

Abstract
Activation of protein kinase G (PKG) Iα in nociceptive neurons induces long-term hyperexcitability that causes chronic pain. Recently, a derivative of the fungal metabolite balanol, N46, has been reported to inhibit PKG Iα with high potency and selectivity and attenuate thermal hyperalgesia and osteoarthritic pain. Here we determined co-crystal structures of the PKG Iα C-domain and cAMP-dependent protein kinase (PKA) Cα, each bound with N46, at 1.98 Å and 2.65 Å, respectively. N46 binds the active site with its external phenyl ring, specifically interacting with the glycine-rich loop and the αC helix. Phe-371 at the PKG Iα glycine-rich loop is oriented parallel to the phenyl ring of N46, forming a strong π-stacking interaction, whereas the analogous Phe-54 in PKA Cα rotates 30° and forms a weaker interaction. Structural comparison revealed that steric hindrance between the preceding Ser-53 and the propoxy group of the phenyl ring may explain the weaker interaction with PKA Cα. The analogous Gly-370 in PKG Iα, however, causes little steric hindrance with Phe-371. Moreover, Ile-406 on the αC helix forms a hydrophobic interaction with N46 whereas its counterpart in PKA, Thr-88, does not. Substituting these residues in PKG Iα with those in PKA Cα increases the IC50 values for N46, whereas replacing these residues in PKA Cα with those in PKG Iα reduces the IC50, consistent with our structural findings. In conclusion, our results explain the structural basis for N46-mediated selective inhibition of human PKG Iα and provide a starting point for structure-guided design of selective PKG Iα inhibitors.
Keywords: cyclic GMP (cGMP), protein kinase G (PKG), inhibitor, crystal structure, drug design, ATP competitive inhibitor, inhibitor specificity, nonopioid analgesics, protein–ligand complex
Introduction
Chronic pain is a debilitating condition that affects nearly 25 million adults in the United States (1). Opioid pain relievers are the most prescribed medication class in the United States (2). The increasing prescription of opioid pain relievers is associated with a dramatic increase in opioid misuse, abuse, overdose, and opioid use disorder, contributing to a $504 billion economic cost in the United States in 2015 and more than 63,600 opioid overdose deaths in 2016 (2–6). Another major category of analgesics, COX inhibitors, has long-term cardiovascular side effects (7). Therefore, a new type of nonopioid-based pain reliever is in demand for effective pain management.
Reversible protein phosphorylation regulates all aspects of cell survival. Consequently, dysregulation of protein kinases is often involved in human diseases such as cancer (8), diabetes (9–11), and chronic pain (12, 13). More than 30 protein kinase inhibitors have been approved by the Food and Drug Administration in the past 23 years, with the majority of them targeting tyrosine kinases for cancer treatment (14).
Beyond its role as a central regulator of smooth muscle tone, cGMP-dependent protein kinase (PKG)3 Iα activation in nociceptive neurons results in long-term hyperexcitability that causes chronic pain (15, 16). PKG Iα is also a crucial modulator of cortical neuronal activity in pathological pain; thus, it represents a novel target for developing analgesic therapeutic agents (17). A recent study demonstrated that N46, a derivative of the fungal metabolite balanol, inhibits PKG Iα with high potency and selectivity, resulting in attenuation of thermal hyperalgesia and osteoarthritic pain in rats (18).
PKG Iα belongs to the AGC kinase family and consists of N-terminal regulatory (R) and C-terminal catalytic (C) domains (Fig. 1A) (19, 20). PKG Iα shares a large degree of sequence similarity with cAMP-dependent protein kinase (PKA). In particular, the PKG Iα C-domain shows 45% sequence identity with the PKA Cα, consistent with their similar structures. The C-domain includes small and large lobes that consist of mostly β strands and α helices, respectively. A highly acidic active site is formed between the two lobes that binds Mg2+, ATP, and substrates. In the absence of cGMP, the activity of PKG Iα is negatively regulated by the interaction between the R- and C-domains (21, 22).
Figure 1.

Domain organizations and overall structures of N46 bound PKG Iα and PKA Cα. A, the domain organizations of PKG Iα and PKA Cα. The catalytic domains used for crystallization are shaded in orange and labeled with the corresponding residue numbers. Phosphorylated residues are indicated (PKG Iα T517 and PKA Cα S139/T197/S338). B and C, overall structures of the PKG Iα C–Ν46 (B) and PKA Cα–N46 (C) complexes. The N-and C termini are labeled with the corresponding residue numbers. The structures are rendered as cartoons, with N46 shown as sticks. The small and large lobes are colored gray and tan, respectively. Atoms in N46 are colored as follows: carbons, yellow; oxygen, red; nitrogen, blue; fluorine, cyan. Enlarged views show Fo-Fc omit maps of N46 (contoured at 3.0 σ level).
Three classes of small-molecule PKG inhibitors have been widely used for functional studies of PKG (23, 24). The first class is the R-diastereomer of the phosphorothioate analogs of cGMP, including Rp-cGMPS (25). This compound binds the R-domain and stabilizes its inactive state without causing conformational changes required for activation (26). The second class consists of small molecules that compete with ATP by directly binding the active site within the C-domain. These reagents include H-89, balanol, and KT-5823 (27–32). The third class includes peptide inhibitors that also bind the active site and prevent substrate binding. However, all of these inhibitors lack potency, specificity, and activity in vivo. For example, Rp-cGMPS is not potent (Ki = 20 μm) and nonselectively inhibits other cyclic nucleotide effectors, such as phosphodiesterase and PKA (23). KT-5823 also inhibits other kinases and may not inhibit PKG in intact cells (33). Despite its high potency in vitro, DT-2 does not inhibit PKG in platelets or in rat mesangial cells (34).
