

Abstract
In response to changes in nutrient conditions, cells rearrange the composition of plasma membrane (PM) transporters to optimize their metabolic flux. Not only transcriptional gene regulation, but also inactivation of specific transporters is important for fast rearrangement of the PM. In eukaryotic cells, endocytosis plays a role in transporter inactivation, which is triggered by ubiquitination of these transporters. The Nedd4 family E3 ubiquitin ligase is responsible for ubiquitination of the PM transporters and requires that a series of α-arrestin proteins are targeted to these transporters. The mechanism by which an α-arrestin recognizes its cognate transporters in response to environmental signals is of intense scientific interest. Excess substrates or signal transduction pathways are known to initiate recognition of transporters by α-arrestins. Here, we identified an endocytic-sorting signal in the monocarboxylate transporter Jen1 from yeast (Saccharomyces cerevisiae), whose endocytic degradation depends on the Snf1-glucose signaling pathway. We found that the C-terminal 20-amino acid-long region of Jen1 contains an amino acid sequence required for association of Jen1 to the α-arrestin Rod1, as well as lysine residues important for glucose-induced Jen1 ubiquitination. Notably, fusion of this region to the methionine permease, Mup1, whose endocytosis is normally induced by excess methionine, was sufficient for Mup1 to undergo glucose-induced, Rod1-mediated endocytosis. Taken together, our results demonstrate that the Jen1 C-terminal region acts as a glucose–responding degron for α-arrestin-mediated endocytic degradation of Jen1.
Keywords: endocytosis, glucose, arrestin, ubiquitylation (ubiquitination), E3 ubiquitin ligase, protein degradation, degron, monocarboxylate transporter, nutrient regulation, plasma membrane transporter, Rsp5
Introduction
The plasma membrane (PM)2 is an organelle responsible for communication with the external environment. To maintain cellular homeostasis, the PM controls both inward and outward flux of compounds according to physiological needs. The PM transporters accomplish this task. To adapt the nutritional change, cells rearrange an optimal set of PM transporters by controlling their expression, localization, and degradation. Endocytosis contributes to the relocation and/or degradation of transporters in eukaryotic cells. In response to environmental cues, internalization of most PM transporters is triggered by ubiquitination catalyzed by the neuronal precursor cell expressed developmentally down-regulated protein 4 (Nedd4) family E3 ubiquitin ligase. Nedd4 proteins contain two to four WW domains that interact with Pro-Pro-X-Tyr (PPXY) substrate protein motifs (1). Because most PM proteins lack PPXY motifs, they require adaptor proteins for their endocytic degradation. Recently, it has been reported that α-arrestin family proteins, which contain PPXY motifs, play a role in recruiting Nedd4 family proteins to target PM transporters (2, 3). Saccharomyces cerevisiae has Rsp5 as the sole E3 ubiquitin ligase of the Nedd4 family. The yeast also has 14 α-arrestins, including 10 arrestin-related trafficking adaptors (ARTs) (2, 3), Bul1, -2, and -3 (4, 5), and Spo23 (6), each of which can selectively recognize a specific set of transmembrane cargos for endocytic degradation in response to a wide variety of external conditions. α-Arrestins confer the Rsp5-binding site on specific cargos, as well as control the timing and endocytic degradation rate of transporters by regulating the interaction between transporters and α-arrestins in response to environmental cues.
Many nutrient transporters undergo endocytic inactivation in response to excess substrates to limit their import. Recent studies demonstrate that conformational changes in transporters induced by substrate binding render the α-arrestin–Rsp5 complex accessible to transporters (7–9). In the absence of substrates (ground state), the amino (N)-terminal cytoplasmic region proximal to the first transmembrane spanning region is buried with the cytoplasmic loops of the main body of transporters. Upon substrate binding, a transporter changes its conformation to expose α-arrestin–Rsp5 complex-binding sites, which are located in the N-terminal region and cytoplasmic loops (7, 9). In the case of methionine-induced endocytosis of Mup1, an acidic patch in Mup1 N-tail plays an important role in the recognition of Mup1 by the Ldb19/Art1–Rsp5 complex, which then ubiquitinates the neighboring lysine residues (9).
Some transporters are down-regulated by challenge with nonsubstrate compounds. Import of pyruvate and lactate into yeast cells is mediated by the plasma membrane transporter Jen1, whose expression and localization at the PM are tightly regulated by glucose availability (10–13). Glucose-induced inactivation of Jen1 requires the α-arrestin, Rod1/Art4. Activity of Rod1 is regulated by the phosphorylation state, which is modulated by Snf1 kinase and type 1 protein phosphatase (Glc7-Reg1) (14, 15). In the absence of glucose, Rod1 is phosphorylated by Snf1 kinase, which facilitates binding to the 14-3-3 proteins (Bmh1/2) to inhibit its interaction with Jen1 (14). On the other hand, replenishment of glucose promotes the dephosphorylation of Rod1 by Glc7-Reg1 phosphatase, and thus dissociation of Bmh1/2 to target Rsp5 to Jen1 for ubiquitination (14, 16). Similarly, several studies reported that other adaptors are also regulated by the phosphorylation state in response to external cues (4, 17, 18). It has recently been shown that endocytosis of the mammalian class I glucose transporters, GLUT1 and GLUT4, are suppressed by phosphorylation of the α-arrestin, thioredoxin-interacting protein (TXNIP) (19, 20). This suggests that phosphoregulation of α-arrestin is conserved from mammals to yeast.
Although both substrate-induced and signaling-dependent endocytosis utilize α-arrestins, the mechanisms of target transporter recognition vary. In this study, we aimed to identify the regions of Jen1 recognized by glucose-activated Rod1 to understand the differences between the two systems. Our results demonstrate that the C-terminal 20-amino acid region of Jen1 contains a site recognized by Rod1 and lysine residues important for ubiquitination. Fusing this region to Mup1, whose endocytosis is tolerant to glucose, is sufficient to induce glucose- and Rod1-dependent endocytosis, suggesting that this region acts as the degron responding to glucose signaling.
