolic diseases is not well-defined. We investigated the role of PRMT5 in promoting high-fat–induced hepatic steatosis. A high-fat diet up-regulated PRMT5 levels in the liver but not in other metabolically relevant tissues such as skeletal muscle or white and brown adipose tissue. This was associated with repression of master transcription regulators involved in mitochondrial biogenesis. In contrast, lentiviral short hairpin RNA–mediated reduction of PRMT5 significantly decreased phosphatidylinositol 3-kinase/AKT signaling in mouse AML12 liver cells. PRMT5 knockdown or knockout decreased basal AKT phosphorylation but boosted the expression of peroxisome proliferator–activated receptor α (PPARα) and PGC-1α with a concomitant increase in mitochondrial biogenesis. Moreover, by overexpressing an exogenous WT or enzyme-dead mutant PRMT5 or by inhibiting PRMT5 enzymatic activity with a small-molecule inhibitor, we demonstrated that the enzymatic activity of PRMT5 is required for regulation of PPARα and PGC-1α expression and mitochondrial biogenesis. Our results suggest that targeting PRMT5 may have therapeutic potential for the treatment of fatty liver.
Keywords: epigenetics, fatty acid oxidation, signaling, mitochondria, Akt PKB, mitochondrial biogenesis, non-alcoholic fatty liver, PGC-1α, PPARα, PRMT5
Introduction
The phenotypic spectrum of nonalcoholic fatty liver disease (NAFLD)3 is broad and may range from simple steatosis to steatohepatitis, advanced fibrosis, and cirrhosis. NAFLD is commonly associated with type 2 diabetes, visceral obesity, and hyperlipidemia (1). There is evidence that increased delivery of nonesterified fatty acid to the liver (mainly from white adipose tissue (WAT)) is fundamental to the development of NAFLD (2, 3). The role of diet in the pathogenesis of NAFLD has been investigated both in humans and in animal models (4–6). Subjects on a high-fat diet develop fatty liver (5, 6); those on a low-fat/high-carbohydrate diet develop fatty liver via increased de novo fatty acid synthesis (4). In addition, high dietary fat intake is associated with obesity and insulin resistance. As a consequence of insulin resistance, suppression of lipolysis by insulin is impaired, leading to increased nonesterified fatty acid delivery to the liver (7, 8). Thus, high dietary fatty acids have profound effects on insulin resistance, NAFLD, and cardiovascular disease (9).
Phosphatidylinositol 3-kinase (PI3K), a lipid kinase, generates the second messenger phosphatidylinositol 1,4,5-trisphosphate (10). The accumulation of phosphatidylinositol 1,4,5-trisphosphate recruits protein kinase B (AKT) to the cell membrane, where it is phosphorylated and activated by phosphoinositide-dependent kinase 1 and 2 (11, 12). Downstream of this PI3K/AKT pathway is mTOR, a major effector that is specifically implicated in the regulation of cell growth via nutrient availability and cellular bioenergetics (13). Activation of the PI3K–AKT–mTOR pathway is accompanied by aberrant lipid metabolism in cancers (14), where it increases the protein levels of sterol regulatory element–binding proteins (SREBPs) and its target, fatty acid synthase (FASN) (15, 16). Inhibition of the mTOR pathway is associated with impaired induction of transcription factors, i.e. peroxisome proliferator activated receptor α (PPARα) and its target genes, such as carnitine palmitoyltransferase 1 (CPT1). Under conditions favoring re-esterification over β-oxidation in the liver, newly synthesized triglycerides are carried on very-low-density lipoproteins (VLDLs) to be redistributed to WAT for storage or to skeletal muscle for utilization (under exercise conditions). Inhibition of PI3K/AKT can abolish insulin-induced suppression of VLDL assembly and secretion by regulating the degradation of apolipoprotein B (ApoB) and the gene expression of hepatic microsomal triglyceride transfer protein (17). Thus, the PI3K–AKT–mTOR signaling pathway plays an integral role in regulating several pathways contributing to lipid homeostasis in the liver, including de novo lipid biosynthesis, fatty acid β-oxidation (18, 19), and VLDL assembly and secretion.
The protein arginine methyltransferase family consists of three subfamilies that differ in their ability to carry out monomethylation or asymmetric dimethylation (type I), monomethylation or symmetric dimethylation (type II), or exclusively monomethylation (type III) (20, 21). PRMT5 functions as the major type II arginine methyltransferase that symmetrically dimethylates arginine residues on histones as well as nonhistone proteins, including RNA splicing factors and cell cycle regulators (22, 23). PRMT5-mediated methylation of histones H2AR3 (24), H4R3 (25), and H3R8 (26) regulates chromatin structure and influences transcription. We have shown that PRMT5 is required for activation of adipogenic gene expression by promoting adipogenic master transcriptional factor PPARγ2 expression during pre-adipocyte differentiation (27, 28). PRMT5 has been shown to methylate SREBP1a on Arg-321 and increases SREBP1a transcriptional activity, thereby promoting de novo lipogenesis and tumor growth (29). Overexpressing exogenous GFP-PRMT5 resulted in elevated levels of phospho-AKT in 293T cells (30). However, little is known about the role of endogenous PRMT5 in regulating lipid homeostasis in the context of NAFLD.
In this study, we investigated the function of PRMT5 in a diet-induced obesity mouse model. We found that a high-fat diet increased PRMT5 expression in the liver but not in other metabolically relevant tissues. Hepatic PRMT5 expression, however, was not altered in the liver of the obese Agouti mouse, suggesting that high-fat diet consumption, but not obesity per se, induced PRMT5 expression. Global signaling pathway analysis showed that knockdown of PRMT5 attenuated PI3K/AKT signaling. In the nontransformed mouse liver cell line model α mouse liver 12 (AML12), reducing PRMT5 levels by shRNA or inhibiting its enzymatic activity with a small-molecule inhibitor led to an increase in PPARα and PGC-1α levels and mitochondrial biogenesis. Our results suggest that inhibiting PRMT5 favors β-oxidation, which would contribute to the reversal of hepatic steatosis.
Results
PRMT5 is ubiquitously expressed in mice
Although PRMT5 is ubiquitously expressed at varying levels in all mouse tissues examined, its expression is most abundant in the spleen and kidneys, and it is expressed in tissues important for metabolic control, such as liver, muscle, brown adipose tissue, and inguinal WAT (Fig. 1A). Similarly, PRMT5 was detected in the cytoplasm as well as in the nucleus of the nontransformed AML12 murine hepatocyte cell line (Fig. 1B).
Figure 1.
