

Abstract
Protein composition at the plasma membrane is tightly regulated, with rapid protein internalization and selective targeting to the cell surface occurring in response to environmental changes. For example, ion channels are dynamically relocalized to or from the plasma membrane in response to physiological alterations, allowing cells and organisms to maintain osmotic and salt homeostasis. To identify additional factors that regulate the selective trafficking of a specific ion channel, we used a yeast model for a mammalian potassium channel, the K+ inward rectifying channel Kir2.1. Kir2.1 maintains potassium homeostasis in heart muscle cells, and Kir2.1 defects lead to human disease. By examining the ability of Kir2.1 to rescue the growth of yeast cells lacking endogenous potassium channels, we discovered that specific α-arrestins regulate Kir2.1 localization. Specifically, we found that the Ldb19/Art1, Aly1/Art6, and Aly2/Art3 α-arrestin adaptor proteins promote Kir2.1 trafficking to the cell surface, increase Kir2.1 activity at the plasma membrane, and raise intracellular potassium levels. To better quantify the intracellular and cell-surface populations of Kir2.1, we created fluorogen-activating protein fusions and for the first time used this technique to measure the cell-surface residency of a plasma membrane protein in yeast. Our experiments revealed that two α-arrestin effectors also control Kir2.1 localization. In particular, both the Rsp5 ubiquitin ligase and the protein phosphatase calcineurin facilitated the α-arrestin–mediated trafficking of Kir2.1. Together, our findings implicate α-arrestins in regulating an additional class of plasma membrane proteins and establish a new tool for dissecting the trafficking itinerary of any membrane protein in yeast.
Keywords: ubiquitin ligase, calcineurin, arrestin, vacuole, Saccharomyces cerevisiae, protein trafficking (Golgi), endocytosis, inward rectifying channel, protein adaptor, protein quality control, vacuole, yeast
Introduction
All eukaryotic cells continually alter the complement of proteins at the cell surface and in intracellular membranes to reflect changing demands on cellular function. Membrane protein redistribution to the cell surface requires that protein trafficking pathways respond to these demands. In many cases, membrane proteins must be selectively packaged into secretory vesicles that transit from one location to another based on their specific function. These intricate changes in protein trafficking and activity are essential on a continual basis in muscle cells and especially in cardiac myocytes, where with every beat of the contracting heart ion channels, transporters, and hormone receptors must be appropriately positioned to ensure proper function. Despite this central role of regulated protein trafficking in muscle function, we know relatively little about regulated protein trafficking in contractile systems, in part because of the inherent difficulties of working in these systems (1, 2).
To begin to identify regulators of ion channel trafficking that may facilitate cardiac function, we previously employed a yeast model in which a mammalian potassium channel, Kir2.1, was ectopically expressed (3). Kir2.1 is encoded by the KCNJ2 gene and is a member of the family of K+ inward rectifying (Kir)3 2 channels. Kir2.1, one of five members of the Kir family that are expressed in heart, skeletal muscle, and neurons, restores and maintains membrane potential after muscle contraction (4, 5). Functional Kir channels are homotetrameric, and each subunit contains two transmembrane spans (6). When the tetramer assembles, which is thought to occur soon after its synthesis on endoplasmic reticulum (ER)–associated ribosomes (6, 7), a potassium-selective pore is generated. Consistent with a role in cardiac myocyte membrane repolarization, gain-of-function or loss-of-function mutations in Kir2.1 cause short or long QT syndromes, respectively, which result in cardiac arrhythmias (8, 9). For example, loss-of-function mutations that reduce Kir2.1 activity at the cell surface lead to Andersen–Tawil syndrome (8, 10, 11). Moreover, several of the disease-causing KCNJ2 alleles are associated with defects in Kir2.1 trafficking. T192A diminishes Kir2.1 binding to plasma membrane phosphatidylinositol 4,5 bisphosphate (PIP2), reducing channel opening at the cell surface (12), and deletion of amino acids 314–315 abrogates Kir2.1 binding to the clathrin adaptin complex 1 (AP-1), which delivers the channel to the plasma membrane from the trans-Golgi complex (13). Other studies indicate that the regulated exocytosis of Kir2.1 from intracellular stores is critical to maintain Kir2.1 activity at the cell surface (13–15).
Consistent with the importance of protein trafficking in the late secretory pathway, we reported that ESCRT regulates Kir2.1 activity at the cell surface in both yeast and mammalian cells (3). The yeast system relies on the ability of an exogenous potassium channel to rescue the growth of yeast lacking two endogenous potassium channels (TRK1 and TRK2) on low-potassium medium (16), and mutations in genes encoding ESCRT components augment growth under these selective conditions (3). However, we were unable to demonstrate directly that mutations in these trafficking complexes increase the amount of Kir2.1 at the plasma membrane. Furthermore, it is likely that other regulators of the secretory pathway contribute to channel trafficking, because the identification of ESCRT and additional components was undertaken by analyzing the yeast deletion collection. Therefore, functionally redundant factors that contribute to Kir2.1 trafficking were most likely missed when the deletion collection was screened.
Here, we used a targeted screen to reveal the role of α-arrestins in regulating Kir2.1 trafficking in yeast. The α-arrestins are a class of conserved protein adaptors that link specific cargo proteins to a ubiquitin ligase, known as Rsp5, and facilitate protein sorting in the late secretory pathway (17–22). From this screen, we determined that three α-arrestins, Aly1, Aly2, and Ldb19 (also known as Art6, Art3, and Art1), promote Kir2.1-dependent growth of yeast on low-potassium medium. To show directly that the overexpression of the α-arrestins increases Kir2.1 residence at the plasma membrane, we implemented an imaging technology that employs a fluorogen-activating protein (FAP) fusion to Kir2.1. Although this FAP reporter was first developed in yeast (23), surprisingly it has not been used to detect the residence of membrane proteins at the cell surface in this organism. Instead, it has been applied only to monitor protein trafficking in mammalian cells (24, 25). As hypothesized, we were able to demonstrate that Ldb19, Aly1, and Aly2 increase the cell-surface pool of Kir2.1, which they do by increasing Kir2.1 delivery to the plasma membrane rather than by reducing Kir2.1 endocytic turnover. Furthermore, our genetic studies indicated that the Rsp5 E3 ubiquitin ligase is required for α-arrestin–mediated trafficking of Kir2.1 to the cell surface and that phosphoregulation of Aly1 also plays important roles during this process. Together, our findings pave the way for future investigations of α-arrestin regulation of Kir2.1 in human cells, expand the role of α-arrestins in regulating the residence of specific protein cargo, and demonstrate the general applicability of a new fluorescence imaging technology that can detect the levels of low abundance proteins at the yeast plasma membrane.
Results
A targeted screen identifies select α-arrestins as Kir2.1 regulators
The localization of the Kir2.1 potassium channel is tightly regulated to ensure the maintenance of potassium homeostasis in cardiac and other cells (8, 9, 13). Defects in protein quality control and trafficking pathways that govern Kir2.1 activity have clinical consequences (8, 9, 13). For example, mutation of Kir2.1, which abrogates its association with AP-1, a clathrin adaptor required for Golgi-to-plasma membrane trafficking, blunts Kir2.1 targeting to the plasma membrane and leads to Andersen–Tawil syndrome, which causes ventricular arrhythmias (13). To define the protein trafficking and quality control pathways that regulate Kir2.1, we used a system developed previously by our group to uncover the regulators of Kir2.1 surface activity (3). Specifically, we utilized the fact that the expression of an exogenous potassium transporter, such as Kir2.1, restores the growth of yeast lacking two endogenous potassium transporters, TRK1 and TRK2, on low-potassium medium (3, 16, 26, 27). In this system, a growth defect is evident when trk1Δ trk2Δ cells are incubated on medium supplemented with either 25 or 10 mm KCl (Fig. S1A; and see Fig. 1A, panel with pRS426 vector), but improved growth is evident on 25 mm KCl only when yeast express Kir2.1 (Fig. S1A and Ref. 3). These data strongly suggested that Kir2.1 forms a functional potassium transporter when expressed in yeast and elevates intracellular potassium. However, it is possible that the expression of an exogenous protein leads to a stress response, allowing cells to grow on low-KCl medium through an unidentified pathway. Therefore, to validate this system, we first assessed intracellular potassium levels using inductively coupled plasma mass spectrometry (ICP-MS) analyses after cells had been grown for 6–8 h in low-potassium medium. ICP-MS allows for total intracellular element composition to be determined. By using this technique, we found that trk1Δ trk2Δ cells had ∼40% less cellular potassium than their WT counterparts, but ectopic expression of mammalian Kir2.1 in trk1Δ trk2Δ cells increased intracellular potassium by ∼20%, (Fig. 1B). In contrast, the expression of Kir2.1-AAA, in which amino acids 144GYG146 of the potassium selectivity filter within Kir2.1 are mutated to Ala (3, 28), failed to increase intracellular potassium levels compared with the vector control (Fig. 1B). This mutant was also unable to improve growth on low-potassium medium (see below), even though the protein is expressed at similar levels to WT Kir2.1 (3). From these analyses, we show for the first time that the heterologous expression of the Kir2.1 potassium channel in trk1Δ trk2Δ yeast directly increases intracellular potassium.
Figure 1.

α-Arrestins Aly1, Aly2, and Ldb19 promote Kir2.1-dependent growth on low-potassium medium. A, growth of serial dilutions of trk1Δ trk2Δ cells containing the indicated plasmids on SC medium lacking uracil and leucine and containing the indicated added concentrations of KCl is shown after 2 days. B, WT (BY4741) or trk1Δ trk2Δ cells containing the indicated plasmids were grown in 10 mm KCl-containing medium, and then cellular potassium levels were measured using ICP-MS. The ppm of KCl for an equivalent number of cells was determined for four replicates, and the mean ± S.D. was calculated (presented as error bars). Pairwise Student's t tests were performed to assess significance (***, p < 0.0001; ns, p > 0.01, not significant) relative to trk1Δ trk2Δ cells with the vector control. C, lysates from cells expressing Kir2.1 and a vector control or overexpressing Aly1 were analyzed by sucrose gradient density fractionation (30–70%) under conditions to optimize separation of the ER and plasma membrane (PM) fractions. Fractions 1 and 14 are at the top and bottom of the gradient, respectively. Immunoblot analyses were used to detect Kir2.1 (α-HA antibody), ER-resident protein Sec61, and the plasma membrane protein Pma1. One of three representative experiments is shown. D, the percentage of total Kir2.1 signal in the plasma membrane (fractions 8–14) was quantified using ImageJ for three replicate sucrose gradients; the mean percent signal ± S.D. are presented along with the individual data points for each replicate. A Student's t test was performed to assess significance (*, p < 0.01).
Although our earlier systematic screen identified multiple regulators of Kir2.1 in the yeast model, including components of the ESCRT pathway (3), one caveat was that only single-gene deletions were examined. Therefore, a gene whose function is redundant with that of a second gene would have been missed. Consequently, we adopted a targeted screening approach in which we selectively overexpressed functionally redundant factors in Kir2.1-expressing trk1Δ trk2Δ yeast. Growth on low-potassium medium was then measured to identify members of the corresponding gene family that might regulate Kir2.1 trafficking. As a first proof of principle for this analysis, we chose the α-arrestin–trafficking adaptors because: 1) α-arrestins are important for the selective trafficking of a wide array of integral membrane cargo proteins, from G protein–coupled receptors to nutrient permeases and metal transporters (17–21), demonstrating their functional plasticity; 2) α-arrestins can act in a functionally redundant manner such that multiple α-arrestins may regulate the localization of a single membrane cargo protein, making it likely that these genes would have been missed in our initial screen (17, 19, 20); 3) α-arrestins can regulate both intracellular sorting and endocytosis (18–20, 29–32), indicating that Kir2.1 localization can be affected through more than one route; and 4) α-arrestins are conserved from yeast to man (33), suggesting that the results of our study have the potential to define previously unknown regulators of Kir2.1 in mammalian cells.