Results
The C-terminal region of Jen1 is important for Rod1-dependent endocytic degradation
Rod1 was previously reported as an ART protein that regulates glucose-induced ubiquitination and endocytosis of Jen1 (14). Additionally, the Rsp5 adaptor protein Bul1 and its paralog Bul2 are also involved in endocytic degradation of Jen1 in response to multiple stimuli, including prolonged incubation in lactate medium and treatment with cycloheximide or rapamycin (21). Bul1/2 also contributes to Jen1 endocytosis in the presence of glucose in the rod1Δ cells (21) (Fig. S1). Therefore, we investigated how these adaptor proteins recognize Jen1 for endocytic degradation. To identify the regions of Jen1 required for adaptor recognition, we carried out a deletion analysis of Jen1. Transmembrane hidden Markov model (TMHMM) analysis (http://www.cbs.dtu.dk/services/TMHMM/)3 predicted that Jen1 has 12 transmembrane domains, and its N and C termini face the cytoplasm. We, therefore, generated Jen1ΔN and Jen1ΔC mutants that lack the N-terminal (2–94 amino acid residues) and C-terminal tails (574–616 amino acid residues), respectively (Fig. 1A). We expressed GFP-fused versions of WT and mutant Jen1 from the JEN1 promoter for analysis. First, we confirmed whether the Jen1 mutants preserved their transport activity by examining the sensitivity of the cells expressing the mutant Jen1 to fluoropyruvate, a toxic pyruvate analog. Because Jen1 is the only transporter responsible for pyruvate uptake, cell growth in a medium containing pyruvate as the sole carbon source depends on Jen1 activity (22). Accordingly, WT cells were sensitive to fluoropyruvate during respiratory growth, whereas cells became tolerant to fluoropyruvate when Jen1 was absent (Fig. S2). Similar to WT cells, cells expressing Jen1ΔN or Jen1ΔC were sensitive to fluoropyruvate, demonstrating reduced growth (Fig. S2). This suggests that deletion of the N-terminal or C-terminal regions of Jen1 did not affect the function of the core domain of Jen1. As expected, mutant Jen1 proteins localized to the PM in the presence of lactate (Fig. 1B, 0 min). Internalization of Jen1 was slightly delayed by deletion of its N-terminal region, and most Jen1ΔN localized to the vacuole after a 120-min incubation with glucose. However, Jen1ΔC was significantly stabilized at the PM even after a 120-min incubation with glucose (Fig. 1B). Immunoblot analysis also demonstrated that the degradation of both mutants was retarded. In addition, a delay in Jen1 endocytosis was more evident for the Jen1ΔC mutant than for the Jen1ΔN mutant (Fig. 1C). Because Jen1 lacking both its N-terminal and C-terminal regions (Jen1ΔNC-GFP) mislocalized to the endoplasmic reticulum, we were unable to evaluate its endocytosis (Fig. S3A). However, we observed that the Jen1 mutant with N-terminal deletion and amino acid substitutions in the C-terminal region (see later) localized to the PM and was highly resistant to glucose-induced endocytosis. This suggests that the membrane-spanning core domain of Jen1 does not carry information for endocytic sorting (Fig. S3B). Altogether, these results indicate that the C-terminal region of Jen1 plays a major role in its glucose-induced endocytosis.
Figure 1.

The cytoplasmic region of Jen1 is necessary for Rod1-dependent internalization of Jen1. A, schematic model of Jen1 deletion mutants lacking the N-terminal or C-terminal cytoplasmic regions. Gray lines show polypeptides of Jen1 deletion mutants. Boxes indicate predicted membrane-spanning α-helices (transmembrane domain). B, fluorescence distribution of GFP-tagged Jen1 or its deletion mutants analyzed in jen1Δ cells. Cells harboring pJen1–GFP (pSF3), pJen1ΔN–GFP (pSF5), or pJen1ΔC–GFP (pDS2) were grown in HC(Lac) medium for 3 h to induce expression of Jen1–GFP and its derivatives. Glucose (a final concentration of 2%) was then added to the culture to terminate their expression and induce their endocytosis. The images were taken at 0, 60, and 120 min after glucose addition. The scale bar indicates 5 μm. C, jen1Δ cells expressing GFP-tagged Jen1 or its deletion mutants were grown as described in B. Equal volumes of the culture were harvested at the indicated times and total protein lysates were immunoblotted with anti-GFP and anti-Pgk1 antibodies. Pgk1 was detected on the same blots as the loading control. D and F, the bul1Δbul2Δjen1Δ (D) or rod1Δjen1Δ (F) cells harboring pJen1–GFP (pSF3), pJen1ΔN–GFP (pSF5), or pJen1ΔC–GFP (pDS2) were subjected to fluorescence microscopic analysis. Induction of expression and endocytosis of Jen1 and its derivatives was performed as described in B. The scale bar indicates 5 μm. E and G, glucose-induced degradation of Jen1–GFP, Jen1ΔN–GFP, and Jen1ΔC–GFP expressed in bul1Δbul2Δjen1Δ (E) and rod1Δjen1Δ (G) cells was analyzed by immunoblot analysis as described in C.
We found that Jen1ΔN-GFP was highly stabilized in rod1Δ cells but not in bul1Δbul2Δ cells (Fig. 1), indicating that endocytosis of Jen1ΔN is almost fully dependent on Rod1. In addition, Jen1ΔC-GFP was highly stabilized in bul1Δbul2Δ cells, in which endocytic degradation of Jen1 is largely dependent on Rod1 (Fig. 1, D and E). Taken together, we conclude that the C-terminal region plays a pivotal role in Rod1-dependent endocytosis of Jen1. Because Jen1ΔC-GFP was slightly stabilized by ROD1 disruption (Fig. 1, F and G), Rod1 could have some minor contribution to the recognition of the N-terminal region. The fact that Jen1 was noticeably stabilized by its N-terminal deletion in rod1Δ cells (Fig. 1, F and G) suggested that adaptors other than Rod1, most likely Bul1 and Bul2, might be also involved in recognition of the N-terminal region.
Rod1 recognizes the C-terminal region of Jen1 for its ubiquitination
We further analyzed whether glucose-induced ubiquitination of Jen1 was affected by these mutations. We used a vrp1Δ strain in which general endocytosis is impaired and thus ubiquitinated endocytic cargos are expected to accumulate at the PM (23). In the immunoblot analysis, the majority of WT Jen1-GFP proteins underwent some modification that reduced their migration on the gel after glucose addition (Fig. 2A). Jen1ΔN-GFP also gave a mobility shift in response to glucose addition, although it was less efficient probably because the N-terminal region is recognized by Bul proteins and to a lesser extent by Rod1 (Fig. 2A). However, the mobility shift was hardly detected for Jen1ΔC-GFP (Fig. 2A).
Figure 2.

The cytoplasmic region of Jen1 is required for glucose-induced ubiquitination of Jen1 and its interaction with Rod1. A, vrp1Δ cells expressing GFP-tagged Jen1 or its derivatives (ΔN and ΔC) were grown and analyzed as described in the legend to Fig. 1C. B, vrp1Δ cells expressing GFP-tagged Jen1 or its derivatives (ΔN and ΔC) were grown in HC(Lac) medium for 3 h, followed by treatment with glucose for 10 min. Total protein extracts were subjected to immunoprecipitation with anti-GFP antibody, followed by immunoblot analysis with anti-GFP and anti-ubiquitin antibodies. C, rod1Δjen1Δ cells harboring the plasmid encoding Rod1-VC (pSF48) and the plasmid encoding either WT Jen1–VN (pSF26), Jen1ΔN–VN (pSF27), or Jen1ΔC–VN (pSF28) were grown in HC medium to an early log phase and then transferred to HC(Lac) medium to induce Jen1 expression. After 3 h incubation, glucose was added to the culture at a final concentration of 2% and cells were incubated for 10 min. Fluorescence from structurally complemented Venus was observed by fluorescence microscopy (BiFC assay). Differential interference contrast (DIC) images were also taken. The scale bar indicates 5 μm.
