

Abstract
In higher eukaryotes, several ATP-utilizing enzymes known as hexokinases activate glucose in the glycolysis pathway by phosphorylation to glucose 6-phosphate. In contrast to canonical hexokinases, which use ATP, ADP-dependent glucokinase (ADPGK) catalyzes noncanonical phosphorylation of glucose to glucose 6-phosphate using ADP as a phosphate donor. Initially discovered in Archaea, the human homolog of ADPGK was described only recently. ADPGK's involvement in modified bioenergetics of activated T cells has been postulated, and elevated ADPGK expression has been reported in various cancer tissues. However, the physiological role of ADPGK is still poorly understood, and effective ADPGK inhibitors still await discovery. Here, we show that 8-bromo–substituted adenosine nucleotide inhibits human ADPGK. By solving the crystal structure of archaeal ADPGK in complex with 8-bromoadenosine phosphate (8-Br-AMP) at 1.81 Å resolution, we identified the mechanism of inhibition. We observed that 8-Br-AMP is a competitive inhibitor of ADPGK and that the bromine substitution induces marked structural changes within the protein's active site by engaging crucial catalytic residues. The results obtained using the Jurkat model of activated human T cells suggest its moderate activity in a cellular setting. We propose that our structural insights provide a critical basis for rational development of novel ADPGK inhibitors.
Keywords: glycolysis, enzyme inhibitor, glucose metabolism, crystal structure, X-ray crystallography, 8-bromo-adenosine monophosphate, 8-bromo-AMP, ADP-dependent glucokinase, reactive oxygen species, T cell activation
Introduction
Glycolysis is a central metabolic process in all living cells. It developed early in evolution and is conserved in almost all known organisms. In general, glycolysis generates energy (ATP) by decomposing glucose to pyruvate. In the presence of oxygen, most mammalian cells strongly reduce glycolytic flux to favor mitochondrial ATP generation by oxidation of pyruvate to CO2 and H2O. Aerobic glycolysis, i.e. conversion of glucose-derived pyruvate to lactic acid in the presence of oxygen, was first described by Otto Warburg and is now broadly known as the “Warburg effect” (1). Increased aerobic glycolysis is a characteristic trait of rapidly proliferating cancer cells but also nontransformed tissues, such as activated immune cells (T cells, B cells, or myeloid cells) (2).
Glycolysis begins with the activation of glucose by phosphorylation to glucose 6-phosphate. In higher eukaryotes, this step is catalyzed by several tissue type–specific isoforms of ATP-utilizing enzymes known as hexokinases (HKs;4 isoforms I to IV in human) (3). An alternative glycolytic pathway involving ADP-dependent glucokinase (ADPGK) (EC 2.7.1.147) was first described in hyperthermophilic Archaea (4), but ADPGK homologs were subsequently discovered and functionally characterized in mouse (mADPGK) (3) and human (hADPGK) (5, 6). ADPGKs are structurally unrelated to HKs and constitute distant homologs of family B of sugar kinases within the ribokinase superfamily (7). The physiological significance of a unique ability of ADPGKs to use ADP instead of ATP as a phosphate donor for glucose 6-phosphate generation remains unknown.
Genome-wide expression profiling data of various human and murine tissues indicate that ADPGK is preferentially expressed in hematopoietic lineage, i.e. macrophages, monocytes, dendritic cells, and T and B cells (8–11). It has been recently demonstrated that ADPGK activity is rapidly and transiently triggered in human T cells upon activation (stimulation of T cell receptor (TCR)) or treatment with phorbol 12-myristate 13-acetate (PMA), a pharmacological mimic of TCR-derived diacylglycerol (DAG)) (10). Elevated ADPGK activity temporarily coincides with TCR-induced glucose uptake and with mitochondrial respiration decrease and thus with a metabolic shift toward aerobic glycolysis similar to the Warburg effect. Importantly, enhanced ADPGK activity contributes to the generation of the mitochondrial “oxidative signal,” a TCR-induced transient increase in mitochondrial reactive oxygen species (ROS) production. ROS, in turn, facilitate activation of transcription factors, including NF-κB, AP-1, and nuclear factor of activated T cells, which results in T cell activation–induced gene expression (10, 12, 13).
Interestingly, ADPGK is highly expressed in tumor tissues (e.g. lymphocytic leukemia, myeloid leukemia, multiple myeloma, and prostate and breast cancers) (5, 8, 14, 15), whereas mutated ADPGK peptides were found as tumor-derived neoantigens inducing strong anti-tumor immune response (16). ADPGK was also postulated to replace hexokinase function under hypoxic conditions (3, 5, 17), because priming glycolysis with ADP instead of ATP might increase the available ATP pool and thus support cellular survival under hypoxia. Nevertheless, this assumption still awaits experimental confirmation.
Although the specific role of ADPGK in cellular metabolism requires further elucidation, its critical function in T cell activation makes ADPGK an attractive target for the development of immunomodulatory compounds. Nucleotide analogs have demonstrated their value as clinically relevant inhibitors of various nucleotide-binding proteins in hematological malignancies (18). Here, we identified a modified adenosine nucleotide, 8-bromoadenosine monophosphate (8-Br-AMP), as an inhibitor of ADPGK activity. By solving a high-resolution crystal structure of ADPGK in complex with 8-Br-AMP, we defined a mechanistic basis for its inhibitory action. We also demonstrate that 8-Br-AMP blocks T cell activation-induced ROS generation (the oxidative signal) and thus ROS-dependent gene expression without significantly influencing the overall glycolytic flux. Our research provides a solid basis for the rational development of specific and more potent ADPGK inhibitors, compounds which are likely to show significant immunomodulatory activity.
Results
Biochemical characterization of hADPGK and identification of inhibitors
hADPGK was expressed using an Escherichia coli recombinant system, and basic enzymatic properties were characterized. ADPGK activity was monitored as the rate of glucose 6-phosphate formation measured in a coupled reaction with glucose-6-phosphate dehydrogenase, as described previously (3, 5, 10). The pH optimum for the hADPGK-driven reaction was found to be in the physiological range (Fig. S1A) (5), and all other experiments were performed at pH 7.4. In our hands, the Km values of hADPGK for ADP and glucose were 0.12 ± 0.01 and 0.15 ± 0.025 mm, respectively (Fig. 1, A and B), thus in the range of previously reported values (0.56 mm for ADP and 0.48 mm for glucose (6)). We also tested the ADPGK specificity toward nucleoside diphosphates other than ADP and established that only GDP could serve as an alternative ADPGK substrate, but with much lower efficiency compared with ADP. The specific activity of ADPGK with GDP as a substrate was only 6% of that determined under the same conditions in the presence of ADP as a phosphate donor (Fig. S1B). We also evaluated the possible product inhibition. Under the experimental conditions, the initial activity of hADPGK in the presence of AMP was ∼85% of the maximal enzymatic activity; however, this slight decrease was independent of AMP concentration up to 5 mm (Fig. 1C) allowing us to conclude that no significant product inhibition was associated with AMP at levels far exceeding the Km values for both substrates.
Figure 1.

Enzymatic characterization of hADPGK. A, Km and Vmax determination for ADP. B, Km and Vmax determination for glucose. C, hADPGK inhibition by adenosine analogs. D, inhibition of hADPGK activity by 8-Br-AMP.
