

Abstract
SMO (Smoothened), the central transducer of Hedgehog signaling, is coupled to heterotrimeric Gi proteins in many cell types, including cardiomyocytes. In this study, we report that activation of SMO with SHH (Sonic Hedgehog) or a small agonist, purmorphamine, rapidly causes a prolongation of the action potential duration that is sensitive to a SMO inhibitor. In contrast, neither of the SMO agonists prolonged the action potential in cardiomyocytes from transgenic GiCT/TTA mice, in which Gi signaling is impaired, suggesting that the effect of SMO is mediated by Gi proteins. Investigation of the mechanism underlying the change in action potential kinetics revealed that activation of SMO selectively reduces outward voltage-gated K+ repolarizing (Kv) currents in isolated cardiomyocytes and that it induces a down-regulation of membrane levels of Kv4.3 in cardiomyocytes and intact hearts from WT but not from GiCT/TTA mice. Moreover, perfusion of intact hearts with Shh or purmorphamine increased the ventricular repolarization time (QT interval) and induced ventricular arrhythmias. Our data constitute the first report that acute, noncanonical Hh signaling mediated by Gi proteins regulates K+ currents density in cardiomyocytes and sensitizes the heart to the development of ventricular arrhythmias.
Keywords: G protein, G protein-coupled receptor (GPCR), heart, Hedgehog signaling pathway, cardiovascular, potassium channel, ion channel, action potential, arrhythmias, noncanonical, Smoothened
Introduction
The vertebrate Hh (Hedgehog) pathway is initiated by binding of SHH (Sonic Hedgehog), Indian Hedgehog, or Desert Hedgehog to Patched1 (1). Ligand binding inhibits Patched1, leading to derepression of the central transducer protein SMO (Smoothened), a member of the G protein–coupled receptor (GPCR)2 superfamily (1). Activation of SMO facilitates conversion of the Gli family of transcriptional regulators into full transcriptional activators. Conversely, in the absence of Hh ligands, GLI2 and GLI3 are processed into transcriptional repressors by cAMP-dependent kinase (PKA), glycogen synthase kinase 3, and casein kinase I phosphorylation of multiple residues, which targets them for partial proteasomal degradation (1–4). SMO has an evolutionarily conserved function to selectively activate the Gi family of heterotrimeric G proteins (5–7). Activation of Gi proteins reduces the intracellular levels of cAMP and the activity of PKA, the main negative regulator of the GLI2 and GLI3 transcription factors. However, the requirement of Gi for activation of the GLI transcription factors is cell type- and context-dependent. This is not surprising because PKA activity is determined by the net activity of a plethora of GPCRs that stimulate or inhibit adenylyl cyclase, their localization, the presence of agonists, and the expression profile of PKA-regulatory proteins. Notwithstanding the role of Gi in GLI activation, the function of SMO as a GPCR is essential for several noncanonical Hh signaling outputs and, as opposed to the canonical pathway, is rapid and independent of GLI-transcriptional activity (8–11).
GLI-dependent and -independent Hh signaling coexist in cells that have primary cilia, mainly cells in interphase or quiescent, like cardiomyocytes. We previously reported that SMO/Gi protein coupling mediates up-regulation of canonical Hh signaling in response to ischemia and reperfusion injury, using animals with Tet-Off controlled cardiomyocyte-specific expression of a Gi inhibitory peptide (the C-terminal domain of the Gαi2 subunit) (GiCT/TTA mice) (12, 13). In addition, we found that SMO activation rapidly reduces cAMP levels elevated in response to β2-adrenergic receptor stimulation with isoproterenol (ISO) in cardiomyocytes in a Gi protein–dependent manner (12). Because Gi proteins regulate a large number of ionic channels that shape the action potential through inhibition of adenylyl cyclase/PKA by Gαi subunits, our previous findings suggested that noncanonical Hh signaling could have important repercussions in cardiac physiology.
In this study, we sought to investigate whether SMO/Gi coupling could regulate cardiac electrophysiology. Our results demonstrate that SMO/Gi coupling selectively reduces outward repolarizing K+ currents mediated by Kv channels, increases the action potential (AP) duration and induces ventricular arrhythmias.
Results
Hedgehog signaling increases cardiomyocyte action potential duration in a Gi-dependent manner
We first evaluated whether activation of SMO with Shh or with a small molecule agonist, purmorphamine (PUR) (14), resulted in acute changes of AP parameters. We recorded APs on ventricular cardiomyocytes isolated from adult WT mice using the whole-cell configuration of the patch-clamp technique. We found that addition of Shh or PUR to cardiomyocytes caused a rapid onset (<5 min) prolongation of the AP duration (APD) measured at 1 Hz, compared with vehicle control (Fig. 1A). No spontaneously triggered activity was observed in cardiomyocytes stimulated with PUR or Shh. Phase-plane plot analysis of the traces show that the repolarization speed is strongly reduced by Shh or PUR (Fig. 1B). A summary of AP characteristics of vehicle and PUR-treated WT cardiomyocytes at a frequency of 1 Hz showed no difference in any other parameter (Fig. S1). The APD was significantly prolonged by Shh or PUR throughout the entire repolarization phase (Fig. 1C), and it was mediated by SMO, because it was prevented by the inverse agonist KAAD-cyclopamine (Fig. 1D) (5). Remarkably, PUR failed to elicit a prolongation of the APD in cardiomyocytes isolated from GiCT/TTA mice (Fig. 1, E and F), indicating that Gi activation downstream of SMO mediates APD prolongation. PUR did not affect the time to achieve 90% repolarization (APD90), nor any other AP parameter, in GiCT/TTA cardiomyocytes (DMSO: 26 ± 4 ms versus PUR: 19 ± 7 ms) (Fig. S2).
Figure 1.

Activation of Smoothened prolongs the action potential duration in isolated mouse ventricular cardiomyocytes. A, representative examples of APs measured from WT myocytes at 1 Hz after 5 min of perfusion with 0.1% DMSO (vehicle), 5 μm PUR or 2.5 μg/ml recombinant Shh. B, representative phase-plane plots of APs individual cardiomyocytes treated as in A. C, APD to reduce the maximal amplitude by 25% (APD25), 50% (APD50), 75% (APD75), or 90% (APD90) in cardiomyocytes stimulated for 5 min as in A. One-way ANOVA was used. *, p < 0.05; **, p < 0.01 (n = 4–6 cardiomyocytes from independent isolation). D, length of the APD90 in cardiomyocytes pretreated or not for 2 h with 0.5 μm KAAD-CP, followed by 5 min of stimulation with 0.1% DMSO (vehicle), 5 μm PUR, or 2.5 μg/ml rShh (Shh). One-way ANOVA was used. *, p < 0.001; **, p < 0.05 (n = 5–6 cardiomyocytes from independent isolations). E, representative examples of APs measured from TTA and GiCT/TTA myocytes at 1 Hz after 5 min of perfusion with 0.1% DMSO (vehicle) or 5 μm PUR. F, representative phase-plane plots of APs individual cardiomyocytes from GiCT/TTA mice, treated as in A. All quantitative data are expressed as means ± S.E.