To test whether these mobility shifts were caused by ubiquitination, we carried out immunoprecipitation of GFP-tagged Jen1 mutants expressed in vrp1Δ cells, followed by immunodetection with an anti-ubiquitin antibody. Ubiquitin-positive bands were clearly detected in WT Jen1 and Jen1ΔN cells but only faint signals were observed in Jen1ΔC cells (Fig. 2B). In addition, the size of these ubiquitin-positive bands was similar to that of slower migrating bands of WT Jen1 and Jen1ΔN probed with an anti-GFP antibody (Fig. 2B), indicating that slower migrating bands correspond to ubiquitinated Jen1. Taken together, these results suggest that the C-terminal region of Jen1 plays an important role in Jen1 ubiquitination.
To test whether the defect in ubiquitination of Jen1ΔC might be due to impaired interaction with Rod1, we sought to analyze the in vivo interaction between Jen1 and Rod1 using a bimolecular fluorescence complementation (BiFC) assay. The BiFC assay enables assessment of the subcellular localization of protein–protein interactions in living cells (24). We constructed strains co-expressing Jen1 proteins fused with the Venus N-terminal fragment (Jen1-VN) and Rod1 fused with the Venus C-terminal fragment (Rod1-VC). As shown in Fig. 2C, interactions between WT Jen1-VN and Rod1-VC were detected at the cell surface after glucose addition, whereas the BiFC signal was almost lost in Jen1ΔC-VN-expressing cells. BiFC dots were also observed on the surface of cells expressing Jen1ΔN-VN, although it was fewer than with WT Jen1-VN (Fig. 2C). These results suggest that the C-terminal region of Jen1, and to a lesser extent the N-terminal region, play an important role in the interaction between Jen1 and Rod1.
His612, Ile613, and Glu614 residues of Jen1 are necessary for its recognition and ubiquitination by Rod1
We next tried to narrow down the region within the C-terminal region to identify the Rod1-dependent sorting signal. To this end, we generated deletion mutants of Jen1 lacking C-terminal 33-, 23-, and 13-amino acid residues, termed as Δ33C, Δ23C, and Δ13C, respectively (Fig. S4A). GFP-fused mutants were used to examine glucose-induced endocytosis by fluorescence microscopy and immunoblot analyses. As shown in Fig. S4, none of the mutants restored efficient endocytosis. Therefore, we decided to further narrow down an endocytosis signal in the distal region of the C-terminal region. Recently, it was reported that an acidic patch in the Mup1 N-terminal cytosolic tail is necessary for Ldb19/Art1-dependent endocytosis of Mup1 (9). Accordingly, we reasoned that a domain rich in acidic amino acids (amino acids 610–614) located within the distal C-terminal region could be involved in the endocytosis of Jen1 (Fig. 3A). To test this hypothesis, we performed alanine-scanning mutagenesis against the acidic domain and its vicinity. We first analyzed mutations E610A,E611A, H612A,I613A,E614A, and T615A,V616A. Fluorescence microscopy and immunoblot analyses revealed that the H612A,I613A,E614A mutation strongly blocked glucose-induced endocytosis, whereas E610A,E611A and T615A,V616A mutants were efficiently sorted to the vacuole (Fig. 3, B and C). Accordingly, we substituted every amino acid within the His612–Ile–Glu sequence to alanine and tested these mutants for endocytosis by fluorescence microscopy. Among the three mutants with single amino acid substitution, only the E614A mutant became resistant to glucose-induced internalization (Fig. S5). Interestingly, the H612A,I613A mutation also blocked internalization (Fig. S5). We therefore concluded that the His612–Ile–Glu region is critical for Jen1 endocytosis, and hereafter refer to the H612A,I613A,E614A mutation as the 3A mutation.
Figure 3.

Mutations in His612, Ile613, and Glu614 of Jen1 resulted in loss of interaction with Rod1. A, the C-terminal 20-amino acid sequence of Jen1 is shown. Acidic amino acids in the 604–616 region are highlighted. The C-terminal sequence of Jen1Δ13C is also shown. B, the jen1Δ cells expressing Jen1(H610A,E611A)-GFP (pSF34), Jen1(H612A,I613A,E614A)-GFP (pSF35), or Jen1(T615A,V616A)-GFP (pSF37) were subjected to fluorescence microscopic analysis as described in the legend to Fig. 1B. The scale bar indicates 5 μm. C, the jen1Δ cells expressing GFP-tagged Jen1 or its derivatives (E610A,E611A, H612A,I613A,E614A, and T615A,V616A) were subjected to immunoblot analysis as described in the legend to Fig. 1C. D, the rod1Δjen1Δ cells harboring the plasmid encoding Rod1–VC (pSF48) and the plasmid encoding either WT Jen1–VN (pSF26) or Jen1(3A)–VN (pSF49) were subjected to BiFC analysis as shown in Fig. 2C. The scale bar indicates 5 μm. E, protein extracts from the vrp1Δ cells expressing WT Jen1–GFP or Jen1(3A)–GFP were analyzed by immunoblot analysis as described in the legend to Fig. 1C.
To test whether the His612–Ile–Glu region is important for ubiquitination of Jen1, we performed Western blot analysis of the 3A mutant expressed in vrp1Δ cells and found that a glucose-responsible migration shift was dramatically decreased by the 3A mutation (Fig. 3D). To test whether the His612–Ile–Glu region is required for recognition by Rod1, we co-expressed the VN-tagged Jen1(3A) mutant and VC-tagged Rod1 to perform a BiFC assay. As shown in Fig. 3E, glucose-induced generation of the BiFC signal for the Rod1–Jen1(3A) interaction was significantly decreased. Altogether, we concluded that the His612–Ile–Glu region is required for glucose-induced recognition of Jen1 by Rod1. We showed that the N-terminal FLAG- or GFP-tagged Jen1 protein also underwent glucose-induced endocytic degradation in a Rod1- and His612–Ile–Glu region-dependent manner (Fig. S6). This fact indicated that the GFP tagging to the C terminus of Jen1 did not affect its degradation properties.