As mentioned, balanol is a potent inhibitor of PKG but also inhibits other serine and threonine kinases such as PKA, most PKC isoforms, and Ca2+-dependent protein kinase (30, 35). To improve inhibitor selectivity for PKG Iα, a homology model of PKG Iα docked with balanol was generated based on the crystal structure of the PKA Cα–balanol complex. Several amino acid differences near their binding pockets were identified, and balanol was modified to preferentially interact with PKG Iα–specific residues (18). In particular, the homology model showed that Thr-88 of PKA Cα corresponds to Ile-406 in PKG Iα (16). To exploit this difference, a propoxy group was added to the external phenyl ring (ring D) of the balanol derivatives to selectively interact with Ile-406 of PKG Iα. Although one such compound, N46, was reported to have a high selectivity and potency for PKG Iα over PKA Cα, the exact molecular basis of its improved affinity and specificity is unknown.
Results and discussion
Several crystal structures have been solved for mammalian PKG I, but these are of various fragments of the R-domains (36–39). Because N46 directly targets the C-domain of PKG Iα, we first obtained an isolated C-domain that is fully active. To understand the molecular basis of N46's high selectivity for human PKG Iα, we determined co-crystal structures of N46 bound to the human PKG Iα C-domain and human PKA Cα at 1.98 Å and 2.65 Å, respectively (Fig. 1 and Fig. S1 and Table S1). The PKG Iα C–N46 complex was crystallized in the P42 space group with one molecule in the asymmetric unit. The molecule shows clear electron density for the bound N46 and the C-domain used for crystallization, excluding the first 10 residues at the N terminus (Fig. 1B). The PKA Cα–N46 complex was crystallized in the P3121 space group with one molecule in the asymmetric unit (Fig. 1C and Fig. S1). The final model shows clear density for the Cα subunit except for the first 10 residues. Unlike previous PKA Cα structures, the N-terminal αA helix disengages from the catalytic core because of unusual crystal packing interactions (Fig. S2). The αA helix of a neighboring symmetry mate occupies the equivalent position seen in previous structures and provides the same set of interactions with the catalytic core.
The overall structure of the PKG Iα C–N46 complex is similar to the AMP-PNP-bound structure.4 It shows a closed conformation with the fully ordered glycine-rich loop and C-terminal tail (Fig. 1B). N46 binds to a pocket that extends from the hinge region to the inner surface of the αC helix and spans ∼20 Å (Fig. 2A). The pocket can be divided into three subsites according to the interaction between the PKG Iα C-domain and AMP-PNP: the adenine, the ribose, and the extended triphosphate subsites. N46 binds to all three subsites in the extended active site of the PKG Iα C-domain (Fig. 2A).
Figure 2.
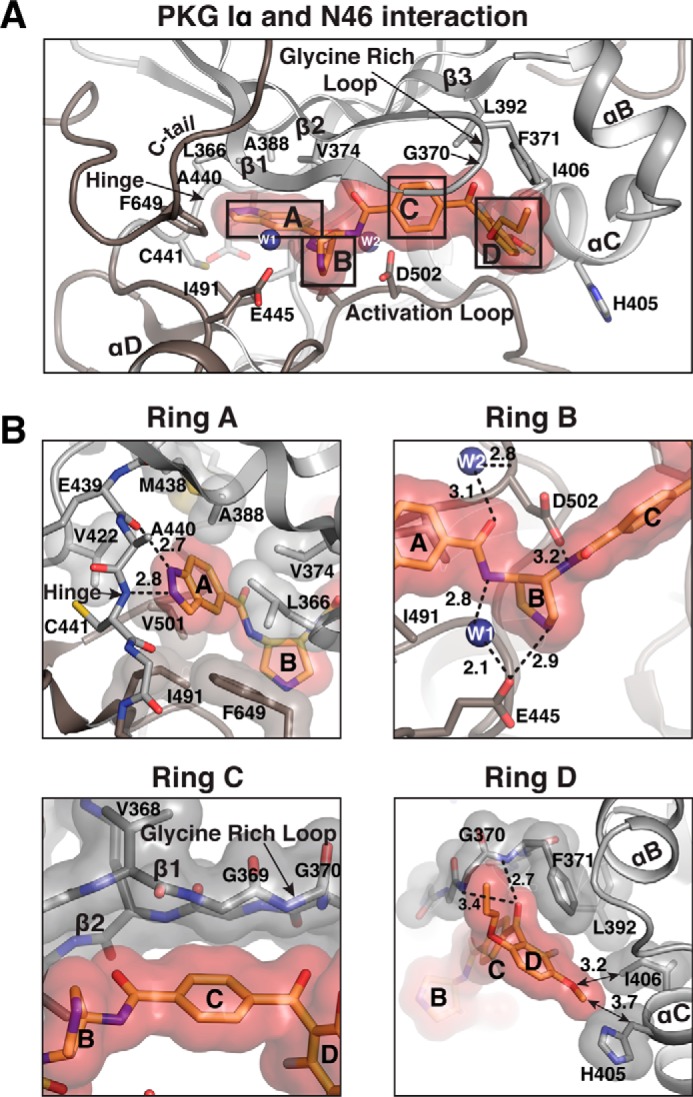
Interactions between the PKG Iα C-domain and N46. A, detailed interactions with N46. Only the regions near the active site are shown. N46 is shown with a transparent surface. Residues contacting N46 are shown as sticks. Water molecules are shown as blue spheres. B, enlarged views for each ring of N46, highlighting its interactions with different regions of the active site. Residues that provide VDW interactions are shown with transparent surfaces. Hydrogen bonds are shown as dotted lines, and arrows indicate key VWD interactions, with distances indicated in angstroms.
The A-ring (indazole ring) binds the adenine subsite consisting of the hinge (loop between β5-αD) and hydrophobic residues from both small and large lobes (Fig. 2B). Specifically, the protonated 1-N binds the backbone carbonyl of Glu-439, whereas the unprotonated 2-N interacts with the backbone amide of Cys-441 through hydrogen bonds. Additionally, the indazole ring is surrounded by several hydrophobic residues that coat the adenine subsite. These residues include Leu-366, Val-374, Ala-388, Val-422, Met-438, Ile-491, Val-501, and Phe-649.