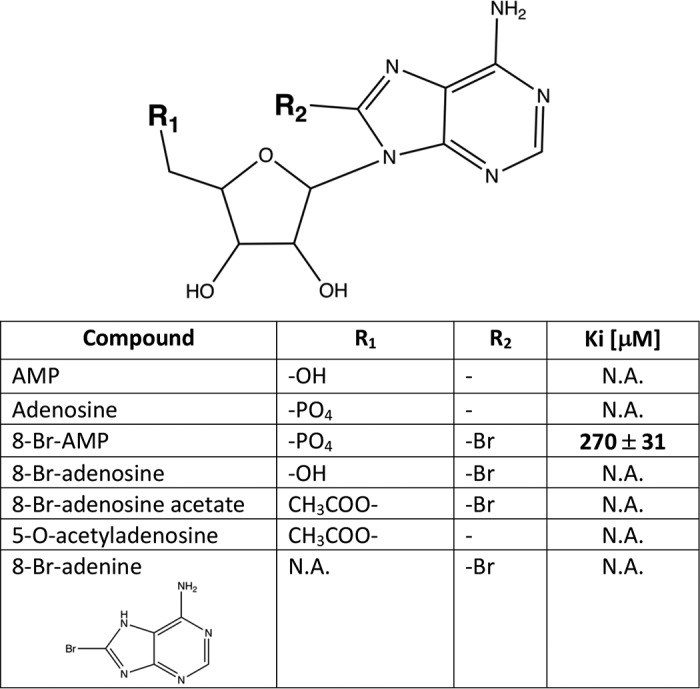
To identify ADPGK inhibitors, several adenosine analogs were tested for their ability to reduce the recombinant hADPGK activity (Fig. 1C and Table 1, and Table S1). First, we evaluated adenosine and 5-O-acetyladenosine for potential product-like inhibition, but none affected the activity of ADPGK. Interestingly, 8-Br-AMP at 1 mm concentration inhibited more than 70% of the initial hADPGK activity. We thus further focused on 8-Br-AMP and performed a limited structure–activity relationship evaluation by testing the activity of other 8-bromo–substituted adenosine analogs, 8-bromoadenine, 8-bromoadenosine, and 8-bromoadenosine acetate. None of the tested analogs of 8-Br-AMP reduced the activity of hADPGK (even at 5 mm concentration) demonstrating the importance of the α-phosphate group in 8-bromo-substituted adenosine binding (Fig. 1C). Our data demonstrate that bromine substitution is essential for hADPGK inhibition (compare AMP and 8-Br-AMP). However, it is only effective when adenosine is phosphorylated (compare 8-Br-AMP and 8-Br-adenosine). Further evaluation demonstrated that 8-Br-AMP inhibition has a competitive nature (Fig. 1D and Fig. S2) and is characterized by Ki of 270 ± 30 μm.
Table 1.
hADPGK inhibition by adenosine nucleotide analogs
N.A. means not applicable (no inhibition observed).
Crystal structure of apo phADPGK
To uncover the structural determinants of ADPGK inhibition by bromine-substituted nucleotide analogs, we attempted to crystallize hADPGK in complex with 8-Br-AMP. Despite significant efforts to crystallize hADPGK in apo- or inhibitor-bound forms (including testing of a large number of crystallization conditions, protein variants, and mutants), our trials remained unsuccessful. Therefore, we cloned and expressed a homolog of hADPGK from the hyperthermophilic archaeon Pyrococcus horikoshii (phADPGK). To use phADPGK as a structural surrogate for hADPGK, we thus verified whether 8-Br-AMP is a phADPGK inhibitor, and we have performed similar kinetic studies as those conducted for human homolog. The Km values of phADPGK for ADP and glucose were 0.13 ± 0.025 and 0.64 ± 0.1 mm, respectively (Fig. S3), and similarly as for hADPGK, 8-Br-AMP competitively inhibits phADPGK with Ki of 160 ± 16 μm (Fig. S4).
phADPGK crystallized readily, which allowed solving the structures of both the apo-form and of the protein in complex with glucose and 8-Br-AMP. These structures are informative regarding the mechanism of inhibition of not only phADPGK but also hADPGK, because both proteins share significant sequence homology at the active site (Fig. S5).
Initial molecular replacement to solve the structure of apo phADPGK using the previously reported P. horikoshii ADPGK structure (PDB code 1L2L; P212121 space group) (19) was unsuccessful suggesting structural differences within the enzyme in the two different crystal forms. The structure was thus solved by molecular replacement using a large domain of phADPGK (1L2L) and refined to Rwork/Rfree values of 0.168/0.221 at 2.0 Å resolution in P3221 space group (Table 2). The asymmetric unit contains a single phADPGK molecule. Residues 4–457 are well-defined by electron density, including two previously unresolved surface loops (residues 157–162 and 171–174, Fig. 2A). However, the nucleotide-binding loop (residues 435–439) was not defined by electron density in our structure. This suggests multiple conformations of this fragment, and it was not included in the final model. phADPGK belongs to the class of α/β (a/b) proteins and adopts the ribokinase-like fold. The topology of the protein is identical to that described previously based on the P212121 structure (19) and consists of a large α/β domain containing 13 α-helices, 11 β-strands, and three 310 helices and a small domain with four α-helices and seven β-strands. The small domain is composed of helices α2–α5 as well as strands β2–β4 and β8–β11, whereas the remaining secondary structure elements form the large domain. The link between the domains is composed of three extended stretches of the polypeptide chain (Fig. 2).
Table 2.
Data collection and refinement statistics
| phADPGK apo | phADPGK/8-Br-AMP | |
|---|---|---|
| Data collection | ||
| Wavelength | 0.9184 | 0.9184 |
| Resolution range | 44.63–2.0 (2.072–2.0)a | 37.36–1.81 (1.875–1.81) |
| Space group | P 32 2 1 | C 1 2 1 |
| Cell dimensions | ||
| a, b, c (Å) | 77.14, 77.14, 133.89 | 15.84, 74.71, 62.95 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 99.46, 90.00 |
| Rmeas | 26.4 (183.5) | 10.9 (69.4) |
| I/σI | 11 (1.9) | 9.17 (1.81) |
| Completeness (%) | 99.9 (99.2) | 97 (96) |
| Multiplicity | 19.9 (20.0) | 3.39 (3.5) |
| CC½ | 0.997 (0.556) | 0.996 (0.732) |
| Refinement | ||
| Resolution (Å) | 2.0 | 1.8 |
| No. of reflections | 31,827 (3092) | 46,878 (4097) |
| Rwork/Rfree | 16.83/22.10 | 16.08/20.19 |
| No. of atoms | 3779 | 4238 |
| Protein | 3562 | 3656 |
| Ligand/ionb | 47 | 60 |
| Ramachandran favored (%) | 98 | 99 |
| Ramachandran allowed (%) | 2 | 1 |
| Ramachandran outliers (%) | 0 | 0 |
| B-factors | ||
| Protein | 33.66 | 27.59 |
| Ligand/ion | 74.16 | 25.62 |
| Water | 37.86 | 38.48 |
| r.m.s.d. | ||
| Bond lengths (Å) | 0.012 | 0.007 |
| Bond angles (°) | 1.15 | 0.95 |
a Values in parentheses are for highest-resolution shell.
b Seven sulfate ions and two ethylene glycol molecules are in the apo structure. Two 8-Br-AMP molecules are in the holo structure.