The Jen1 C-terminal region is a Rod1-dependent degron
We next asked whether the Jen1 C-terminal region is enough for recognition by Rod1. To address this, we fused the C-terminal 20-amino acid region of Jen1 (597–616 amino acid residues), hereafter referred to as the Jen1 C-tail, to the methionine permease Mup1, whose endocytosis is not under the control of Rod1 (Fig. 4A, Fig. S7). Previous reports showed that endocytic degradation of Mup1 is regulated by Ldb19/Art1 in response to methionine excess (2, 9). In the presence of methionine, the GFP-tagged fusion protein, termed Mup1–Jen1C–GFP, was internalized from the PM in an Ldb19-dependent manner as well as native Mup1–GFP, implying that fusion of the Jen1 C-tail to Mup1 does not affect its methionine-induced degradation (Fig. S7). As expected, methionine-induced endocytosis of Mup1–Jen1C–GFP was independent of Rod1 (Fig. S7). As shown in Fig. 4B, Mup1–Jen1C–GFP was transiently expressed by the JEN1 promoter localized at the cell surface in medium depleted of both glucose and methionine. Upon addition of glucose, Mup1–Jen1C–GFP underwent endocytic degradation, whereas Mup1–GFP remained at the PM (Fig. 4, B and C). Notably, glucose-induced endocytic degradation of Mup1–Jen1C–GFP was significantly impaired with deletion of the ROD1 gene or introduction of the 3A mutation to the Jen1 C-tail (Fig. 4, B and C). These results suggest that glucose-induced Mup1–Jen1C degradation occurs through recognition of the Jen1 C-tail by Rod1. We therefore conclude that the Jen1 C-tail is enough to induce Rod1-mediated endocytosis of PM transporters.
Figure 4.

Transplanting the Jen1 C-tail to Mup1 leads to Rod1-dependent endocytosis of Mup1. A, schematic depictions of Mup1 and Mup1–Jen1C. The C-terminal 20-amino acid region (Jen1 C-tail) was fused to the C terminus of Mup1. B and C, the wildtype (WT) or rod1Δ cells expressing Mup1–GFP (pSF43), Mup1–Jen1C–GFP (pSF45), or Mup1–Jen1C(3A)–GFP (pSF47) under the control of the JEN1 promoter were grown in HC medium to an early log phase, and then transferred to HC(Lac) medium lacking methionine. After 3 h incubation, glucose was added to the culture to chase the localization shift (B) and degradation (C) of GFP-fused proteins. Subcellular localization of Mup1–GFP or Mup1–Jen1C–GFP was observed by fluorescence microscopy. The scale bar indicates 5 μm. Degradation of GFP-fused proteins was monitored by immunoblot analysis using an anti-GFP antibody.
A recent study reported that the Ldb19–Rsp5 complex recognizes the Mup1 N-tail region to ubiquitinate Lys27 and Lys28 within the N-tail region in response to methionine signals (9). Accordingly, we asked whether these lysine residues of Mup1–Jen1C are also involved in glucose- and Rod1-mediated ubiquitination. Interestingly, Mup1(K27R,K28R)–Jen1C–GFP was efficiently endocytosed in response to glucose treatment (Fig. 5, A and B), although its methionine-induced endocytosis was blocked (Fig. S8, A and B). In support of these findings, ubiquitination of Mup1(K27R,K28R)–Jen1C–GFP was observed in vrp1Δ cells after glucose stimulation, but not after methionine stimulation (Fig. 5C and Fig. S8C). These results indicate that the Rod1–Rsp5 complex ubiquitinates lysine residue(s) other than those of methionine-induced and Ldb19-mediated ubiquitination. The Jen1 C-tail (597–616 amino acid residues) contains two lysine residues (Lys599 and Lys607). Accordingly, we substituted these residues of Mup1–Jen1C–GFP to arginine to determine whether they are involved in the down-regulation of the fusion permease. We found that Mup1–Jen1C(K599R,K607R)–GFP lost its capacity for glucose-induced internalization and ubiquitination (Fig. 5, B and C). In agreement with this result, this mutation caused loss of glucose-induced internalization and ubiquitination of Jen1 (Fig. 6, A–C). Genome-wide proteomic analysis revealed that the Lys388 residue located in the cytoplasmic loop of Jen1 is ubiquitinated, and substitution of this residue to arginine decreases glucose-induced turnover of Jen1 (13, 25). However, mutant Jen1 still underwent ubiquitination similar to the WT, indicating that lysine residues other than Lys388 may also be ubiquitinated (13). Our findings therefore suggest that these residues are also ubiquitinated, or alternatively, recognition of the Jen1 degron by Rod1 is compromised by the mutation, thereby reducing ubiquitination of Jen1.
Figure 5.

Lysine residues in the Jen1 C-tail are important for Rod1-dependent ubiquitination of Mup1–Jen1C. A, schematic depiction of Mup1–Jen1C. Lysine residues that replaced arginine are highlighted. B, internalization of Mup1(K27R,K28R)–GFP (pSF51), Mup1(K27R,K28R)–Jen1C–GFP (pSF53), and Mup1–Jen1C(K599R,K607R)–GFP (pSF55) expressed from the JEN1 promoter in WT cells was analyzed as described in the legend to Fig. 4B. The scale bar indicates 5 μm. C, protein extracts of vrp1Δ cells expressing Mup1–GFP, Mup1(K27R,K28R)–GFP, Mup1–Jen1C–GFP, Mup1(K27R,K28R)–Jen1C–GFP, or Mup1–Jen1C(K599R,K607R)–GFP from the JEN1 promoter were grown as described in the legend to Fig. 4B and analyzed by immunoblot as described in the legend to Fig. 1C. Arrowheads indicate ubiquitinated species.
Figure 6.