The B-ring (pyrrolidine ring), which connects the A-ring to the C-ring, interacts with the acidic ribose subsite directly and indirectly through water molecules (Fig. 2B). The ribose subsite consists of the hinge and activation loop residues. The side chains of Glu-445 at the hinge and Asp-502 at the activation loop form hydrogen bonds with the amine groups on either side. Two water molecules bridge the interaction with N46 at this subsite. These water molecules are located adjacent to the amide connecting the B-ring to the A-ring, bridging them to the side chains of Glu-445 and Asp-502 through hydrogen bonds.
The C-ring (phenyl ring) interacts with β1 and the glycine-rich loop through van der Waals (VDW) contacts (Fig. 2B). In particular, Val-368, Gly-369, and Gly-370 are within 3.4–3.8 Å from the C-ring, providing VDW interactions. Because these interactions are through backbone atoms, this region does not provide any PKG-selective contacts.
The D-ring (external phenyl ring) with the propoxy and methoxy groups provides two interactions that are PKG-specific and may explain its high selectivity for PKG Iα over PKA Cα (Fig. 2B). In designing N46, the propoxy group was added to the phenyl ring to provide a preferential interaction with Ile-406 of PKG Iα over PKA Cα, which has a threonine (Thr-88) at the analogous position (Fig. S3) (18). However, the structure shows that the methoxy group points toward the side chain of Ile-406 instead, whereas the propoxy group points toward the glycine-rich loop, each providing hydrophobic interactions. Additionally, the D-ring, along with the carbonyl group that connects the D-ring to the C-ring, docks to the tip of the glycine-rich loop through hydrogen bonds and VDW interactions. The interconnecting carbonyl group hydrogen bonds with the backbone amide of Phe-371 and uniquely forms a lone pair–π interaction with its side chain. The D-ring and the side chain of Phe-371 are off-centered, and they interact through a parallel displaced π interaction.
The overall interactions between the PKA Cα subunit and N46 are similar to those in the PKG Iα C–N46 complex because most of the contact residues are highly conserved between the two kinases (Fig. 3A). However, the structure shows differences that may explain a higher IC50 value for the PKA Cα subunit.
Figure 3.
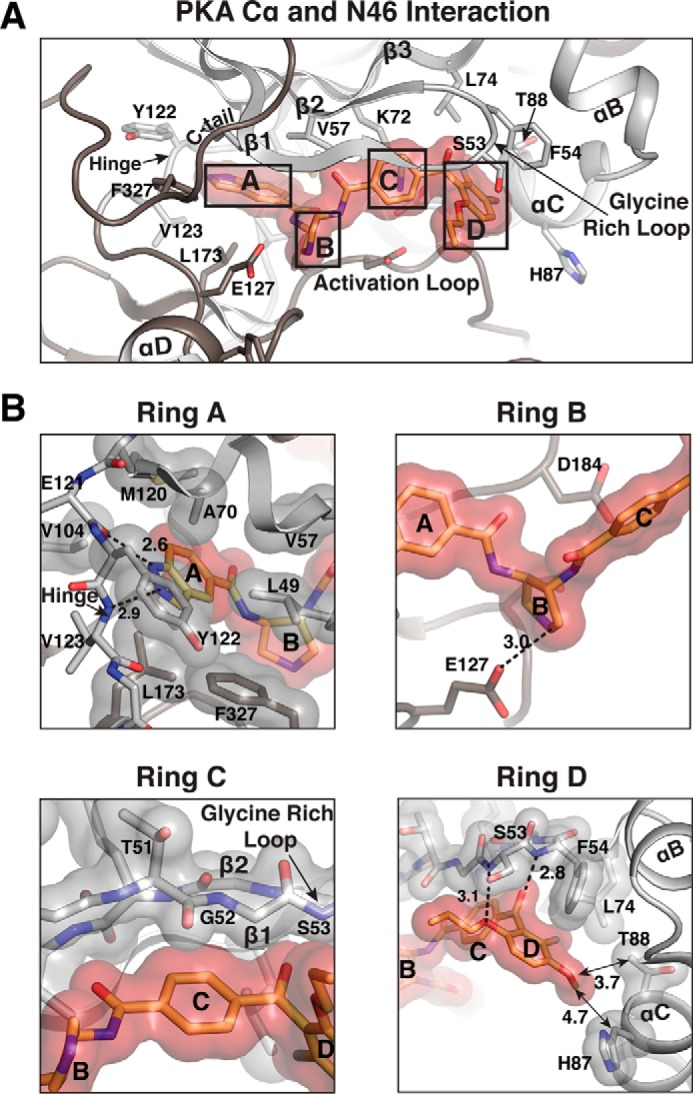
Interactions between PKA Cα and N46. A, detailed interactions with N46. Only the regions near the active site are shown. Residues contacting N46 are shown as sticks. B, enlarged views for each ring of N46, highlighting its interactions with different regions of the active site. Residues that provide VDW interactions are shown with transparent surfaces. Hydrogen bonds are shown as dotted lines, with their distances given in angstroms.
The A-ring binds the adenine subsite, and the interactions in this region are essentially the same as in PKG Iα. These include hydrogen bonds between the A-ring and the backbone atoms of Glu-121 and Val-123 at the hinge and VDW contacts with a hydrophobic pocket consisting of Leu-49, Val-57, Ala-70, Val-104, Met-120, Leu-173, and Phe-327 (Fig. 3B). Tyr-122 at the hinge region provides an additional hydrophobic contact unseen in PKG Iα because Tyr-122 replaces Ala-440 of PKG Iα. Although the B-ring similarly docks onto the ribose subsite, its amine group interacts only with the hinge residue Glu-127 through a hydrogen bond, not with the activation loop residue Asp-184 (Fig. 3B). Unlike Asp-502 of PKG I, which forms a hydrogen bond with N46 (Fig. 2B), the side chain of Asp-184 points away and no longer interacts with N46 in PKA. The C-ring similarly docks to β1 and the glycine rich loop and interacts with the backbone atoms of Thr-51, Gly-52, and Ser-53.