We overexpressed 12 of the 14 known yeast α-arrestins in trk1Δ trk2Δ cells constitutively expressing Kir2.1 or the Kir2.1-AAA potassium channel mutant (3, 28) as a negative control (Figs. 1A and S1B). In this experiment, yeast containing an empty vector provided another control, and in all cases growth was essentially identical between the vector and Kir2.1-AAA–expressing cells. In contrast, overexpression of one of three α-arrestins, Aly1, Aly2, and Ldb19, strongly improved growth on low-potassium medium when Kir2.1 was expressed, but their expression had no effect on the growth of cells containing the vector control or the Kir2.1-AAA mutant (Fig. 1A). Additionally, ICP-MS analyses demonstrated that overexpression of Aly1, Aly2, and Ldb19 significantly increased intracellular potassium levels compared with the vector control (see below). Four of the other α-arrestins tested (Bul2, Rog3, Art5, and Rim8) modestly improved growth on low potassium, whereas the other five α-arrestins tested (Rod1, Bul1, Bul2, Ecm21, Csr2, and Art10) showed no improvement of growth on low-potassium medium (Fig. S1B). Based on these data, we suggest that Aly1, Aly2, and Ldb19 promote Kir2.1 activity and/or localization at the plasma membrane, and we chose these three α-arrestins as the focus of further study.
Previously, we used indirect immunofluorescence microscopy and biochemical fractionation experiments to determine that most Kir2.1 is retained in the ER and only a small pool of total Kir2.1 reaches the plasma membrane (3). These data align with the fact that the screen for growth on low potassium is quite sensitive and that Kir2.1 exhibits a high open probability (5, 16, 26, 34), so only a small population of cell surface–resident protein is needed for growth. Nevertheless, we fractionated trk1Δ trk2Δ cells expressing Kir2.1 that either contained a vector control or overexpressed ALY1. After sucrose gradient density centrifugation, we resolved the ER and plasma membrane–enriched fractions and examined Kir2.1 residence (Fig. 1C). Consistent with our previous study, the majority of Kir2.1 co-migrated with the Sec61-marked ER fractions (Fig. 1C, fractions 1–6), and only ∼12% of Kir2.1 co-migrated with the Pma1-marked plasma membrane fractions (fractions 8–14) when lysates from cells containing the vector control were examined (Fig. 1, C and D). However, in extracts from cells overexpressing ALY1, the amount of Kir2.1 co-migrating with the Pma1-marked plasma membrane fractions (fractions 8–14) rose significantly when gradients from three independent experiments were analyzed (Fig. 1, C and D). This increase in the plasma membrane fraction of Kir2.1 in cells overexpressing Aly1 is not because of an overall elevated abundance of Kir2.1 in the cell. Specifically, when whole-cell extracts were examined by immunoblotting, overexpression of Aly1, Aly2, or Ldb19 did not significantly alter Kir2.1 abundance compared with the vector control (Fig. S1C). Taken together, these data support a model in which the overexpression of select α-arrestins promotes Kir2.1 trafficking to the plasma membrane, thereby increasing intracellular potassium. Although this function is counter to the more frequently reported role of α-arrestins in regulating the endocytosis of plasma membrane proteins, the results are consistent with other studies showing that α-arrestins can also play important roles in intracellular protein sorting and recycling (29, 30, 35, 36).
FAP-tagged Kir2.1 measures surface channel abundance
We next sought to better characterize Kir2.1 residence in cells overexpressing ALY1, ALY2, or LDB19. However, the large fraction of ER-localized Kir2.1 occludes analyses of the plasma membrane pool of Kir2.1, which is of low abundance, when using traditional imaging approaches. We therefore implemented an alternate strategy that employs a fluorogen-activating protein. This strategy was first described by Waggoner and co-workers (37) and makes use of single-chain antibodies (SCA) selected for their ability to bind to nonfluorescent malachite green (MG)–derived dyes (Fig. 2A). Neither the SCA nor the MG-derived dye fluoresce in solution, but when the dye binds the SCA, a >10,000-fold increase in fluorescence is detected (23, 38). Under these conditions, the SCA and the MG-derived dye comprise a FAP pair. Moreover, by using a cell-permeant form of the dye (referred to hereafter as MG-ESTER), or a cell-impermeant form of the dye (referred to hereafter as MG-B-TAU), the localization of the total cellular pool versus the plasma membrane–resident pool of the fusion protein can be detected and quantified (Fig. 2B). Although the dye-binding SCA modules were first identified in yeast (23, 37, 38), this technology has not been used to monitor the cellular residence of proteins expressed in this model organism.
Figure 2.
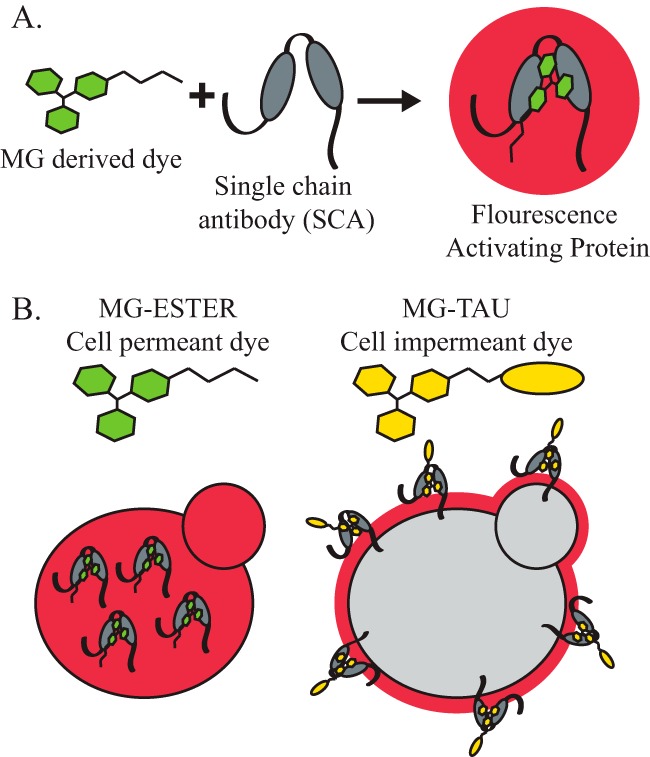
The use of a fluorogen-activating protein to report on cellular residence. A, the FAP technique makes use of a MG-derived dye that binds an SCA. Neither the MG nor the SCA is fluorescent, but when the MG dye is bound by an SCA, fluorescence is detected. B, the MG-derived dye can be conjugated to a membrane-soluble side chain (MG-ESTER, cell-permeant dye) that freely passes through the yeast cell wall and the plasma membrane, where it can be bound by intracellular SCAs to activate fluorescence and monitor the intracellular levels of a tagged protein. The MG-derived dye can also be conjugated to a membrane-impermeant side chain, as is the case with the MG-B-TAU dye. This dye can no longer enter the cell, so only the SCAs on the external face of the cell surface will be bound by dye and fluoresce.
To make use of the cell-excluded dye, the FAP tag must reside on the extracellular side of the plasma membrane. Because both the N- and C termini of Kir2.1 are cytosolic (6), we appended an N-terminal transmembrane (TM) domain to Kir2.1 and inserted the FAP tag upstream of the TM. This generated FAP-TM-Kir2.1, which positions the FAP tag in the extracellular space where it will be accessible to MG-B-TAU (Fig. 3A). We then placed the gene encoding this fusion protein under the control of the strong, constitutive TEF1 promoter. To verify that the MG-B-TAU dye was cell-impermeant in yeast and thus unable to activate the fluorescence of the intracellular pool of Kir2.1, we generated plasmids expressing C-terminal FAP-tagged versions of Sec61 and Sec63 from the constitutive TEF1 promoter. Sec61 and Sec63 are components of the protein translocation machinery in the yeast ER and stably reside in this compartment (39). Next, the FAP-TM-Kir2.1, Sec61-TM, and Sec63-TM expression plasmids were transformed into trk1Δ trk2Δ cells and imaged by confocal microscopy after a 30-min incubation with either the cell-permeant (MG-ESTER) or cell-impermeant (MG-B-TAU) dyes. As anticipated, Sec61-FAP– or Sec63-FAP–containing cells incubated with the MG-ESTER dye showed a localization pattern consistent with ER residence, with clear perinuclear and cortical ER rings (40). In contrast, FAP-TM-Kir2.1 incubated with MG-ESTER had a punctate distribution with cortical patches and a region of intracellular exclusion consistent with the nucleus (Fig. 3B, top row). Under these conditions, it was impossible to discern plasma membrane–localized FAP-TM-Kir2.1 from ER-localized FAP-TM-Kir2.1 using the cell-permeant dye. However, when the same concentration of MG-B-TAU was added for 30 min and images were acquired using identical parameters, a fluorescent signal was absent in the Sec63-FAP–containing cells, yet dim fluorescence was evident for FAP-TM-Kir2.1 (Fig. 3B, second and third rows). This putative plasma membrane pool of FAP-TM-Kir2.1 was enhanced when images were adjusted using post-acquisition modifications (Fig. 3B, third row). Although the intensity of the cell-surface pool was modest, clear puncta at the cell periphery corresponding to FAP-TM-Kir2.1 were reproducibly observed, whereas no signal was detected for Sec63-FAP (Fig. 3B, bottom row). When the fluorescent signals for FAP-TM-Kir2.1, Sec61-FAP, and Sec63-FAP were quantified, the signals with the permeant dye were stronger compared with the signals with the impermeant dye (Fig. 3, C and D). In addition, although the fluorescent signals with the impermeant dye for both FAP-TM-Kir2.1 and Sec63-FAP increased with longer incubation times, suggesting that some cell-impermeant dye may enter the cells over time via bulk endocytosis, the fluorescent signal for FAP-TM-Kir2.1 was significantly brighter than that of the Sec63-FAP control (Fig. 3D). Importantly, the signal intensity of FAP-TM-Kir2.1 was ∼10-fold higher when it was detected with the permeant versus the impermeant dyes (Fig. 3, C and D). These results are consistent with the biochemical fractionation data indicating that only ∼10% of Kir2.1 is found at the cell surface (Fig. 1, C and D).
Figure 3.