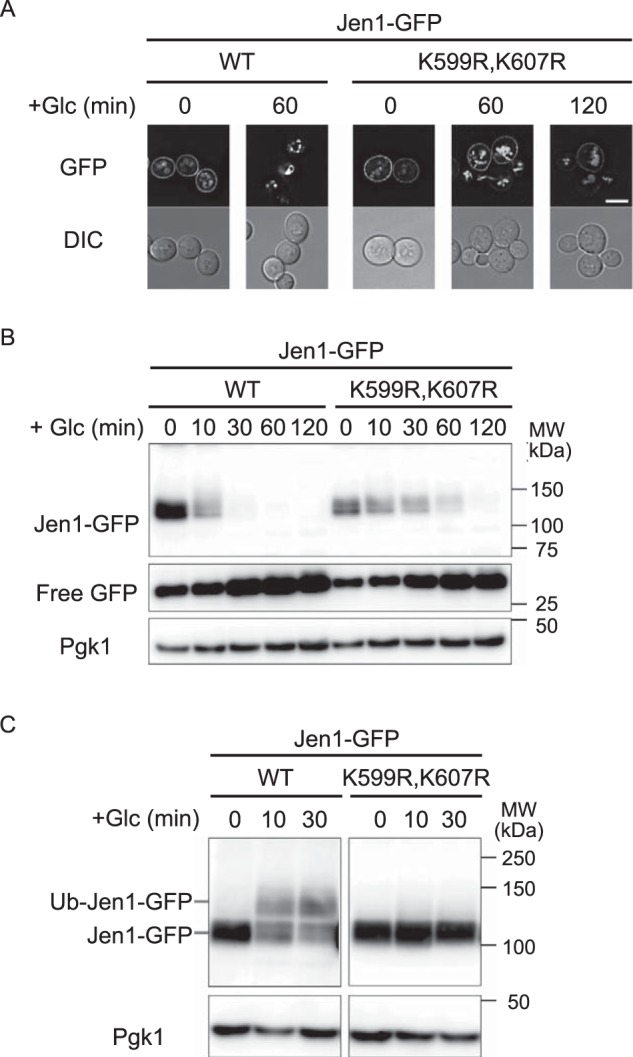
Mutations of Lys599 and Lys607 in the Jen1 C-tail render Jen1 resistant to glucose-induced endocytosis. A and B, jen1Δ cells expressing WT Jen1–GFP or Jen1(K599R,K607R)–GFP (pSF54) were subjected to fluorescence microscopic (A) and immunoblot analyses (B) as described in the legend to Fig. 1. The scale bar indicates 5 μm. C, vrp1Δ cells expressing WT Jen1–GFP or Jen1(K599R,K607R)–GFP were subjected to immunoblot analysis as described in the legend to Fig. 1C.
Discussion
In yeast, the α-arrestin–Rsp5 network executes the quality and quantity control of PM nutrient transporters through endocytic degradation. One key question of this system is how α-arrestins recognize specific transporters for ubiquitination. Many transporters are rapidly inactivated by excess corresponding substrates. It has been proposed that conformational changes induced by substrate transport trigger exposure of degron, which contains α-arrestin–Rsp5 complex-binding site(s) and lysine residues to be ubiquitinated (7–9, 26). However, Jen1 differs from these transporters in that its substrates, lactate and pyruvate, do not trigger rapid degradation; instead, it is inactivated by a fermentable carbon source including glucose. In this study, we demonstrate that the Jen1 C-tail functions as a Rod1-dependent degron sufficient to mediate substrate-independent endocytosis of PM transporters.
Previous reports on Fur4 (uracil permease) and Mup1 demonstrated that in the ground state (substrate-unbound state), the cytoplasmic loops of the core domain of transporters could associate with the N-terminal cytoplasmic region proximal to the first transmembrane domain (7, 9). Upon substrate binding, transporters change their conformation to the active state, thereby exposing the N-terminal region, where the α-arrestin–Rsp5 complex associates for ubiquitination of the neighboring lysine residues. In addition to the N-terminal α-arrestin-binding domain, the exposed cytoplasmic loop(s) is likely required for interaction with α-arrestin proteins (9). In contrast to this substrate-dependent N-terminal degron system, Jen1 has a degron in the C-terminal cytoplasmic tail. Several lines of evidence indicate that the Jen1 degron may be permanently presented for Rod1 recognition, regardless of the presence or absence of the substrates. First, Rod1 does not require the specific cytoplasmic loop of transporters for recognition. We transplanted the Jen1 degron to Mup1, rendering this permease sensitive to Rod1- and glucose-dependent endocytosis. Second, the endocytosis of Jen1 is independent of its substrates. We confirmed that Jen1–GFP expressed in the presence of raffinose was normally degraded by glucose replenishment (Fig. S9). Moreover, Mup1 fused with the Jen1 degron (Mup1–Jen1C) endocytosed in the absence of methionine in response to a glucose stimulus. According to the current paradigm that transporters stay in the ground state without substrates, these transporters may not change their conformations but are endocytosed by a signal other than their substrates. Third, the Jen1 degron is located in a region distal to the core domain of Jen1. This enables Rod1 to easily access the degron. Fourth, the Jen1 degron may be self-contained. Although it remains unproven whether Lys599 and Lys607 of Jen1 are actually ubiquitinated, these residues are important for Rod1-dependent ubiquitination of Jen1 and Mup1–Jen1C. Rod1–Rsp5 may recognize the His612–Ile–Glu sequence for ubiquitination of the neighboring Lys residues, or otherwise Rod1–Rsp5 may recognize these Lys residues together with the His–Ile–Glu sequence and ubiquitinate them. Activity of Rod1 is regulated through its phosphorylation state via the glucose signaling pathway composed of Snf1 kinase and Glc7–Reg1 phosphatase (14). It is proposed that dephosphorylated Rod1 is competent to associate with Jen1. Accordingly, we speculate that the continuously presented degron may help Jen1 to be quickly degraded in response to glucose.
Talaia et al. (21) demonstrated that Jen1 mutants lacking transport activity are degraded by glucose-dependent (i.e. Rod1-dependent) endocytosis, whereas they are resistant to Bul1/2-dependent endocytosis. These observations suggest that Bul1/2, but not Rod1, may require a conformational change of Jen1 for its recognition. Our study shows that Bul1/2 proteins can recognize the N-terminal cytoplasmic region of Jen1, which is similar to Mup1 in that the Mup1 N-tail, recognized by Ldb19, plays important roles for its substrate-induced endocytosis. Thus, we speculate that a transport-dependent conformational change might expose the Jen1 N-tail where Bul1/2 could then associate. Identification of the N-terminal degron will help us to understand the mechanism of Bul1/2-dependent inactivation of Jen1.
A degron of the yeast general amino acid permease Gap1 shows properties similar to those of the Jen1 degron. Although Gap1 is down-regulated by excess substrates, it also undergoes ubiquitination and endocytosis in response to ammonium, a preferred nitrogen source in yeast. Incorporation of ammonium into the cells activates the target of rapamycin complex 1 (TORC1) kinase (4, 8). This renders the Rsp5 adaptors Bul1 and Bul2 dephosphorylated through regulation of the activities of Npr1 kinase and Sit4 phosphatase. Dephosphorylated Bul1/2 may recognize the N-terminal region distal to the first transmembrane domain of Gap1 to ubiquitinate its Lys9 and Lys16 residues (4). Because the TORC1 signaling pathway is enough to down-regulate Gap1 mutants deficient in a substrate transport, it is proposed that a conformational change is dispensable for the TORC1-dependent endocytosis of Gap1 (8). These findings, together with our results, demonstrate that the substrate-independent endocytosis of transporters utilizes degrons located in the distal region of the cytoplasmic tail of transporters.