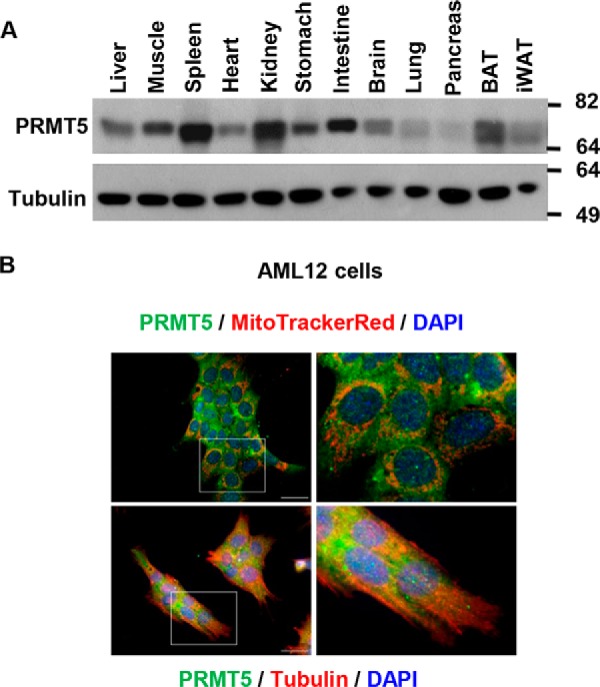
PRMT5 is ubiquitously expressed in mouse tissues. A, PRMT5 protein levels in mouse tissues were determined by Western blot analysis. BAT, brown adipose tissue; iWAT, inguinal WAT. B, localization of PRMT5 in the mouse AML12 liver cell line. PRMT5 expression was visualized by immunofluorescent staining (green), the cytoskeleton was labeled by immunofluorescent staining of tubulin (red), and nuclei were stained by DAPI (blue). Scale bars = 200 μm. All images represent three independent experiments.
A high-fat diet increases hepatic PRMT5 expression
PRMT5 mRNA and protein levels were significantly increased in the livers of mice fed a high-fat diet (HFD) compared with those fed a regular diet (RD), as assessed by Western blot analysis (Fig. 2A), quantitative PCR (Fig. 2B), and immunofluorescent staining of liver sections (Fig. 2C). In contrast, the high-fat diet did not modify PRMT5 expression in other metabolically relevant tissues, such as muscle and brown and inguinal white adipose tissue (Fig. 2D). To further elucidate the role of PRMT5 in hepatic steatosis, we asked whether PRMT5 up-regulation in hepatocytes is associated with obesity by examining PRMT5 levels in the livers of Agouti mice. Yellow Agouti mice with dominant mutations at the Agouti locus develop obesity, hyperinsulinemia, insulin resistance, hyperglycemia, hyperleptinemia, and hepatic steatosis in adulthood (31–33). Young Agouti mice are lean, but adult animals are obese even when they are fed regular chow (32). Therefore, the obesity in adult Agouti mice is independent of a high-fat diet. Unlike the diet-induced obesity model, PRMT5 protein levels were not different in the livers of obese 5-month-old versus normal weight 1-month-old Agouti mice (Fig. 2E). This suggested that the increase of PRMT5 was not associated with obesity per se but with high-fat diet–induced obesity.
Figure 2.
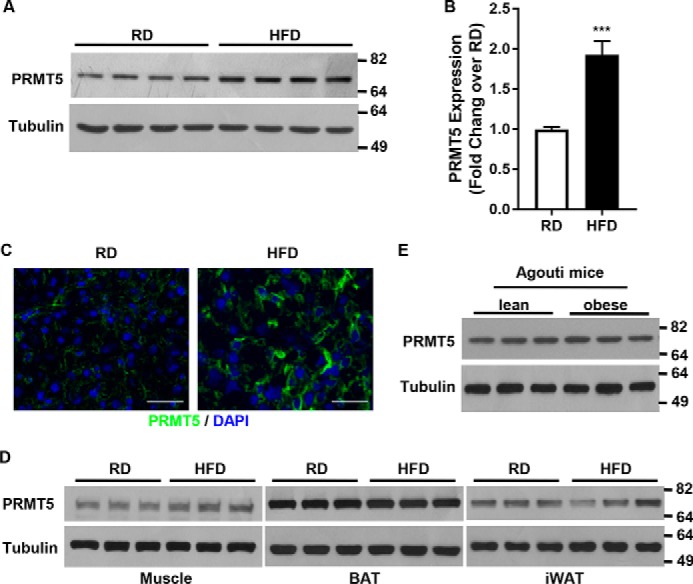
A high-fat diet increases hepatic PRMT5 expression. A and B, PRMT5 protein (A) and mRNA (B) levels in livers of mice fed RD or HFD were examined by Western blot analysis or qRT-PCR. Total protein or RNA was extracted from livers of 4-month-old male mice after 12 weeks of feeding with RD or HFD, respectively. C, PRMT5 expression in liver sections of the above samples were analyzed by immunofluorescent staining of PRMT5 (green) and DAPI nuclear staining (blue). Scale bars = 200 μm. D, PRMT5 levels in muscle, brown, or white adipose tissues from mice fed a normal diet or high-fat diet were analyzed by Western blotting. BAT, brown adipose tissue; iWAT, inguinal WAT. E, PRMT5 expression in the livers of lean or obese Agouti mice was measured by Western blotting. Total protein was extracted from livers of RD-fed 1- or 5-month-old Agouti mice. ***, p < 0.001, two-tailed Student's t test.
PRMT5 gene silencing decreases PI3K/AKT signaling
We sought to dissect the molecular mechanism underlying the regulation of hepatic lipid homeostasis by PRMT5. Because PRMT5 is overexpressed in many cancer cell lines (29, 34–36), we generated PRMT5 knockdown in the nontransformed mouse liver cell line AML12 to avoid confounding metabolic effects of cancer-related factors in transformed cell lines. As shown in Fig. 3A, reduction of PRMT5 expression was achieved by shRNA-mediated gene silencing. A global signaling network analysis measuring 70 key proteins involved in more than 20 signaling pathways showed that PRMT5 knockdown decreased the PI3K–AKT pathway (Fig. 3B). The reduction of phospho-AKT Ser-473 was detected in the array (Fig. S1), which was confirmed by a Western blot analysis that also showed decreased Thr-308 phosphorylation on AKT in PRMT5 knockdown cells (Fig. 3C). To confirm this finding, we utilized the inducible PRMT5 knockout mouse embryonic fibroblast (MEF) cell line, which harbors an estrogen receptor (ER)-Cre directed PRMT5flox/flox allele (37). When treated with 4-hydroxytamoxifen (4-OHT), the Cre recombinase translocates to the nucleus and excises an exon of PRMT5 to disrupt its expression. The PRMT5flox/flox MEF cells were cultured in the presence of 2 μm 4-OHT for 10 days, followed by DMEM for 3 days without 4-OHT. The efficiency of PRMT5 knockout was confirmed by Western blot analysis (Fig. 3D). In this PRMT5 knockout model, complete loss of PRMT5 led to a decrease in phospho-AKT Thr-308 and Ser-473. In contrast, up-regulation of phospho-AKT Ser-473 and Thr-308 was detected in the livers of mice fed a high-fat diet (Fig. 3E).
Figure 3.