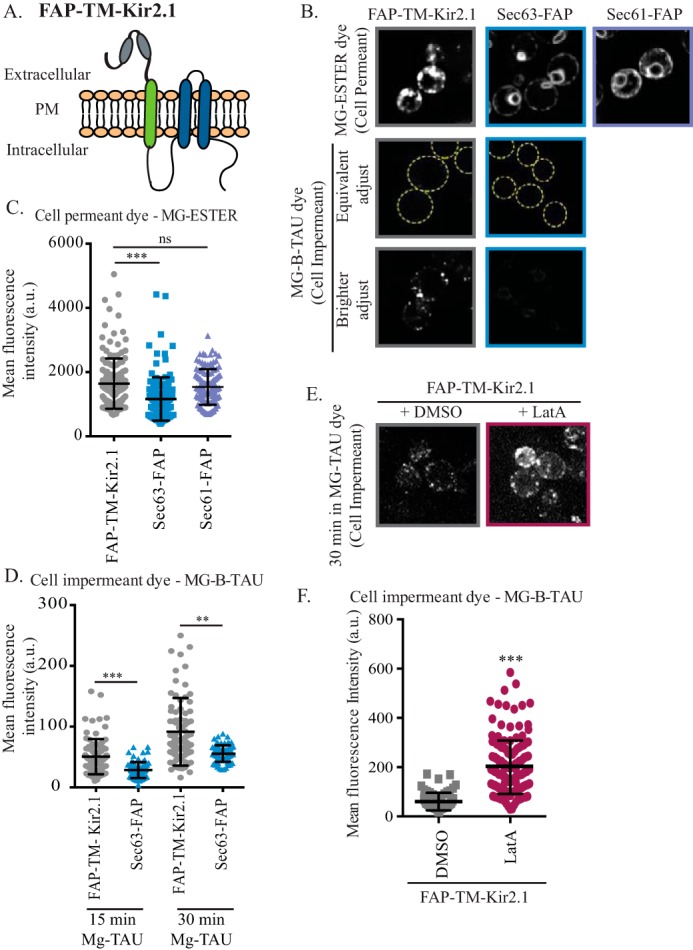
FAP-tagged Kir2.1 can be detected at the cell surface. A, schematic of FAP-tagged Kir2.1. Kir2.1 transmembrane domains are shown in blue, and the platelet-derived growth factor transmembrane domain is shown in green. The FAP tag extends from the N terminus and is shown as gray ovals. B, cells expressing FAP-TM-Kir2.1, Sec63-FAP, or Sec61-FAP were incubated with the MG-ESTER or MG-B-TAU dye to activate intracellular or cell-surface FAP-tag fluorescence, respectively. Confocal microscopy images of medial sections of cells incubated with dye for 30 min are shown. In the top two rows, cells are adjusted equivalently to show that the signal from intracellular FAP-TM-Kir2.1 or Sec63-FAP is lost when the MG-B-TAU dye is used. Yellow dashed-line circles are used in the second row to demark the cell. The bottom row of images are the same images as shown in the second row but are adjusted to allow the cell-surface fluorescence of the MG-B-TAU–stained FAP-TM-Kir2.1–expressing cells to be seen. C and D, total cellular fluorescence for n > 70 cells, imaged as in B, was measured, and mean fluorescence intensities for all cells (in a.u.) are presented as scatter plots. The horizontal midline in each plot represents the mean fluorescence intensity, and the error bars represent ±S.D. In D, the cellular fluorescence was quantified at both 15 and 30 min post-MG-B-TAU addition. Kruskal–Wallis statistical analyses with Dunn's post hoc test were performed, and the statistical significance is indicated (**, p < 0.001; ***, p < 0.0001; ns, p > 0.01, not significant). E, confocal microscope images of cells expressing FAP-TM-Kir2.1 that were incubated for 90 min with LatA or DMSO and stained with MG-B-TAU for 30 min are shown. F, the total cellular fluorescence for n > 60 cells, imaged as in E, was measured, and mean fluorescence intensities for all cells (in a.u.) are presented as scatter plots. A Student's t test was performed to assess significance (***, p < 0.0001).
Next, we set out to establish that the punctae at the cell periphery represent the plasma membrane pool of FAP-TM-Kir2.1. Specifically, we found that the MG-B-TAU–dependent fluorescence signal rose significantly when the cells were pretreated with latrunculin A (LatA) (Fig. 3, E and F). LatA blocks actin polymerization and thereby prevents endocytosis, yet it leaves intracellular sorting intact (41). Thus, treatment with LatA allowed FAP-TM-Kir2.1 to accumulate at the cell surface, increasing the FAP-TM-Kir2.1 pool that can be stained with the impermeant dye. Together, these data demonstrate that the MG-B-TAU dye is cell-impermeant in yeast, consistent with what has been reported for MG-B-TAU in mammalian cells (38). Further, when used with a FAP-tagged Kir2.1 chimera, the cell-surface population of this protein can be quantified.
The localization and relative cellular distribution of FAP-TM-Kir2.1 appeared consistent with what we observed previously when the residence of HA-tagged Kir2.1 was queried using indirect immunofluorescence microscopy (Figs. 1C and 3, B–D) (3). Nevertheless, we wanted to assess the function, stability, and regulation of this new FAP-TM–tagged construct. As evident with the WT and HA-tagged proteins, we found that FAP-TM-Kir2.1 improved growth on low-potassium medium compared with the vector control; however, FAP-TM-Kir2.1 rescued growth less effectively compared with the HA-tagged protein (Fig. 4A). Indeed, immunoblot analyses of whole-cell extracts revealed that the steady-state levels of the FAP-TM-Kir2.1 protein were dramatically lower than Kir2.1 (Fig. 4B). The reduction in FAP-TM-Kir2.1 was unlikely to arise from altered gene expression because FAP-TM-Kir2.1 and Kir2.1 are expressed from the same promoter. Instead, when the degradation rate of FAP-TM-Kir2.1 was compared with HA-tagged Kir2.1, we found that FAP-tagged Kir2.1 was significantly less stable that the HA-tagged Kir2.1 (Fig. 4, C and D). Inhibition of the proteasome via incubation of cells with MG132 significantly stabilized FAP-tagged Kir2.1 (Fig. 4, C and D), demonstrating that the FAP-tagged Kir2.1 is subject to proteasome-mediated degradation. Next, to determine whether the degradation of FAP-TM-Kir2.1 was regulated by the same factors acting in the late secretory pathway as reported previously for Kir2.1 (3), we examined FAP-TM-Kir2.1 activity and localization in cells lacking the retromer subunit Vps35. Consistent with our previous findings, Kir2.1-dependent growth on low-potassium medium was greatly enhanced in a vps35Δ trk1Δ trk2Δ strain compared with trk1Δ trk2Δ cells, and growth rescue mediated by FAP-TM-Kir2.1 in this strain background was similarly improved on low-potassium medium (Fig. 4A). Moreover, we found that FAP-TM-Kir2.1 levels at the cell surface, as assessed with MG-B-TAU, increased in cells lacking Vps35 (Fig. 4, E and F). Although the protein is less stable than HA-Kir2.1, these combined data indicate that FAP-TM-Kir2.1 is a functional potassium channel whose trafficking itinerary is consistent with that of the active HA-tagged protein (3).
Figure 4.

The plasma membrane residence and activity of Kir2.1 and FAP-tagged Kir2.1 are regulated similarly. A, growth of serial dilutions of trk1Δ trk2Δ or trk1Δ trk2Δ vps35Δ cells containing the indicated plasmids on SC medium lacking leucine and containing the indicated amount of KCl is shown. Growth shown is at 4 days. B, whole-cell extracts from trk1Δ trk2Δ cells expressing Kir2.1 or FAP-TM-Kir2.1 were analyzed by immunoblotting. Red dots indicate bands of the correct molecular mass for Kir2.1 and FAP-TM-Kir2.1 (α-HA). Molecular masses are denoted in kilodaltons. Whole-cell extracts were loaded at two different concentrations (lanes 1 and 2) as shown in the loading control (α-G6PDH). Two different exposures of the same blot are shown to facilitate comparisons. C, yeast cells (trk1Δ trk2Δ pdr5Δ) expressing either HA-tagged or FAP-tagged Kir2.1 were grown to mid-logarithmic phase, treated with either MG132 or DMSO (vehicle control), and dosed with cycloheximide (CHX), and samples were taken at the indicated time points. Kir2.1 protein abundance was assessed by immunoblotting with the indicated antibodies. Molecular masses are denoted in kilodaltons. D, immunoblots as in C were quantified for a minimum of four replicate experiments, and the means are plotted. Data are normalized to 100% for t = 0 time point in each case. Error bars represent ±S.D., and a Student's t test was performed to assess significance (**, p < 0.001; ***, p < 0.0001; ns = p > 0.01, not significant). E, trk1Δ trk2Δ or trk1Δ trk2Δ vps35Δ cells expressing FAP-TM-Kir2.1 were treated with LatA or DMSO for 90 min and then incubated with MG-B-TAU dye for 30 min and imaged by confocal microscopy. F, total cellular fluorescence for n > 50 cells, imaged as in C, was measured, and mean fluorescence intensities for all cells (in a.u.) is presented as scatter plots. A Student's t test was performed to assess significance (***, p < 0.0001).
α-Arrestins increase Kir2.1 residence at the plasma membrane in a Rsp5-dependent manner
Based on the data presented in Fig. 1, α-arrestin overexpression should augment cell-surface residence of Kir2.1 at the plasma membrane. Therefore, we employed the FAP-TM-Kir2.1 chimera to determine whether increased staining with MG-B-TAU dye was apparent when the α-arrestins were overexpressed. As hypothesized, cells overexpressing α-arrestins Aly1, Aly2, or Ldb19 had significantly brighter FAP-TM-Kir2.1–dependent cell-surface fluorescence compared with cells with a vector control (Fig. 5, A and B). To ensure that increased fluorescence intensity was not caused by a reduced rate of endocytosis, but rather corresponded to an elevated rate of delivery to the plasma membrane, we pretreated cells with LatA and then added MG-B-TAU before conducting image analysis by confocal microscopy. At the concentrations used (200 μm), LatA resulted in loss of both filamentous actin and cortical actin patches, the latter of which is associated with yeast endocytosis. Consequently, this concentration completely prevents fluid phase endocytosis of lucifer yellow (41–43). Thus, in our experiments, endocytosis was completely blocked but delivery to the cell surface remained intact. As shown in Fig. 5, C and D, greater FAP-TM-Kir2.1 cell-surface fluorescence was detected when ALY1, ALY2, or LDB19 was overexpressed compared with the vector control in the LatA-treated cells. These data demonstrate that the α-arrestins facilitate Kir2.1 delivery to the cell surface and do not require Kir2.1 endocytosis to impact Kir2.1 abundance at the cell surface.
Figure 5.

α-Arrestin regulation of Kir2.1 requires the Rsp5 ubiquitin ligase. A, maximum Z-projections of confocal microscopy images were acquired in trk1Δ trk2Δ cells expressing FAP-TM-Kir2.1 and in the presence of a vector control or plasmids overexpressing the indicated α-arrestins. The cells were incubation with MG-B-TAU dye for 30 min. B, total cellular fluorescence for the maximum Z-projections of n > 30 cells, imaged as in A, was measured, and mean fluorescence intensities for all cells (in a.u.) are presented as scatter plots. The horizontal midline in each plot represents the mean fluorescence intensity, and the error bars represent ±S.D. Kruskal–Wallis statistical analyses with Dunn's post hoc test were performed, and the statistical significance compared with the vector control is indicated (p values are assigned as listed below). C, maximum Z-projections of confocal microscopy images were acquired in trk1Δ trk2Δ cells expressing FAP-TM-Kir2.1 and containing the vector control or plasmids overexpressing the indicated α-arrestins. The cells were incubation with LatA for 90 min and the MG-B-TAU dye for 30 min. D, total cellular fluorescence for the maximum Z-projections of n > 50 cells, imaged as in C, was measured, and mean fluorescence intensities for all cells (in a.u.) are presented as scatter plots. The horizontal midline in each plot represents the mean fluorescence intensity, and the error bars represent ±S.D. Kruskal–Wallis statistical analyses with Dunn's post hoc test were performed (*, p < 0.01; **, p < 0.001; ***, p < 0.0001; ns, p > 0.01, not significant).
All α-arrestins in yeast contain (L/P)PXY motifs. This motif binds WW-domains in a ubiquitin ligase, Rsp5, which is involved in the trafficking of several α-arrestin–dependent cargo proteins (19, 20, 31, 44, 45). However, exceptions have been noted in which Rsp5 function during the sorting of α-arrestin–dependent cargo proteins was dispensable (17). We previously showed that Aly1, Aly2, and Ldb19 mutants where each of the (L/P)PXY motifs is altered to (L/P)PXG (referred to hereafter as Aly1PPXY-less, Aly2PPXY-less, and Ldb19PPXY-less) were unable to bind Rsp5. These PPXY-less mutants of Aly1, Aly2, and Ldb19 also failed to regulate clathrin-mediated or clathrin-independent endocytosis of the G protein–coupled receptors Ste2 and Ste3 (17, 46). To investigate the role of the Rsp5-binding motif in Kir2.1 trafficking, we examined the PPXY-less mutants of Aly1 and Aly2 and found that sensitivity to azetidine-2-carboxylic acid (AzC) is lost, a phenotype associated with a defect in Gap1 sorting to the plasma membrane (Fig. S2C) (22, 30). Consistent with these data, even though overexpression of Aly1, Aly2, or Ldb19 promoted Kir2.1-dependent growth on low-potassium medium (Figs. 1A and 6A), the PPXY-less mutant forms of these α-arrestins were dramatically less effective than their WT counterparts (Fig. 6A). It should be noted that overexpression of any of these α-arrestins, either the PPXY-less mutants or their WT counterparts, did not significantly alter the steady-state abundance of Kir2.1 compared with the vector control. This result was evident regardless of whether the TEF1pr-Kir2.1 expression system or the ZWF1pr-Kir2.1 expression system was used (data not shown and Fig. S1C). Consistent with an important role for Rsp5 in regulating Kir2.1 activity at the cell surface, there was less Kir2.1 at the cell surface in each of the α-arrestin PPXY-less mutants than in cells expressing their WT counterpart (Fig. 5). In almost all cases, the PPXY-less mutants did not significantly increase cell-surface Kir2.1 abundance compared with the vector control; only a modest increase in cell-surface abundance was uncovered with Aly2PPXY-less. Further evidence supporting Rsp5-dependent trafficking was apparent because PPXY-less α-arrestins failed to elevate intracellular potassium compared with the vector control when assessed by ICP-MS. In contrast, the WT Aly1, Aly2, and Ldb19 α-arrestins significantly increased intracellular potassium (Fig. 6B). These data strongly suggest that the binding of select α-arrestins to Rsp5 promotes Kir2.1 trafficking to the cell surface.