Figure 2.

Crystal structures of phADPGK. A, structural alignment of apo phADPGK, PDB code 1L2L (gray), and from this study (blue); previously missing loops are marked (green). B, superposition of phADPGK (apo) and phADPGK–8-Br-AMP complex (orange): nucleotide-binding loop is indicated by an arrow (NBD). C, stereo view of molecular architecture of phADPGK active site with bound 8-Br-AMP. Simulated annealing Fc − Fo omit maps contoured at 3σ are shown as blue mesh. D, close-up view of glucose-binding site. Extensive water network (gray spheres) is present within a putative β-phosphate site. E, molecular details of second 8-Br-AMP (8BR2) molecule-binding site. F, relative orientation of 8-Br-AMP molecules.
Despite the overall architectural similarity between our structure and the structure of phADPGK solved previously in the space group P212121, the respective orientation of the small and large domains differs between the structures. The calculated r.m.s.d. is 1.742 Å for the whole molecules (0.179 and 0.847 Å for the small and large domains respectively). This reorientation explains the difficulties encountered in molecular replacement using the whole-protein structure as a search model.
Crystal structure of phADPGK–8-Br-AMP–glucose complex
The structure of phADPGK–8-Br-AMP–glucose ternary complex was determined by molecular replacement using the same search model as for the apoprotein. The structure was refined to Rwork/Rfree values of 0.16/0.20 at 1.81 Å resolution in space group C2 (Table 2). The asymmetric unit contains a single phADPGK molecule. The protein model encompasses residues 5–457, all of which are unambiguously defined by the electron density. The structure has 8-Br-AMP and glucose bound at the active site. The nucleotide-binding loop (435–439), which was disordered in the apo structure, is stabilized in the ligand-containing complex and well-defined by the electron density.
Both small molecule ligands (8-Br-AMP and glucose) were visible in the electron density even before their introduction into the model, which was done at the late stages of refinement. Final electron density maps unambiguously define the conformations and the binding pockets for both ligands (Fig. 2, C and D, and Fig. S6A).
8-Br-AMP is located at the nucleotide-binding site between the small and the large domains in the proximity of the nucleotide-binding loop, which isolates the binding pocket from the solvent (Fig. 2C). A prominent hydrophobic surface, which provides significant interactions to the adenosine moiety of 8-Br-AMP, is composed of residues Ala-376, Lys-430, Val-431, and Pro-435. Moreover, residues Thr-346, Ile-441, and Ile-445 are responsible for further hydrophobic contacts with the ribose moiety. The inhibitor is also stabilized by multiple hydrogen bonds (Fig. 2C). The amine group at position 6 of the adenine ring donates hydrogen bonds to the side-chain carboxyl of Glu-198 and main-chain carbonyl of Val-432. The amine at position 1 contributes a hydrogen bond with the Val-432 backbone amide. The bromine atom at position 8 is involved in water molecule–mediated interactions with Tyr-347 hydroxyl, Asn-199 side–chain amide, Arg-200 guanidine group, and α-phosphate of 8-Br-AMP. The ribose moiety is stabilized by an extensive network of water molecules mediating interactions with the side chains of Glu-296 and Thr-429 and the main-chain amide of Gly-348. The phosphate group contributes a number of hydrogen bonds to the side chains of Asn-295 and His-345 and water molecule–mediated interactions with side-chain carboxyls of Glu-296 and Glu-298, main-chain amide of Gly-442, Tyr-347 hydroxyl, and the main-chain carboxyl of Thr-438. The binding of the phosphate group is further stabilized by interaction with the guanidine moiety of Arg-200.
The glucose moiety is bound in a groove located at the hinge region between the large and small domains. The binding site is composed of a number of residues assembling an amphipathic pocket (Asp-35, Asp-37, Glu-91, Gly-114, Gly-115, Gln-116, Ile-119, His-179, Ile-181, Ile-202, Asp-443, and Val-439). Glucose is stabilized by hydrogen bond interactions contributed by the side chains of Asp-37, Glu-91, and His-179 as well as the Gly-115 backbone amide. Prominent hydrophobic interactions with glycine residues within a conserved GG motif (residues 114–115) further stabilize glucose in its binding pocket. The hydroxyl at position 6 is located exactly at the active site being coordinated by hydrogen bonds contributed by the side chains of Gln-116 and Asp-443 and further bonds mediated by two water molecules. The water molecules occupy the presumed positions of phosphonyl oxygens of the incoming phosphate group providing insight into the mechanism of catalysis (Fig. 2D).
A prominent electron density cloud in a solvent-exposed region of the protein remained unexplained by the completed model containing the above-described ligands. This density unambiguously describes another 8-Br-AMP molecule (Fig. S6B). The noncanonical binding site is located between small and large domains and partially overlaps with the entrance to the canonical nucleotide-binding site. The inhibitor fits between two hydrophobic residues, namely Trp-82 and Phe-272 (Figs. 2E and 3A). The adenosine moiety is positioned by π–π interaction with Phe-272 side chain and hydrophobic interaction with Trp-82. Additionally, a salt bridge connects the α-phosphate group of the inhibitor and the guanidine group of Arg-86. Because the second inhibitor-binding site is located in the proximity of the entrance to the catalytic cleft (Fig. 2F), one could envision an allosteric action of the inhibitors described in this study or alternatively the possible physiological role of the site in participation of substrate/product exchange. To rule out this possibility, we disrupted the binding site by mutating Phe-272 into alanine. No significant difference in the inhibition profile of 8-Br-AMP was observed when phADPGK(F272A) was compared with the WT protein (Fig. 3B and Fig. S7). The same was true for glucose 6-phosphate production kinetics (Fig. S7). To further assess the 8-Br-AMP binding event and compare the WT and F272A phADPGKs, we performed a label-free microscale thermophoresis (MST) experiment. MST has been previously shown to be of great use for quantification of the biomolecular interactions, and its label-free variant allows us to measure affinities and discriminate binding sites for protein–ligand interaction (20). We have tested the binding of 8-Br-AMP to phADPGK, and its F272A variant and the Kd values were determined as 16.85 ± 4.12 and 17.44 ± 3.51 μm, respectively (Fig. 3C). The measured affinities were corresponding to those of hADPGK, where 8-Br-AMP binds with a Kd value of 16.55 ± 4.78 μm (Fig. S8).
Figure 3.

Comparison of archaeal ADPGK structures in closed conformation. A, superposition of phADPGK–8-Br-AMP (orange and gray) with tlADPGK–Mg-AMP (green, PDB code 4B8S). The inset shows the localization of an additional 8-Br-AMP molecule in the proximity of the active-site entrance. The distance measured between Gly-87 and Ile-316 (phADPGK) and the corresponding Gly-92 and Leu-326 (tlADPGK) are indicated by dashed lines. B, phADPGK F272A substitution does not impact protein activity and inhibition by 8-Br-AMP. C, 8-Br-AMP binding analysis by MST. D, superposition of phADPGK (gray) and tlADPGK (green) active sites. 8-Br-AMP induces prominent side-chain rearrangements.