Silencing PRMT5 decreases PI3K/AKT signaling. A, Western blot analysis of PRMT5 in AML12 cells following lentiviral shRNA knockdown of PRMT5. B, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially expressed proteins with PRMT5 knockdown. Total protein extracted from scramble or PRMT5 knockdown cells was analyzed for the expression of key proteins involved in more than 20 signaling pathways. Signals were normalized to α- and β-tubulin. Results were obtained from two independent experiments performed in duplicate. C and D, reduction of phospho-AKT Thr-308 and Ser-473 was observed in PRMT5 knockdown liver cells (C) as well as PRMT5 knockout MEF cells (D). KO, knockout. E, a high-fat diet up-regulated the hepatic expression of phospho-AKT Thr-308 and Ser-473. Total protein was extracted from livers of 4-month-old male mice fed either a normal or high-fat diet for 12 weeks.
PRMT5 regulates the expression of master transcription factors of mitochondrial biogenesis
PPARα has a central role in fatty acid β-oxidation, lipid and lipoprotein metabolism, the inflammatory response, and oxidative stress in the liver. Activation of PPARα induces expression of mitochondrial genes in the oxidation pathways (38). Peroxisome proliferator–activated receptor γ, coactivator 1α (PGC-1α) is a transcriptional co-activator that regulates the expression of genes involved in energy metabolism. It is the master regulator of mitochondrial biogenesis (39–41). We surveyed the expression of these transcription factors in PRMT5 knockdown cells. Silencing PRMT5 significantly increased the expression of PPARα and PGC-1α (Fig. 4, A and B). These results were confirmed in PRMT5 knockout MEF cells (Fig. 4C). Consistent with increased expression of PPARα and PGC-1α, the expression of mitochondrial biogenesis genes such as CytoC and Cox7a-1 was elevated in PRMT5 knockdown cells (Fig. 4D). These results were replicated in PRMT5 knockout MEF cells, where expression of these genes was up-regulated (Fig. 4E) and mitochondrial staining was significantly increased (Fig. 4F). Activation of PI3K/AKT signaling has been shown to induce the expression of SREBP1(42, 43). Liu et al. (29) showed that, in the liver cancer cell line HepG2, silencing PRMT5 decreased SREBP1a translation but not transcription. We examined, in PRMT5 knockdown cells, the expression of acetyl-CoA carboxylase and FASN, the direct downstream targets of SREBP1 in de novo fatty acid synthesis. The levels of these proteins were not affected upon silencing PRMT5 (Fig. S2), which suggested that de novo fatty acid synthesis is not perturbed by PRMT5 silencing. It has been shown that insulin regulates lipid deposition in the goose liver by activating PI3K/AKT/mTOR signaling, which suppresses VLDL assembly and secretion (44). VLDL assembly involves transfer of lipids by the microsomal triglyceride transfer protein (MTP) to ApoB and fusion of ApoB-containing precursor particles with triglyceride droplets to form mature VLDL (45). Although PRMT5 knockdown in mouse liver cells reduced PI3K/AKT signaling, it did not affect phospho-mTOR or ApoB and MTP gene expression (Fig. S2). In contrast, in PRMT5 knockout MEF cells, loss of PRMT5 significantly decreased the levels of phospho-AKT but also had no effect on the expression of phospho-mTOR (Fig. S3).
Figure 4.

PRMT5 regulates master transcription factors of fatty acid metabolism and mitochondrial biogenesis. A, qRT-PCR analysis of the mRNA levels of PRMT5, PPARα, and PGC-1α in AML12 cells expressing either a scrambled sequence shRNA or shRNA-PRMT5. B, Western blot analysis of PPARα in scramble and shRNA-PRMT5 AML12 cells. C, Western blot analysis of PRMT5 and PPARα in WT MEF and PRMT5 knockout MEF cells. KO, knockout. D, expression of mitochondrial biosynthesis genes by qRT-PCR in scramble or shRNA-PRMT5 cells. E, relative mRNA expression of mitochondrial biosynthesis genes measured by qRT-PCR in WT MEF and PRMT5 knockout MEF cells. F and G, mitochondria in PRMT5flox/flox WT or knockout MEF (F) and AML12 cells with scramble or PRMT5 knockdown (G) were stained with the mitochondrion-selective dye MitoTrackerTM Red CMXRos and nuclei were labeled with DAPI (blue). Scale bars = 200 μm. Each column represents the mean of three independent experiments performed in triplicate. Error bars represent standard deviations. **, p < 0.01; ***, p < 0.001; two-tailed Student's t test.
PRMT5 enzymatic activity is required for the regulation of PGC-1α and PPARα expression
Next we asked whether the enzymatic activity of PRMT5 is required for its regulation of PGC-1α and PPARα expression. To address this question, a WT PRMT5 or a catalytically dead PRMT5 (G367A,R368A) double mutant (46) was introduced into AML12 cells by transient transfection (Fig. 5A). As opposed to the enzyme-dead mutant, the expression of exogenous WT PRMT5 elevated phospho-AKT Thr-308 and Ser-473 and markedly reduced PPARα (Fig. 5B). EPZ015666 (GSK3235025) is a potent and selective small-molecule inhibitor targeting the enzymatic activity of PRMT5 with an IC50 of 22 nm in biochemical assays (47). Although EPZ015666 decreases cell proliferation in various cancer lines (48, 49), it did not inhibit proliferation in AML12 cells, not even in the presence of high-dose EPZ015666 for 8 weeks (Fig. S4). On the contrary, EPZ015666 treatment markedly decreased phospho-AKT Thr-308 and Ser-473 but increased PPARα in a dose-dependent manner (Fig. 5C). The expression of PPARα and PGC-1α, the master regulators of mitochondrial biogenesis, was elevated in the presence of EPZ015666 in a dose-dependent manner (Fig. 5D) with concomitantly increased density in mitochondrial staining (Fig. 5E). Inhibition of PRMT5 enzymatic activity did not change the phosphor-mTOR levels in AML12 cells (Fig. S5). As a biological consequence of increasing mitochondrial biogenesis and promoting fatty acid β-oxidation, lipid accumulation in cells treated with the PRMT5 inhibitor was significantly reduced (Fig. 5F). Collectively, these results suggest that the enzymatic activity of PRMT5 is required to execute its function in metabolic pathways.
Figure 5.

PRMT5 enzymatic activity is required for its regulation of PPARα expression. A, a FLAG-tagged WT or enzyme-dead (G367A,R368A) double PRMT5 mutant was introduced to AML12 cells by transient transfection. The expression of exogenous proteins was examined by Western blot analysis using anti-FLAG antibody. The total levels of PRMT5 were measured by Western blotting against anti-PRMT5 antibody. B, AKT phosphorylation and expression of PPARα in WT or mutant PRMT5-expressing cells were analyzed by Western blotting. C, protein levels of phospho-AKT and PPARα were determined by Western blotting in AML12 cells treated with DMSO or increasing doses of the PRMT5 inhibitor EPZ015666. D, the relative mRNA of PPARα and PGC-1α was measured by qRT-PCR in AML12 cells treated with DMSO or increasing doses of EPZ015666. E, AML12 cells treated with DMSO or 100 nm EPZ015666 were stained by MitoTrackerTM Red CMXRos (red), and nuclei were labeled with DAPI (blue). Scale bars = 200 μm. ***, p < 0.001; two-tailed Student's t test. F, AML12 cells pretreated with DMSO or 100 nm EPZ015666 were exposed to medium containing 1 μm palmitic acid and 100 μm oleic acid for 5 days. Intracellular lipids were stained with Oil Red O.