Figure 6.

Overexpression of Aly1, Aly2, and Ldb19 increases both Kir2.1 residence at the plasma membrane and intracellular potassium. A, growth of serial dilutions of trk1Δ trk2Δ cells containing the indicated plasmids on SC medium lacking leucine and uracil and containing the indicated added amounts of KCl is shown after 2 days. B, trk1Δ trk2Δ cells expressing Kir2.1 and the indicated overexpressed α-arrestin plasmids were grown in 10 mm KCl, and cellular potassium levels were measured using ICP-MS. The ppm corresponding to KCl for an equivalent number of cells was determined for four replicates, and the mean ± S.D. was calculated (presented as error bars). Kruskal–Wallis statistical analyses with Dunn's post hoc test were performed, and the statistical significance compared with the vector control for each is indicated (**, p < 0.001; ***, p < 0.0001; ns, p > 0.01, not significant). C, schematic of the Aly1 and Aly2 protein coding regions, where the numbers indicate the amino acid position, dark red boxes denote the N-terminal arrestin-fold, the light red boxes denote the C-terminal arrestin-fold, green ovals indicate the position of the (L/P)PXY motifs, and blue circles indicate the sites of ubiquitin (Ub) modification identified via MS. D, GST-fused α-arrestins were isolated from WT (BJ5459) cells after α-arrestin expression (under control of the CUP1 promoter) was induced with copper sulfate. Isolated proteins were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. Molecular masses are denoted in kilodaltons. E, growth of serial dilutions of trk1Δ trk2Δ cells containing the indicated plasmids on SC medium lacking leucine and uracil and containing the indicated added amounts of KCl is shown after 4 days.
One hypothesis for why Rsp5 is needed to increase Kir2.1 abundance at the cell surface is that Rsp5 ubiquitination of the α-arrestins is required for their role in intracellular protein sorting. For many α-arrestins, monoubiquitination is linked to the regulation of protein trafficking, and loss of monoubiquitination impairs α-arrestin recruitment to membranes and interactions with key trafficking factors (18, 19, 47). To test this hypothesis for Aly1 and Aly2, we performed LC-MS analysis of isolated GST-tagged Aly1 and Aly2 from WT yeast. Spectra corresponding to peptides covering 60 and 56% of Aly1 and Aly2, respectively, were obtained (Fig. S2, B and C), and diglycine-modified lysines, a signature of ubiquitination in MS spectra (48–50), were identified at the positions indicated (Figs. 6C and S2, A and B). Notably, modification of Lys-392 in Aly2 was identified in our analyses and also in three other proteomics studies (51–53), lending strong support for this position as a valid ubiquitination site. We mapped the ubiquitination sites onto the Phyre-predicted secondary structures of Aly1 and Aly2 (Fig. 6C) (30, 54, 55) and found that Lys-392 in Aly2 corresponds to the conserved Lys-379 site in Aly1, which also had been identified as being ubiquitinated in a proteomic screen (52). In addition, based on secondary structure predictions, the Lys-392 and Lys-379 sites in Aly1 and Aly2, respectively, map to the same region within the C-terminal arrestin-fold as one of the four monoubiquitination sites in Rod1 (data not shown and Ref. 18). Based on these observations, we mutated Lys-392 in Aly2 and the analogous Lys-379 residue in Aly1 to arginine using site-directed mutagenesis and examined the ubiquitination status of the resulting mutant α-arrestins via immunoblot analysis. Although ubiquitination was clearly detected for the WT Aly1 and Aly2 species, Aly1K379R and Aly2K392R lacked a discernible ubiquitin signal (Fig. 6D). As a control in these experiments, Aly1PY-less and Aly2PY-less were also employed, and a similar loss of ubiquitination (Fig. 6D), as anticipated based on previous findings (22), was observed. These results confirmed that select lysines are ubiquitinated in Aly1 and Aly2.
We then assessed the impact of the Lys-to-Arg mutations in Aly1 and Aly2 on known phenotypes associated with overexpression of WT Aly1 and Aly2, such as resistance to rapamycin and sensitivity to AzC (see above and Refs. 22 and 30). We found that loss of these ubiquitination sites resulted in hypomorphic Aly1 and Aly2 alleles that were less effective at conferring sensitivity to AzC, suggesting a reduced ability to promote Gap1 trafficking to the plasma membrane (Fig. S2C). Although ubiquitination was dispensable for Aly1-induced resistance to rapamycin, the modification was required for Aly2-induced resistance to rapamycin (Fig. S2C). These data suggest that ubiquitination at these sites is important for some but not all Aly1- or Aly2-mediated activities. Finally, we examined the ability of the ubiquitination-deficient Aly1 and Aly2 mutants to promote Kir2.1 activity by monitoring their ability to promote growth on low-potassium medium. Interestingly, both ALY1K392R and ALY2K379R were hypomorphic to their respective WT alleles (Fig. 6E), showing a reduced capacity for growth on low-potassium medium. As observed above, the PPXY-less mutants were also unable to stimulate Kir2.1-depenent growth on low potassium. Taken together, these findings suggest that ubiquitination of Aly1 and Aly2 is essential for optimal Kir2.1 trafficking to the cell surface. In addition, binding of Aly1, Aly2, and Ldb19 to the Rsp5 ubiquitin ligase also appears to be required for α-arrestin–dependent delivery of Kir2.1 to the plasma membrane.
α-Arrestin phosphorylation impacts Kir2.1 trafficking
α-Arrestins are heavily phosphorylated. Thus far, more than 24 and 13 phosphorylated residues have been identified in Aly1 and Ldb19, respectively, using MS (22, 52, 56–58). In many, but not all instances, phosphorylated α-arrestins fail to stimulate endocytosis of the associated membrane proteins, whereas dephosphorylated α-arrestins are more potent regulators of endocytosis (18, 22, 58–60). Previously, we found that Aly1 is dephosphorylated by the protein phosphatase calcineurin (22) and that dephosphorylation stimulates Aly1-mediated endocytosis of the aspartic acid permease Dip5. However, the calcineurin-mediated dephosphorylation of Aly1 had no effect on Aly1-dependent recycling of the Gap1 permease (22). These data indicate that the action of calcineurin on a given α-arrestin can affect the trafficking of specific cargo proteins. Therefore, to uncover the potential role of α-arrestin phosphorylation on Kir2.1 trafficking, we mutated the calcineurin-binding site in Aly1, PILKIN (aa 832–837), to a series of alanines (ALY1AAAAAA) (22), and the protein was overexpressed. In this strain, Kir2.1-dependent growth on low potassium was enhanced (Fig. 7A). Consistent with these data, intracellular potassium as well as FAP-TM-Kir2.1 residence at the cell surface was also enhanced when the Aly1AAAAAA protein was overexpressed (Fig. 7, B–D).
Figure 7.

Calcineurin negatively regulates Aly1-mediated Kir2.1 trafficking to the plasma membrane. A, growth of serial dilutions of trk1Δ trk2Δ cells containing the indicated plasmids on SC medium lacking leucine and uracil and containing the indicated amount of KCl is shown after 2 days. B, maximum Z-projections (Max Z) of confocal microscopy images acquired in trk1Δ trk2Δ cells expressing FAP-TM-Kir2.1 and containing a vector control or plasmids overexpressing the indicated α-arrestins. Cells were incubated with MG-B-TAU dye for 30 min. C, total cellular fluorescence for the maximum Z-projections of n > 45 cells, imaged as in B, was measured, and mean fluorescence intensities for all cells (in a.u.) are presented as scatter plots. The horizontal midline in each plot represents the mean fluorescence intensity, and the error bars represent ±S.D. Kruskal–Wallis statistical analyses with Dunn's post hoc test were performed, and the statistical significance compared with the vector control is indicated. D, trk1Δ trk2Δ cells expressing Kir2.1 and the indicated overexpression α-arrestin plasmids were grown in 10 mm KCl and cellular potassium levels were measured by ICP-MS. The PPM of KCl for an equivalent number of cells was determined for 4 replicates, and the mean and S.D. were calculated (presented as error bars). Kruskal–Wallis statistical analyses with Dunn's post hoc test were performed, and the statistical significance compared with the vector control for each is indicated (*, p < 0.01; **, p < 0.001; ***, p < 0.0001; ns, p > 0.01, not significant). E, growth of serial dilutions of trk1Δ trk2Δ or trk1Δ trk2Δ cnb1Δ cells containing the indicated plasmids on SC medium lacking leucine and uracil and containing the indicated amount of KCl is shown after 2 days. F, model for calcineurin (CN) regulation of Aly1-mediated trafficking of Kir2.1 Red dashed lines indicate exocytosis or endocytosis of Kir2.1. Gray circles on Aly1 indicate phosphorylation sites, with fewer phosphorylation sites in the version of Aly1 that has been dephosphorylated by calcineurin.
To further assess the role of calcineurin in regulating Aly1, we deleted the gene encoding the calcineurin regulatory subunit CNB1, which is required for catalytic activity (61), in the trk1Δ trk2Δ strain background. We then measured the ability of Aly1 to promote Kir2.1-dependent growth on low potassium. As shown in Fig. 7E, we found that the absence of calcineurin activity augmented Aly1-mediated growth on low-potassium medium, consistent with the phosphorylated form favoring Kir2.1 residence at the cell surface. Based on these combined data, we propose a model whereby calcineurin-mediated dephosphorylation of Aly1 impairs the ability of an α-arrestin to stimulate Kir2.1 trafficking to the cell surface (Fig. 7F). We further propose that phosphorylation serves as a molecular switch: dephosphorylated Aly1 stimulates endocytosis, whereas phosphorylated Aly1 recognizes the intracellular pool of select integral membrane proteins and stimulates their trafficking to the cell surface (Fig. 7F).
Discussion
By applying a genetic analysis in a yeast model, we demonstrated that select α-arrestins regulate Kir2.1 cell-surface abundance by facilitating channel trafficking to the plasma membrane. Confirmation that the α-arrestins directly affect Kir2.1 function at the plasma membrane, and consequently an increase in intracellular potassium, was uncovered by applying a spectroscopic technique that is rarely used in yeast, ICP-MS. Our results confirm the general applicability of this method for future studies on the function of any plasma membrane resident ion channel in this organism. Consistent with previous studies, we also found that the α-arrestin–mediated effect on Kir2.1 requires α-arrestin phosphorylation and interaction with a ubiquitin ligase, Rsp5, that plays an important role in regulating the trafficking of myriad transporters and other plasma membrane cargo (18–20, 22, 31, 44, 58, 59). In addition, for the first time we also applied the FAP-tagging system to demonstrate that this technique can differentiate a cell surface–localized protein pool from the intracellular pool in yeast. Consequently, this technology may now become widely applicable in protein trafficking studies in this organism, which thus far have relied on cell-surface biotinylation, which is of low efficiency and may permeabilize cells (62), or on GFP-tagging methods, which may not easily quantify or differentiate between intracellular versus cell-surface protein pools (63, 64). We envision that the FAP system will next allow for fine spatial and temporal endocytic dynamics to be resolved for any endogenous yeast protein without interference from the competing intracellular fluorescence signal, which is inherent in this organism (63–65).