The D-ring interacts less strongly with PKA Cα compared with PKG Iα because of two PKA-specific residues, Phe-54 and Thr-88 (Fig. 3B). The structure shows that the side chain of Phe-54 at the tip of the glycine-rich loop rotates ∼30° and provides a weaker T-shaped π interaction with the D-ring. Because of this rotation, the interconnecting carbonyl no longer forms a lone pair–π interaction with the aromatic Phe-54. In addition, the side chain of Thr-88 of the αC helix is smaller and less hydrophobic than that of Ile-406 of PKG Iα, thus providing a much weaker hydrophobic interaction with the methoxy group (3.7 Å) (Fig. 3B). The structural alignment with the PKG Iα C–N46 complex suggests that a steric clash between the side chain of the preceding residue, Ser-53, and the propoxy moiety causes the rotation of the Phe-54 side chain. As seen in Fig. S3, N46 moves away slightly from the active site because of the steric clash. This allows more room between the D-ring and the glycine-rich loop, causing the rotation of the Phe-54 side chain.
The reported inhibition constant of balanol for PKA Cα is 1.6 nm, whereas N46 inhibits PKA with an IC50 of 1.0 μm, showing an over 600-fold increase (18). Comparing the PKA Cα–N46 complex with the PKA Cα–balanol complex reveals that this reduction is mostly due to loss of hydrogen bonds (Fig. 4). The PKA Cα–balanol complex shows 12 nonsolvent mediated hydrogen bonds and large numbers of VDW interactions between the extended active site and balanol. The PKA Cα–N46 complex shows that, although the most of the VDW contacts are preserved, N46 forms only 6 direct hydrogen bonds because of the modifications on the C and D rings.
Figure 4.
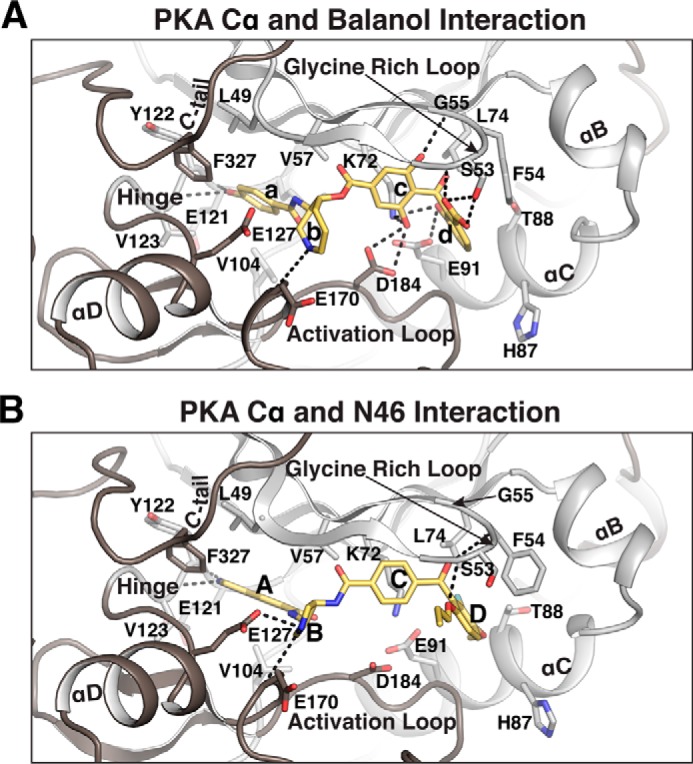
Structural comparison between PKA Cα bound with balanol and N46. A and B, detailed interactions between PKA Cα and balanol (PDB code 1BX6) (A) orN46 (B). Only the regions near the active site are shown. Residues contacting balanol or N46 are shown as sticks. Direct hydrogen bonds are shown as dotted lines. The rings of balanol and N46 are labeled a–d and A–D, respectively.
Substituting the phenol of balanol (Fig. 4A, ring a) with the indazole ring of N46 (Fig. 4B, ring A) does not reduce the number of hydrogen bonds and VDW contacts with the adenine subsite. In the PKA–balanol complex, the phenol forms hydrogen bonds with the same backbone atoms of Glu-121 and Val-123 at the hinge region (Fig. 4A) as the indazole. However, replacing a more puckered azepane ring of balanol (Fig. 4A, ring b) with a less puckered pyrrolidine of N46 (Fig. 4B, ring B) results in one additional hydrogen bond at the ribose subsite. The puckered azepane ring interacts mainly with a conserved catalytic loop residue, Glu-170, through its backbone (Fig. 4A). In the PKA–N46 complex, the less puckered pyrrolidine ring brings its amine group within a hydrogen bonding distance of the Glu-127 side chain, forming a new hydrogen bond (Fig. 4B).
Removing two hydroxyl groups from the c-ring of balanol (Fig. 4A) disrupts all four hydrogen bonds with the triphosphate subsite. In the PKA Cα–balanol complex, two hydroxyl groups on the c-ring interact with Gly-55, Lys-72, and Asp-184 through four hydrogen bonds. In major contrast, the C-ring of N46 (Fig. 4B) no longer binds these residues and interacts with the glycine-rich loop through VDW contacts.