These results advocate for an insignificant role of the second inhibitor-binding site, and they suggest that binding of the second 8-Br-AMP molecule may be the result of high inhibitor concentration applied under the crystallization condition.
8-Br-AMP binding induces major conformational changes
The previously reported structure of apo phADPGK (PDB code 1L2L) was found in an extended open conformation, unusual for this class of proteins. Our structure of apo phADPGK demonstrates a more closed conformation, similar to other apo ADPGK structures reported to date. The small domain is shifted toward the large domain by 8° in our structure compared with a previous structure (PDB code 1L2L). This shift corresponds to the relative domain movement of 8 Å on average (Fig. 2A). The structure of phADPGK–8-Br-AMP–glucose complex demonstrates even more compact conformation that we will term “closed” in further discussions. Comparison with our apoprotein structure revealed the relative shift of the small domain toward the large domain by 11°, which translates into an average 9 Å of relative domain movement (Fig. 2B).
Binding of the inhibitor seems to promote the closed conformation of the protein. To determine whether this is related to the particular inhibitor utilized in our study or rather a general mechanism related to the substrate/substrate analog binding and catalysis in general, we compared our structure to that of ADPGK from Thermococcus litoralis (tlADPGK) in complex with AMP (21). Both structures are found in a closed conformation; however, the domain arrangement is tighter in the phADPGK–8-Br-AMP–glucose complex. This more compact protein architecture is reflected by a relative shift of a small domain toward a large domain by 6°, which corresponds to the relative domain movement of 4 Å (Fig. 3A), when the structures of phADPGK and tlADPGK are compared. It remains to be determined whether this difference is related to the bromine substituent present in our inhibitor or to a general difference between analyzed proteins.
The amino acid residues comprising the hydrophobic surface within the nucleotide-binding site found in the tlADPGK structure are almost identical as those of phADPGK, and the only difference is the substitution of Val-431 by Ile-439 in the case of tlADPGK. This allows deciphering the contribution of bromine substitution in 8-Br-AMP binding by comparing the binding modes within the two structures. No prominent differences between AMP and 8-Br-AMP hydrophobic interaction pattern were observed. However, the inhibitor binding still induces major conformational changes within the active site as compared with the AMP-containing structure, all of which are related to polar interactions (Fig. 3D). The most notable rearrangement involves a Glu–Asn–Arg motif (residues 198–200 and 203–205 in phADPGK and tlADPGK, respectively) within which the main chain shifts toward the inhibitor molecule by 1–2 Å compared with the structure containing AMP. The hydrogen bond between the side chain of Glu-198 and the amine group at position 6 of the 8-Br-AMP adenine ring is not present in the AMP-containing structure. Moreover, the bromine moiety induces a 3–4 Å shift in side chains of Asn-199 and Arg-200 in the direction of the α-phosphate compared with the AMP-containing structure (Fig. 3D). Remarkably, the single water molecule mediates interactions between bromine moiety with side chains of Asn-199 and Arg-200 as well as with α-phosphate. Further differences between AMP and 8-Br-AMP structures involve stabilization of the Tyr-347 side chain, which was found in two alternative conformations in tlADPGK but is rigid in the structure containing 8-Br-AMP. This stabilization is also induced by a water-mediated interaction of Tyr-347 side–chain hydroxyl and the bromine atom.
Next, the Glu-296 side chain, which does not interact with AMP in the tlADPGK structure, shifts toward the inhibitor molecule and forms a water-mediated interaction with the O3 atom of the ribose moiety, Tyr-347 side chain, and the α-phosphate. To further assess the impact of these interactions, we designed several phADPGK mutants. The substitutions of Arg-200 and Glu-298 for alanine led to inactivation of phADPGK. Remarkably, the substitution of Tyr-347 did not cause any significant changes either in the protein activity or inhibition by 8-Br-AMP (Fig. S9).
Taken together, the bromine atom engages a number of interactions essential for the described side-chain rearrangements. The majority of 8-Br-AMP–induced changes within the active site involve, directly or indirectly (via water molecules), the α-phosphate group. This observation provides hints why only a hydrogen bond–rich moiety (phosphonate) is accepted at that position of the inhibitor, whereas analogs not able to form these important interactions (8-bromoadenosine and 8-bromoadenosine acetate) display no inhibitory activity (Fig. 1D).
8-Br-AMP inhibits T cell activation–induced ROS release and gene expression
Activation of ADPGK in human T cells is an important step among metabolic/signaling events in TCR-induced mitochondrial ROS release (oxidative signal generation) and consequently ROS-dependent gene expression (10–12). Using a well-established model of PMA (DAG)–mediated activation of the human Jurkat T cell line, we tested the effect of 8-Br-AMP on these cellular events. Treatment of Jurkat T cells with 8-Br-AMP, at concentrations which did not result in any noticeable cytotoxicity (Fig. S10C), dose-dependently reduced the oxidative signal generation and, consequently, diminished transcription of IL-2 and IκBα. The highest 8-Br-AMP concentration tested (100 μm) decreased the PMA-triggered generation of ROS to around half of the initial value (Fig. 4A). Interestingly, the obtained extent of inhibition corresponds to that obtained upon siRNA- or shRNA-mediated ADPGK knockdown (50–65%) (10). The PMA/ionomycin–induced expression of classical T cell activation-responsive genes, IL-2 and IκBα, was decreased in the presence of 8-Br-AMP (Fig. 4B). Moreover, 8-Br-adenosine, which was ineffective as a hADPGK inhibitor in vitro (Fig. 1D), was also ineffective in decreasing the PMA-induced oxidative signal (Fig. S10A) and PMA/ionomycin-induced downstream gene expression when used at a concentration corresponding with that of 8-Br-AMP (Fig. S10B).
Figure 4.

8-Br-AMP inhibits T cell activation-induced ROS generation and subsequent NF-κB–dependent gene expression but has no significant influence on basal glycolytic flux. A, Jurkat T cells stained with H2DCF-DA and pre-treated (30 min) with respective concentrations of 8-Br-AMP were activated by PMA treatment (1 h), and the oxidative signal was measured by FACS. Cumulative results of 3 (25 μm), 4 (50 μm), or 5 (100 μm) independent, triplicated experiments are shown (mean values ± S.D.). Statistical significance was calculated by single sample t test (control (ctr) set up to 100%). B, 8-Br-AMP-pre-treated Jurkat T cells (30 min) were activated by PMA/ionomycin treatment for 1 h. Next, IL-2 and IκBα gene expressions were assayed by RT-PCR. C, Jurkat T cells were pre-treated with 8-Br-AMP (30 min) and treated continuously during the monitoring of glycolytic flux by ECAR with Seahorse Extracellular Flux XFe24 analyzer, see under “Experimental procedures” for details. Left side, original ECAR plot; right side, mean values ± S.D. of ECAR measured after glucose or oligomycin injections. Statistical significance was calculated by two-sided, unpaired t test.