Because the repolarization phase is faster in the mouse heart and lacks the characteristic plateau of human cardiomyocytes, we also investigated the effect of acute SMO activation with PUR on ventricular cardiomyocytes isolated from guinea pigs, which have an AP kinetics similar to human cardiomyocytes. Consistent with the mouse data, PUR increased repolarization time in guinea pig cardiomyocytes by ∼50%, as shown in a representative trace (Fig. 2A) and in APD measurements (Fig. 2B). These findings demonstrate that activation of SMO/Gi signaling with either Shh or PUR rapidly increases repolarization time of single cardiomyocytes by a noncanonical signaling pathway, because the response time is incompatible with Gli-dependent transcription changes.
Figure 2.
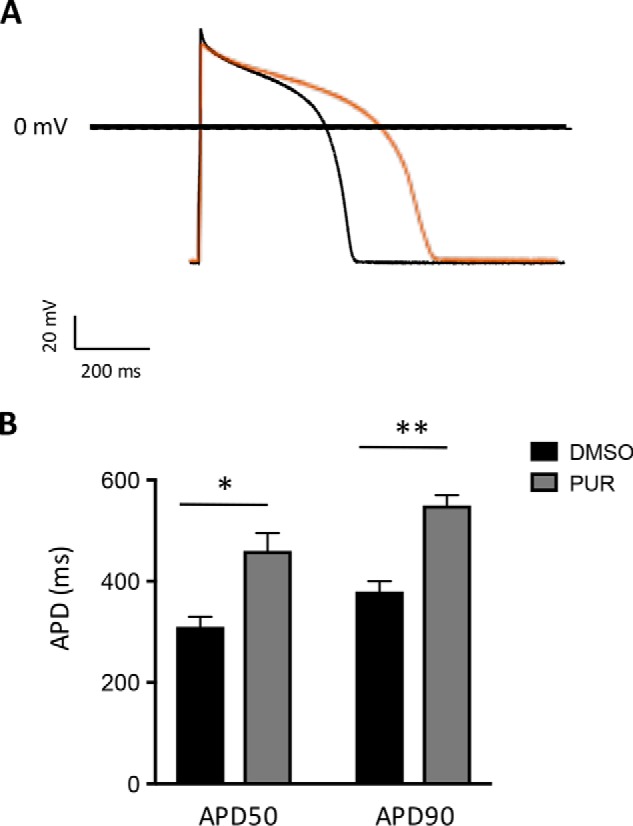
Activation of Smoothened prolongs the action potential duration in guinea pig ventricular cardiomyocytes. A, representative AP elicited in isolated guinea pig ventricular cardiomyocytes incubated with 0.1% DMSO (black trace) or 5 μm purmorphamine (red trace) for 1 h. B, mean APD to reduce the maximal amplitude by 50% (APD50) and 90% (APD90) in guinea pig cardiomyocytes treated as in A. The data are expressed as means ± S.E. Student's t test was used. *, p < 0.005; **, p < 0.0001 (n = 7–8 cardiomyocytes).
Smoothened activation causes a reduction in outward repolarizing K+ currents
The repolarization phase of the AP is governed by several Ca2+-, Na+-, and K+-permeable channels. Among the latter, voltage-gated K+ currents are the main determinants of AP repolarization kinetics (15). Prolongation of the APD can be caused by an increase in inward rectifying K+ currents and/or a reduction of outward K+ currents (IK). We recorded whole-cell Kv currents in ventricular myocytes isolated from WT mice treated with PUR or DMSO (vehicle). With voltage-gated Ca2+ and Na+ currents blocked, outward IK currents were recorded during 4.5-s depolarizing voltage steps to potentials between −40 and +40 mV from a holding potential of −70 mV; representative outward K+ current waveforms recorded in WT adult mouse cardiomyocytes are shown in Fig. 3A. As expected for this current, the rate of rise and the amplitude increased with depolarization; the largest and most rapidly activating current in Fig. 3A was evoked at +40 mV. Thus, the currents analyzed here were assumed to reflect only the activation of Ca2+-independent, depolarization-activated Kv channels. These currents were markedly reduced by PUR (Fig. 3A). Peak outward K+ current densities at all tested positive potentials were lower in PUR-treated cells than in vehicle-treated controls (Fig. 3B), reaching the maximal difference at +40 mV (PUR = 9.9 ± 1.1 pA/pF, n = 13 versus DMSO = 16.9 ± 1.5 pA/p, n = 15). Conversely, PUR did not modify inward rectifying K+ currents (Fig. S3), ruling them out as mediators of the AP remodeling by SMO/Gi signaling.
Figure 3.

Outward repolarizing K+ currents in ventricular myocytes are reduced after SMO activation. A, representative whole-cell Kv currents recorded at room temperature from vehicle control (DMSO) and 5 μm PUR-treated ventricular myocytes in response to 4.5-s depolarizing voltage steps to test potentials between −40 and +40 mV (10-mV increments) from a holding potential of −70 mV. Recorded currents were normalized for differences in cell size (whole-cell membrane capacitance) to obtain current densities. B, peak outward K+ current densities as a function of voltage in vehicle-treated versus purmorphamine-treated cardiomyocytes (means ± S.E., n = 11–12 cardiomyocytes). C, depolarization-induced Kv inactivation to investigate the contribution of Ito,f to the total outward IK currents. Panel a, whole-cell Kv currents were recorded in response to voltage steps to potentials ranging from −40 to +40 mV (in 10-mV increments) from a holding potential of −70 mV in vehicle control (DMSO) or 5 μm PUR-treated cardiomyocytes. Panel b, outward Kv currents evoked at the same test potentials following a brief prepulse to −20 mV from a holding potential of −30 mV to inactivate Ito,f. Panel a–b, the amplitudes of Ito,f in individual cells of each group were then obtained by digital off-line subtraction of the recordings with the prepulse (panel b) from the recordings without the prepulse (panel a).
The decay phase of the outward Kv currents can be described by the sum of two exponentials, reflecting the fast components of inactivation (Ito,f), the slow components of inactivation (IK,slow), and a noninactivating current (Iss) (16). Fitting and kinetic analyses of the total Kv currents revealed that Ito,f and IK,slow densities were reduced in PUR-treated cells by 48 and 29% at +40 mV, respectively (Table 1). The mean whole-cell membrane capacitance (Cm) was not affected by PUR, indicating that the reduced Ito,f and IK,slow current densities are not the result of nonspecific cellular enlargement. No significant differences in the densities of the steady-state outward K+ currents, Iss, determined as the currents remaining at the end of 4.5-s voltage steps, were observed.
Table 1.
Modulation of outward K+ current in isolated cardiomyocytes from wildtype mice stimulated with purmorphamine
The results were determined by Student's t test and indicate the number of cardiomyocytes tested in each condition, isolated from seven different hearts.
| Treatment | Density |
τk,slow | τto,f | Ito,f/IK,total | ||
|---|---|---|---|---|---|---|
| IK,slow | Ito,f | Iss | ||||
| pA/pF | pA/pF | pA/pF | ms | ms | ||
| DMSO (n = 15) | 9.7 ± 0.8 | 4.2 ± 0.7 | 4.1 ± 0.4 | 1455 ± 152 | 113 ± 9 | 0.22 ± 0.1 |
| PUR (n = 13) | 6.9 ± 0.8a | 2.2 ± 0.4a | 5.1 ± 0.8 | 1507 ± 177a | 93 ± 8a | 0.16 ± 0.1a |
a p < 0.05, PUR versus DMSO.