Rsp5-dependent and ligand-induced endocytosis of the G protein–coupled receptor Ste2 also requires Rod1 (27). Binding of pheromone (α-factor) to Ste2 induces a conformational change, which triggers its phosphorylation and the exposure of its C-terminal tail, which interacts with Rod1. Because the 135-residue C-terminal tail itself has the ability to interact with Rod1 in vitro, the cytoplasmic loops in the core domain of Ste2 are unlikely to contribute to the association between Ste2 and Rod1 (27). In addition, it was demonstrated that recognition of a signal sequence for ligand-mediated endocytosis of Ste2 is independent of the ligand-induced conformational change (28). These findings suggest that recognition of the degron by Rod1 may not be coupled with a ligand-induced conformational change. Because phosphorylation of Ste2 is prerequisite for its ubiquitination, we cannot exclude the possibility that the phosphorylation may change a conformation of the cytoplasmic domain of Ste2 to promote binding with Rod1. Although Jen1 and Ste2 possess Rod1-dependent degrons in their C-terminal tails, there is no significant sequence similarity in their C-terminal tails. This allows us to speculate that the sorting signal may be three-dimensionally coded. Alternatively, Rod1 with altered phosphorylation states may discern specific sequences of target transmembrane proteins, because Ste2 endocytosis requires dephosphorylation of Rod1 mediated by calcineurin, which is triggered by calcium influx due to pheromone response (27).
Rod1 is also involved in down-regulation of the high-affinity glucose transporter Hxt6, whose internalization is triggered by high concentrations of glucose (3, 16). It will be interesting to assess whether a conformational change associated with glucose binding is required for recognition of the Hxt6 degron with Rod1, specifically, whether Rod1 operates in both substrate-dependent and substrate-independent ways. In mammalian cells, endocytosis of the glucose transporter GLUT4 is mediated by the α-arrestin, TXNIP, whose expression is up-regulated by glucose but suppressed by insulin (19, 20, 29). Insulin stimulation also inactivates TXNIP by protein kinase B-mediated phosphorylation of its Ser308 residue, thereby preventing GLUT4 from endocytic degradation (20). Our findings and further analysis will help to understand the mechanism of substrate- and signaling-dependent endocytosis of glucose transporters in mammalian cells.
Experimental procedures
Strains, plasmid constructions, and growth conditions
The S. cerevisiae strains used in this study are listed in Table 1. Gene disruptions were performed as described by Gueldener et al. (30) and Janke et al. (31) using disruption cassettes containing drug-resistance genes or recovering auxotrophic genes such as kanMX, natNT2, Kluyveromyces lactis LEU2, or Schizosaccharomyces pombe his5+.
Table 1.
Yeast strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| BY4742 | MATα; ura3Δ0, leu2Δ0, his3Δ1, lys2Δ0 | Brachmann et al. (34) |
| jen1Δ | BY4742; jen1Δ::KanMX | Open Biosystems |
| rod1Δ | BY4742; rod1Δ::KanMX | Open Biosystems |
| vrp1Δ | BY4742; vrp1Δ::KanMX | Open Biosystems |
| YSF10 | BY4742; rod1Δ::KanMX jen1Δ::natNT2 | This study |
| YSF11 | BY4742; bul1Δ::LEU2 bul2Δ::KanMX jen1Δ::his5+ | This study |
| HKY52 | BY4742; bul1Δ::KanMX rod1Δ::loxP jen1Δ::his5+ | This study |
| YSF12 | BY4742; bul1Δ::LEU2 bul2Δ::KanMX rod1Δ::his5+ jen1Δ::natNT2 | This study |
The plasmids and oligonucleotides used in this study are listed in Tables S1 and S2, respectively. To express C terminally GFP-tagged proteins, we first constructed pRS316–GFP–ADHt. The DNA fragment containing GFP ORF followed by ADH1 terminator (GFP–ADHt) was PCR-amplified with the oligonucleotide DNAs oTAKA145 and oTAKA138 using pFA6a–GFP(S65T)–kanMX6 as a template. The resulting GFP–ADHt fragment was digested with EcoRI and SalI and cloned into pRS316 to generate pRS316–GFP–ADHt. To generate pRS316–Jen1–GFP (pSF3), the DNA fragment of JEN1, including its promoter region, was PCR-amplified using oligonucleotides ST13 and ST25 from BY4742 genomic DNA as a template, followed by digestion with SpeI and BamHI, and then ligated to SpeI and BamHI-digested pRS316–GFP–ADHt. To generate N terminally GFP-tagged Jen1 plasmid, we first constructed pRS316–JEN1p–BamHI–JEN1 (pSF56), which contained a BamHI site just after the first codon. The DNA fragment of the JEN1 promoter region was PCR-amplified using oligonucleotides ST25 and DS16-r from the BY4742 genomic DNA as a template, followed by digestion with SpeI and BamHI. The DNA fragment of JEN1, including its terminator region, was PCR-amplified using oligonucleotides DS16 and DS15 from the BY4742 genomic DNA as a template, followed by digestion with BamHI and EcoRI. These fragments were ligated to SpeI- and EcoRI-digested pRS316 vector to generate pSF56. The GFP (S65T variant) DNA cassette with BamHI sites on both sides was ligated to BamHI-digested pSF56 and then a plasmid with properly oriented GFP was selected as pRS316–GFP–Jen1 (pSF57). Plasmids expressing Jen1, Mup1, and Rod1, which have truncations, point mutations, and/or fusion tags were constructed using an in vivo DNA assembly method (32). Combinations of PCR products and vector DNA used for in vivo assembly are shown in Table S3.
Yeast cells were grown in YPD (1% yeast extract, 2% peptone and 2% glucose) medium or Hartwell's complete (HC) medium (33). The carbon source of HC medium was replaced from 2% glucose to 2% raffinose (HC(Raf)) or 0.5% lactic acid (pH 5.5) (HC(Lac)) as necessary. HC(EG) medium was identical to the HC medium except that a mixture of 3% ethanol and 2% glycerol was used as the carbon source. Cells were always harvested during the exponential growth phase. HC(Lac) medium was used to induce genes from the JEN1 promoter after growing cells to mid-log phase in HC medium. For expressing genes from the MUP1 promoter, HC(Raf) medium lacking methionine was used. To initiate glucose repression and endocytosis, glucose was added to the culture at a final concentration of 2%.