Last, substituting a carboxyl group and a hydroxyl group on the d-ring of balanol (Fig. 4A) with a bulky and hydrophobic propoxy group and a fluorine atom, respectively (Fig. 4B, ring D of N46), significantly weakened the interaction with the glycine-rich loop and the αC helix. In the PKA–balanol complex, the carboxyl group on the d-ring forms strong hydrogen bonds with both the side chain and backbone of Ser-53 at the glycine-rich loop, whereas the 3-hydroxyl group binds the side chains of Glu-91 and Lys-72 through hydrogen bonds. Additionally, the d-ring is oriented parallel to the side chain of Phe-54, allowing a parallel π-stacking interaction between them as well as a lone pair–π interaction between the carbonyl group and Phe-54. None of these interactions are preserved in the PKA–N46 complex, although a new hydrogen bond forms between the propoxy group and the backbone amide of Ser-53.
We noticed that the side chain of Phe-54 remains parallel to the d-ring when bound to balanol and rotates when bound to N46 (35). The balanol-bound PKA structure shows that this is because balanol binds deeper into the pocket, allowing a parallel π-stacking interaction with Phe-54 (Fig. S4A). In contrast, N46 cannot bind as deeply because of its bulky methoxy group, resulting in enough space between the D-ring and Phe-54, which allows Phe-54 to rotate to provide VDW contact with the D-ring (Fig. S4B).
To test the molecular basis for N46's PKG Iα-selective inhibition over PKA, we mutated the unique contact residues in PKG Iα to those in PKA and vice versa. Specifically, for PKG Iα, we mutated Gly-370 and Ile-406 to the corresponding residues in PKA Cα (i.e. G370S and I406T). We also mutated these two PKA Cα residues into the corresponding PKG Iα residues (S53G and T88I). For PKG Iα, we generated two single mutants (G370S and I406T) and a double mutant (G370S/I406T). For PKA Cα, we only generated a double mutant (S53G/T88I). We then measured IC50 values using in vitro kinase assays (Fig. 5). N46 showed an IC50 of 43 nm for WT PKG Iα, whereas it inhibited PKA Cα with an IC50 of 1030 nm, showing an ∼24-fold difference in selectivity. The PKG Iα single mutants were inhibited with higher IC50 values of 90 nm and 142 nm for G370S and I406T, respectively. The double mutant PKG Iα showed an IC50 value of 301 nm, demonstrating a synergistic effect of the two mutations. In contrast, the PKA Cα double mutant showed an IC50 of 552 nm, which is almost half of that seen in WT Cα. The higher IC50 values seen in the PKG Iα mutants and the lower value of the PKA Cα double mutant compared with their respective WT are consistent with our structural findings.
Figure 5.

N46 inhibition of PKG Iα and PKA Cα. A, we performed in vitro kinase inhibition assays using purified WT and mutant PKG Iα and PKA Cα in the presence of increasing concentrations of N46, as described under “Experimental procedures.” B, IC50 values based on the curves shown in A.
Despite lack of data on inhibition constants of N46 for other kinases, our model of a PKCα isoform (PDB code 3IW4) docked with N46 suggests that N46 is a poor inhibitor for the PKCα isoform (Fig. S5) (40). The model shows that the tip of the glycine-rich loop curls in toward the active site and clashes with the C-ring. In particular, Phe-350 at the glycine-rich loop occupies the part of the pocket the C-ring binds, suggesting that N46 would interact poorly with PKCα. Consistent with the model, Sung et al. (18) reported that, at 0.75 μm of N46, PKCδ had 68% residual activity, whereas PKG Iα was completely inhibited with 0% residual activity .
Our structural and biochemical data suggest new strategies for generating N46 derivatives with higher selectivity for PKG Iα over PKA Cα. Amino acid sequences at the hinge region and β7 that make up the left edge and the base of the adenine pocket are different in PKG Iα compared with PKA Cα. PKG Iα has Ala-Cys-Leu (residues 440–442) at the hinge, whereas PKA has Tyr-Val-Pro (residues 122–124) (Fig. S6). This causes PKG Iα to have a wider adenine pocket compared with the PKA Cα subunit (Fig. 6). Additionally, at the base of the adenine pocket, PKG Iα has an isoleucine (Ile-491 at β7) replacing a leucine (Leu-173) of PKA Cα, providing a slightly deeper pocket. Thus, to improve selectivity for PKG Iα, bulkier heterocyclic rings could be engineered in N46 to fill this unique pocket. Also, a reactive group can be placed here to covalently link to Cys-441 because PKA lacks a cysteine residue at the analogous position (Fig. S6). During the initial design of N46, the propoxy group was added to increase its interaction with Ile-406 at the αC helix. However, our structures revealed that this group points in an opposite direction (toward the glycine-rich loop) and interacts with Gly-370 instead. Thus, it may be possible to add an additional ethyl or propyl group here to improve interaction with PKG Iα. This modification should cause steric hindrance with Ser-53 of PKA at the glycine-rich loop while providing additional nonpolar interactions with Gly-370 in PKG Iα. In conclusion, our structural and biochemical data in part explain N46's selectivity for PKG Iα and provide a starting point for structure-guided design of selective PKG Iα inhibitors.
Figure 6.

Adenine pockets of PKG and PKA. The surfaces of the active site pockets for PKG Iα C-domain and PKA Cα are colored in red. Enlarged views show the adenine pockets. The active site pockets were calculated using Hollow (46).
Experimental procedures
Expression and purification of the hPKG Iα C-domain
The sequence encoding the human PKG Iα C-domain (327–671) was cloned into the pBlueBacHis2A vector. The vector was modified to put a tobacco etch virus (TEV) protease site just before the PKG coding sequence. The protein was expressed in High Five cells. The cells were grown at 28 °C and infected at a multiplicity of infection of 3.0 for 32 h. All cells were lysed in buffer A (25 mm Tris (pH 7.5), 500 mm NaCl, and 1 mm β-mercaptoethanol) with a Constant Systems TS cell disrupter (Daventry Northants, UK) and cleared via ultracentrifugation. The supernatant was loaded onto a Bio-Rad Nuvia nickel affinity column, washed with buffer A and eluted with buffer A containing 300 mm imidazole. The His tag was removed by incubating the sample with TEV protease at 4 °C overnight. TEV was removed from the protein sample by performing a second nickel affinity chromatography and collecting the flow-through fractions. The sample was further purified by anion exchange chromatography (Mono Q 10/100 GL, GE Healthcare) in buffer B (25 mm Tris (pH 7.5) and 1 mm β-mercaptoethanol) with and without 1 m sodium chloride. This was followed by size exclusion chromatography (Hiload 16/60 Superdex 75, GE Healthcare) in buffer C (25 mm Tris (pH 7.5), 150 mm sodium chloride, and 1 mm tris(2-carboxyethyl)phosphine (TCEP)).