PRMT5 regulates PGC-1α and PPARα through phospho-AKT signaling
We sought to dissect whether PRMT5 directly regulates the expression of PPARα and PGC-1α or indirectly mediates this through phospho-AKT signaling. By analyzing the published PRMT5 ChIP-seq data (GEO: GSE75739) (50), we failed to identify significant PRMT5 binding above the IgG control at these gene loci (Fig. S6). Introducing an exogenous activated AKT (Myr-AKT) to PRMT5 WT or knockout cells attenuated the increased expression of PPARα and PGC-1α because of the loss of PRMT5 (Fig. 6A). This observation was confirmed in AML12 hepatocytes, where the up-regulation of PPARα and PGC-1α in PRMT5 knockdown cells was blocked by overexpression of Myr-AKT (Fig. 6B). These results suggested that the regulation of PPARα and PGC-1α by PRMT5 was mediated through phospho-AKT signaling, although we could not completely rule out the possibility that PRMT5 might directly regulate these genes.
Figure 6.
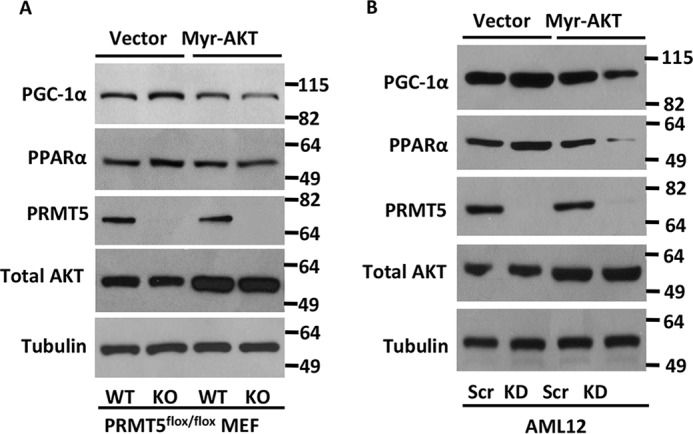
PRMT5 regulates PPARα and PGC-1α expression via AKT signaling. A, PRMT5 flox/flox WT or knockout MEF cells were transfected with vector or Myr-AKT and harvested 48 h post-transfection. Protein samples were analyzed by Western blotting for PPARα and PGC-1α expression. KO, knockout. B, AML12 cells were transduced with scramble or shPRMT5 viral supernatant for 24 h and then selected in medium containing 2 μg/ml puromycin for 48 h. Then cells were transfected with vector or Myr-AKT and harvested 48 h post-transfection. Protein samples were analyzed by Western blotting for PPARα and PGC-1α expression. KD, PRMT5 knockdown.
Discussion
In this study we showed that high-fat diet–induced obese mice have increased levels of PRMT5 in the liver but not in other metabolically active organs, such as adipose tissues and muscle. We found that this elevated hepatic PRMT5 is associated with a high-fat diet rather than obesity per se. It has been shown that HFD dramatically changed chromatin structure, transcription factor binding, and the gene expression landscape in the mouse liver. Many genes associated with lipid, steroid, cholesterol, and amino acid metabolism, gluconeogenesis, the inflammatory response, oxidative stress, and oxidoreductase activity were altered. It is not clear whether up-regulation of PRMT5 is one of the early events that further shape the chromatin environment and impact gene expression under HFD or whether it is regulated by other key transcription factors involved in this process.
PI3K/AKT signaling plays a critical role in regulating diverse cellular functions, including metabolism, growth, proliferation, cell survival, transcriptional activity, and protein synthesis. Dysregulation of the PI3K/AKT pathway is implicated in cancer, type 2 diabetes, insulin resistance, inflammatory and autoimmune disorders, and cardiovascular and neurological diseases (12). The comprehensive understanding of the AKT signaling network has been expanded with the identification of numerous downstream pathways (49–51). In contrast, our knowledge of the upstream regulator or activator of AKT is limited. Wei et al. (30) reported that overexpressing GFP-PRMT5 led to an increase in phospho-AKT levels in 293T cells. However, this observation was not further validated with the endogenous protein (30). In this study, we identified endogenous PRMT5, an epigenetic modifier, as the upstream regulator of AKT activation in the context of high fat diet-induced fatty liver. Wei et al. (30) also showed that overexpressing GFP-PRMT5 increased mTOR Ser-2448 phosphorylation, which might be the mechanism by which PRMT5 regulates phospho-AKT. We did not detect any change in phospho-mTOR Ser-2448 levels in PRMT5 knockout MEF cells or in AML12 cells with PRM5 knockdown or treated with EPZ015666 (Figs. S2, S3, and S5). In this study, PRMT5 regulation of phospho-AKT appears to be independent of mTOR signaling. AKT activation involves the phosphorylation of two residues, Thr-308 in the activation loop and Ser-473 in the C-terminal hydrophobic motif. Phosphorylation of Ser-473 has been extensively studied in cancers as a marker for full AKT activity (52–54). Other studies also showed that Thr-308 is predominantly phosphorylated in activated AKT kinase in tumor cells (55–57). In our study, knockdown of PRMT5 or treatment with the PRMT5 enzymatic inhibitor EPZ015666 in hepatocytes attenuated both phospho-AKT Thr-308 and Ser-473. Our results indicate that targeting PRMT5 blocks the activation of AKT, which may have a profound effect on its downstream signaling networks.
Mitochondrial dysfunction is associated with many diseases, such as type 2 diabetes and neurodegenerative diseases (58, 59). PGC-1α is a master regulator of mitochondrial biogenesis by activating the transcription factors nuclear respiratory factor 1 and nuclear respiratory factor 2. These induce mitochondrial transcription factor A, which drives the transcription and replication of mitochondrial DNA (59, 60). PGC-1α expression levels are regulated in response to a plethora of stimuli, such as exercise, feeding, fasting, and cold exposure, and modulated by numerous factors, such as Ca2+/calmodulin–dependent protein kinase IV (CaMKIV) and calcineurin A, AMP-activated protein kinase (AMPK), and p38 mitogen-activated protein kinase (p38 MAPK) (61, 62). We here report that PRMT5 repressed the expression of PGC-1α in hepatocytes. Silencing or complete loss of PRMT5 increased the expression of PGC-1α and mitochondrial biogenesis. AKT was found to phosphorylate and inhibit PGC-1α (63). PRMT5 ChIP-seq in MDA-MB-231 cells did not identify PRMT5 binding sites at the gene loci of PPARα and PGC-1α (50). With no PRMT5 ChIP-seq data from hepatocytes, we cannot completely dismiss the possibility that PRMT5 might have a direct role in regulating these genes. Introducing activated Myr-AKT to PRMT5 knockout MEF or knockdown AML12 cells decreased the up-regulation PPAR α and PGC-1α. These results suggest that it is more likely that PRMT5 regulates the expression of PPARα and PGC-1α via AKT signaling.