As opposed to the well-document role of α-arrestins in regulating endocytosis (18–21, 31, 44), we have shown that Aly1, Aly2, and Ldb19 promote Kir2.1 localization to the cell surface. However, distinct from their function during endocytosis, the role of α-arrestins in regulating the intracellular recycling pathways is still emerging. Indeed, our discovery that α-arrestins augment Kir2.1 cell-surface abundance is reminiscent of a related activity observed for Aly1 and Aly2 in increasing the abundance of the general amino acid permease Gap1 at the cell surface after nutrient deprivation (30). Aly2-dependent recycling of Gap1 requires both phosphoregulation of Aly2 by the Npr1 kinase, which is negatively regulated by TORC1 and has long been known to be a positive regulator of Gap1 trafficking to the cell surface (30, 35, 58, 66, 67), and also AP-1, the clathrin adaptor essential for endosome-to-Golgi retrieval of select cargo proteins (68–70). Our current model for Aly2-mediated recycling of Gap1 is that Aly2 stimulates Gap1 packaging into AP-1 and clathrin-coated vesicles that shuttle Gap1 from endosomes to the trans-Golgi network. Aly2 is a substrate for Npr1 in vitro, and Npr1is required for Aly2-dependent regulation of Gap1 trafficking, making it likely that Npr1 phosphorylation of Aly2 in vivo stimulates Aly2-mediated Gap1 recycling (30). Therefore, we propose that Aly2 operates similarly to increase Kir2.1 cell-surface localization by enhancing endosome-to-Golgi trafficking of Kir2.1, ultimately increasing Kir2.1 localization at the plasma membrane.
We also describe here a novel role for calcineurin-associated Aly1 dephosphorylation as an inhibitory signal during Kir2.1 trafficking to the cell surface. This result suggests that phosphorylation/dephosphorylation of Aly1 may serve as a functional switch in which dephosphorylated Aly1 stimulates cargo endocytosis (as seen for another cargo, Dip5 (22, 44)), whereas phosphorylated Aly1 promotes anterograde cargo trafficking (as seen for Kir2.1 in this study). Future studies will delineate the intracellular sorting pathway(s) controlled by Aly1 via this switch and establish whether a conformational change in Aly1 triggers these distinct responses. Interestingly, the calcineurin-mediated negative regulation of Kir2.1, as shown here, may be conserved in more complex organisms (71–73). For example, in mammalian cells, Kir2.1 interacts with the protein kinase A–anchoring protein (AKAP), which scaffolds multiple kinases and phosphatases (including calcineurin). Association with AKAP increases Kir2.1 channel function when calcineurin and other phosphatases are inhibited. Consequently, Kir2.1 may be negatively regulated in a similar fashion by calcineurin in higher organisms (72). In addition, pharmacological inhibition of calcineurin decreases Kir2.1 channel activity at the cell surface in Xenopus oocytes (71), and calcium influx as an indirect result of Kir2.1 hyperpolarization activates calcineurin to stimulate myocyte development (73).
In contrast to Aly1 and Aly2 (30), Ldb19-mediated protein recycling has not been described previously, and thus our findings add a new dimension to the functional repertoire for this α-arrestin. Nevertheless, Aly1, Aly2, and Ldb19 have overlapping functions in regulating the clathrin-independent and clathrin-mediated endocytosis of the yeast mating pheromone receptor Ste3 (46). In support of a broader role for α-arrestins in regulating intracellular sorting, another α-arrestin, Rod1, controls a complex intracellular trafficking itinerary for the lactate permease Jen1. In this case, Rod1 regulates the glucose-induced, Rsp5-dependent endocytosis of Jen1, but after internalization Jen1 traffics from endosomes to the Golgi rather than being routed from endosomes to the vacuole for degradation (29). Subsequently, Rod1 is recruited to Golgi membranes and facilitates Jen1 retrieval and return to endosomes, which subsequently induces Jen1 trafficking to and degradation in the vacuole. The role of Rod1 in regulating this two-step protein “shuffle” demonstrates both the functional plasticity of α-arrestin–mediated trafficking and the complex itinerary of intracellular trafficking. In fact, a similar role has been described for two other α-arrestins, Bul1 and Bul2, which stimulate Gap1 endocytosis as well as Golgi-to-endosome sorting (32, 36, 60, 74). A more detailed definition of the mechanism by which Aly1, Aly2, and Ldb19 regulate Kir2.1 trafficking in yeast and mammalian cells represents an important area of study and is in progress. However, it should be noted that it is not trivial to definitively link a given α-arrestin in yeast with a homolog or even a paralog in human cells, as members of the α-arrestin family are defined by a structural motif as well as by their functional association with ubiquitin ligases and cargo proteins (33, 75). Yet, the amino acid sequence conservation between α-arrestins within a given organism and even more so between different organisms is exceeding low (33, 75).
Our earlier study defined a role for the ESCRT machinery, retromer, and AP-1, as well as other Rsp5 adaptor proteins (e.g. Bsd2 and Tre1) in regulating Kir2.1 activity at the cell surface (3). For example, loss of an ESCRT-1 component, Vps23, improves yeast growth on low-potassium medium, consistent with Kir2.1 trafficking into the MVB pathway and on to the vacuole for degradation (3). The regulation of Kir2.1 by ESCRT is conserved, as both the knockdown of ESCRT components or pharmacological inhibition of lysosomal degradation increases Kir2.1 abundance in HEK293 cells. Consistent with Kir2.1 trafficking to the MVB pathway in yeast, the loss of Bsd2 or Tre1, which represent other Rsp5 adaptors that stimulate cargo ubiquitination and trafficking to the vacuole (76, 77), augments growth of Kir2.1-expressing yeast on low-potassium medium (3). Thus, Bsd2 and Tre1 may help direct Rsp5 ubiquitination onto Kir2.1 to promote vacuolar trafficking, whereas the α-arrestins act instead as trafficking adaptors to stimulate the recycling of Kir2.1 from endosomes back to the Golgi or directly to the cell surface. It is interesting that loss-of-function mutations in any one of four other factors important for endosome-to-Golgi sorting (retromer, GARP, AP-1, and Ypt6) were also identified in our yeast screen. We posit that the loss of these endosome-to-Golgi recycling components impairs the MVB-dependent sorting of Kir2.1, based on the model described above for Jen1 regulation by an α-arrestin (29). Alternatively, loss of these endosome-to-Golgi sorting factors may lead to compensatory changes in protein trafficking that either allow for direct transit of Kir2.1 from endosomes to the cell surface or improve the activity of other intracellular recycling pathways. Defining how these intracellular sorting pathways contribute to α-arrestin–mediated trafficking will also be a topic for future efforts.
In addition to defining a role for α-arrestins in stimulating Kir2.1 trafficking to the cell surface and demonstrating that Aly1 dephosphorylation negatively regulates this pathway, our results strongly suggest that the α-arrestins bind Rsp5 to enhance Kir2.1 cell-surface localization. One of the hallmarks of α-arrestins is that they contain (L/P)PXY motifs and thus bind WW domains in the Rsp5 ubiquitin ligase, whose ortholog in mammals is Nedd4-2 (19–21, 33, 45, 78). During endocytosis, α-arrestins bridge plasma membrane cargo proteins and Rsp5 (18–21). Rsp5 can then ubiquitinate the cargo proteins, which triggers endocytosis but also ubiquitinates the α-arrestin (18, 19, 31). In some instances, α-arrestin lysine mutants, which cannot be ubiquitinated, are nonfunctional (17–19, 31), and it has been suggested that monoubiquitination is needed to activate an α-arrestin during protein trafficking (79). For example, monoubiquitination of Rod1 is required to regulate Jen1 trafficking (18), and similarly, monoubiquitination of Ldb19 is critical for Can1 trafficking (19). Interestingly, the loss of Ldb19 monoubiquitination impairs the recruitment of this α-arrestin to the Golgi and plasma membranes (19), suggesting that monoubiquitination may play an important role during intracellular sorting as well as endocytosis. Loss of α-arrestin Rim8 monoubiquitination reduces binding to the ESCRT subunit Vps23 and diminishes Rim8 function (80), suggesting that monoubiquitination can influence α-arrestin binding to key protein-trafficking regulators. In this study, we identified the site of Aly1 and Aly2 monoubiquitination and showed that mutant forms of Aly1 and Aly2, which cannot be modified, are unable to effectively promote Kir2.1-dependent growth on low potassium. Thus, as is the case for many other α-arrestins, monoubiquitination of Aly1 and Aly2 facilitates the trafficking of distinct cargo proteins. Based on these data, we propose that our PPXY-less Aly1, Aly2, and Ldb19 mutants may exhibit compromised function, not because of their inability to recruit Rsp5 to Kir2.1 but perhaps as a result of their inability to be recruited to membranes or bind auxiliary factors that promote intracellular Kir2.1 sorting. Here, too, future in-depth studies will be needed to establish whether this principle applies to the sorting of Kir2.1 in yeast and mammalian cells.
Experimental procedures
Yeast strains and growth conditions
Yeast strains used in this study and their construction are described in Table 1. Yeast were grown in synthetic complete (SC) medium prepared as described (81). SC low-potassium medium was also prepared as described with the use of monosodium glutamate as a nitrogen source, the addition of 20 mm MES to maintain the pH, and the indicated amount of KCl (3). Liquid medium was filter-sterilized, and for plated medium, 2% (w/v) agar was added prior to autoclaving. Plasmids were transformed into yeast via the lithium-acetate method and selected for on the appropriate SC medium. Yeast cells were grown at 30 °C unless otherwise indicated. For growth assays on solid medium, 5-fold serial dilutions of saturated, overnight cultures (starting concentration of 1.0 × 107 cells/ml) were plated onto the indicated medium and grown for 3–6 days at 30 °C.
Table 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | (89) |
| BY4743 | MATa/MATa his3Δ1/his3Δ1 leu2Δ0/ leu2Δ0 ura3Δ0/ura3Δ0 met15Δ0/MET15 LYS2/lys2Δ0 | (89) |
| trk1Δtrk2Δ | MATα trk1Δ::HIS3 trk2Δ::HIS3 his3Δ200 leu2Δ1 ura3–52 trp1–1 ade2 | (16, 90) |
| Query strain trk1Δtrk2Δ (YAK01) | MATα trk1Δ::URA3 trk2Δ::NAT his3Δ1 leu2Δ0 ura3Δ0 lyp1Δ0 lysΔ0 can1Δ:: STE2pr-HIS3 | (3) |
| trk1Δtrk2Δvps35Δ | MATα trk1Δ::URA3 trk2Δ::NAT vps35Δ::KAN his3Δ1 leu2Δ0 ura3Δ0 lyp1Δ0 lysΔ0 can1Δ::STE2pr-HIS3 | This study. This strain was derived from YAK01. VPS35 coding region was replaced by the KAN cassette as described in Ref. 91. |
| trk1Δtrk2Δcnb1Δ | MATα trk1Δ::HIS3 trk2Δ::HIS3 cnb1Δ::HPH his3Δ200 leu2Δ1 ura3–52 trp1–1 ade2 | This study. This strain was derived from the trk1Δtrk2Δ strain described in Refs. 16 and 89. The CNB1 coding region was replaced by the hygromycin resistance cassette as described in 90. |
| HRD1/DOA10 (WT) | MATα ade2 his3 leu2 ura3 trp1 | (3, 92) |
| pdr5Δ | MATα pdr5Δ::KAN his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 | (93) |
| BJ5459 | MATa his3Δ200 leu2Δ1 ura3–52 trp1 lys2–801 pep4Δ::HIS3 prb1Δ1.6R can1 GAL | (94) |
Plasmids and DNA manipulations
Plasmids used in this study and details of their construction are described in Table S1. PCR amplifications were performed using Phusion High Fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, MA), and all constructs generated were verified using Sanger sequencing (Genewiz, South Plainfield, NJ).