T cell activation–induced mitochondrial ROS release and the resulting gene expression rely on TCR (PMA/DAG)-mediated, transient up-regulation of glucose metabolism (10). Because Br-AMP was reported to inhibit AMPK activity (22–24) and bind to HKs (25), it could have a significant influence on the overall glucose metabolism. To evaluate the influence of 8-Br-AMP on the overall glucose flux of proliferating Jurkat T cells, we employed the “glycolysis stress test” method and Seahorse extracellular flux analyzer. 8-Br-AMP at 100 μm only slightly decreased the basal (determined after addition of glucose) and the maximal (determined in the presence of oligomycin) glycolytic flux indicating no significant influence of the inhibitor on the general glucose metabolism (Fig. 4C).
Discussion
Development of inhibitory strategies against highly glycolytic metabolism of activated immune cells or proliferating cancer cells as well as against T cell activation–induced release of pro-inflammatory cytokines may have a great impact on autoimmunity or malignancies. Although the general role of ADPGK in cellular metabolism awaits characterization, the involvement of TCR (DAG)–induced ADPGK activity in the metabolic shift toward aerobic glycolysis of activated T cells and in the regulation of T cell function makes ADPGK a putative immunomodulatory target (10). We report here that the structural basis of ADPGK inhibition by 8-bromo–substituted adenosine nucleotide constitutes the first step for pharmacologically relevant inhibitor identification.
8-Br-AMP is a relatively promiscuous inhibitor. It was reported to inhibit AMPK (22–24) but only the 5-aminoimidazole-4-carboxamide ribonucleotide–stimulated or AMP-stimulated enzyme and not the constitutive AMPK activity (22, 26, 27). Although the affinity was not reported, the high concentration (1 mm) of 8-Br-AMP used suggests low affinity. Apart from AMPK inhibition, 8-Br-AMP was crystallized at the active sites of yeast hexokinase IIIB (HK IIIB) (25) (the structure is not deposited at the PDB), phosphodiesterase 4B (PDB code 1RO9) (28), and histidine triad nucleotide-binding protein (HINT, PDB code 5RHN) (29). Because of its promiscuity, 8-Br-AMP by itself is an unlikely starting point for direct inhibitor optimization, but it serves well to define the pharmacophore and the binding pocket for virtual screening.
8-Br-AMP is a competitive inhibitor of ADPGK, but the bromine substitution induces prominent changes within the protein-active site by engaging crucial catalytic residues. In particular, the side-chain position of the conserved arginine residue (Arg-200) is affected. During glucose phosphorylation, Arg-200 is responsible for the positioning of β-phosphate and stimulates the phosphate transfer from ADP (21, 30, 31). In our structure, the Arg-200 side chain is involved in water-mediated interaction with the bromine and shifts toward α-phosphate of 8-Br-AMP compared with the AMP-containing structure (Fig. 3D). This induced disposition may be utilized in the rational design of more potent and selective inhibitors.
We identified a second inhibitor binding site outside of the ADP-binding pocket. Secondary sites are relatively frequent crystallization artifacts related to high concentrations of ligands utilized in the experiments. Occasionally, however, high-ligand concentrations allow for identifying weak secondary binding sites involved in substrate access/product egress of the binding pocket. Alternatively, secondary sites may be responsible for allosteric regulation (32, 33). To evaluate the relevance (if any) of a secondary 8-Br-AMP–binding site, we first analyzed the known structures of ADPGK homologs from T. litoralis and Mus musculus, which indicated little conservancy in the region of interest. tlADPGK has tyrosine in the position equivalent to Phe-272 of phADPGK, whereas mADPGK is characterized by methionine. These are residues of comparable hydrophobic character that might suggest conservation (and thus functionality) of the site. However, mutating the major residue within the site (phADPGK-F272A) had no significant effect on enzyme activity, 8-Br-AMP inhibition, and inhibitor binding. This allowed us to conclude that the additional inhibitor-binding site seen in our structure represents an experimental artifact resulting from high 8-Br-AMP concentration used for protein crystallization.
Prior data demonstrated that Thermococcus ADPGK oscillates between different conformational states (21, 34). The enzyme either adopts an open conformation, which is characteristic for the apoprotein, or a closed conformation seen in a protein containing AMP and glucose at the active site. Strikingly, Ito et al. (34) demonstrated that tlADPGK containing ADP, but in the absence of glucose, is found in the open state. Similarly, binding of AMP does not induce any prominent domain rearrangement in mADPGK (6). This implies that the protein adopts closed conformation only when both substrates occupy the active site (or alternatively, although less likely, that glucose induces the closed conformation). This study adds further complexity to conformational transitions related to ADPGK catalysis/inhibitor binding. The crystal structure of phADPGK reported by Tsuge et al. (19) contains the apoprotein found in an unusually extended conformation. In contrast, the structure of apo phADPGK presented in our work adopts a more compact conformation concerning the relative arrangement of small and large domains (Fig. 2A). In our second structure, binding of a substrate (glucose) and substrate analog (8-Br-AMP) induces domain closure, which in fact is even tighter than the one described for tlADPGK in the presence of glucose and AMP. This phenomenon coincides with prominent rearrangements of amino acids within the substrate-binding pocket when 8-Br-AMP- and AMP-bound structures are compared (Fig. 3D). Clearly, ADPGK is a dynamic protein present in multiple open conformations in the absence of both ligands to facilitate their recognition. Ligand binding stabilizes the closed conformation capable of catalysis. The same is observed for competitive inhibitors occupying the ADP-binding pocket in the presence of a second substrate.
Prior to this study, the only attempt to determine the crystal structure of ADPGK in complex with the inhibitor was that for tlADPGK (21). tlADPGK was crystallized in the presence of Mg-ADPβS (PDB code 4B8S), but the electron density was explained by the authors with the AMP molecule, and no density for the β-phosphate or magnesium ion was observed, most probably because of the hydrolysis of the inhibitor. Therefore, the structure reflects a product-containing structure of ADPGK in the closed conformation. Upon comparing the tlADPGK–AMP and phADPGK–8-Br-AMP structures, the r.m.s.d. is 1 Å for the whole molecules and 0.62 Å for the large domains only. However, the crevice dimensions between the small and large domain differ significantly between both proteins. Compared with the tlADPGK structure, the small and large domains of phADPGK reported in our study are much closer together, which is reflected by the measured distances between Gly-87 and Ile-316 residues being 11 and 16 Å for phADPGK and tlADPGK, respectively (Fig. 3A). Because the second 8-Br-AMP molecule, which binds phADPGK, is located on the protein surface in the proximity of the described crevice entrance, this interaction could be additionally responsible for a tighter arrangement of large and small domains in the case of phADPGK compared with the tlADPGK–AMP complex.
Our data also demonstrate that 8-Br-AMP treatment down-regulates cellular processes that were earlier described to depend on ADPGK activity (10–12). Using the Jurkat model of human T cell activation, we demonstrated that 8-Br-AMP diminishes DAG/PMA-induced ROS release and therefore ROS-dependent pro-inflammatory gene expression. Because Br-AMP likely acts promiscuously on several molecular targets, including sugar and protein kinases, it is currently unclear whether the inhibitory action is solely ADPGK-dependent. However, it is remarkable that the extent of the inhibitory effect of 8-Br-AMP on ROS generation and ROS-dependent gene expression parallels that previously reported for ADPGK knockdown experiments (10). In addition, corresponding concentrations of 8-Br-AMP induced no significant reduction in the overall glycolytic flux of proliferating Jurkat T cells. This observation seems to preclude the inhibitory effect of tested concentrations of 8-Br-AMP on major metabolic hubs, like ATP-dependent HKs or AMPK. The observation also advocates that ADPGK activity is not essential for the overall cellular glucose metabolism. This is consistent with prior studies on ADPGK knockdown, knockout, and overexpression in several cancer cell lines cultured under normoxic, hypoxic, or anoxic conditions (5, 17). These conclusions are, however, largely speculative, and further investigation is necessary.