To further confirm the finding of the fitting analysis, we performed a two-step voltage inactivation protocol to isolate Ito,f. Inactivation of the fast component by delivery of a prepulse of −30 mV revealed a significant decrease in Ito,f of 25% at +40 mV after incubation with PUR (Fig. 3C). Thus, acute stimulation of SMO reduces outward K+ current density in cardiomyocytes.
In adult mouse ventricular myocytes, Ito,f is mediated Kv4.2/Kv4.3 pore-forming α subunits and KChiP auxiliary subunits, whereas IK,slow is mediated by Kv1.4, Kv1.5, and Kv2.1 (15, 16). To test the idea that Ito,f remodeling by SMO is mediated by altered distribution of Kv channels, we immunostained several Kv channel subunits on isolated ventricular myocytes treated with vehicle or PUR for 15 min. Mean fluorescence signal intensity of Kv4.3 at or near the cell surface quantified on confocal Z-scan images was ∼79% lower in PUR-treated cells (35 ± 9, n = 8) compared with vehicle control (165 ± 34, n = 8) (Fig. 4A). Remarkably, no difference in Kv4.3 staining was observed in cardiomyocytes from GiCT/TTA mice (vehicle: 97.3 ± 14.0 versus PUR: 131.6 ± 8.9, n = 8) (Fig. 4A). Thus, immunofluorescence experiments yielded data consistent with the whole-cell patch-clamping results. In contrast, Kv4.2, Kv2.1, Kv1.5, Kv1.4, Kir2.1, and Kir2.2 staining was not affected by PUR (Fig. S4). Next, we perfused hearts from TTA or GiCT/TTA mice with vehicle, PUR, or PUR after 30 min of perfusion with KAAD-cyclopamine. Upon perfusion, we prepared left ventricle homogenates and separated the membrane and soluble fractions. Western blotting analysis revealed a specific ∼40% reduction of Kv4.3 by PUR in the membrane fraction of control hearts, which was prevented by KAAD-cyclopamine, but the decrease was not evident in hearts from GiCT/TTA mice (Fig. 4B). Altogether, these data suggest that a reduction in plasmalemmal Kv4.3 levels underlies the decrease in Ito,f density induced by SMO agonists.
Figure 4.

Down-regulation of Kv4.3 membrane levels by SMO activation is Gi-dependent. A, immunofluorescent staining of Kv4.3 in fixed cardiomyocytes isolated from TTA or GiCT/TTA mice after 15 min of incubation with 0.1% DMSO or 5 μm PUR. B, 20 mg of protein in crude membrane preparations from hearts of TTA and GiCT/TTA mice, perfused as described with vehicle (DMSO), 5 μm PUR, or 0.5 μm KAAD-cyclopamine followed by 5 μm PUR were separated by SDS-PAGE and blotted using anti-Kv4.3 antibody. The graph shows densitometric quantification of Kv4.3 levels in ventricle membrane preparations from TTA mice (black circles) or GiCT/TTA mice (red circles) treated as indicated. Student's t test was used. *, p < 0.05 (n = 4 hearts).
Smoothened reduces PKA activity, increases repolarization time, and induces arrhythmias in intact hearts in a Gi-dependent manner
We next evaluated whether SMO activation had global effects on PKA activity in the intact heart as it does in isolated neonatal cardiomyocytes (12). Hearts from WT mice were perfused ex vivo using the Langendorff technique for 20 min in Tyrode's buffer, followed by 10 min with ISO to stimulate Gs-dependent cAMP production and PKA activation. As a measure of PKA activity, we evaluated phospholamban phosphorylation at Ser-16 (S16-PLN), a well-characterized PKA site (17), in whole-tissue homogenates prepared at the end of perfusion. As expected, ISO increased S16-PLN phosphorylation by 4-fold (Fig. 5A). Simultaneous perfusion with PUR prevented Ser-16 phosphorylation (Fig. 5A). Addition of KAAD-cyclopamine (KAAD-CP), a SMO inverse agonist, during the equilibration period, prevented the reduction in phospho-S16-PLN by PUR (Fig. 5A), indicating that the effect of PUR is specifically mediated by activation of SMO. These findings demonstrate that PUR opposes PKA activation specifically through SMO and, together with the findings in isolated cardiomyocytes, suggested that acute activation of SMO in the heart could modulate events relevant to cardiac physiology. In agreement with this hypothesis, perfusion of WT mouse hearts with PUR or Shh resulted in a transient depression of the autonomic heart rate (HR) (Fig. S5) and increased the HR-corrected QT (QTc) interval, which reflects the repolarization phase of the myocardial AP, as determined using volume-conducted electrocardiography (ECG) (Fig. 5B). To investigate whether this was mediated by activation of Gi proteins by SMO, we tested the effect of PUR on hearts from GiCT/TTA mice. This mouse model is superior to the use of pertussis toxin to inhibit Gi because it allows the analysis of reduced Gi activity exclusively in cardiomyocytes, avoids the need to pretreat the animals with the toxin prior to excision of the heart and the risk of partial efficacy of Gi ADP-ribosylation, and eliminates the co-occurrence of stress triggered signals caused by acute Gi inhibition. Remarkably, although the baseline ex vivo ECG parameters in hearts from GiCT/TTA mice were comparable with TTA control hearts, with exception of a lower HR (1 S1), perfusion with PUR did not increase the QTc interval in GiCT/TTA hearts, whereas it did in littermate TTA controls (Fig. 5C). Prolongation of the QTc interval is strongly associated with development of arrhythmias. Thus, we analyzed the ECG recordings for signs of arrhythmic episodes. Remarkably, perfusion with SMO agonists in Tyrode's solution induced ventricular arrhythmias in hearts from control mice, but not from GiCT/TTA animals (Fig. 5, D and E, and Tables 2 and 3). Arrhythmic episodes were never detected in any genotype perfused with vehicle for up to 1 h (Fig. 5, D and E). Activation of SMO in control hearts resulted in a very high incidence of ventricular premature beats (VPBs) (100% of PUR-perfused and 80% of Shh-perfused) and ventricular tachycardia (VT) (82% of PUR-perfused and 22% of Shh-perfused hearts, respectively) (Fig. 5E and Table 2). Perfusion with the SMO inhibitor KAAD-CP before addition of either Hh agonist greatly reduced the number of VPBs and completely prevented the occurrence of VT (Fig. 5E and Table 2). These findings demonstrate that SMO-dependent activation of Gi induces rapid electrophysiological changes that are conducive to ventricular arrhythmias.
Figure 5.