Expression and purification of hPKA Cα
The pET15b plasmid encoding human PKA Cα was transformed into BL21 (DE3) Escherichia coli cells. The cells were grown at 37 °C until OD600 = 1.0 was reached. The expression was induced by 0.5 mm isopropyl β-d-thiogalactopyranoside at 18 °C for 18 h. The cells were then lysed by the Constant Systems TS cell disruptor in buffer A. The lysate was then cleared by ultracentrifugation and membrane filtration. The supernatant was applied onto a GE His-Trap column for nickel affinity purification. The protein was eluted by buffer A containing 300 mm imidazole. The His tag was removed by incubating the protein with TEV protease at 4 °C overnight, followed by a second nickel affinity chromatography. The protein was then further purified by anion exchange chromatography (anion exchange chromatography, Mono Q 10/100 GL, GE Healthcare) in buffer D (25 mm potassium phosphate (pH 7.0) and 1 mm β-mercaptoethanol) with and without 1 m sodium chloride. This was followed by size exclusion chromatography (Hiload 16/60 Superdex 75, GE Healthcare) in buffer C.
Crystallization and structure determination
To obtain crystals of the PKG Iα C-domain–N46 complex, 14 mg ml−1 of the PKG Iα C-domain was incubated with 1 mm N46 for 30 min at room temperature. Crystals were obtained by mixing 1 μl of the C-domain–N46 complex solution with 1 μl of well solution (24% w/v PEG 1500 and 20% v/v glycerol) and 0.2 μl of additive (30% w/v trimethylamine N-oxide dihydrate) at 22 °C. To obtain crystals of the PKA Cα–N46 complex, 12 mg ml−1 of PKA Cα was incubated with 1 mm of N46 for 30 min at room temperature. Crystals were obtained by mixing 0.2 μl of the C-domain–N46 complex solution with 16% (w/v) PEG 8000, 0.04 m potassium phosphate (monobasic), and 20% (v/v) glycerol. PKG Iα C-domain and PKA Cα crystals were cryoprotected by paratone, and diffraction images were collected at the Advanced Light Source (Berkeley, CA). Data were processed using CCP4.iMosflm (41). The structures of the PKG Iα C-domain–N46 and PKA Cα–N46 complexes were determined by Phaser-MR using AMP-PNP–bound PKG Iα C-domain (PDB code 6BG2) and balanol-bound PKA Cα (PDB code 1BX6) as molecular replacement probes (42). Both final structures were manually built using Coot and refined using Phenix.Refine (43, 44). The figures were generated using PyMOL (Schrödinger, LLC).
In vitro kinase assays
FLAG-tagged WT and mutant PKG Iα proteins were purified from transiently transfected 293T cells as described previously (45). PKA Cα WT and its mutant were purified as described above. The purified kinases were diluted in kinase dilution buffer (10 mm potassium phosphate (pH 7.0), 1 mm EDTA, 35 mm β-mercaptoethanol, and 0.1% BSA) so that the reactions produced ∼105 counts per reaction (corresponding to about 36 pmol phosphate incorporation). Reactions were initiated by adding 10 μl of diluted kinase to 5 μl of 3× kinase reaction mixture (120 mm HEPES (pH 7.4), 1.56 mg/ml Kemptide, 30 mm MgCl2, 300 μm ATP, 360 μCi/ml [γ-32P]ATP and 30 μm cGMP) containing variable amounts of the N46 inhibitor diluted in DMSO (control assays contained DMSO alone). Reactions were run for 1.5 min at 30 °C and stopped by spotting on P81 phosphocellulose paper. Unincorporated [γ-32P]ATP was removed by washing P81 paper 4 × 2 liters in 0.45% O-phosphoric acid. 32P incorporation was measured by liquid scintillation counting. The data were analyzed using GraphPad Prism 7.
Author contributions
L. Q. and C. K. conceptualization; C. K. resources; L. Q., B. S., S. A., D. C., and C. K. data curation; L. Q., B. S., D. C., and C. K. formal analysis; C. K. supervision; C. K. funding acquisition; L. Q. and B. S. validation; L. Q., B. S., and C. K. investigation; L. Q., B. S., and C. K. visualization; L. Q., B. S., and C. K. methodology; L. Q. and C. K. writing-original draft; C. K. project administration; L. Q., B. S., D. C., and C. K. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Andrey Kovalevsky and Friedrich W. Herberg for critical reading of the manuscript. We also thank Paul Leonard (MD Anderson Cancer Center) for assistance with the initial screening of the PKG Iα–N46 complex crystals and Ying-Ju Sung (Geisinger Commonwealth School of Medicine) for kindly providing N46. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health; the NIGMS, National Institutes of Health; and the Howard Hughes Medical Institute. The ALS-ENABLE beamlines are supported in part by NIGMS, National Institutes of Health National Grant P30 GM124169-01. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract DE-AC02-05CH11231. The Pilatus detector was funded under National Institutes of Health Grant S10OD021832.
This project was supported in part by the Protein and Monoclonal Antibody Production Shared Resource at Baylor College of Medicine with funding from National Institutes of Health Cancer Center Support Grant P30 CA125123. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Table S1.
The atomic coordinates and structure factors (codes 6C0T and 6C0U) have been deposited in the Protein Data Bank (http://wwpdb.org/).