In summary, our study demonstrates that the bromine-substituted AMP analog inhibits ADPGK. However, in the case of the investigation of the role of ADPGK in cellular metabolism and regulation of T cell activation, caution must be taken in the interpretation of the results due to 8-Br-AMP promiscuity. The provided crystal structure explains the binding mode and constitutes a starting point for further inhibitor development.
Experimental procedures
Molecular cloning and expression
The hADPGK-encoding gene (residues 29–497) was codon-optimized for the E. coli expression system, synthesized, and cloned into the pET24d plasmid (Genscript). For expression, the gene was subcloned into pGEX-6p-1 plasmid resulting in a construct bearing the N-terminal GST tag followed by the PreScission protease cleavage site. The gene encoding full-length phADPGK (residues 2–457) was synthesized and cloned into pET15b plasmid (Genescript). The final expression construct bearing the N-terminal hexahistidine tag followed by TEV protease recognition site was generated by subcloning into pETM11 vector (EMBL). Site-directed mutagenesis of phADPGK has been performed as described previously (35).
For protein expression, E. coli BL21 (DE3) competent cells were transformed with either pGEX6p1-hADPGK or pETM11-phADPGK plasmids and cultured at 37 °C in LB Lenox medium (Bioshop) or Terrific Broth (Bioshop), respectively. When the A600 reached 0.6, the expression was induced by addition of isopropyl β-d-1-thiogalactopyranoside at final concentration of 0.4 mm, and the culture was continued for 20 h at 20 °C. Cells were harvested by centrifugation at 4000 × g. Pellets were flash-frozen in liquid nitrogen and stored at −80 °C until further use.
hADPGK purification
Frozen pellets were thawed, resuspended in lysis buffer (50 mm Tris-HCl, pH 8.0, 500 mm NaCl), and lysed by sonication. The lysate was clarified by centrifugation (20 min at 50,000 × g; 4 °C), and the protein of interest was recovered using GSH-Sepharose (GE Healthcare). Overnight on-column proteolysis with PreScission protease facilitated the GST-tag removal, and the related hADPGK was further purified by size-exclusion chromatography on Superdex 75 (GE Healthcare) using the same buffer. Purified hADPGK was concentrated to 1 mg/ml and stored at −80 °C for further analysis.
phADPGK purification
Frozen pellets were thawed, resuspended in lysis buffer (20 mm Hepes, pH 8.0, 350 mm NaCl, 10 mm MgCl2, 2.5% glycerol, 20 mm imidazole), and lysed by sonication. The lysate was clarified by centrifugation (20 min at 50,000 × g; 4 °C), heat shocked at 90 °C for 10 min, and again clarified by centrifugation (20 min at 50,000 × g; 4 °C). The protein of interest was recovered by metal affinity chromatography using chelating-Sepharose (GE Healthcare) pre-equilibrated with the lysis buffer. Protein was eluted using imidazole gradient (up to 500 mm). Fractions containing phADPGK were incubated at 4 °C with TEV protease (DNA Gdansk) for 12 h. The protein of interest was separated from the released tag and the remaining uncleaved forms by size-exclusion chromatography (Superdex 200, GE Healthcare) in GFB buffer (20 mm Hepes, pH 8.0, 150 mm NaCl, 5 mm MgCl2). Purified phADPGK was either directly used for crystallization trials or concentrated to 1 mg/ml and stored at −80 °C for further analysis.
ADPGK activity assay and inhibitor screening
ADPGK activity was assessed by monitoring the level of one of the reaction products (glucose 6-phosphate) in a coupled glucose-6-phosphate dehydrogenase (G6PD) reaction essentially as described previously (6, 10). The reaction was initiated by addition of ADPGK substrates, ADP and d-glucose (0.5 mm), and NADP reduction by G6PD was monitored spectrophotometrically at 340 nm. Activity assays were performed in 96-well format spectrophotometric plates using TECAN microplate UV-visible reader. Unless stated otherwise, the assays were performed at 37 °C in reaction buffer (20 mm Tris-HCl, pH 7.4, 250 mm sucrose, 50 mm KCl, 5 mm MgCl2) supplemented with glucose-6-phosphate dehydrogenase (1 unit), NADP (0.5 mm). Potential inhibitors were screened at an initial concentration of 1 mm. Km or Ki values were determined by varying glucose, ADP, and inhibitor concentrations, and analysis was done using nonlinear regression and competitive inhibition model using GraphPad Prism 6 software as described previously (6). The 8-bromo–substituted analogs were purchased from Sigma (Germany) except for 8-Br-AMP, which was obtained from Jena Bioscience (Jena, Germany).
Crystallization and structural determination
phADPGK was concentrated up to 10 mg/ml and crystallized using a sitting drop vapor diffusion method. 2-μl drops of protein solution containing glucose (5 mm), Hepes (20 mm at pH 8.0), sodium chloride (150 mm), and magnesium chloride (5 mm) were mixed with an equal volume of the reservoir solution containing 15% (w/v) PEG 6000, 0.2 m LiSO4, and 0.1 m citrate buffer, pH 3.6. The plates were incubated at 20 °C and crystals appeared after 3–5 days.
Crystals of phADPGK–glucose–8-Br-AMP complex were obtained using a similar procedure as those of the apoprotein, except that the protein solution was supplemented with 8-Br-AMP (1 mm) and d-glucose (5 mm) and mixed with an equal volume of the reservoir solution containing ammonium nitrate (0.2 m) and PEG 3350 (20%). Co-crystals of the complex appeared after approximately 1 week.
Crystals were soaked in the reservoir solution supplemented with glycerol (20%) and flash-cooled in liquid nitrogen. Diffraction data were collected on beamline BL14.1 operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II (Berlin-Adlershof, Germany) (36). The data were indexed, integrated, scaled, and merged using XDS package (37) with XDSAPP2.0 GUI (38). Molecular replacement was performed in Phaser (39, 40) using the large domain of phADPGK (PDB code 1L2L) as a search model. Structures were refined using Phenix (41), and model building was done with Coot (42, 43).
Structure coordinates were deposited in Protein Data Bank with the accession codes 5O0I (phADPGK apo) and 5O0J (phADPGK/8-Br-AMP).
Binding analysis by microscale thermophoresis
MonolithNT.LabelFree instrument (NanoTemper Technologies GmbH, Munich, Germany) was used to measure the binding interactions between purified phADPGK or phADPGK F272A and 8-Br-AMP. A range of concentrations of 8Br-AMP from 30.5 nm to 1 mm was incubated for 5 min with each protein variant at a concentration of 500 nm in assay buffer (20 mm Tris-HCl, pH 7.4, 50 mm NaCl, 5 mm MgCl2, 2 mm glucose). The samples were loaded into the MonolithNT.LabelFree standard-treated glass capillaries, and initial fluorescence measurement followed by thermophoresis measurement were carried out using 40% LED power and 40% MST power, respectively. All measurements were performed at a fixed temperature of 37 °C. Kd values were calculated using the MO.Affinity Analysis software. Each experiment was performed in triplicate.