Activation of Smoothened in intact hearts inhibits PKA signaling and induces ventricular arrhythmias in a Gi protein–dependent manner. A, WT mice hearts were retrogradely perfused for 20 min using the Langendorff technique with 1 μm isoproterenol (ISO), 5 μm PUR, or 0.5 μm KAAD-CP together with purmorphamine. Tissue homogenates were separated by SDS-PAGE and immunoblotted using anti-Ser(P)-16 phospholambdan (PS16)-PLN and β-actin for loading control. For densitometric quantification of the fractional PS16-PNL, the data represent means ± S.E. Students' t test was used. *, p < 0.05 (n = 3). B, length of the QT interval corrected by heart rate (QTc) in WT mice hearts perfused with 0.1% DMSO (black circles), 5 μm purmorphamine (red circles), 20% DMEM (vehicle, open black circles), or Shh in 20% DMEM (Shh, open red circles) for 20 min. The data represent means ± S.E. One-way ANOVA was used. *, p < 0.05, PUR versus DMSO (n = 8); **, p < 0.001, Shh versus vehicle (n = 5). C, effect of perfusion with 0.1% DMSO (black circles) or 5 μm purmorphamine (red circles) on QTc interval prolongation in control mice (TTA) versus transgenic GiCT/TTA mice. One-way ANOVA was used. *, p < 0.01 (n = 4–8). D, representative ECG traces in WT mice hearts perfused with 0.1% DMSO or with 5 μm PUR. Arrows indicate examples of VPB and VT. E, incidence of VPBs and VTs in hearts from control TTA mice or from GiCT/TTA animals during 20 min of perfusion with vehicle (combined data from 0.1% DMSO and 20% DMEM), 5 μm PUR, Shh in 20% DMEM (Shh), or the latter with inclusion of 0.5 μm KAAD-CP during the equilibration phase (n = 5–8). The numbers inside the pie charts indicate the percentages of hearts with said type of arrhythmia.
Table 2.
Incidence and characteristics of arrhythmias in hearts from adult TTA mice in response to acute perfusion with Hedgehog agonists
Hearts from 3–5-month-old TTA mice were perfused in Tyrode's buffer for 20 min followed by addition of vehicle (DMSO or 20% DMEM (vehicle)) or the agonists PUR or Shh for another 20 min or were perfused with Tyrode's buffer containing KAAD-CP for 20 min followed by perfusion for another 20 min with KAAD-CP plus PUR or KAAD-CP plus Shh.
| Parameter | DMSO (n = 6) | PUR (n = 8) | KAAD-CP + PUR (n = 8) | Vehicle (n = 5) | Shh (n = 5) | KAAD-CP+ Shh (n = 5) |
|---|---|---|---|---|---|---|
| VPB (events/20 min) | 0 | 268a | 9b | 0 | 46a | 8b |
| VPB incidence (% hearts) | 0 | 100a | 37.5b | 0 | 80a | 40b |
| VT (events/20 min) | 0 | 184a | 0b | 0 | 1a | 0 |
| VT incidence (% hearts) | 0 | 87.5a | 0b | 0 | 20a | 0 |
| Mean VT duration (ms) | 0 | 1,020 ± 670a | 0b | 0 | 740 | 0 |
a p < 0.05, PUR versus DMSO and Shh versus vehicle.
b p < 0.05, PUR versus KAAD-CP + PUR and Shh versus KAAD-CP + Shh.
Table 3.
Incidence and characteristics of arrhythmias in hearts from adult GiCT/TTA mice in response to acute perfusion with Hedgehog agonists
Hearts from 3–5-month-old GiCT/TTA mice were perfused in Tyrode's buffer for 20 min followed by addition of vehicle (DMSO or 20% DMEM (vehicle)) or by PUR or Shh for another 20 min.
| Parameter | DMSO (n = 6) | PUR (n = 6) | Vehicle (n = 5) | Shh (n = 6) |
|---|---|---|---|---|
| VPB (events/20 min) | 0 | 0 | 0 | 1 |
| VPB incidence (% hearts) | 0 | 0 | 0 | 16.7 |
| VT (events/20 min) | 0 | 0 | 0 | 0 |
| VT incidence (% hearts) | 0 | 0 | 0 | 0 |
Discussion
Since our initial report of coupling of SMO to Gi proteins (5), mounting evidence suggested that although it contributes to Gli activation in some cell types but not all (10, 18), the most important function of SMO/Gi coupling is in acute noncanonical signaling events. This study shows the physiological relevance of SMO-Gi coupling in mediating a novel acute effect of Hh signaling in the myocardium. First, we show that activation of Gi by SMO in the myocardium is strong enough to oppose PKA activation by adrenergic stimulation, because of a decrease of cAMP production in single cardiomyocytes (reported in our previous study (12)). We observed a rapid-onset prolongation of the AP duration in isolated cardiomyocytes by stimulation of the Hh pathway with Shh or the small molecule agonist PUR, which was completely prevented by impairment of Gi signaling in cells from GiCT/TTA mice and was specifically reduced by a SMO inhibitor. SMO/Gi coupling was also required for increased turnover of Kv4.3 pore-forming subunits of Kv channels, which underlies a decrease in total and fast outward repolarizing K+ currents and provides an explanation for the prolongation of the AP. Remarkably, the same effect is observed in the AP of guinea pig cardiomyocytes, which have an AP kinetics and channel function more similar to human myocardial cells.
An important consequence of the acute effect of Hh signaling on the AP of individual cardiomyocytes is the high incidence of ventricular arrhythmias, which require active Gi signaling. Collectively, our findings demonstrate the existence of a noncanonical Hh pathway in the myocardium that could trigger dangerous arrhythmias. Therefore, the proposed development of small molecule agonists of the Hh pathway for therapeutic use after ischemic cardiac events, mostly to improve revascularization (19), should be carefully reconsidered in the light of our findings, because the beneficial proangiogenic effects of Shh could be attained by other growth factors that do not trigger electrophysiological changes.
Future studies will be necessary to investigate whether the effect of Hh on myocardial function changes over time as the effect of parallel signaling outputs kicks into effect, such as reduction of caspase activity and induction of GLI transcriptional targets. In this regard, during the completion of our studies, an article was published that concludes that Shh reduces AP duration based on evaluation of the effect of Shh after 6 h (20). A potential explanation for the contradictory findings is that desensitization of noncanonical SMO/Gi signaling after the rapid onset effects could result in a late distinct remodeling phenotype or, as mentioned above, other effectors of the Hh signaling pathway that are activated with a slower kinetics could have a different effect on the AP kinetics. Electrophysiological evaluation of healthy adult cardiomyocytes at longer times postisolation was not possible because of a decrease in cell viability, and the use of neonatal cardiomyocytes for longer term experiments is discouraged because they express a different set of ionic channels. Nevertheless, our exhaustive analysis using the physiological agonist Shh and an agonist and an antagonist of SMO prove the connection between SMO signaling and acute changes in repolarization kinetics. Moreover, the fact that cells from GiCT/TTA mice are refractory to the SMO agonists also serves as a control of specificity of the agonists used, which do not appear to have nonspecific effects on the ionic channels that govern the AP phases.