Overexpression of PRMT5 in various cancers has been linked to cancer progression and poor prognosis (34, 64). Therefore, PRMT5 is well-recognized as a viable therapeutic target in cancer treatment, and a number of inhibitors have been developed within the past couple of years to target cancer, β-thalassemia, or sickle cell anemia. EPZ015666 is a potent PRMT5 inhibitor that has been shown to be active in vivo (47). A first time in human, open-label, dose escalation study of GSK3326595, a potent and selective PRMT5 inhibitor similar to EPZ015666, in subjects with advanced or recurrent solid tumors as well as subjects with a subset of solid tumors and non-Hodgkin's lymphoma was approved by the Food and Drug Administration and started in August 2016 (ClinicalTrials.gov identifier NCT02783300). We now report that inhibiting PRMT5 by EPZ015666 markedly decreased both phospho-AKT Thr-308 and phospho-AKT Ser-473, up-regulated PPARα and PGC-1α expression, and significantly increased mitochondrial biogenesis. Excessive intake of dietary lipids was shown to decrease hepatic PPARα and PGC-1α expression with a reduced numerical density of hepatic mitochondria (65). A number of PPARα agonists have been shown to enhance hepatic mitochondrial biogenesis and increase fatty acid β-oxidation in diet-induced obese mice (66, 67). As an upstream regulator of AKT, PPARα, and PGC-1α, PRMT5 might be a valuable target to treat fatty liver via multiple mechanisms or may be even more broadly applied to other diseases that are associated with mitochondrial dysfunction. Chen et al. (68) reported that PRMT5 methylates GATA 4 and inhibits its transcriptional activity in cardiomyocytes, which is important in prevention of cardiac hypertrophy and heart failure. Thus, the balance of PRMT5 levels in the context of different diseases needs to be considered when developing PRMT5 inhibition–based therapies.
Experimental procedures
Animals
C57BL/6 (BL6) mice were housed in a temperature-controlled environment with a 12-h light, 12-h dark cycle and fed a standard chow ad libitum. Starting at 8 weeks of age, male mice were fed ad libitum either an RD (12% fat content) or HFD (45% fat content, catalog no. D12451, Research Diets, New Brunswick, NJ) for 3 months (69). Animals were then euthanized, and tissue samples were collected to be either snap-frozen in liquid nitrogen or embedded in optimal cutting temperature (OCT) compound blocks. The Agouti mice were housed in a temperature-controlled environment with a 12-h light, 12-h dark cycle and fed a standard chow ad libitum. All procedures were approved by the Institutional Animal Care and Utilization Committee at the University of Toledo.
Cell culture
The nontransformed α mouse liver 12 (AML12) cell line was provided by Dr. Anthony Imbalzano (University of Massachusetts Medical School). This cell line was established from hepatocytes from a mouse (CD1 strain, line MT42) transgenic for human transforming growth factor α (70). AML12 cells retain the capacity to express high levels of serum- and liver-specific proteins (70). These cells were maintained in monolayers, as described previously (70). They were cultured in DMEM/F12 medium (Invitrogen) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 μg/ml streptomycin, insulin (5 μg/ml), transferrin (5 μg/ml) and sodium selenite (5 ng/ml) (ITS) (Invitrogen). Human embryonic kidney 293T cells were cultured in DMEM (Invitrogen) with 10% fetal bovine serum and 100 μg/ml streptomycin. PRMT5flox/flox MEFs were immortalized PRMT5flox/flox mouse embryonic fibroblasts and obtained from Dr. Mark Bedford (University of Texas MD Anderson Cancer Center).
Lentiviral shRNA construction and transduction
PRMT5 shRNA knockdown lentiviral constructs were prepared using the psp-108 vector (Addgene), and the lentivirus particles were produced by co-transfection with plasmids pMD2.G (Addgene) and psPAX2 (Addgene) into human embryonic kidney 293T cells. Titers were determined, and the same number of virus was used, as indicated. PRMT5 knockdown targeting sequences were as follows: shRNA1, 5′GCACAGTTTGAGATGCCTTAT3′; shRNA2, 5′CCTCTTGTGAATGCGTCTCTT3′. The FLAG-PRMT5 and FLAG-PRMT5 (G367A,R368A) double mutant were from Dr. Said Sif (Qatar University) (46).
Transient transfection
Cells were plated in 6-well plates or 60-mm dishes to 60–70% confluency and then transfected with 6 or 15 μg of plasmid DNA, respectively, using Lipofectamine 2000 transfection reagent, following the manufacturer's protocol. Cells were harvested 48 h post-transfection for protein analysis by Western blotting.
Immunofluorescence
Cells were seeded on glass coverslips and incubated overnight. For mitochondrion staining, cells were incubated with MitoTrackerTM Red CMXRos (250 nm, Thermo Fisher, catalog no. M7512) at 37 °C for 30 min before being fixed with 4% paraformaldehyde for 20 min at room temperature, followed by permeabilization in ice-cold methanol for 10 min at −20 °C. Cells were then incubated in blocking buffer (5% BSA in PBS) for 1 h at room temperature, followed by incubation with anti-PRMT5 antibody (Santa Cruz Biotechnology, catalog no. sc-376937) and anti-β-tubulin antibody (Cell Signaling Technology, catalog no. 15115) at 4 °C overnight. Cells were washed three times for 5 min in PBS and then incubated for 2 h with Alexa Fluor 488–conjugated goat anti-mouse secondary antibody against PRMT5 and Alexa Fluor 555–conjugated goat anti-rabbit secondary antibody for β-tubulin (diluted 1:100 in blocking buffer, Invitrogen) at room temperature. Finally, the nuclei were stained with DAPI for 30 min at room temperature before visualization.
Formalin-fixed and paraffin-embedded tissue sections were deparaffinized and rehydrated through graded ethanol solutions. The slides were incubated with blocking buffer (PBS containing 5% normal goat serum and 0.3% Triton X-100) for 60 min and then the slides were incubated with PRMT5 antibody at 4 °C overnight. Subsequently, the slides were washed with PBS, and incubated with Alexa Fluor 488-conjugated goat anti-mouse secondary antibody and DAPI for 2 h before observation using a Leica fluorescence microscope.
RT-qPCR
Total RNA was extracted from AML12 cells or MEF cells using TRIzol regent (Invitrogen), and cDNA was synthesized with the SuperScript Reverse Transcriptase III kit (Invitrogen) according to the manufacturer's instructions. The cDNA was amplified in 96-well reaction plates with SYBR Green PCR Master Mix (Applied Biosystems) on an ABI 7500 real-time PCR thermocycler. The sequences of forward and reverse primers are listed in Table S1. The relative levels of target transcripts were calculated from duplicate samples after normalization against the U36 housekeeping gene. Dissociation curve analysis was performed after PCR amplification to confirm primer specificity. Relative mRNA expression was calculated using the ΔΔCT method.