Determination of ROS generation and changes in gene expression and cell death
Jurkat T cells (clone E6-1, human acute T cell leukemia cell line) were cultured at standard conditions in RPMI medium supplemented with 10% FBS, 1 mm glutamine, and penicillin/streptomycin (10 units/ml). All chemicals were from Sigma, unless stated otherwise. Cells were stained with H2DCF-DA (Life Technologies, Inc., 5 μm) for 30 min (±8-Br-AMP), stimulated with PMA (10 ng/ml, 1 h) without inhibitor removal, washed with ice-cold PBS, and analyzed by FACS. Generation of the oxidative signal was calculated as described previously (10, 12).
To analyze activation-induced gene expression, Jurkat T cells were pre-treated ± 8-Br-AMP for 30 min and activated with PMA (10 ng/ml) and ionomycin (1 μm) for 1 h in the presence/absence of inhibitor. Next, total cellular RNA was isolated with RNeasy mini kit (Qiagen) and reverse-transcribed. Gene expression was analyzed by SYBR Green monitored RT-PCR with IL-2- and IκBα-specific primers. The measurements were normalized at the actin transcript level. Conditions of reverse transcription and RT-PCR and primer sequences were identical to those reported before (10, 12). Cell death was measured by propidium iodide (PI) exclusion method. Cells were stained with PI solution (4 μg/ml in PBS) for 30 min, and PI+ cells were assayed by FACS.
Seahorse measurement of glycolytic flux
Total glycolytic flux was determined as extracellular acidification rate (ECAR) using Seahorse XFe 24 extracellular flux analyzer (Seahorse, Agilent). Jurkat T cells were pre-treated with ±100 μm 8-Br-AMP for 30 min. Next, 700 × 103 cells/well were plated using Cell Tak cell adhesive (Corning) and glucose- and FBS-free standard Seahorse Dulbecco's modified Eagle's medium supplemented with 2 mm glutamine and 1 mm sodium pyruvate ± 100 μm 8-Br-AMP. “Glycolysis stress tests” were performed according to manufacturer's procedure, with serial injections of glucose (10 mm), oligomycin (1 μm), and 2-deoxyglucose (20 mm).
Author contributions
P. G., M. M. K., and G. D. conceptualization; P. G. formal analysis; P. G. supervision; P. G. and G. D. funding acquisition; P. G., M. M. K., K. P. R., K. K., and M. M. investigation; P. G. visualization; P. G. methodology; P. G., M. M. K., and G. D. writing-original draft; P. G. and G. D. project administration; P. G., M. M. K., and G. D. writing-review and editing; J. P., M. D., and G. D. resources.
Supplementary Material
Acknowledgments
We thank Urszula Nowak for help during the initial phase of the project, Klaudia Woś from the MCB Structural Biology Core Facility for help in crystallization, and Helmholtz-Zentrum Berlin for the allocation of beamtime. The Faculty of Biochemistry, Biophysics and Biotechnology of Jagiellonian University is a partner of the Leading National Research Center (KNOW) supported by the Ministry of Science and Higher Education.
This work was supported in part by National Science Center Research Grants UMO-2015/19/D/NZ1/02009 (to P. G.) and UMO-2012/07/E/NZ1/01907 (to G. D.) and funding from the European Union's Horizon 2020 Research and Innovation Program under Grant Agreement No 730872. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S10 and Table S1.
The atomic coordinates and structure factors (codes 5O0I and 5O0J) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HK
- hexokinase
- ADPGK
- ADP-dependent glucokinase
- mADPGK
- mouse, ADPGK
- hADPGK
- human ADPGK
- TCR
- T cell receptor
- DAG
- diacylglycerol
- PMA
- phorbol 12-myristate 13-acetate
- ROS
- reactive oxygen species
- MST
- microscale thermophoresis
- PDB
- Protein Data Bank
- AMPK
- 5′ AMP-activated protein kinase
- G6PD
- glucose 6-phosphate dehydrogenase
- PI
- propidium iodide
- ECAR
- extracellular acidification rate
- r.m.s.d.
- root mean square deviation
- 8-Br-AMP
- 8-bromoadenosine monophosphate
- TEV
- tobacco etch virus
- Mg-ADPβS
- Mg-adenosine 5′-[β-thio]diphosphate
- H2DCF-DA
- 2′,7′-dichlorodihydrofluorescein diacetate.
References
- 1. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 2. O'Neill L. A., Kishton R. J., and Rathmell J. (2016) A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 10.1038/nri.2016.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ronimus R. S., and Morgan H. W. (2004) Cloning and biochemical characterization of a novel mouse ADP-dependent glucokinase. Biochem. Biophys. Res. Commun. 315, 652–658 10.1016/j.bbrc.2004.01.103 [DOI] [PubMed] [Google Scholar]
- 4. Kengen S. W., Tuininga J. E., de Bok F. A., Stams A. J., and de Vos W. M. (1995) Purification and characterization of a novel ADP-dependent glucokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 270, 30453–30457 10.1074/jbc.270.51.30453 [DOI] [PubMed] [Google Scholar]
- 5. Richter S., Richter J. P., Mehta S. Y., Gribble A. M., Sutherland-Smith A. J., Stowell K. M., Print C. G., Ronimus R. S., and Wilson W. R. (2012) Expression and role in glycolysis of human ADP-dependent glucokinase. Mol. Cell. Biochem. 364, 131–145 10.1007/s11010-011-1212-8 [DOI] [PubMed] [Google Scholar]
- 6. Richter J. P., Goroncy A. K., Ronimus R. S., and Sutherland-Smith A. J. (2016) The structural and functional characterization of mammalian ADP-dependent glucokinase. J. Biol. Chem. 291, 3694–3704 10.1074/jbc.M115.679902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merino F., and Guixé V. (2008) Specificity evolution of the ADP-dependent sugar kinase family: in silico studies of the glucokinase/phosphofructokinase bifunctional enzyme from Methanocaldococcus jannaschii. FEBS J. 275, 4033–4044 10.1111/j.1742-4658.2008.06544.x [DOI] [PubMed] [Google Scholar]
- 8. Hruz T., Laule O., Szabo G., Wessendorp F., Bleuler S., Oertle L., Widmayer P., Gruissem W., and Zimmermann P. (2008) Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinformatics 2008, 420747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C. L., Haase J., Janes J., Huss J. W. 3rd., and Su A. I. (2009) BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10, R130 10.1186/gb-2009-10-11-r130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamiński M. M., Sauer S. W., Kamiński M., Opp S., Ruppert T., Grigaravičius P., Grudnik P., Gröne H. J., Krammer P. H., and Gülow K. (2012) T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2, 1300–1315 10.1016/j.celrep.2012.10.009 [DOI] [PubMed] [Google Scholar]
- 11. Kaminski M. M., Roth D., Krammer P. H., and Gulow K. (2013) Mitochondria as oxidative signaling organelles in T-cell activation: physiological role and pathological implications. Arch. Immunol. Ther. Exp. 61, 367–384 10.1007/s00005-013-0235-0 [DOI] [PubMed] [Google Scholar]
- 12. Kaminski M. M., Sauer S. W., Klemke C. D., Süss D., Okun J. G., Krammer P. H., and Gülow K. (2010) Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J. Immunol. 184, 4827–4841 10.4049/jimmunol.0901662 [DOI] [PubMed] [Google Scholar]