Our data point at Kv4.3 and/Kv4.2 channels, the molecular determinants of Ito,f, as targets of acute SMO signaling. Phosphorylation of Kv channels by PKA, protein kinase C (PKC), and calmodulin-dependent protein kinase II (CAMKII) kinases regulates their gating properties and their membrane localization (21, 22), and both PKA and PKC are known to be engaged by SMO (23). Nevertheless, because of the central role of Gi proteins as strict mediators of SMO in the heart and the pleiotropic effect of PKA signaling in the regulation of the cardiac AP, we believe that changes in other ionic currents, beyond those analyzed in this study, might also contribute to the repolarization phenotype. In conclusion, we report for the first time a regulatory effect of acute, noncanonical cardiac Hh signaling in the rodent heart that results in remodeling of Kv channels and an increased incidence of arrhythmias. Given the up-regulation of Shh in response to hypoxia, it will be relevant to investigate in the future a potential link between the Hh pathway and the occurrences of life-threatening arrhythmias in patients surviving myocardial infarction.
Experimental procedures
Animals
The mice were housed, handled, and euthanized according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Thomas Jefferson University Committee on Animal Care (Institutional Animal Care and Use Committee protocol no. 831D). GiCT/TTA mice, their TTA littermates, and WT mice were used for the study. Transgenic cardiomyocyte-specific inducible GiCT mice (GiCT/TTA) were obtained by breeding mice encoding a doxycycline-sensitive TTA transgene (α-myosin heavy chain (MHC)–TTA) with mice encoding a TTA-driven α-MHC–GiCT transgene, as previously described (13). GiCT/TTA animals showed normal size, appearance, and survival when compared with WT littermates as reported previously (13). No sudden death was observed in either WT or GiCT/TTA mice (age ≤ 5 months). Hearts from GiCT/TTA animals were also normal in appearance and weight. Electrophysiological analysis using volumetric ECG recordings indicated ECG baseline parameters in GiCT/TTA mice were similar to WT littermates. None of the transgenic animals showed any overt phenotype under nonstress conditions, but GiCT/TTA mice did have a reduced recovery of cardiac function after ischemia/reperfusion injury, as previously published. Guinea pigs (Dunkin Hartley, 300–350 g) were purchased from Charles River (Kent, UK) and housed at the University of Leeds following approved institutional guidelines.
Isolation of adult mouse cardiomyocytes
Adult WT and GiCT/TTA mice were anesthetized with tribromoethanol (Avertin; 100 mg/kg, intraperitoneally; Sigma), anticoagulated with heparin sodium (0.5 unit/g, intraperitoneally). The hearts were surgically removed via a thoracotomy. Although fully immersed in Ca2+ free Tyrode's (135 mmol/liter NaCl, 4 mmol/liter KCl, 0.33 mmol/liter NaH2PO4, 1 mmol/liter MgCl2, 10 mmol/liter HEPES, 10 mmol/liter glucose, and 10 mmol/liter 2,3-butanedione monoxime), the aorta was cannulated and mounted on a Langendorff apparatus. Following a 10–12-min enzymatic digestion with 0.5 mg/ml collagenases B and D (Roche) and 0.02 mg/ml protease XIV (Sigma), the heart was submersed in Ca2+ free Tyrode's, whereas the atria and aorta were removed. The myocytes were then extracted from the heart in Ca2+-free Tyrode's with BSA. Isolated myocytes were resuspended in 4% fetal bovine serum minimum Eagle's medium (Sigma), plated on laminin-coated coverslips, and placed in a 95% air, 5% CO2 incubator at 37 °C. Approximately 1 h after plating, Ca2+-tolerant ventricular myocytes adhered to the laminin, and damaged cells were removed by replacing the medium with fresh serum-free minimum Eagle's medium, supplemented with antibiotics (penicillin/streptomycin). The cells were examined electrophysiologically within 6 h of isolation.
Isolation of guinea pig myocytes
Guinea pig ventricular myocytes were isolated according to methods described in detail elsewhere (24). The use of guinea pigs was approved by the UK Home Office. The guineas pigs (Dunkin Hartley, 300–350 g) were euthanized by cervical dislocation following the UK Home Office Guidance on the Operation of Animals (Scientific Procedures) Act 1986 and University of Leeds institutional guidelines. Cervical dislocation was performed by a highly skilled operator, followed by quick thoracotomy to remove the hearts, which were then perfused for 5 min at 37 °C as a Langendorff preparation, with a Ca2+-free, oxygenated isolation solution containing 135 mmol/liter NaCl, 6 mmol/liter KCl, 0.33 mmol/liter NaH2PO4, 5 mmol/liter sodium pyruvate, 1 mmol/liter MgCl2, 10 mmol/liter HEPES, and 10 mmol/liter glucose (adjusted with NaOH) with collagenase type II (Worthington, LORNE; 100 units/ml) protease (Sigma; 0.66 mg/ml) and BSA (Sigma; 1.66 mg/ml) for 5 min and then washed with 1 mmol/liter Ca2+-containing isolation solution for an extra 5 min. The ventricles were cut free and minced in fresh isolation solution with 1 mmol/liter CaCl2, and myocytes were isolated and stored at room temperature for up to 6 h after isolation.
Hedgehog pathway agonists and antagonists
Experiments using isolated cardiomyocytes were performed using recombinant Shh, purified as previously described (25). Experiments of intact heart perfusion, because of the large amount of perfusion medium needed, were performed using Shh-containing or control-conditioned medium prepared as follows. HEK 293 (CRL-1573) cells were obtained from American Type Culture Collection (Manassas, VA). The cells were maintained in DMEM with 10% fetal bovine serum at 37 °C with 5% carbon dioxide. Shh- and pcDNA-conditioned media were prepared by transfecting 80% confluent 100-mm plates of HEK 293 cells with 24 μg of pRK5 full-length mouse Shh or pcDNA3 using Lipofectamine (Life Technologies). The cells were grown for 24 h; then they were washed with serum-starvation medium comprising DMEM and 0.5% FBS and further cultured with the above medium for another 2 days. The medium was harvested after centrifugation at 1000 rpm for 5 min. The supernatant was then sterile-filtered with a 0.22-μm syringe filter and kept at −80 °C. Prior to use, conditioned medium was diluted 5× in the experimental solution. The activity of Shh, both recombinant and in the conditioned medium, was measured by a Gli-luciferase assay in NIH-3T3 cells, as previously described (25). Purmorphamine and KAAD-cyclopamine were obtained from EMD Millipore (Billerica, MA) and dissolved in DMSO.