Protein extraction and immunoprecipitation
Protein extracts were prepared as described by Becuwe et al. (14). Briefly, 100 μl of 100% (w/v) TCA was added to 1 ml of cell culture and incubated on ice for 10 min. TCA precipitates were harvested by centrifugation for 5 min at 4 °C and 15,000 × g, and then lysed by mixing with glass beads in 100 μl of 10% (w/v) TCA for 10 min at 4 °C. The disrupted pellets were centrifuged for 5 min at 4 °C and 15,000 × g. The pellets were resuspended in TCA sample buffer (50 mm Tris-HCl, pH 6.8, 100 mm DTT, 2% SDS, 0.1% bromphenol blue, and 10% glycerol, containing an additional 200 mm unbuffered Tris solution) at a concentration of 50 μl/initial OD unit, and treated at 37 °C. The samples were subjected to immunoblot analysis after resolving by SDS-PAGE.
For immunoprecipitation, 10 A600 units of glucose-treated yeast cells were harvested by centrifugation. The resulting cell pellets were suspended in 10% (w/v) TCA and incubated on ice for 10 min for inactivation. TCA precipitates were then collected by centrifugation for 5 min at 4 °C and 15,000 × g, and cracked by mixing with glass beads in 200 μl of 10% (w/v) TCA for 10 min at 4 °C. The disrupted pellets were collected by centrifugation at 4 °C and 15,000 × g for 5 min. The resulting pellets were lysed in 200 μl of TCA sample buffer lacking DTT, and denatured at 37 °C for 15 min. After centrifugation, 20 μl (1 A600 eq) of supernatants were taken as total lysates, and diluted with 30 μl of TCA sample buffer. The remaining supernatants (170 μl) were diluted with 1.8 ml of TWIP buffer (50 mm Tris-HCl, pH 7.6, 150 mm sodium chloride, 0.5% Tween 20, and 0.1 mm EDTA) and incubated with 2 μg of anti-GFP antibody (Roche Diagnostics, Rotkreuz, Switzerland). After incubation for 2 h at 4 °C, antigen–antibody complexes were isolated by being incubated with DynabeadsTM Protein G (Invitrogen) for 1 h at 4 °C. Beads were then washed 3 times with 1 ml of TWIP buffer before eluting the immunoprecipitate with TCA sample buffer. The total lysate and immunoprecipitates were treated at 37 °C for 15 min before SDS-PAGE.
Immunoblot analysis
The samples were run on polyacrylamide gels of appropriate concentrations and transferred to polyvinylidene difluoride membranes (Immobilon-P Membrane, EMD Millipore, Temecula, CA). The membranes were probed with anti-GFP (mFX75, Wako Pure Chemical Industries, Osaka, Japan), anti-DYKDDDDK (1E6, Wako Pure Chemical Industries), anti-3-phosphoglycerate kinase (Pgk1) (22C5D8, Abcam, Cambridge, United Kingdom), or anti-Ub (P4D1, Santa Cruz Biotechnology, Dallas, TX) antibodies. Immunoreactive protein bands were detected with horseradish peroxidase-conjugated goat anti-mouse IgG (Bio-Rad) and chemiluminescence with ImmobilonTM Western Chemiluminescent HRP Substrate (ECL) (Millipore Sigma, Burlington, MA). For the detection of protein in the immunoprecipitation, mouse IgG TrueBlot® ULTRA (eBioscience, San Diego, CA) was used as secondary antibodies. Signal detection was performed using the ImageQuant LAS-4000 image analyzer (GE Healthcare, Little Chalfont, UK).
Fluorescence microscopy
Cells expressing fluorescent proteins were grown in HC medium to the mid-log phase and further incubated in HC(Lac), HC(Raf), or HC(EG) medium for 3 h to induce their expression. For fluorescence microscopy analysis in all figures, cells were visualized under ×100 oil immersion objective (1.40 NA; Olympus, Japan) using an Olympus IX71 fluorescence microscope equipped with ORCA-Flash2.8 (Hamamatsu Photonics K.K., Hamamatsu, Japan) and processed using MetaMorph version 7.8.2.0 imaging software (Molecular Devices, Sunnyvale, CA). Images of GFP-fused proteins were represented by maximum intensity projections of 15 step Z-stacks (step size; 0.2 μm) and deconvolved using MetaMorph version 7.8.2.0 imaging software. Particularly, images acquired for the BiFC assay were shown on the same intensity scale.
Fluoropyruvate sensitivity assay
Fluoropyruvate sensitivity was assessed by growth on HC(EG) solid medium containing 1.0 or 10 mm fluoropyruvate. Yeast cells grown in HC(EG) liquid medium were resuspended in sterile water at a concentration of 1.0 A600/ml and their serial dilutions were spotted on plates with or without fluoropyruvate.
Author contributions
S. F. data curation; S. F. and T. S. funding acquisition; S. F., D. S., H. K., M. O., S. T., Y. T., K. G., and T. S. investigation; S. F. writing-original draft; K. G. formal analysis; K. G. and T. S. supervision; T. S. conceptualization; T. S. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Dr. Fumiyoshi Abe for the helpful discussion.
This work was supported by Japan Society for the Promotion of Science (JSPS) Core-to-Core Program (A. Advanced Research Networks entitled “Establishment of international agricultural immunology research-core for a quantum improvement in food safety”) and JSPS KAKENHI Grants JP26450084 (to T. S.) and JP17J02369 (to S. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S9 and Tables S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PM
- plasma membrane
- Nedd4
- neuronal precursor cell expressed developmentally down-regulated protein 4
- ART
- arrestin-related trafficking adaptor
- TXNIP
- thioredoxin-interacting protein
- TMHMM
- transmembrane hidden Markov model
- BiFC
- bimolecular fluorescence complementation
- TORC1
- target of rapamycin complex 1
- DIC
- differential interference contrast
- HC
- Hartwell's complete.