L. Qin, B. Sankaran, D. E. Casteel, and C. Kim, unpublished data.
- PKG
- cGMP-dependent protein kinase
- PKA
- protein kinase A
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- VDW
- van der Waals
- TEV
- tobacco etch virus
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Walitt B., Nahin R. L., Katz R. S., Bergman M. J., and Wolfe F. (2015) The prevalence and characteristics of fibromyalgia in the 2012 national health interview survey. PLoS ONE 10, e0138024 10.1371/journal.pone.0138024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. U.S. Department of Health and Human Services (HHS), Office of the Surgeon General (2016) Facing addiction in America: The Surgeon General's report on alcohol, drugs, and health. U.S. Department of Health and Human Services (HHS), Office of the Surgeon General, Washington, D.C. [PubMed] [Google Scholar]
- 3. Rummans T. A., Burton M. C., and Dawson N. L. (2018) How good intentions contributed to bad outcomes: the opioid crisis. Mayo Clin. Proc. 93, 344–350 10.1016/j.mayocp.2017.12.020 [DOI] [PubMed] [Google Scholar]
- 4. The Council of Economic Advisers (2017) The Underestimated Cost of the Opioid Crisis. The Council of Economic Advisers, Washington, D.C. [Google Scholar]
- 5. Ryan S. A. (2018) Calculating the real costs of the opioid crisis. Pediatrics 141, e20174129 10.1542/peds.2017-4129 [DOI] [PubMed] [Google Scholar]
- 6. Hedegaard H., Warner M., and Minino A. M. (2017) Drug Overdose Deaths in the United States, 1999–2015. NCHS Data Brief 1–8 [PubMed] [Google Scholar]
- 7. Marsico F., Paolillo S., and Filardi P. P. (2017) NSAIDs and cardiovascular risk. J. Cardiovasc. Med. (Hagerstown) 18, Suppl. 1, e40–e43 10.2459/JCM.0000000000000443 [DOI] [PubMed] [Google Scholar]
- 8. Arora A., and Scholar E. M. (2005) Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 315, 971–979 10.1124/jpet.105.084145 [DOI] [PubMed] [Google Scholar]
- 9. Koya D., and King G. L. (1998) Protein kinase C activation and the development of diabetic complications. Diabetes 47, 859–866 10.2337/diabetes.47.6.859 [DOI] [PubMed] [Google Scholar]
- 10. Boric M., Jelicic Kadic A., and Puljak L. (2014) Cutaneous expression of calcium/calmodulin-dependent protein kinase II in rats with type 1 and type 2 diabetes. J. Chem. Neuroanat. 61–62, 140–146 [DOI] [PubMed] [Google Scholar]
- 11. Fountas A., Diamantopoulos L. N., and Tsatsoulis A. (2015) Tyrosine kinase inhibitors and diabetes: a novel treatment paradigm? Trends Endocrinol. Metab. 26, 643–656 10.1016/j.tem.2015.09.003 [DOI] [PubMed] [Google Scholar]
- 12. Tsuda M., Mizokoshi A., Shigemoto-Mogami Y., Koizumi S., and Inoue K. (2004) Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 45, 89–95 10.1002/glia.10308 [DOI] [PubMed] [Google Scholar]
- 13. Milligan E. D., and Watkins L. R. (2009) Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36 10.1038/nrn2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berndt N., Karim R. M., and Schönbrunn E. (2017) Advances of small molecule targeting of kinases. Curr. Opin. Chem. Biol. 39, 126–132 10.1016/j.cbpa.2017.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo C., Gangadharan V., Bali K. K., Xie R. G., Agarwal N., Kurejova M., Tappe-Theodor A., Tegeder I., Feil S., Lewin G., Polgar E., Todd A. J., Schlossmann J., Hofmann F., Liu D. L., et al. (2012) Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol. 10, e1001283 10.1371/journal.pbio.1001283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sung Y. J., Chiu D. T., and Ambron R. T. (2006) Activation and retrograde transport of protein kinase G in rat nociceptive neurons after nerve injury and inflammation. Neuroscience 141, 697–709 10.1016/j.neuroscience.2006.04.033 [DOI] [PubMed] [Google Scholar]
- 17. Gangadharan V., Wang X., and Luo C. (2017) [EXPRESS] Cyclic GMP-dependent protein kinase-I localized in nociceptors modulates nociceptive cortical neuronal activity and pain hypersensitivity. Mol. Pain 13, 1744806917701743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sung Y. J., Sofoluke N., Nkamany M., Deng S., Xie Y., Greenwood J., Farid R., Landry D. W., and Ambron R. T. (2017) A novel inhibitor of active protein kinase G attenuates chronic inflammatory and osteoarthritic pain. Pain 158, 822–832 10.1097/j.pain.0000000000000832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hofmann F., and Wegener J. W. (2013) in Guanylate Cyclase and Cyclic GMP: Methods and Protocols (Krieg T., and Lukowski R., eds.), pp. 17–50, Humana Press, Totowa, NJ [Google Scholar]
- 20. Francis S. H., Busch J. L., Corbin J. D., and Sibley D. (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 10.1124/pr.110.002907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alverdi V., Mazon H., Versluis C., Hemrika W., Esposito G., van den Heuvel R., Scholten A., and Heck A. J. (2008) cGMP-binding prepares PKG for substrate binding by disclosing the C-terminal domain. J. Mol. Biol. 375, 1380–1393 10.1016/j.jmb.2007.11.053 [DOI] [PubMed] [Google Scholar]
- 22. Wall M. E., Francis S. H., Corbin J. D., Grimes K., Richie-Jannetta R., Kotera J., Macdonald B. A., Gibson R. R., and Trewhella J. (2003) Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 100, 2380–2385 10.1073/pnas.0534892100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Butt E. (2009) in cGMP: Generators, Effectors and Therapeutic Implications (Schmidt H. H. H. W., Hofmann F., and Stasch J.-P., eds.) pp. 409–421, Springer, Berlin: [PubMed] [Google Scholar]