Western blot analysis
Tissues or cells were washed with cold PBS and lysed in 1% NP40 lysis buffer containing a protease and phosphatase inhibitor mixture (Roche Diagnostics). Proteins were analyzed by SDS-PAGE, followed by immunoprobing with the following antibodies: PPARα, sc-398394; PRMT5, sc-376937; acetyl-CoA carboxylase α, sc-137104; FASN, sc-48357; mTOR, sc-293089; and p-mTOR, sc-293133. For normalization, monoclonal antibodies against β-tubulin (Developmental Studies Hybridoma Bank, E7-S) were used. Blots were incubated with horseradish peroxidase–conjugated anti-goat IgG (Santa Cruz Biotechnology) and anti-mouse or anti-rabbit IgG (GE Healthcare) antibodies, and proteins were detected by ECL (GE Healthcare).
Fatty acid treatments
Cells were pretreated with increasing doses of EPZ015666 for 6 days, seeded at a density of 10,000 cells/well in 24-well plates, and cultured for 24 h. Palmitic acid (Sigma-Aldrich, catalog no. P0500) and oleic acid (Sigma-Aldrich, catalog no. O1008) were prepared by conjugation with nonfat BSA (Sigma-Aldrich, catalog no. A8806). Cells at 50% confluency were exposure to 1 μm palmitic acid and 100 μm oleic acid in the presence of EPZ015666 for 5 days. Lipid droplets were stained with Oil Red O.
Oil Red O staining
To assess intracellular neutral lipid, dietary fatty acids-treated cells were fixed using 10% formalin and stained using Oil Red O (Sigma-Aldrich) for 30 min at room temperature. Stained cells were washed with water and rinsed with 60% isopropanol. Lipid accumulation was visualized using light microscopy.
MTT assay
Cell proliferation was measured using the MTT assay. Three thousand cells from each treated group were plated in a 96-well plate. Cell proliferation was measured at 24h intervals by adding MTT solution (final concentration, 5 μg/ml) to each well and incubating for 4 h. After removal of the medium, the plate was air-dried, and 100 μl of DMSO was added. The plate was incubated at room temperature for 30 min with gentle shaking. Absorbance was measured at A540 in a Synergy H4 Hybrid microplate reader (Bio Tek, Winooski, VT).
ActivSignal IPAD assay
Cells were lysed in PBS + 1% NP40 lysis buffer and submitted to ActivSignal for further processing. The expression or phosphorylation of 70 key proteins involved in more than 20 signaling pathways was analyzed by antibodies with high specificity and sensitivity in combination with two distinct antibodies per target. The signal was normalized to the expression of housekeeping genes. Each pathway was covered by multiple targets. The experiment was performed twice.
KEGG pathway enrichment analysis
Proteins with more than a 1.5-fold change after PRMT5 knockdown in immuno-paired–antibody detection (IPAD) assay were fetched to perform pathway enrichment analysis using DAVID (71). Pathway annotations from the KEGG pathway database with a p value smaller than 0.05 were defined as significantly enriched (72).
Author contributions
L. H., J. L., X.-O. Z., and Q. W. data curation; L. H., X.-O. Z., and Q. W. formal analysis; L. H., J. L., K. S., S. M. N., and Q. W. investigation; L. H., J. L., and Q. W. methodology; S. M. N. and M. M. L. funding acquisition; S. M. N. and Q. W. project administration; S. M. N., M. M. L., and Q. W. writing-review and editing; M. M. L. and Q. W. supervision; Q. W. conceptualization; Q. W. writing-original draft.
Supplementary Material
Acknowledgments
We thank Drs. Anthony Imbalzano, Scott Leblanc, Mark Bedford, and Yu Liu for invaluable comments to improve this manuscript.
This work was supported by University of Massachusetts Medical School internal funds and National Institutes of Health Grants R01-DK054254, R01-DK083850, and R01HL112248. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Table S1.
- NAFLD
- nonalcoholic fatty liver disease
- WAT
- white adipose tissue
- PI3K
- phosphatidylinositol 3-kinase
- SREBP
- sterol regulatory element–binding protein
- FASN
- fatty acid synthase
- PPAR
- peroxisome proliferator–activated receptor
- VLDL
- very-low-density lipoprotein
- shRNA
- short hairpin RNA
- HFD
- high-fat diet
- RD
- regular diet
- MEF
- mouse embryonic fibroblast
- 4-OHT
- 4-hydroxytamoxifen
- DMEM
- Dulbecco's modified Eagle's medium
- ChIP-seq
- ChIP sequencing
- cDNA
- complementary DNA
- DAPI
- 4′,6-diamidino-2-phenylindole
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolum bromide
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- RT-qPCR
- quantitative real-time PCR.
References
- 1. Byrne C. D., Olufadi R., Bruce K. D., Cagampang F. R., and Ahmed M. H. (2009) Metabolic disturbances in non-alcoholic fatty liver disease. Clin. Sci. 116, 539–564 10.1042/CS20080253 [DOI] [PubMed] [Google Scholar]
- 2. Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., and Parks E. J. (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 10.1172/JCI23621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lewis G. F., Carpentier A., Adeli K., and Giacca A. (2002) Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 23, 201–229 10.1210/edrv.23.2.0461 [DOI] [PubMed] [Google Scholar]
- 4. Hudgins L. C., Hellerstein M., Seidman C., Neese R., Diakun J., and Hirsch J. (1996) Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J. Clin. Invest. 97, 2081–2091 10.1172/JCI118645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim S. P., Ellmerer M., Van Citters G. W., and Bergman R. N. (2003) Primacy of hepatic insulin resistance in the development of the metabolic syndrome induced by an isocaloric moderate-fat diet in the dog. Diabetes 52, 2453–2460 10.2337/diabetes.52.10.2453 [DOI] [PubMed] [Google Scholar]
- 6. Westerbacka J., Lammi K., Häkkinen A. M., Rissanen A., Salminen I., Aro A., and Yki-Järvinen H. (2005) Dietary fat content modifies liver fat in overweight nondiabetic subjects. J. Clin. Endocrinol. Metab. 90, 2804–2809 10.1210/jc.2004-1983 [DOI] [PubMed] [Google Scholar]
- 7. Luyckx F. H., Lefebvre P. J., and Scheen A. J. (2000) Non-alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab. 26, 98–106 [PubMed] [Google Scholar]
- 8. Marceau P., Biron S., Hould F. S., Marceau S., Simard S., Thung S. N., and Kral J. G. (1999) Liver pathology and the metabolic syndrome X in severe obesity. J. Clin. Endocrinol. Metab. 84, 1513–1517 10.1210/jcem.84.5.5661 [DOI] [PubMed] [Google Scholar]
- 9. Zivkovic A. M., German J. B., and Sanyal A. J. (2007) Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 86, 285–300 10.1093/ajcn/86.2.285 [DOI] [PubMed] [Google Scholar]
- 10. Fresno Vara J. A., Casado E., de Castro J., Cejas P., Belda-Iniesta C., and González-Barón M. (2004) PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 30, 193–204 10.1016/j.ctrv.2003.07.007 [DOI] [PubMed] [Google Scholar]
- 11. Osaki M., Oshimura M., and Ito H. (2004) PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 9, 667–676 10.1023/B:APPT.0000045801.15585.dd [DOI] [PubMed] [Google Scholar]