References
- 1. Boase N. A., and Kumar S. (2015) NEDD4: the founding member of a family of ubiquitin-protein ligases. Gene 557, 113–122 10.1016/j.gene.2014.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin C. H., MacGurn J. A., Chu T., Stefan C. J., and Emr S. D. (2008) Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 135, 714–725 10.1016/j.cell.2008.09.025 [DOI] [PubMed] [Google Scholar]
- 3. Nikko E., and Pelham H. R. (2009) Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic 10, 1856–1867 10.1111/j.1600-0854.2009.00990.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merhi A., and André B. (2012) Internal amino acids promote Gap1 permease ubiquitylation via TORC1/Npr1/14-3-3-dependent control of the Bul arrestin-like adaptors. Mol. Cell. Biol. 32, 4510–4522 10.1128/MCB.00463-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Novoselova T. V., Zahira K., Rose R. S., and Sullivan J. A. (2012) Bul proteins, a nonredundant, antagonistic family of ubiquitin ligase regulatory proteins. Eukaryot. Cell 11, 463–470 10.1128/EC.00009-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aubry L., and Klein G. (2013) True arrestins and arrestin-fold proteins: a structure-based appraisal. in Progress in Molecular Biology and Translational Science (Louis M. L., ed) pp. 21–56, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 7. Keener J. M., and Babst M. (2013) Quality control and substrate-dependent downregulation of the nutrient transporter Fur4. Traffic 14, 412–427 10.1111/tra.12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghaddar K., Merhi A., Saliba E., Krammer E. M., Prévost M., and André B. (2014) Substrate-induced ubiquitylation and endocytosis of yeast amino acid permeases. Mol. Cell. Biol. 34, 4447–4463 10.1128/MCB.00699-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guiney E. L., Klecker T., and Emr S. D. (2016) Identification of the endocytic sorting signal recognized by the Art1-Rsp5 ubiquitin ligase complex. Mol. Biol. Cell 27, 4043–4054 10.1091/mbc.e16-08-0570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andrade R. P., and Casal M. (2001) Expression of the lactate permease gene JEN1 from the yeast Saccharomyces cerevisiae. Fungal Genet. Biol. 32, 105–111 10.1006/fgbi.2001.1254 [DOI] [PubMed] [Google Scholar]
- 11. Lodi T., Fontanesi F., and Guiard B. (2002) Co-ordinate regulation of lactate metabolism genes in yeast: the role of the lactate permease gene JEN1. Mol. Genet. Genomics 266, 838–847 10.1007/s00438-001-0604-y [DOI] [PubMed] [Google Scholar]
- 12. Paiva S., Kruckeberg A. L., and Casal M. (2002) Utilization of green fluorescent protein as a marker for studying the expression and turnover of the monocarboxylate permease Jen1p of Saccharomyces cerevisiae. Biochem. J. 363, 737–744 10.1042/bj3630737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paiva S., Vieira N., Nondier I., Haguenauer-Tsapis R., Casal M., and Urban-Grimal D. (2009) Glucose-induced ubiquitylation and endocytosis of the yeast Jen1 transporter: role of lysine 63-linked ubiquitin chains. J. Biol. Chem. 284, 19228–19236 10.1074/jbc.M109.008318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Becuwe M., Vieira N., Lara D., Gomes-Rezende J., Soares-Cunha C., Casal M., Haguenauer-Tsapis R., Vincent O., Paiva S., and Léon S. (2012) A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J. Cell Biol. 196, 247–259 10.1083/jcb.201109113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alvaro C. G., Aindow A., and Thorner J. (2016) Differential phosphorylation provides a switch to control how α-arrestin Rod1 down-regulates mating pheromone response in Saccharomyces cerevisiae. Genetics 203, 299–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Llopis-Torregrosa V., Ferri-Blázquez A., Adam-Artigues A., Deffontaines E., van Heusden G. P., and Yenush L. (2016) Regulation of the yeast Hxt6 hexose transporter by the Rod1 α-Arrestin, the Snf1 protein kinase, and the Bmh2 14-3-3 protein. J. Biol. Chem. 291, 14973–14985 10.1074/jbc.M116.733923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. MacGurn J. A., Hsu P. C., Smolka M. B., and Emr S. D. (2011) TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147, 1104–1117 10.1016/j.cell.2011.09.054 [DOI] [PubMed] [Google Scholar]
- 18. O'Donnell A. F., Huang L., Thorner J., and Cyert M. S. (2013) A calcineurin-dependent switch controls the trafficking function of alpha-arrestin Aly1/Art6. J. Biol. Chem. 288, 24063–24080 10.1074/jbc.M113.478511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu N., Zheng B., Shaywitz A., Dagon Y., Tower C., Bellinger G., Shen C. H., Wen J., Asara J., McGraw T. E., Kahn B. B., and Cantley L. C. (2013) AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 49, 1167–1175 10.1016/j.molcel.2013.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waldhart A. N., Dykstra H., Peck A. S., Boguslawski E. A., Madaj Z. B., Wen J., Veldkamp K., Hollowell M., Zheng B., Cantley L. C., McGraw T. E., and Wu N. (2017) Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell Rep. 19, 2005–2013 10.1016/j.celrep.2017.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Talaia G., Gournas C., Saliba E., Barata-Antunes C., Casal M., André B., Diallinas G., and Paiva S. (2017) The α-arrestin Bul1p mediates lactate transporter endocytosis in response to alkalinization and distinct physiological signals. J. Mol. Biol. 429, 3678–3695 10.1016/j.jmb.2017.09.014 [DOI] [PubMed] [Google Scholar]
- 22. Akita O., Nishimori C., Shimamoto T., Fujii T., and Iefuji H. (2000) Transport of pyruvate in Saccharomyces cerevisiae and cloning of the gene encoded pyruvate permease. Biosci. Biotechnol. Biochem. 64, 980–984 10.1271/bbb.64.980 [DOI] [PubMed] [Google Scholar]
- 23. Becuwe M., and Leon S. (2014) Integrated control of transporter endocytosis and recycling by the arrestin-related protein Rod1 and the ubiquitin ligase Rsp5. eLife 3, 10.7554/eLife.03307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu C. D., and Kerppola T. K. (2003) Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539–545 10.1038/nbt816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., and Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 10.1038/nbt849 [DOI] [PubMed] [Google Scholar]
- 26. Cain N. E., and Kaiser C. A. (2011) Transport activity-dependent intracellular sorting of the yeast general amino acid permease. Mol. Biol. Cell 22, 1919–1929 10.1091/mbc.e10-10-0800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alvaro C. G., O'Donnell A. F., Prosser D. C., Augustine A. A., Goldman A., Brodsky J. L., Cyert M. S., Wendland B., and Thorner J. (2014) Specific α-arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein-coupled receptor Ste2. Mol. Cell. Biol. 34, 2660–2681 10.1128/MCB.00230-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang C. I., Schandel K. A., and Jenness D. D. (2014) Interaction among Saccharomyces cerevisiae pheromone receptors during endocytosis. Biol. Open 3, 297–306 10.1242/bio.20146866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parikh H., Carlsson E., Chutkow W. A., Johansson L. E., Storgaard H., Poulsen P., Saxena R., Ladd C., Schulze P. C., Mazzini M. J., Jensen C. B., Krook A., Bjornholm M., Tornqvist H., Zierath J. R., et al. (2007) TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 4, e158 10.1371/journal.pmed.0040158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gueldener U., Heinisch J., Koehler G. J., Voss D., and Hegemann J. H. (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23 10.1093/nar/30.6.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 10.1002/yea.1142 [DOI] [PubMed] [Google Scholar]
- 32. Oldenburg K. R., Vo K. T., Michaelis S., and Paddon C. (1997) Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 25, 451–452 10.1093/nar/25.2.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burke D., Dawson D., and Stearns T. (2000) Methods in yeast genetics 2000: a Cold Spring Harbor Laboratory Course Manual, 2000 Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 34. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 10.1002/(SICI)1097-0061(19980130)14:2%3C115::AID-YEA204%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.