Mouse cardiomyocyte electrophysiological recordings
The whole-cell patch clamp recordings were obtained from myocytes isolated from the ventricles of adult (2–4 months old) WT, TTA, and GiCT/TTA mouse hearts. Action potential and voltage-gated potassium currents recordings were conducted using an Axopatch 200A amplifier (Molecular Devices, Union City, CA), interfaced to a Dell microcomputer with a Digidata 1322A series analog/digital interface (Molecular Devices), using pClamp 8 software (Molecular Devices). Whole-cell patch-clamp experiments were conducted at room temperature (22–24 °C). Recording pipettes contained 135 mmol/liter KCl, 1 mmol/liter MgCl2, 10 mmol/liter EGTA, 10 mmol/liter HEPES, and 5 mmol/liter glucose (pH 7.2; 310 mOsm). The bath solution contained 136 mmol/liter NaCl, 4 mmol/liter KCl, 1 mmol/liter CaCl2, 2 mmol/liter MgCl2, 5 mmol/liter CoCl2, 10 mmol/liter HEPES, 0.04 mmol/liter tetrodotoxin, and 10 mmol/liter d-glucose (pH 7.4; 300 mOsm). For current-clamp experiments, the tetrodotoxin and CoCl2 were omitted from the bath. Patch electrodes were fabricated and polished by heating. We used pipettes with resistance of 2–4 MΩ. Whole-cell membrane capacitances and series resistances were compensated electronically prior to recording voltage-clamp currents. Voltage errors resulting from the uncompensated series resistance were always ≤8mV and were not corrected. The experimental data were sampled at 5 kHz; current and voltage signals were low-pass–filtered at 1 kHz prior to digitization and storage. For current-clamp experiments, a series of 1-ms-long current steps from 10 to 250 pA in 20-pA increments were delivered at 1-Hz frequency to cells in each group.
For action potential analysis, AP overshoot (peak depolarization) and resting membrane potential were determined and the difference between them yielded the spike height. AP duration was measured as the time it took to cross the voltage at 25% (APD25), 50% (APD50), 75% (APD75), and 90% (APD90) of the spike height. Rheobase is the magnitude of a 1-ms current injection pulse sufficient to elicit an AP, and threshold was defined as the voltage at which the dV/dt deviated from 0 mV ms−1 as determined from the phase-plane plot.
Depolarization-activated outward K+ (Kv) currents were recorded in response to 4.5-s voltage steps to potentials between −40 and + 40 mV from a holding potential (HP) of −70 mV; voltage steps were presented in 10-mV increments at 15-s intervals. Inwardly rectifying K+ currents, IK1, were recorded in response to 350-ms voltage steps to test potentials between −40 and −120 mV (in 10-mV increments) from the same HP. Voltage-clamp data were compiled and analyzed using Clampfit (Molecular Devices) and Origin 7.0 (OriginLab Corp., Northampton, MA) software. Whole-cell membrane capacitances (Cm) were acquired from Clampex (Molecular Devices). Leak currents were always <10 pA and were not corrected. Peak Kv currents at each test potential were defined as the maximal outward K+ current recorded during the 4.5-s voltage steps. The decay phases of the outward K+ currents evoked during 4.5-s depolarizing voltage steps are described by the sum of two exponentials using previously described methods: y(t) = A1exp(−t/τ1) + A2exp(−t/τ2) + B, where t is time, τ1 and τ2 are the decay time constants, A1 and A2 are the amplitudes of the inactivating current components (Ito,fast or Ito,f and IK,slow), and B is the amplitude of the noninactivating current component, Iss (26). Fitting residuals and correlation coefficients were determined to assess the quality of fits; only fits with correlation coefficients >0.980 were used in this study. In all cells, IK1 densities were determined from the amplitudes of the currents measured at the end of 350-ms hyperpolarizing voltage steps (−90 to −120 mV) from a HP of −70 mV. Current amplitudes in each cell were normalized to whole-cell membrane capacitances (in the same cell), and current densities (pA/pF) are reported.
The rapidly activating and rapidly inactivating Kv current, Ito,f, was isolated by a two-step voltage protocol, using previously described procedures (27). Briefly, total whole-cell Kv currents were first evoked in response to 4.5-s depolarizing voltage steps to potentials between −40 and +40 mV (in 10-mV increments) from a holding potential of −70 mV. A prepulse paradigm that included a brief (60 ms) step to −10 mV before the 4.5-s depolarizing voltage steps to potentials between −40 and +40 mV (in 10-mV increments) was then used. Off-line subtractions of the currents evoked with the prepulse from the currents evoked without the prepulse were performed to isolate Ito,f.
All electrophysiological recordings were conducted in the whole-cell configuration. Pipette resistance was maintained within the range of 1.5–1.8 mΩ. Recording pipettes were filled with a solution containing 5 mmol/liter NaCl, 135 mmol/liter CsF, 10 mmol/liter EGTA, 5 mmol/liter MgATP, and 5 mmol/liter HEPES (pH 7.2) with CsOH. The cells were maintained in a solution containing 5 mmol/liter NaCl, 132.5 mmol/liter CsCl, 1 mmol/liter CaCl2, 1 mmol/liter MgCl2, 0.1 mmol/liter CdCl2, 20 mmol/liter HEPES, and 11 mmol/liter glucose (pH 7.35) with CsOH.
Guinea pig cardiomyocyte action potential recording. Isolated single ventricular myocytes were transferred to a static bath recording chamber mounted on an Olympus CK40 inverted microscope. The cells were allowed to settle in the bath for 10–15 min before starting continuous perfusion (3–5 ml/min) with extracellular solution containing the following: 140 mmol/liter NaCl, 4 mmol/liter KCl, 1.8 mmol/liter CaCl2, 1 mmol/liter MgCl2, 10 mmol/liter HEPES, 10 mmol/liter d-glucose (pH adjusted to 7.4 with NaOH). Whole-cell patch-clamp recordings were made in current clamp using patch pipettes of 4–7 MΩ resistance at room temperature (21–23 °C). To evaluate the effect of purmorphamine on guinea pig myocytes action potential, the cells were preincubated with purmorphamine (5 μmol/liter) for 1 h at 37 °C. Control measurements were taken from the same untreated myocytes populations. The intracellular solution consisted of 110 mmol/liter KCl, 10 mmol/liter NaCl, 10 mmol/liter EGTA, 1 mmol/liter MgCl2, 10 mmol/liter HEPES, 5 mmol/liter MgATP (pH 7.2, KOH). The signals were acquired using a Axopatch 200B controlled by Clampex 10 software via a Digidata 1322A interface (Axon Instruments, Inc., Foster City, CA). Action potentials were evoked using current injections with the smallest values used to reduce stimulus artifacts, and the cell was stimulated every 10 s with an averaged 10 APs used to measure action potential durations in both controls and test conditions. Offline analysis was carried out using the data analysis package Clampfit 10 (Axon Instruments), and the data are expressed as means ± S.E. Statistical analysis was performed using unpaired Student's t tests, where p < 0.05 was considered statistically significant.
Volume-conducted ECG
Adult mice were heparinized (0.5 unit/g intraperitoneally) and then anesthetized with 2,2,2-tribromoethanol (Avertin; 425 mg/kg, intraperitoneal administration). Anesthesia was confirmed by lack of the pinch reflex. The hearts were quickly removed through a thoracotomy and rinsed in Tyrode's solution (130 mmol/liter NaCl, 24 mmol/liter NaHCO, 1.2 mmol/liter NaH2PO4, 1.0 mmol/liter MgCl2, 5.6 mmol/liter glucose, 4.0 mmol/liter KCl, and 1.8 mmol/liter CaCl2) and equilibrated with a 95% O2, 5% CO2 gas mixture. Hearts were then rapidly cannulated, perfused in a retrograde fashion via an aortic cannula with 37 °C oxygenated Tyrode's solution at the rate of 2–3 ml/min by using minipulse 3 peristaltic pump (Gilson Inc., Middleton, WI), and placed in a chamber. The solution in the chamber was used as a volume conductor for recording ECGs. ECGs were recorded with a Cyber Amp380 amplifier (Axon Instruments) and digitized at 10 kHz with a Digidata 1400A and AxoScope 10.2 (Molecular Devices) after equilibration for 20 min as reported previously (28). In each record, the QT interval was determined as the time from the start of ventricular activity (the initiation of the QRS complex) to the time of minimum voltage (29). QT intervals were corrected for heart rate using the Bazzett's formula QTc = QT/RR1/2, were RR is the interval duration between two consecutive QRS complexes.