Yeast protein extraction and immunoblot analysis
Whole-cell extracts of yeast proteins were made by growing cells in low-potassium SC medium supplemented with the indicated amount of KCl to mid-exponential phase at 30 °C (A600 = 0.6–0.8) and harvesting equivalent numbers of cells by centrifugation. Cell pellets were flash-frozen in liquid nitrogen and stored at −80 °C until processing. Cells were then lysed using sodium hydroxide, and proteins were precipitated using trichloroacetic acid (TCA) as described (82). The protein precipitates were solubilized in SDS/urea sample buffer (22) and heated to 37 °C for 15 min. Extracts were resolved by SDS-PAGE, and proteins were identified by immunoblotting with mouse anti-HA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) to detect Kir2.1 and rabbit anti-Zwf1 (glucose-6-phosphate dehydrogenase, referred to as G6PDH) antibody (Sigma) as a protein loading control. Anti-Sec61 antibody (83) and anti-Pma1 (40B7 Abcam, Cambridge, MA) were also used where indicated. Anti-mouse or anti-rabbit secondary antibodies conjugated to IRDye 800 or IRDye 680 (Li-Cor Biosciences, Lincoln, NE) were detected using an OdysseyTM Fc IR imaging system (LI-COR Biosciences), or where indicated, horseradish peroxidase-conjugated anti-rabbit immunoglobulin secondary antibody was applied (Jackson ImmunoResearch, West Grove, PA) and was detected with the Supersignal chemiluminescent substrate (Pierce). In this case, images were processed on a Bio-Rad ChemiDoc XRS+ image station with Image Lab 5.2.1 software (Hercules, CA).
Protein stability assays
Yeast cells lacking PDR5 and transformed with Kir2.1-HA or FAP-tagged Kir2.1 (expressed under the constitutive TEF1 promoter) were grown overnight to saturation, inoculated at an A600 of 0.25 into low-potassium SC medium supplemented with 100 mm KCl, and allowed to grow to mid-log phase at 30 °C. To inhibit the proteasome, cultures were treated with 100 μm MG132 (Selleck Chemicals, Houston, TX) in DMSO or an equivalent amount of the DMSO vehicle as a control, and incubated with agitation for 30 min at 37 °C. At this point, 150 μg/ml cycloheximide (Sigma) was added to each culture to halt protein translation, and cells were further incubated with agitation at 37 °C. Total 1-ml aliquots were withdrawn at 0, 30, 60, and 90 min after the addition of cycloheximide, 17.5 mm NaN3 was added, and cells were harvested by centrifugation and flash-frozen in liquid nitrogen. Whole-cell extracts were prepared by the TCA precipitation method described above and analyzed via SDS-PAGE and immunoblotting. Immunoblotting used the same antibodies and chemiluminescent methodology as described above for horseradish peroxidase-conjugated antibodies.
Biochemical fractionation
The subcellular distribution of Kir2.1 was assessed by sucrose gradient sedimentation assays as described (84). In brief, 40 ml of the indicated yeast cultures were grown to an A600 of 0.8, harvested by centrifugation, and lysed by glass-bead agitation as described (3). Clarified cell lysates were layered onto a 30–70% sucrose gradient cushion and centrifuged at 100,000 × g in a Beckman SW41 rotor for 14 h at 4 °C. Equal fractions were then collected by pipetting from the top of the tube, and proteins were assessed by SDS-PAGE and immunoblotting. Signal intensity was quantified by obtaining densitometry measurements of bands within the linear range of detection using ImageJ software (National Institutes of Health, Bethesda, MD).
Biochemical purification and MS analyses
GST-fused Aly1 or Aly2 was expressed from pKK212-derived plasmids under the control of the CUP1 promoter in BJ5459 cells as described (22). In brief, cells were grown to mid-exponential phase, and expression was induced from the CUP1 promoter by the addition of 200 μm CuSO4 for 1 h. Protein extracts from these cells were generated by glass-bead lysis with vigorous agitation in co-immunoprecipitation buffer (50 mm Tris-HCl, pH 7.4, 15 mm EGTA, 100 mm NaCl, 0.2% Triton X-100, 5 mm N-ethylmaleimide, and phosphatase inhibitors). GST fusion proteins were purified from equal concentrations of clarified lysates by incubation with GSH-Sepharose beads (GE Healthcare). The harvested proteins were washed in co-immunoprecipitation buffer and eluted in Laemmli buffer (85). Proteins were then either resolved by SDS-PAGE and immunoblotted with α-ubiquitin and α-GST antibodies exactly as described in (22) to assess ubiquitination, or they were resolved by SDS-PAGE and stained with Coomassie Blue with the band corresponding to Aly1 or Aly2 excised for MS analyses. Proteins were excised from the gel and digested, and peptides were separated and analyzed by LC-MS/MS as described elsewhere (86). In brief, gel slices were destained with 50% acetonitrile and 25 mm ammonium bicarbonate before incubation with 10 mm DTT at 56 °C for 1 h and a 1-h incubation with 55 mm iodoacetamide to reduce and alkylate proteins. Trypsin was added to the gel pieces, and proteins were digested overnight at 37 °C. Proteolytic peptides were extracted with 70% acetonitrile and 5% formic acid, vacuum-dried, and reconstituted in 0.1% formic acid for LC-MS/MS analyses. Peptides were analyzed by nano LC-MS/MS with a Dionex HPLC system (Dionex Ultimate 3000, Thermo Fisher Scientific) interfaced with a linear ion trap MS (LTQ-XL, Thermo Fisher Scientific). Peptides were separated on a C18 column (PicoChip column, New Objective, Inc., Woburn, MA). The mass spectrometer was run in data-dependent MS/MS mode so that each full MS spectrum was followed by MS/MS scans of the eight most abundant molecular ions, and dynamic exclusion was enabled to reduce the selection of previously analyzed peptides for CID (collusion-induced dissociation). MS/MS spectra were searched against the Saccharomyces Genome Database using the MASCOT search engine (version 2.4.0, Matrix Science Ltd.). Oxidation of methionine and carboxyaminomethylation of cysteines were set as variable and static modifications, respectively. Identification of the results were filtered with Scaffold (Proteome Software, Portland, OR).
Inductively coupled plasma mass spectrometry
Total potassium levels in yeast cells was measured using ICP-MS. Samples for these analyses were prepared essentially as described (87, 88) with the following modifications. In brief, yeast cells containing the indicated plasmids were grown to saturation in low phosphate SC medium supplemented with 100 mm KCl. These cells were then washed in low phosphate SC medium with no KCl supplementation, inoculated at an A600 of 0.2 into low-phosphate SC medium supplemented with 10 mm KCl, and grown for 6–8 h, which is when the cultures reached an approximate A600 of 1.0. One A600 unit's worth of cells was then harvested by filtration on isopore membrane filters (Thermo Fisher) and washed twice in 1 μm EDTA and twice in double distilled water. The cells collected on the membrane filters were dissolved overnight in 1.5 ml of 30% nitric acid at 65 °C. Samples were then diluted 15-fold to a final concentration of 2% nitric acid, and an internal standard of beryllium, germanium, and thallium was added to all samples to evaluate signal strength and instrument drift. Sample metal concentrations were measured with a PerkinElmer NexION 300X ICP-MS, calibrated with a 5-point calibration curve. A blank sample consisting of 2% nitric acid was run after every 7–10 samples to ensure that there were no signal memory effects. At least three biological replicates were run for each sample to ensure a robust set of ICP-MS measurements.
FAP staining and confocal microscopy imaging
To assess Kir2.1-FAP localization, yeast cells containing the pRS415-TEF1pr-FAP-TM-Kir2.1 plasmid, where the FAP corresponded to the dL5 version of this protein motif (23, 38), were grown overnight to saturation in low-potassium SC medium supplemented with 100 mm KCl. The cells were then diluted to an A600 of 0.2 in low-potassium SC medium with 100 mm KCl and grown for 4–5 h at 30 °C until cells reached mid-exponential phase growth (A600 = 0.5–0.7). Where indicated, the cells were then treated with 200 μm LatA (Molecular Probes, Eugene, OR) or with an equal volume of DMSO vehicle for 2 h at 30 °C prior to imaging. To allow the FAP tag to fluoresce, yeast cells were next incubated with 1 μm MG-B-TAU (cell-impermeant dye) or MG-ESTER (cell-permeant dye) for 15 min at room temperature and imaged. To this end, cells were plated onto 35-mm glass-bottom microwell dishes coated with poly-d-lysine (MatTek Corp., Ashland, MA) to ensure that the yeast remained stationary. Images were acquired using a Nikon Eclipse Ti inverted microscope outfitted with a Prairie-swept field confocal scan head, an Agilent monolithic laser launch, and an Andor iXon3 camera. NIS-Elements software was used to control the imaging parameters, and all images within an experiment were captured using identical settings. Fluorescence of the FAP-bound MG-B-TAU and MG-ESTER dyes was excited using 636-nm light and the fluorescent emission was detected at 664 nm (23, 38), which was achieved using the 640-nm laser line. Maximum projections of 0.2 μm, Z-stacks through the entire cell were generated in ImageJ software (National Institutes of Health). All images within an experiment were adjusted equivalently, and an equal Unsharp Mask was applied to each image using Photoshop software (Adobe Systems Inc., San Jose, CA).
Image quantification and statistical analyses
The fluorescence intensity for all images was quantified using ImageJ software. To quantify total fluorescence with the Mg-B-TAU cell-impermeant dye, maximum projections of 0.2-μm Z-stacks through an entire cell were generated, and cells were outlined manually in ImageJ. Mean pixel intensities were measured in arbitrary units (a.u.) for each cell, and the mean background pixel intensity was subtracted. The distribution of mean pixel intensities in a.u. and the distribution of pixel intensities for each group of cells are presented as box-and-whisker plots, where the horizontal midline represents the median, the box is bounded by the upper and lower quartiles, and the whiskers denote maximal and minimal fluorescence intensities. A nonparametric Kruskal–Wallis test and Dunn's multiple-comparison post hoc analyses were performed using Prism software (GraphPad, La Jolla, CA). Statistically significant differences are indicated in the figures by asterisks with the associated p values provided in the figure legends.
Author contributions
N. A. H., C. J. K., T. D. M., A. R. K., M. P. B., D. J. B., A. F. O., and J. L. B. conceptualization; N. A. H., T. D. M., A. R. K., P. G. N., A. A. A., D. J. B., A. V. K., and A. F. O. data curation; N. A. H., C. J. K., T. D. M., A. R. K., P. G. N., A. A. A., A. D., C. S.-G., D. J. B., A. V. K., and A. F. O. formal analysis; N. A. H., M. P. B., D. J. B., A. F. O., and J. L. B. supervision; M. P. B., D. J. B., A. V. K., A. F. O., and J. L. B. funding acquisition; N. A. H., C. J. K., T. D. M., A. R. K., P. G. N., A. A. A., A. D., C. S.-G., M. P. B., and A. F. O. investigation; N. A. H. and A. F. O. visualization; N. A. H., C. J. K., T. D. M., A. R. K., P. G. N., A. A. A., A. D., C. S.-G., M. P. B., D. J. B., A. V. K., and A. F. O. methodology; N. A. H., C. J. K., T. D. M., P. G. N., and A. F. O. writing-original draft; A. F. O., and J. L. B. project administration; N. A. H., C. J. K., T. D. M., A. R. K., P. G. N., A. A. A., C. S.-G., M. P. B., D. J. B., A. V. K., A. F. O., and J. L. B. writing-review and editing.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL127711 (to A. V. K.), R01 GM075061 (to J. L. B. and A. F. O.), P30 DK079307 (to J. L. B.), and U54-GM103529 (to A. D., C. S.-G., and M. P. B). This work also was supported by National Science Foundation (NSF) MCB CAREER Grant 1553143 (to A. F. O.) and NSF EAR-IF 0948366 (to D. J. B.), a Pilot Grant from P30 DK079307 (to A. F. O.), and a grant from the Shurl and Kay Curci Foundation (to M. P. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2, Table S1, and additional references.