- 13. Sena L. A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D. A., Wang C. R., Schumacker P. T., Licht J. D., Perlman H., Bryce P. J., and Chandel N. S. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 10.1016/j.immuni.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu D., Sunkel B., Chen Z., Liu X., Ye Z., Li Q., Grenade C., Ke J., Zhang C., Chen H., Nephew K. P., Huang T. H., Liu Z., Jin V. X., and Wang Q. (2014) Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 42, 3607–3622 10.1093/nar/gkt1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee K. E., Spata M., Bayne L. J., Buza E. L., Durham A. C., Allman D., Vonderheide R. H., and Simon M. C. (2016) Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov. 6, 256–269 10.1158/2159-8290.CD-15-0822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yadav M., Jhunjhunwala S., Phung Q. T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T. K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J. R., and Delamarre L. (2014) Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 10.1038/nature14001 [DOI] [PubMed] [Google Scholar]
- 17. Richter S., Morrison S., Connor T., Su J., Print C. G., Ronimus R. S., McGee S. L., and Wilson W. R. (2013) Zinc finger nuclease mediated knockout of ADP-dependent glucokinase in cancer cell lines: effects on cell survival and mitochondrial oxidative metabolism. PLoS ONE 8, e65267 10.1371/journal.pone.0065267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robak T. (2011) New nucleoside analogs for patients with hematological malignancies. Expert Opin. Investig. Drugs 20, 343–359 10.1517/13543784.2011.554822 [DOI] [PubMed] [Google Scholar]
- 19. Tsuge H., Sakuraba H., Kobe T., Kujime A., Katunuma N., and Ohshima T. (2002) Crystal structure of the ADP-dependent glucokinase from Pyrococcus horikoshii at 2.0-A resolution: a large conformational change in ADP-dependent glucokinase. Protein Sci. 11, 2456–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seidel S. A., Wienken C. J., Geissler S., Jerabek-Willemsen M., Duhr S., Reiter A., Trauner D., Braun D., and Baaske P. (2012) Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding. Angew. Chem. Int. Ed. Engl. 51, 10656–10659 10.1002/anie.201204268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rivas-Pardo J. A., Herrera-Morande A., Castro-Fernandez V., Fernandez F. J., Vega M. C., and Guixé V. (2013) Crystal structure, SAXS and kinetic mechanism of hyperthermophilic ADP-dependent glucokinase from Thermococcus litoralis reveal a conserved mechanism for catalysis. PLoS ONE 8, e66687 10.1371/journal.pone.0066687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies S. P., Carling D., and Hardie D. G. (1989) Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur. J. Biochem. 186, 123–128 10.1111/j.1432-1033.1989.tb15185.x [DOI] [PubMed] [Google Scholar]
- 23. Yang J., Craddock L., Hong S., and Liu Z. M. (2009) AMP-activated protein kinase suppresses LXR-dependent sterol regulatory element-binding protein-1c transcription in rat hepatoma McA-RH7777 cells. J. Cell. Biochem. 106, 414–426 10.1002/jcb.22024 [DOI] [PubMed] [Google Scholar]
- 24. Alba G., El Bekay R., Alvarez-Maqueda M., Chacón P., Vega A., Monteseirín J., Santa María C., Pintado E., Bedoya F. J., Bartrons R., and Sobrino F. (2004) Stimulators of AMP-activated protein kinase inhibit the respiratory burst in human neutrophils. FEBS Lett. 573, 219–225 10.1016/j.febslet.2004.07.077 [DOI] [PubMed] [Google Scholar]
- 25. Shoham M., and Steitz T. A. (1980) Crystallographic studies and model building of ATP at the active site of hexokinase. J. Mol. Biol. 140, 1–14 10.1016/0022-2836(80)90353-8 [DOI] [PubMed] [Google Scholar]
- 26. Musi N., Hayashi T., Fujii N., Hirshman M. F., Witters L. A., and Goodyear L. J. (2001) AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 280, E677–E684 10.1152/ajpendo.2001.280.5.E677 [DOI] [PubMed] [Google Scholar]
- 27. Jäger S., Handschin C., St-Pierre J., and Spiegelman B. M. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022 10.1073/pnas.0705070104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu R. X., Rocque W. J., Lambert M. H., Vanderwall D. E., Luther M. A., and Nolte R. T. (2004) Crystal structures of the catalytic domain of phosphodiesterase 4B complexed with AMP, 8-Br-AMP, and rolipram. J. Mol. Biol. 337, 355–365 10.1016/j.jmb.2004.01.040 [DOI] [PubMed] [Google Scholar]
- 29. Brenner C., Garrison P., Gilmour J., Peisach D., Ringe D., Petsko G. A., and Lowenstein J. M. (1997) Crystal structures of HINT demonstrate that histidine triad proteins are GalT-related nucleotide-binding proteins. Nat. Struct. Biol. 4, 231–238 10.1038/nsb0397-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guixé V., and Merino F. (2009) The ADP-dependent sugar kinase family: kinetic and evolutionary aspects. IUBMB Life 61, 753–761 10.1002/iub.217 [DOI] [PubMed] [Google Scholar]
- 31. Tokarz P., Wiśniewska M., Kamiński M. M., Dubin G., and Grudnik P. (2018) Crystal structure of ADP-dependent glucokinase from Methanocaldococcus jannaschii in complex with 5-iodotubercidin reveals phosphoryl transfer mechanism. Protein Sci. 27, 790–797 10.1002/pro.3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hughes T. S., Giri P. K., de Vera I. M., Marciano D. P., Kuruvilla D. S., Shin Y., Blayo A. L., Kamenecka T. M., Burris T. P., Griffin P. R., and Kojetin D. J. (2014) An alternate binding site for PPARγ ligands. Nat. Commun. 5, 3571 10.1038/ncomms4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ludlow R. F., Verdonk M. L., Saini H. K., Tickle I. J., and Jhoti H. (2015) Detection of secondary binding sites in proteins using fragment screening. Proc. Natl. Acad. Sci. U.S.A. 112, 15910–15915 10.1073/pnas.1518946112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ito S., Fushinobu S., Yoshioka I., Koga S., Matsuzawa H., and Wakagi T. (2001) Structural basis for the ADP-specificity of a novel glucokinase from a hyperthermophilic archaeon. Structure 9, 205–214 10.1016/S0969-2126(01)00577-9 [DOI] [PubMed] [Google Scholar]
- 35. Edelheit O., Hanukoglu A., and Hanukoglu I. (2009) Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9, 61 10.1186/1472-6750-9-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., and Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 10.1107/S0909049512006395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sparta K. M., Krug M., Heinemann U., Mueller U., and Weiss M. S. (2016) XDSAPP2.0. J. Appl. Crystallogr. 49, 1085–1092 10.1107/S1600576716004416 [DOI] [Google Scholar]
- 39. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 43. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.