Immunofluorescence
Detection of Kv channel pore forming subunits (Kv4.2, Kv4.3, Kv2.1, Kv1.5, Kv1.4, Kir2.1, and Kir2.2) was performed as described previously (5). Briefly, freshly isolated myocytes were fixed in 4% paraformaldehyde for 20 min and subsequently suspended in PBS for immunostaining. The cells were collected from 3–6 animals for each group. The cells were permeabilized and blocked with 0.2% Triton X-100 (Sigma) and 5% goat serum (Invitrogen) in PBS and then incubated in primary antibody overnight at 4 °C. The monoclonal anti-mouse Kv4.3 channels (1:10, NeuroMab), anti-mouse Kv 4.2 (1:10, NeuroMab), anti-rabbit Kv 1.5 (1:10, Alomone Labs), anti-mouse Kv 2.1 (1:10, NeuroMab), anti-mouse Kv 1.4 (1:10, NeuroMab), anti-mouse Kir 2.1(1:50, NeuroMab), and anti-mouse Kir2.2 (1:50, NeuroMab) primary antibodies were used. After washes with 0.1% Tween 20 in PBS, the cells were incubated with goat anti-mouse Alexa Fluor 488 (Molecular Probes), goat anti-mouse Alexa Fluor 555 (Molecular Probes), and secondary antibodies for 1 h at room temperature and mounted using ProLong® Gold antifade reagents (Invitrogen). Stained slides were viewed with a Zeiss LMS510 Meta confocal microscope in Bioimaging Shared Resource of the Sidney Kimmel Cancer Center (funded under NCI, National Institutes of Health Grant 5 P30 CA-56036) at Thomas Jefferson University.
To quantitatively assess Kv-channel distribution in myocytes, eight Z-scan images were obtained at 0.25-μm intervals from middle of each cell, and a projection image was assembled using National Institutes of Health Image 1.48v software. Data from the lines at the outermost positions along the length of the cell were used to get an average estimate of the fluorescence distribution at or near the cell surface in each cell. For analysis of fluorescence intensities, images were produced using identical acquisition settings. Pixel intensities at the cell periphery of cardiomyocytes were determined using the histogram function in NIH ImageJ software v1.48.
Western blotting
Western blots were performed on fractionated ventricular membrane proteins prepared from adult WT and GiCT/TTA mice hearts as previously described (28). Briefly, hearts were homogenized on ice in a protease (Roche Applied Science) and phosphatase inhibitors (Sigma) containing buffer consisting of 25 mmol/liter Tris-HCl (pH 7.5), 5 mmol/liter EDTA (pH 8), and 5 mmol/liter EGTA (pH 8). The nuclei and debris were pelleted by centrifugation at 1,000 × g for 10 min. This procedure was repeated, and the supernatants from both low-speed spins were pooled and centrifuged at 80,000 × g for 30 min. The pellets were resuspended in the above solution and centrifuged at 80,000 g for another 10 min. The final pellets were sonicated and solubilized in protease and phosphatase inhibitors containing radioimmune precipitation assay buffer consisting of 50 mmol/liter Tris-HCl (pH 7.5), 5 mmol/liter EDTA (pH 8), 150 mmol/liter NaCl, 0.1% SDS, 1% NP40, 0.5% sodium deoxycholate for 1 h. Insoluble material was centrifuged at 13,000 rpm for 10 min. Solubilized membranes were aliquoted and stored frozen at −80 °C until used. The protein content of each of the solubilized membrane preparations was determined using a protein assay kit (Thermo Scientific). Proteins were separated on a NuPAGE-Novex 4–12% bis-Tris gel (Invitrogen) and transferred onto nitrocellulose membranes. The membranes were then blocked and incubated overnight at 4 °C with primary antibodies. The blots were then incubated with secondary antibody conjugated to horseradish peroxidase (1:5000), and visualized using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). For normalization of signals, blotting was performed with an anti-β-actin mAb, followed by incubation with an IRDye 680- or IRDye 800CW-conjugated secondary antibody (Li-Cor). The membranes were imaged with the Odyssey IR imaging system (Li-Cor), and quantitative densitometric analysis was performed by applying Odyssey version 1.2 IR imaging software.
Statistics
The results are expressed as means ± S.E. Statistical significance was examined with Student's t test, one-way ANOVA, or chi-squared test. p < 0.05 was considered significant.
Author contributions
L. C. and M. A.-O. data curation; L. C., M. A.-O., and C. P. formal analysis; L. C., M. A.-O., and N. R.-D. G. investigation; L. C. writing-original draft; M. L. C. and N. R.-D. G. conceptualization; M. C., D. R. M., C. P., and N. R.-D. G. supervision; M. C. and N. R.-D. G. writing-review and editing; W. J. K., C. P., and N. R.-D. G. resources; D. R. M. and N. R.-D. G. funding acquisition.
Supplementary Material
Acknowledgments
We thank Glenn Radice for the use of the volume-conducted ECG equipment and Shalini Maitra for technical assistance.
This work was supported in part by National Institutes of Health Grant 2RO1GM080396 (to D. R. M. and N. R.-D. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We dedicate this article to the memory of Prof. Chris Peers, who sadly passed away during the review of the manuscript.
This article contains Table S1 and Figs. S1–S7.
- GPCR
- G protein–coupled receptor
- PKA
- cAMP-dependent kinase
- ISO
- isoproterenol
- AP
- action potential
- APD
- AP duration
- PUR
- purmorphamine
- HR
- heart rate
- ECG
- electrocardiogram
- VPB
- ventricular premature beat
- VT
- ventricular tachycardia
- DMEM
- Dulbecco's modified Eagle's medium
- HP
- holding potential
- ANOVA
- analysis of variance
- CP
- cyclopamine
- KAAD-CP
- 3-keto-N-(aminoethyl-aminocaproyl-dihydrocinnamoyl)cyclopamine.