- 12. Manning B. D., and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fingar D. C., Richardson C. J., Tee A. R., Cheatham L., Tsou C., and Blenis J. (2004) mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell Biol. 24, 200–216 10.1128/MCB.24.1.200-216.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lien E. C., Lyssiotis C. A., and Cantley L. C. (2016) Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. Recent Results 207, 39–72 [DOI] [PubMed] [Google Scholar]
- 15. Jeon T. I., and Osborne T. F. (2012) SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 23, 65–72 10.1016/j.tem.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krycer J. R., Sharpe L. J., Luu W., and Brown A. J. (2010) The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 21, 268–276 10.1016/j.tem.2010.01.001 [DOI] [PubMed] [Google Scholar]
- 17. Sidiropoulos K. G., Meshkani R., Avramoglu-Kohen R., and Adeli K. (2007) Insulin inhibition of apolipoprotein B mRNA translation is mediated via the PI-3 kinase/mTOR signaling cascade but does not involve internal ribosomal entry site (IRES) initiation. Arch. Biochem. Biophys. 465, 380–388 10.1016/j.abb.2007.06.029 [DOI] [PubMed] [Google Scholar]
- 18. Sipula I. J., Brown N. F., and Perdomo G. (2006) Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism 55, 1637–1644 10.1016/j.metabol.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 19. Soliman G. A. (2011) The integral role of mTOR in lipid metabolism. Cell Cycle 10, 861–862 10.4161/cc.10.6.14930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bedford M. T., and Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 10.1016/j.molcel.2008.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bedford M. T., and Richard S. (2005) Arginine methylation an emerging regulator of protein function. Mol. Cell 18, 263–272 10.1016/j.molcel.2005.04.003 [DOI] [PubMed] [Google Scholar]
- 22. Friesen W. J., Paushkin S., Wyce A., Massenet S., Pesiridis G. S., Van Duyne G., Rappsilber J., Mann M., and Dreyfuss G. (2001) The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell Biol. 21, 8289–8300 10.1128/MCB.21.24.8289-8300.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scoumanne A., Zhang J., and Chen X. (2009) PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 37, 4965–4976 10.1093/nar/gkp516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tee W. W., Pardo M., Theunissen T. W., Yu L., Choudhary J. S., Hajkova P., and Surani M. A. (2010) Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 24, 2772–2777 10.1101/gad.606110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fabbrizio E., El Messaoudi S., Polanowska J., Paul C., Cook J. R., Lee J. H., Negre V., Rousset M., Pestka S., Le Cam A., and Sardet C. (2002) Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645 10.1093/embo-reports/kvf136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pal S., Vishwanath S. N., Erdjument-Bromage H., Tempst P., and Sif S. (2004) Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol. 24, 9630–9645 10.1128/MCB.24.21.9630-9645.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. LeBlanc S. E., Konda S., Wu Q., Hu Y. J., Oslowski C. M., Sif S., and Imbalzano A. N. (2012) Protein arginine methyltransferase 5 (Prmt5) promotes gene expression of peroxisome proliferator-activated receptor γ2 (PPARγ2) and its target genes during adipogenesis. Mol. Endocrinol. 26, 583–597 10.1210/me.2011-1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. LeBlanc S. E., Wu Q., Lamba P., Sif S., and Imbalzano A. N. (2016) Promoter-enhancer looping at the PPARγ2 locus during adipogenic differentiation requires the Prmt5 methyltransferase. Nucleic Acids Res. 44, 5133–5147 10.1093/nar/gkw129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu L., Zhao X., Zhao L., Li J., Yang H., Zhu Z., Liu J., and Huang G. (2016) Arginine methylation of SREBP1a via PRMT5 promotes de novo lipogenesis and tumor growth. Cancer Res. 76, 1260–1272 10.1158/0008-5472.CAN-15-1766 [DOI] [PubMed] [Google Scholar]
- 30. Wei T. Y., Juan C. C., Hisa J. Y., Su L. J., Lee Y. C., Chou H. Y., Chen J. M., Wu Y. C., Chiu S. C., Hsu C. P., Liu K. L., and Yu C. T. (2012) Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 103, 1640–1650 10.1111/j.1349-7006.2012.02367.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Millar S. E., Miller M. W., Stevens M. E., and Barsh G. S. (1995) Expression and transgenic studies of the mouse agouti gene provide insight into the mechanisms by which mammalian coat color patterns are generated. Development 121, 3223–3232 [DOI] [PubMed] [Google Scholar]
- 32. Moussa N. M., and Claycombe K. J. (1999) The yellow mouse obesity syndrome and mechanisms of agouti-induced obesity. Obes. Res. 7, 506–514 10.1002/j.1550-8528.1999.tb00440.x [DOI] [PubMed] [Google Scholar]
- 33. Wolff G. L., Roberts D. W., and Mountjoy K. G. (1999) Physiological consequences of ectopic agouti gene expression: the yellow obese mouse syndrome. Physiol. Genomics 1, 151–163 10.1152/physiolgenomics.1999.1.3.151 [DOI] [PubMed] [Google Scholar]
- 34. Blanc R. S., and Richard S. (2017) Arginine methylation: the coming of age. Mol. Cell 65, 8–24 10.1016/j.molcel.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 35. Cantor J. R., and Sabatini D. M. (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898 10.1158/2159-8290.CD-12-0345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanamaluru D., Xiao Z., Fang S., Choi S. E., Kim D. H., Veenstra T. D., and Kemper J. K. (2011) Arginine methylation by PRMT5 at a naturally occurring mutation site is critical for liver metabolic regulation by small heterodimer partner. Mol. Cell Biol. 31, 1540–1550 10.1128/MCB.01212-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bezzi M., Teo S. X., Muller J., Mok W. C., Sahu S. K., Vardy L. A., Bonday Z. Q., and Guccione E. (2013) Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 27, 1903–1916 10.1101/gad.219899.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kersten S. (2014) Integrated physiology and systems biology of PPARα. Mol. Metab. 3, 354–371 10.1016/j.molmet.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dorn G. W. 2nd, Vega R. B., and Kelly D. P. (2015) Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29, 1981–1991 10.1101/gad.269894.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanchis-Gomar F., García-Giménez J. L., Gómez-Cabrera M. C., and Pallardó F. V. (2014) Mitochondrial biogenesis in health and disease: molecular and therapeutic approaches. Curr. Pharm. Des. 20, 5619–5633 10.2174/1381612820666140306095106 [DOI] [PubMed] [Google Scholar]
- 41. Valero T. (2014) Mitochondrial biogenesis: pharmacological approaches. Curr. Pharm. Des. 20, 5507–5509 10.2174/138161282035140911142118 [DOI] [PubMed] [Google Scholar]
- 42. Laplante M., and Sabatini D. M. (2010) mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 3281–3282 10.1073/pnas.1000323107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamauchi Y., Furukawa K., Hamamura K., and Furukawa K. (2011) Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 71, 4989–4997 10.1158/0008-5472.CAN-10-4108 [DOI] [PubMed] [Google Scholar]