- Kir
- K+ inward rectifying
- ER
- endoplasmic reticulum
- ICP-MS
- inductively coupled plasma mass spectrometry
- FAP
- fluorogen-activating protein
- MVB
- multivesicular body
- ESCRT
- endosomal sorting complexes required for transport
- SCA
- single-chain antibody
- MG
- malachite green
- LatA
- latrunculin A
- a.u.
- arbitrary units
- TM
- transmembrane
- AzC
- azetidine-2-carboxylic acid
- GST
- glutathione S-transferase
- SC
- synthetic complete.
References
- 1. Brette F., and Orchard C. (2003) T-tubule function in mammalian cardiac myocytes. Circ. Res. 92, 1182–1192 10.1161/01.RES.0000074908.17214.FD [DOI] [PubMed] [Google Scholar]
- 2. Curran J., Makara M. A., Little S. C., Musa H., Liu B., Wu X., Polina I., Alecusan J. S., Wright P., Li J., Billman G. E., Boyden P. A., Gyorke S., Band H., Hund T. J., and Mohler P. J. (2014) EHD3-dependent endosome pathway regulates cardiac membrane excitability and physiology. Circ. Res. 115, 68–78 10.1161/CIRCRESAHA.115.304149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kolb A. R., Needham P. G., Rothenberg C., Guerriero C. J., Welling P. A., and Brodsky J. L. (2014) ESCRT regulates surface expression of the Kir2.1 potassium channel. Mol. Biol. Cell 25, 276–289 10.1091/mbc.e13-07-0394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Boer T. P., Houtman M. J., Compier M., and van der Heyden M. A. (2010) The mammalian K(IR)2.x inward rectifier ion channel family: Expression pattern and pathophysiology. Acta Physiol. (Oxf.) 199, 243–256 [DOI] [PubMed] [Google Scholar]
- 5. Nichols C. G., and Lopatin A. N. (1997) Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191 10.1146/annurev.physiol.59.1.171 [DOI] [PubMed] [Google Scholar]
- 6. Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., and Kurachi Y. (2010) Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 10.1152/physrev.00021.2009 [DOI] [PubMed] [Google Scholar]
- 7. Clarke O. B., Caputo A. T., Hill A. P., Vandenberg J. I., Smith B. J., and Gulbis J. M. (2010) Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell 141, 1018–1029 10.1016/j.cell.2010.05.003 [DOI] [PubMed] [Google Scholar]
- 8. Plaster N. M., Tawil R., Tristani-Firouzi M., Canún S., Bendahhou S., Tsunoda A., Donaldson M. R., Iannaccone S. T., Brunt E., Barohn R., Clark J., Deymeer F., George A. L. Jr., Fish F. A., Hahn A., et al. (2001) Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 105, 511–519 10.1016/S0092-8674(01)00342-7 [DOI] [PubMed] [Google Scholar]
- 9. Priori S. G., Pandit S. V., Rivolta I., Berenfeld O., Ronchetti E., Dhamoon A., Napolitano C., Anumonwo J., di Barletta M. R., Gudapakkam S., Bosi G., Stramba-Badiale M., and Jalife J. (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 96, 800–807 10.1161/01.RES.0000162101.76263.8c [DOI] [PubMed] [Google Scholar]
- 10. Andersen E. D., Krasilnikoff P. A., and Overvad H. (1971) Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies: A new syndrome? Acta Paediatr. Scand. 60, 559–564 10.1111/j.1651-2227.1971.tb06990.x [DOI] [PubMed] [Google Scholar]
- 11. Tristani-Firouzi M., and Etheridge S. P. (2010) Kir 2.1 channelopathies: The Andersen-Tawil syndrome. Pflugers Arch. 460, 289–294 10.1007/s00424-010-0820-6 [DOI] [PubMed] [Google Scholar]
- 12. Ai T., Fujiwara Y., Tsuji K., Otani H., Nakano S., Kubo Y., and Horie M. (2002) Novel KCNJ2 mutation in familial periodic paralysis with ventricular dysrhythmia. Circulation 105, 2592–2594 10.1161/01.CIR.0000019906.35135.A3 [DOI] [PubMed] [Google Scholar]
- 13. Ma D., Taneja T. K., Hagen B. M., Kim B. Y., Ortega B., Lederer W. J., and Welling P. A. (2011) Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell 145, 1102–1115 10.1016/j.cell.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stockklausner C., and Klocker N. (2003) Surface expression of inward rectifier potassium channels is controlled by selective Golgi export. J. Biol. Chem. 278, 17000–17005 10.1074/jbc.M212243200 [DOI] [PubMed] [Google Scholar]
- 15. Yoo D., Fang L., Mason A., Kim B. Y., and Welling P. A. (2005) A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J. Biol. Chem. 280, 35281–35289 10.1074/jbc.M504836200 [DOI] [PubMed] [Google Scholar]
- 16. Nakamura R. L., and Gaber R. F. (1998) Studying ion channels using yeast genetics. Methods Enzymol. 293, 89–104 10.1016/S0076-6879(98)93009-9 [DOI] [PubMed] [Google Scholar]
- 17. Alvaro C. G., O'Donnell A. F., Prosser D. C., Augustine A. A., Goldman A., Brodsky J. L., Cyert M. S., Wendland B., and Thorner J. (2014) Specific α-arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein-coupled receptor Ste2. Mol. Cell. Biol. 34, 2660–2681 10.1128/MCB.00230-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Becuwe M., Vieira N., Lara D., Gomes-Rezende J., Soares-Cunha C., Casal M., Haguenauer-Tsapis R., Vincent O., Paiva S., and Léon S. (2012) A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J. Cell Biol. 196, 247–259 10.1083/jcb.201109113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin C. H., MacGurn J. A., Chu T., Stefan C. J., and Emr S. D. (2008) Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 135, 714–725 10.1016/j.cell.2008.09.025 [DOI] [PubMed] [Google Scholar]
- 20. Nikko E., and Pelham H. R. (2009) Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic 10, 1856–1867 10.1111/j.1600-0854.2009.00990.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nikko E., Sullivan J. A., and Pelham H. R. (2008) Arrestin-like proteins mediate ubiquitination and endocytosis of the yeast metal transporter Smf1. EMBO Rep. 9, 1216–1221 10.1038/embor.2008.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Donnell A. F., Huang L., Thorner J., and Cyert M. S. (2013) A calcineurin-dependent switch controls the trafficking function of α-arrestin Aly1/Art6. J. Biol. Chem. 288, 24063–24080 10.1074/jbc.M113.478511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szent-Gyorgyi C., Stanfield R. L., Andreko S., Dempsey A., Ahmed M., Capek S., Waggoner A. S., Wilson I. A., and Bruchez M. P. (2013) Malachite green mediates homodimerization of antibody VL domains to form a fluorescent ternary complex with singular symmetric interfaces. J. Mol. Biol. 425, 4595–4613 10.1016/j.jmb.2013.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pratt C. P., He J., Wang Y., Barth A. L., and Bruchez M. P. (2015) Fluorogenic green-inside red-outside (GIRO) labeling approach reveals adenylyl cyclase-dependent control of BKα surface expression. Bioconjug. Chem. 26, 1963–1971 10.1021/acs.bioconjchem.5b00409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shiwarski D. J., Darr M., Telmer C. A., Bruchez M. P., and Puthenveedu M. A. (2017) PI3K class IIα regulates δ-opioid receptor export from the trans-Golgi network. Mol. Biol. Cell 28, 2202–2219 10.1091/mbc.e17-01-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang W., Ruknudin A., Yang W. P., Shaw S. Y., Knickerbocker A., and Kurtz S. (1995) Functional expression of a vertebrate inwardly rectifying K+ channel in yeast. Mol. Biol. Cell 6, 1231–1240 10.1091/mbc.6.9.1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Donnell B. M., Mackie T. D., Subramanya A. R., and Brodsky J. L. (2017) Endoplasmic reticulum-associated degradation of the renal potassium channel, ROMK, leads to type II Bartter syndrome. J. Biol. Chem. 292, 12813–12827 10.1074/jbc.M117.786376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tinker A., Jan Y. N., and Jan L. Y. (1996) Regions responsible for the assembly of inwardly rectifying potassium channels. Cell 87, 857–868 10.1016/S0092-8674(00)81993-5 [DOI] [PubMed] [Google Scholar]
- 29. Becuwe M., and Leon S. (2014) Integrated control of transporter endocytosis and recycling by the arrestin-related protein Rod1 and the ubiquitin ligase Rsp5. eLife 3, e03307 10.7554/eLife.03307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Donnell A. F., Apffel A., Gardner R. G., and Cyert M. S. (2010) α-Arrestins Aly1 and Aly2 regulate intracellular trafficking in response to nutrient signaling. Mol. Biol. Cell 21, 3552–3566 10.1091/mbc.e10-07-0636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Donnell A. F., McCartney R. R., Chandrashekarappa D. G., Zhang B. B., Thorner J., and Schmidt M. C. (2015) 2-Deoxyglucose impairs Saccharomyces cerevisiae growth by stimulating Snf1-regulated and α-arrestin-mediated trafficking of hexose transporters 1 and 3. Mol. Cell. Biol. 35, 939–955 10.1128/MCB.01183-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Risinger A. L., and Kaiser C. A. (2008) Different ubiquitin signals act at the Golgi and plasma membrane to direct GAP1 trafficking. Mol. Biol. Cell 19, 2962–2972 10.1091/mbc.e07-06-0627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alvarez C. E. (2008) On the origins of arrestin and rhodopsin. BMC Evol. Biol. 8, 222 10.1186/1471-2148-8-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Picones A., Keung E., and Timpe L. C. (2001) Unitary conductance variation in Kir2.1 and in cardiac inward rectifier potassium channels. Biophys. J. 81, 2035–2049 10.1016/S0006-3495(01)75853-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Craene J. O., Soetens O., and Andre B. (2001) The Npr1 kinase controls biosynthetic and endocytic sorting of the yeast Gap1 permease. J. Biol. Chem. 276, 43939–43948 10.1074/jbc.M102944200 [DOI] [PubMed] [Google Scholar]
- 36. Soetens O., De Craene J. O., and Andre B. (2001) Ubiquitin is required for sorting to the vacuole of the yeast general amino acid permease, Gap1. J. Biol. Chem. 276, 43949–43957 10.1074/jbc.M102945200 [DOI] [PubMed] [Google Scholar]
- 37. Szent-Gyorgyi C., Schmidt B. F., Creeger Y., Fisher G. W., Zakel K. L., Adler S., Fitzpatrick J. A., Woolford C. A., Yan Q., Vasilev K. V., Berget P. B., Bruchez M. P., Jarvik J. W., and Waggoner A. (2008) Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26, 235–240 10.1038/nbt1368 [DOI] [PubMed] [Google Scholar]
- 38. Yan Q., Schmidt B. F., Perkins L. A., Naganbabu M., Saurabh S., Andreko S. K., and Bruchez M. P. (2015) Near-instant surface-selective fluorogenic protein quantification using sulfonated triarylmethane dyes and fluorogen activating proteins. Org. Biomol. Chem. 13, 2078–2086 10.1039/C4OB02309A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Corsi A. K., and Schekman R. (1996) Mechanism of polypeptide translocation into the endoplasmic reticulum. J. Biol. Chem. 271, 30299–30302 10.1074/jbc.271.48.30299 [DOI] [PubMed] [Google Scholar]
- 40. Preuss D., Mulholland J., Kaiser C. A., Orlean P., Albright C., Rose M. D., Robbins P. W., and Botstein D. (1991) Structure of the yeast endoplasmic reticulum: Localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast 7, 891–911 10.1002/yea.320070902 [DOI] [PubMed] [Google Scholar]
- 41. Ayscough K. R., Stryker J., Pokala N., Sanders M., Crews P., and Drubin D. G. (1997) High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 137, 399–416 10.1083/jcb.137.2.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coué M., Brenner S. L., Spector I., and Korn E. D. (1987) Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213, 316–318 10.1016/0014-5793(87)81513-2 [DOI] [PubMed] [Google Scholar]
- 43. Kaksonen M., Sun Y., and Drubin D. G. (2003) A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115, 475–487 10.1016/S0092-8674(03)00883-3 [DOI] [PubMed] [Google Scholar]
- 44. Hatakeyama R., Kamiya M., Takahara T., and Maeda T. (2010) Endocytosis of the aspartic acid/glutamic acid transporter Dip5 is triggered by substrate-dependent recruitment of the Rsp5 ubiquitin ligase via the arrestin-like protein Aly2. Mol. Cell. Biol. 30, 5598–5607 10.1128/MCB.00464-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rotin D., Staub O., and Haguenauer-Tsapis R. (2000) Ubiquitination and endocytosis of plasma membrane proteins: Role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J. Membr. Biol. 176, 1–17 10.1007/s00232001079,10.1007/s002320001079 [DOI] [PubMed] [Google Scholar]