- 24. Wolfertstetter S., Huettner J. P., and Schlossmann J. (2013) cGMP-dependent protein kinase inhibitors in health and disease. Pharmaceuticals 6, 269–286 10.3390/ph6020269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Butt E., van Bemmelen M., Fischer L., Walter U., and Jastorff B. (1990) Inhibition of cGMP-dependent protein kinase by (Rp)-guanosine 3′,5′-monophosphorothioates. FEBS Lett. 263, 47–50 10.1016/0014-5793(90)80702-K [DOI] [PubMed] [Google Scholar]
- 26. Campbell J. C., VanSchouwen B., Lorenz R., Sankaran B., Herberg F. W., Melacini G., and Kim C. (2017) Crystal structure of PKG Iβ CNB-B:Rp-cGMPS complex reveals an apo-like, inactive conformation. FEBS Lett. 591, 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Engh R. A., Girod A., Kinzel V., Huber R., and Bossemeyer D. (1996) Crystal structures of catalytic subunit of cAMP-dependent protein kinase in complex with isoquinolinesulfonyl protein kinase inhibitors H7, H8, and H89: structural implications for selectivity. J. Biol. Chem. 271, 26157–26164 10.1074/jbc.271.42.26157 [DOI] [PubMed] [Google Scholar]
- 28. Gustafsson A. B., and Brunton L. L. (1999) Differential and selective inhibition of protein kinase A and protein kinase C in intact cells by balanol congeners. Mol. Pharmacol. 56, 377–382 10.1124/mol.56.2.377 [DOI] [PubMed] [Google Scholar]
- 29. Ono-Saito N., Niki I., and Hidaka H. (1999) H-series protein kinase inhibitors and potential clinical applications. Pharmacol. Ther. 82, 123–131 10.1016/S0163-7258(98)00070-9 [DOI] [PubMed] [Google Scholar]
- 30. Setyawan J., Koide K., Diller T. C., Bunnage M. E., Taylor S. S., Nicolaou K. C., and Brunton L. L. (1999) Inhibition of protein kinases by balanol: specificity within the serine/threonine protein kinase subfamily. Mol. Pharmacol. 56, 370–376 10.1124/mol.56.2.370 [DOI] [PubMed] [Google Scholar]
- 31. Koide K., Bunnage M. E., Gomez Paloma L., Kanter J. R., Taylor S. S., Brunton L. L., and Nicolaou K. C. (1995) Molecular design and biological activity of potent and selective protein kinase inhibitors related to balanol. Chem. Biol. 2, 601–608 10.1016/1074-5521(95)90124-8 [DOI] [PubMed] [Google Scholar]
- 32. Hidaka H., and Kobayashi R. (1992) Pharmacology of protein kinase inhibitors. Annu. Rev. Pharmacol. Toxicol. 32, 377–397 10.1146/annurev.pa.32.040192.002113 [DOI] [PubMed] [Google Scholar]
- 33. Burkhardt M., Glazova M., Gambaryan S., Vollkommer T., Butt E., Bader B., Heermeier K., Lincoln T. M., Walter U., and Palmetshofer A. (2000) KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 275, 33536–33541 10.1074/jbc.M005670200 [DOI] [PubMed] [Google Scholar]
- 34. Bain J., McLauchlan H., Elliott M., and Cohen P. (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204 10.1042/bj20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Narayana N., Diller T. C., Koide K., Bunnage M. E., Nicolaou K. C., Brunton L. L., Xuong N. H., Ten Eyck L. F., and Taylor S. S. (1999) Crystal structure of the potent natural product inhibitor balanol in complex with the catalytic subunit of cAMP-dependent protein kinase. Biochemistry 38, 2367–2376 10.1021/bi9820659 [DOI] [PubMed] [Google Scholar]
- 36. Osborne B. W., Wu J., McFarland C. J., Nickl C. K., Sankaran B., Casteel D. E., Woods V. L. Jr., Kornev A., Taylor S. S., and Dostmann W. R. (2011) Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure 19, 1317–1327 10.1016/j.str.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qin L., Reger A. S., Guo E., Yang M. P., Zwart P., Casteel D. E., and Kim C. (2015) Structures of cGMP-dependent protein kinase (PKG) Iα leucine zippers reveal an interchain disulfide bond important for dimer stability. Biochemistry 54, 4419–4422 10.1021/acs.biochem.5b00572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim J. J., Lorenz R., Arold S. T., Reger A. S., Sankaran B., Casteel D. E., Herberg F. W., and Kim C. (2016) Crystal structure of PKG I:cGMP complex reveals a cGMP-mediated dimeric interface that facilitates cGMP-induced activation. Structure 24, 710–720 10.1016/j.str.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Campbell J. C., Henning P., Franz E., Sankaran B., Herberg F. W., and Kim C. (2017) Structural basis of analog specificity in PKG I and II. ACS Chem. Biol. 12, 2388–2398 10.1021/acschembio.7b00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner J., von Matt P., Sedrani R., Albert R., Cooke N., Ehrhardt C., Geiser M., Rummel G., Stark W., Strauss A., Cowan-Jacob S. W., Beerli C., Weckbecker G., Evenou J. P., Zenke G., and Cottens S. (2009) Discovery of 3-(1 H-indol-3-yl)-4-[2-(4-methylpiperazin-1-yl) quinazolin-4-yl] pyrrole-2, 5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes. J. Med. Chem. 52, 6193–6196 10.1021/jm901108b [DOI] [PubMed] [Google Scholar]
- 41. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 44. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kalyanaraman H., Zhuang S., Pilz R. B., and Casteel D. E. (2017) The activity of cGMP-dependent protein kinase Iα is not directly regulated by oxidation-induced disulfide formation at cysteine 43. J. Biol. Chem. 292, 8262–8268 10.1074/jbc.C117.787358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ho B. K., and Gruswitz F. (2008) HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 8, 49 10.1186/1472-6807-8-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.