References
- 1. Robbins D. J., Fei D. L., and Riobo N. A. (2012) The Hedgehog signal transduction network. Sci. Signal. 5, re6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang B., Fallon J. F., and Beachy P. A. (2000) Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 10.1016/S0092-8674(00)80678-9 [DOI] [PubMed] [Google Scholar]
- 3. Riobó N. A., Lu K., Ai X., Haines G. M., and Emerson C. P. Jr, (2006) Phosphoinositide-3 kinase and Akt are essential for Sonic Hedgehog signalling. Proc. Natl. Acad. Sci. U.S.A. 103, 4505–4510 10.1073/pnas.0504337103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riobo N. A., Lu K., and Emerson C. P. Jr. (2006) Hedgehog signal transduction: signal integration and cross talk in development and cancer. Cell Cycle 5, 1612–1615 10.4161/cc.5.15.3130 [DOI] [PubMed] [Google Scholar]
- 5. Riobo N. A., Saucy B., Dilizio C., and Manning D. R. (2006) Activation of heterotrimeric G proteins by SMOothened. Proc. Natl. Acad. Sci. U.S.A. 103, 12607–12612 10.1073/pnas.0600880103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ogden S. K., Fei D. L., Schilling N. S., Ahmed Y. F., Hwa J., and Robbins D. J. (2008) G protein Galphai functions immediately downstream of SMOothened in Hedgehog signalling. Nature 456, 967–970 10.1038/nature07459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen F., Cheng L., Douglas A. E., Riobo N. A., and Manning D. R. (2013) SMOothened is a fully competent activator of the heterotrimeric G protein G(i). Mol. Pharmacol. 83, 691–697 10.1124/mol.112.082511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brennan D., Chen X., Cheng L., Mahoney M., and Riobo N. A. (2012) Noncanonical Hedgehog signalling. Vitam. Horm. 88, 55–72 10.1016/B978-0-12-394622-5.00003-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chinchilla P., Xiao L., Kazanietz M. G., and Riobo N. A. (2010) Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signalling pathways. Cell Cycle 9, 570–579 10.4161/cc.9.3.10591 [DOI] [PubMed] [Google Scholar]
- 10. Polizio A. H., Chinchilla P., Chen X., Kim S., Manning D. R., and Riobo N. A. (2001) Heterotrimeric Gi proteins link Hedgehog signalling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 286, 19589–19596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Belgacem Y. H., and Borodinsky L. N. (2011) Sonic Hedgehog signalling is decoded by calcium spike activity in the developing spinal cord. Proc. Natl. Acad. Sci. U.S.A. 108, 4482–4487 10.1073/pnas.1018217108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carbe C. J., Cheng L., Addya S., Gold J. I., Gao E., Koch W. J., and Riobo N. A. (2014) Gi proteins mediate activation of the canonical hedgehog pathway in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 307, H66–H72 10.1152/ajpheart.00166.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeGeorge B. R. Jr, Gao E., Boucher M., Vinge L. E., Martini J. S., Raake P. W., Chuprun J. K., Harris D. M., Kim G. W., Soltys S., Eckhart A. D., and Koch W. J. (2008) Targeted inhibition of cardiomyocyte Gi signalling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation 117, 1378–1387 10.1161/CIRCULATIONAHA.107.752618 [DOI] [PubMed] [Google Scholar]
- 14. Sinha S., and Chen J. K. (2006) Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2, 29–30 10.1038/nchembio753 [DOI] [PubMed] [Google Scholar]
- 15. Nerbonne J. M. (2016) Molecular basis of functional myocardial potassium channel diversity. Card. Electrophysiol. Clin. 8, 257–273 10.1016/j.ccep.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niwa N., and Nerbonne J. M. (2010) Molecular determinant of cardiac transient outward potassium current (Ito) expression and regulation. J. Mol. Cell Cardiol. 48, 12–25 10.1016/j.yjmcc.2009.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karczwewski P., Kuschel M., Baltas L. G., Bartel S., and Krause E. G. (1997) Site-specific phosphorylation of a phospholamban peptide by cyclic nucleotide- and Ca2+/calmodulin-dependent protein kinases of cardiac sarcoplasmic reticulum. Basic Res. Cardiol. 92, 37–43 10.1007/BF00794066 [DOI] [PubMed] [Google Scholar]
- 18. Low W. C., Wang C., Pan Y., Huang X. Y., Chen J. K., and Wang B. (2008) The decoupling of Smoothened from Galphai proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev. Biol. 321, 188–196 10.1016/j.ydbio.2008.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kusano K. F., Pola R., Murayama T., Curry C., Kawamoto A., Iwakura A., Shintani S., Ii M., Asai J., Tkebuchava T., Thorne T., Takenaka H., Aikawa R., Goukassian D., von Samson P., Hamada H., Yoon Y. S., Silver M., Eaton E., Ma H., Heyd L., Kearney M., Munger W., Porter J. A., Kishore R., and Losordo D. W. (2005) Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signalling. Nat. Med. 11, 1197–1204 10.1038/nm1313 [DOI] [PubMed] [Google Scholar]
- 20. Paulis L., Fauconnier J., Cazorla O., Thireau J., Soleti R., Vidal B., Ouillé A., Bartholome M., Bideaux P., Roubille F., Le Guennec J. Y., Andriantsitohaina R., Martínez M. C., and Lacampagne A. (2015) Activation of Sonic hedgehog signalling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci. Rep. 5, 7983 10.1038/srep07983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin L., Sun W., Wikenheiser A. M., Kung F., and Hoffman D. A. (2010) KChiP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol. Cell Neurosci. 43, 315–325 10.1016/j.mcn.2009.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Birnbaum S. G., Varga A. W., Yuan L. L., Anderson A. E., Sweatt J. D., and Schrader L. A. (2004) Structure and function of Kv4-family transient potassium channels. Physiol. Rev. 84, 803–833 10.1152/physrev.00039.2003 [DOI] [PubMed] [Google Scholar]
- 23. Riobo N. A., Haines G. M., and Emerson C. P. Jr, (2006) Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signalling. Cancer Res. 66, 839–845 10.1158/0008-5472.CAN-05-2539 [DOI] [PubMed] [Google Scholar]
- 24. Louch W. E., Sheehan K. A., and Wolska B. M. (2011) Methods for cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol. 51, 288–298 10.1016/j.yjmcc.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martinez-Chinchilla P., and Riobo N. A. (2008) Purification and bioassay of hedgehog ligands for the study of cell death and survival. Methods Enzymol. 446, 189–204 10.1016/S0076-6879(08)01611-X [DOI] [PubMed] [Google Scholar]
- 26. Cheng L., Yung A., Covarrubias M., and Radice G. L. (2011) Cortactin is required for N-cadherin regulation of Kv1.5 channel function. J. Biol. Chem. 286, 20478–20489 10.1074/jbc.M111.218560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Granados-Fuentes D., Norris A. J., Carrasquillo Y., Nerbonne J. M., and Herzog E. D. (2012) I(A) channels encoded by Kv1.4 and Kv4.2 regulate neuronal firing in the suprachiasmatic nucleus and circadian rhythms in locomotor activity. J. Neurosci. 32, 10045–10052 10.1523/JNEUROSCI.0174-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swope D., Cheng L., Gao E., Li J., and Radice G. L. (2012) Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol. Cell. Biol. 32, 1056–1067 10.1128/MCB.06188-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y., Wu J., King J. H., Huang C. L., and Fraser J. A. (2014) Measurement and interpretation of electrocardiographic QT intervals in murine hearts. Am. J. Physiol. Heart Circ. Physiol. 306, H1553–H1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.