- 44. Han C., Wei S., He F., Liu D., Wan H., Liu H., Li L., Xu H., Du X., and Xu F. (2015) The regulation of lipid deposition by insulin in goose liver cells is mediated by the PI3K-AKT-mTOR signaling pathway. PLoS ONE 10, e0098759 10.1371/journal.pone.0098759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shelness G. S., and Sellers J. A. (2001) Very-low-density lipoprotein assembly and secretion. Curr. Opin. Lipidol. 12, 151–157 10.1097/00041433-200104000-00008 [DOI] [PubMed] [Google Scholar]
- 46. Pal S., Yun R., Datta A., Lacomis L., Erdjument-Bromage H., Kumar J., Tempst P., and Sif S. (2003) mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol. Cell Biol. 23, 7475–7487 10.1128/MCB.23.21.7475-7487.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chan-Penebre E., Kuplast K. G., Majer C. R., Boriack-Sjodin P. A., Wigle T. J., Johnston L. D., Rioux N., Munchhof M. J., Jin L., Jacques S. L., West K. A., Lingaraj T., Stickland K., Ribich S. A., Raimondi A., et al. (2015) A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 11, 432–437 10.1038/nchembio.1810 [DOI] [PubMed] [Google Scholar]
- 48. Gullà A., Hideshima T., Bianchi G., Fulciniti M., Kemal Samur M., Qi J., Tai Y. T., Harada T., Morelli E., Amodio N., Carrasco R., Tagliaferri P., Munshi N. C., Tassone P., and Anderson K. C. (2018) Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 32, 996–1002 10.1038/leu.2017.334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kryukov G. V., Wilson F. H., Ruth J. R., Paulk J., Tsherniak A., Marlow S. E., Vazquez F., Weir B. A., Fitzgerald M. E., Tanaka M., Bielski C. M., Scott J. M., Dennis C., Cowley G. S., Boehm J. S., et al. (2016) MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 10.1126/science.aad5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rengasamy M., Zhang F., Vashisht A., Song W. M., Aguilo F., Sun Y., Li S., Zhang W., Zhang B., Wohlschlegel J. A., and Walsh M. J. (2017) The PRMT5/WDR77 complex regulates alternative splicing through ZNF326 in breast cancer. Nucleic Acids Res. 45, 11106–11120 10.1093/nar/gkx727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Franke T. F., Kaplan D. R., Cantley L. C., and Toker A. (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275, 665–668 10.1126/science.275.5300.665 [DOI] [PubMed] [Google Scholar]
- 52. David O., Jett J., LeBeau H., Dy G., Hughes J., Friedman M., and Brody A. R. (2004) Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin. Cancer Res. 10, 6865–6871 10.1158/1078-0432.CCR-04-0174 [DOI] [PubMed] [Google Scholar]
- 53. Pérez-Tenorio G., Stål O., and Southeast Sweden Breast Cancer Group (2002) Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br. J. Cancer 86, 540–545 10.1038/sj.bjc.6600126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tang J. M., He Q. Y., Guo R. X., and Chang X. J. (2006) Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer 51, 181–191 10.1016/j.lungcan.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 55. Gallay N., Dos Santos C., Cuzin L., Bousquet M., Simmonet Gouy V., Chaussade C., Attal M., Payrastre B., Demur C., and Récher C. (2009) The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23, 1029–1038 10.1038/leu.2008.395 [DOI] [PubMed] [Google Scholar]
- 56. Tsurutani J., Fukuoka J., Tsurutani H., Shih J. H., Hewitt S. M., Travis W. D., Jen J., and Dennis P. A. (2006) Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. J. Clin. Oncol. 24, 306–314 10.1200/JCO.2005.02.4133 [DOI] [PubMed] [Google Scholar]
- 57. Vincent E. E., Elder D. J., Thomas E. C., Phillips L., Morgan C., Pawade J., Sohail M., May M. T., Hetzel M. R., and Tavaré J. M. (2011) Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 104, 1755–1761 10.1038/bjc.2011.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Herst P. M., Rowe M. R., Carson G. M., and Berridge M. V. (2017) Functional mitochondria in health and disease. Front. Endocrinol. 8, 296 10.3389/fendo.2017.00296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chan D. C. (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252 10.1016/j.cell.2006.06.010 [DOI] [PubMed] [Google Scholar]
- 60. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., and Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 10.1016/S0092-8674(00)81410-5 [DOI] [PubMed] [Google Scholar]
- 61. Handschin C., and Spiegelman B. M. (2006) Peroxisome proliferator-activated receptor γ coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 27, 728–735 10.1210/er.2006-0037 [DOI] [PubMed] [Google Scholar]
- 62. Puigserver P., and Spiegelman B. M. (2003) Peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24, 78–90 10.1210/er.2002-0012 [DOI] [PubMed] [Google Scholar]
- 63. Li X., Monks B., Ge Q., and Birnbaum M. J. (2007) Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature 447, 1012–1016 10.1038/nature05861 [DOI] [PubMed] [Google Scholar]
- 64. Stopa N., Krebs J. E., and Shechter D. (2015) The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell. Mol. Life Sci. 72, 2041–2059 10.1007/s00018-015-1847-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Samuel V. T., and Shulman G. I. (2018) Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27, 22–41 10.1016/j.cmet.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Veiga F. M. S., Graus-Nunes F., Rachid T. L., Barreto A. B., Mandarim-de-Lacerda C. A., and Souza-Mello V. (2017) Anti-obesogenic effects of WY14643 (PPAR-α agonist): hepatic mitochondrial enhancement and suppressed lipogenic pathway in diet-induced obese mice. Biochimie 140, 106–116 10.1016/j.biochi.2017.07.003 [DOI] [PubMed] [Google Scholar]
- 67. Kim S. M., Lee B., An H. J., Kim D. H., Park K. C., Noh S. G., Chung K. W., Lee E. K., Kim K. M., Kim D. H., Kim S. J., Chun P., Lee H. J., Moon H. R., and Chung H. Y. (2017) Novel PPARα agonist MHY553 alleviates hepatic steatosis by increasing fatty acid oxidation and decreasing inflammation during aging. Oncotarget 8, 46273–46285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen M., Yi B., and Sun J. (2014) Inhibition of cardiomyocyte hypertrophy by protein arginine methyltransferase 5. J. Biol. Chem. 289, 24325–24335 10.1074/jbc.M114.577494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Al-Share Q. Y., DeAngelis A. M., Lester S. G., Bowman T. A., Ramakrishnan S. K., Abdallah S. L., Russo L., Patel P. R., Kaw M. K., Raphael C. K., Kim A. J., Heinrich G., Lee A. D., Kim J. K., Kulkarni R. N., et al. (2015) Forced hepatic overexpression of CEACAM1 curtails diet-induced insulin resistance. Diabetes 64, 2780–2790 10.2337/db14-1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu J. C., Merlino G., and Fausto N. (1994) Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. U.S.A. 91, 674–678 10.1073/pnas.91.2.674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kanehisa M., Furumichi M., Tanabe M., Sato Y., and Morishima K. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 10.1093/nar/gkw1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.