- 46. Prosser D. C., Pannunzio A. E., Brodsky J. L., Thorner J., Wendland B., and O'Donnell A. F. (2015) α-Arrestins participate in cargo selection for both clathrin-independent and clathrin-mediated endocytosis. J. Cell Sci. 128, 4220–4234 10.1242/jcs.175372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Herrador A., Léon S., Haguenauer-Tsapis R., and Vincent O. (2013) A mechanism for protein monoubiquitination dependent on a trans-acting ubiquitin-binding domain. J. Biol. Chem. 288, 16206–16211 10.1074/jbc.C113.452250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Udeshi N. D., Mani D. R., Eisenhaure T., Mertins P., Jaffe J. D., Clauser K. R., Hacohen N., and Carr S. A. (2012) Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell. Proteomics 11, 148–159 10.1074/mcp.M111.016857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., and Peng J. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 10.1016/j.cell.2009.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Starita L. M., Lo R. S., Eng J. K., von Haller P. D., and Fields S. (2012) Sites of ubiquitin attachment in Saccharomyces cerevisiae. Proteomics 12, 236–240 10.1002/pmic.201100166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swaney D. L., Beltrao P., Starita L., Guo A., Rush J., Fields S., Krogan N. J., and Villén J. (2013) Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 10, 676–682 10.1038/nmeth.2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ziv I., Matiuhin Y., Kirkpatrick D. S., Erpapazoglou Z., Leon S., Pantazopoulou M., Kim W., Gygi S. P., Haguenauer-Tsapis R., Reis N., Glickman M. H., and Kleifeld O. (2011) A perturbed ubiquitin landscape distinguishes between ubiquitin in trafficking and in proteolysis. Mol. Cell. Proteomics 10, M111.009753 10.1074/mcp.M111.009753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bennett-Lovsey R. M., Herbert A. D., Sternberg M. J., and Kelley L. A. (2008) Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70, 611–625 [DOI] [PubMed] [Google Scholar]
- 55. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 4, 363–371 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 56. Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., and Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 10.1074/mcp.M700468-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., and Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 10.1126/science.1172867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. MacGurn J. A., Hsu P. C., Smolka M. B., and Emr S. D. (2011) TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147, 1104–1117 10.1016/j.cell.2011.09.054 [DOI] [PubMed] [Google Scholar]
- 59. Crapeau M., Merhi A., and André B. (2014) Stress conditions promote yeast Gap1 permease ubiquitylation and down-regulation via the arrestin-like Bul and Aly proteins. J. Biol. Chem. 289, 22103–22116 10.1074/jbc.M114.582320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Merhi A., and André B. (2012) Internal amino acids promote Gap1 permease ubiquitylation via TORC1/Npr1/14–3-3-dependent control of the Bul arrestin-like adaptors. Mol. Cell. Biol. 32, 4510–4522 10.1128/MCB.00463-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cyert M. S., and Thorner J. (1992) Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol. Cell. Biol. 12, 3460–3469 10.1128/MCB.12.8.3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shin B. K., Wang H., Yim A. M., Le Naour F., Brichory F., Jang J. H., Zhao R., Puravs E., Tra J., Michael C. W., Misek D. E., and Hanash S. M. (2003) Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 278, 7607–7616 10.1074/jbc.M210455200 [DOI] [PubMed] [Google Scholar]
- 63. Prosser D. C., Wrasman K., Woodard T. K., O'Donnell A. F., and Wendland B. (2016) Applications of pHluorin for quantitative, kinetic and high-throughput analysis of endocytosis in budding yeast. J. Vis. Exp. 10.3791/54587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tsien R. Y. (1998) The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 10.1146/annurev.biochem.67.1.509 [DOI] [PubMed] [Google Scholar]
- 65. Mateus C., and Avery S. V. (2000) Destabilized green fluorescent protein for monitoring dynamic changes in yeast gene expression with flow cytometry. Yeast 16, 1313–1323 10.1002/1097-0061(200010)16:14%3C1313::AID-YEA626%3E3.0.CO%3B2-O [DOI] [PubMed] [Google Scholar]
- 66. Boeckstaens M., Llinares E., Van Vooren P., and Marini A. M. (2014) The TORC1 effector kinase Npr1 fine tunes the inherent activity of the Mep2 ammonium transport protein. Nat. Commun. 5, 3101 10.1038/ncomms4101 [DOI] [PubMed] [Google Scholar]
- 67. O'Donnell A. F. (2012) The running of the Buls: control of permease trafficking by α-arrestins Bul1 and Bul2. Mol. Cell. Biol. 32, 4506–4509 10.1128/MCB.01176-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Foote C., and Nothwehr S. F. (2006) The clathrin adaptor complex 1 directly binds to a sorting signal in Ste13p to reduce the rate of its trafficking to the late endosome of yeast. J. Cell Biol. 173, 615–626 10.1083/jcb.200510161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Valdivia R. H., Baggott D., Chuang J. S., and Schekman R. W. (2002) The yeast clathrin adaptor protein complex 1 is required for the efficient retention of a subset of late Golgi membrane proteins. Dev. Cell 2, 283–294 10.1016/S1534-5807(02)00127-2 [DOI] [PubMed] [Google Scholar]
- 70. Valdivia R. H., and Schekman R. (2003) The yeasts Rho1p and Pkc1p regulate the transport of chitin synthase III (Chs3p) from internal stores to the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 100, 10287–10292 10.1073/pnas.1834246100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen H., Kubo Y., Hoshi T., and Heinemann S. H. (1998) Cyclosporin A selectively reduces the functional expression of Kir2.1 potassium channels in Xenopus oocytes. FEBS Lett. 422, 307–310 10.1016/S0014-5793(98)00028-3 [DOI] [PubMed] [Google Scholar]
- 72. Dart C., and Leyland M. L. (2001) Targeting of an A kinase-anchoring protein, AKAP79, to an inwardly rectifying potassium channel, Kir2.1. J. Biol. Chem. 276, 20499–20505 10.1074/jbc.M101425200 [DOI] [PubMed] [Google Scholar]
- 73. Konig S., Béguet A., Bader C. R., and Bernheim L. (2006) The calcineurin pathway links hyperpolarization (Kir2.1)-induced Ca2+ signals to human myoblast differentiation and fusion. Development 133, 3107–3114 10.1242/dev.02479 [DOI] [PubMed] [Google Scholar]
- 74. Helliwell S. B., Losko S., and Kaiser C. A. (2001) Components of a ubiquitin ligase complex specify polyubiquitination and intracellular trafficking of the general amino acid permease. J. Cell Biol. 153, 649–662 10.1083/jcb.153.4.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aubry L., Guetta D., and Klein G. (2009) The arrestin fold: Variations on a theme. Curr. Genomics 10, 133–142 10.2174/138920209787847014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hettema E. H., Valdez-Taubas J., and Pelham H. R. (2004) Bsd2 binds the ubiquitin ligase Rsp5 and mediates the ubiquitination of transmembrane proteins. EMBO J. 23, 1279–1288 10.1038/sj.emboj.7600137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stimpson H. E., Lewis M. J., and Pelham H. R. (2006) Transferrin receptor-like proteins control the degradation of a yeast metal transporter. EMBO J. 25, 662–672 10.1038/sj.emboj.7600984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gupta R., Kus B., Fladd C., Wasmuth J., Tonikian R., Sidhu S., Krogan N. J., Parkinson J., and Rotin D. (2007) Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol. Syst. Biol. 3, 116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ho H. C., MacGurn J. A., and Emr S. D. (2017) Deubiquitinating enzymes Ubp2 and Ubp15 regulate endocytosis by limiting ubiquitination and degradation of ARTs. Mol. Biol. Cell 28, 1271–1283 10.1091/mbc.e17-01-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Herrador A., Herranz S., Lara D., and Vincent O. (2010) Recruitment of the ESCRT machinery to a putative seven-transmembrane-domain receptor is mediated by an arrestin-related protein. Mol. Cell. Biol. 30, 897–907 10.1128/MCB.00132-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Johnston G. C., Pringle J. R., and Hartwell L. H. (1977) Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp. Cell Res. 105, 79–98 10.1016/0014-4827(77)90154-9 [DOI] [PubMed] [Google Scholar]
- 82. Volland C., Galan J. M., Urban-Grimal D., Devilliers G., and Haguenauer-Tsapis R. (1994) Endocytose and degradation of the uracil permease of S. cerevisiae under stress conditions: Possible role of ubiquitin. Folia Microbiol. (Praha) 39, 554–557 [DOI] [PubMed] [Google Scholar]
- 83. Brodsky J. L., Hamamoto S., Feldheim D., and Schekman R. (1993) Reconstitution of protein translocation from solubilized yeast membranes reveals topologically distinct roles for BiP and cytosolic Hsc70. J. Cell Biol. 120, 95–102 10.1083/jcb.120.1.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sullivan M. L., Youker R. T., Watkins S. C., and Brodsky J. L. (2003) Localization of the BiP molecular chaperone with respect to endoplasmic reticulum foci containing the cystic fibrosis transmembrane conductance regulator in yeast. J. Histochem. Cytochem. 51, 545–548 10.1177/002215540305100417 [DOI] [PubMed] [Google Scholar]
- 85. Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Qureshi-Emili A., Li Y., Godwin B., Conover D., Kalbfleisch T., et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627 10.1038/35001009 [DOI] [PubMed] [Google Scholar]
- 86. Tan R., Nakajima S., Wang Q., Sun H., Xue J., Wu J., Hellwig S., Zeng X., Yates N. A., Smithgall T. E., Lei M., Jiang Y., Levine A. S., Su B., and Lan L. (2017) Nek7 protects telomeres from oxidative DNA damage by phosphorylation and stabilization of TRF1. Mol. Cell 65, 818–831 e815 10.1016/j.molcel.2017.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Eide D. J., Clark S., Nair T. M., Gehl M., Gribskov M., Guerinot M. L., and Harper J. F. (2005) Characterization of the yeast ionome: a genome-wide analysis of nutrient mineral and trace element homeostasis in Saccharomyces cerevisiae. Genome Biol. 6, R77 10.1186/gb-2005-6-9-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Minear S., O'Donnell A. F., Ballew A., Giaever G., Nislow C., Stearns T., and Cyert M. S. (2011) Curcumin inhibits growth of Saccharomyces cerevisiae through iron chelation. Eukaryot. Cell 10, 1574–1581 10.1128/EC.05163-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 10.1002/(SICI)1097-0061(19980130)14:2%3C115::AID-YEA204%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]
- 90. Ko C. H., and Gaber R. F. (1991) TRK1 and TRK2 encode structurally related K+ transporters in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 4266–4273 10.1128/MCB.11.8.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Longtine M. S., McKenzie A. 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 10.1002/(SICI)1097-0061(199807)14:10%3C953::AID-YEA293%3E3.0.CO%3B2-U [DOI] [PubMed] [Google Scholar]
- 92. Pagant S., Kung L., Dorrington M., Lee M. C., and Miller E. A. (2007) Inhibiting endoplasmic reticulum (ER)-associated degradation of misfolded Yor1p does not permit ER export despite the presence of a diacidic sorting signal. Mol. Biol. Cell 18, 3398–3413 10.1091/mbc.e07-01-0046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., Chu A. M., Connelly C., Davis K., Dietrich F., Dow S. W., et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- 94. Jones E. W. (1991) Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 194, 428–453 10.1016/0076-6879(91)94034-A [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.