

Abstract
Pro-Pro endopeptidases (PPEPs) belong to a recently discovered family of proteases capable of hydrolyzing a Pro–Pro bond. The first member from the bacterial pathogen Clostridium difficile (PPEP-1) cleaves two C. difficile cell-surface proteins involved in adhesion, one of which is encoded by the gene adjacent to the ppep-1 gene. However, related PPEPs may exist in other bacteria and may shed light on substrate specificity in this enzyme family. Here, we report on the homolog of PPEP-1 in Paenibacillus alvei, which we denoted PPEP-2. We found that PPEP-2 is a secreted metalloprotease, which likewise cleaved a cell-surface protein encoded by an adjacent gene. However, the cleavage motif of PPEP-2, PLP↓PVP, is distinct from that of PPEP-1 (VNP↓PVP). As a result, an optimal substrate peptide for PPEP-2 was not cleaved by PPEP-1 and vice versa. To gain insight into the specificity mechanism of PPEP-2, we determined its crystal structure at 1.75 Å resolution and further confirmed the structure in solution using small-angle X-ray scattering (SAXS). We show that a four-amino-acid loop, which is distinct in PPEP-1 and -2 (GGST in PPEP-1 and SERV in PPEP-2), plays a crucial role in substrate specificity. A PPEP-2 variant, in which the four loop residues had been swapped for those from PPEP-1, displayed a shift in substrate specificity toward PPEP-1 substrates. Our results provide detailed insights into the PPEP-2 structure and the structural determinants of substrate specificity in this new family of PPEP proteases.
Keywords: metalloprotease, bacterial adhesion, cell wall, structural biology, substrate specificity, virulence factor, Paenibacillus alvei, PPEP, Pro-Pro endopeptidase
Introduction
Proteolytic processing by proteases is an important post-translational modification involved in the regulation of the activity, stability, localization, and function of target proteins (1, 2). Proteases can be promiscuous, having activity toward many protein substrates, e.g. proteases involved in protein catabolism such as those found in the digestive tract (3). In contrast, other proteases demonstrate high specificity, often related to the tight control of a cellular or physiological process (4). Because of their unique activity, proteases have a wide range of applications in biotechnology and the food industry (5–7).
The susceptibility of a protein substrate toward proteolytic cleavage is determined by the primary, secondary, and tertiary structures. In general, protein stretches enriched in prolines are highly resistant to endo-proteolytic cleavage (8–10), due to the cyclic structure of this amino acid. As a result, the N-terminal amine of proline lacks a hydrogen, and the side chains are typically in the cis conformation. These properties impair the susceptibility of Xaa-Pro bonds to proteolytic cleavage. For example, the widely used protease in proteomics, trypsin, cleaves after arginine and lysine residues except when they are followed by a proline (11).
Proteases with specificity for prolines can have therapeutic applications. For example, it has been demonstrated that peptides involved in gluten intolerance contain a high percentage of proline residues. Hence, in recent years a proline-specific endopeptidase effective in degrading immunogenic gluten epitopes has been developed as a possible therapy for celiac disease patients (12–15). These enzymes are specific for cleavage after proline residues (Pro-Xaa bonds, Xaa≠Pro).
In prokaryotes, proteases play an important role, and secreted proteases from pathogenic bacteria have been shown to be important for virulence by directly being toxic to cells (16) or by modulating important processes related to adhesion and immune evasion (17, 18). We have recently characterized a novel secreted protease in Clostridium difficile, Pro-Pro endopeptidase (PPEP3-1, 3.4.24.89, previously known as Zmp1 (19) (MER0494994)), which has a remarkable preference for hydrolyzing a Pro–Pro bond (20, 21). PPEP-1 cleaves two cell C. difficile surface adhesion proteins (CD2831 and CD3246) and as such is thought to play a role in switching from an adhesive to a motile phenotype (22). The two substrates of PPEP-1 contain multiple consecutive cleavage sites (in total 13), and a consensus motif analysis showed that in addition to the prolines surrounding the scissile bond, highly conserved residues are found, and cleavage preferentially occurs at sites that are composed of (VLI)NPPVP (P3–P3′). The co-crystal structure of a catalytically inactive mutant of PPEP-1 with a substrate peptide has provided some insights in the unusual specificity of PPEP-1 (23), demonstrating that the proline-rich peptide is arranged in a double-kink conformation supported by a unique aromatic–aliphatic side–chain stack. Part of the specificity appears to be determined by the kinked shape rather than substrate side–chain hydrogen bonds.
In this study, we demonstrate that PPEP-1 was in fact only one representative of a novel, broader group of proteases with similar specificities and functions. Interestingly, the closest homologs of PPEP-1 were not identified in other Clostridium species but in Paenibacillus. Bacteria belonging to this genus have been detected in a variety of heterogeneous environments, such as soil, water, rhizosphere, vegetable matter, forage, insect larvae, and clinical samples (24). One of the PPEP-1 homologs, which we named here PPEP-2, is found in Paenibacillus alvei, a bacterium that has been identified as a secondary invader associated with European foulbrood (24). Here, we demonstrate that this is a genuine Pro-Pro endopeptidase but with a very different specificity for the P2 and P3 position relative to the cleavage site compared with PPEP-1. We characterized the PPEP-2 structure in crystallo and in solution. The obtained atomic resolution data shed light on the unique specificity of PPEP-2 compared with the recently reported PPEP-1. Similar to the situation in C. difficile, the PPEP-2 cleavage sites were found in a cell-surface protein, with putative extracellular matrix–binding domains, encoded by the adjacent gene. This suggests a similar role of PPEP-2 in controlling bacterial adhesion.
Results
PPEP-1 homologs are found in Paenibacillus species
To find close homologs of C. difficile PPEP-1 (UniProt ID, Q183R7; NCBI RefSeq, YP_001089343), we performed a database search using BLASTp and TBLASTn. Surprisingly, the top hits (excluding PPEP-1 from C. difficile strains) all belong to the genus Paenibacillus and not to other Clostridium species. Twenty one genome sequences from Paenibacillus species with a PPEP-1 homolog were found in the NCBI database (Table S1), and a multiple sequence alignment showed an amino acid sequence identity of the Paenibacillus PPEPs with C. difficile PPEP-1 ranging from 43 to 49% (Fig. S1). All PPEP-1 homologs in Paenibacillus contain a predicted secretion signal sequence and a HEXXH motif, characteristic of zinc metalloproteases (2). We randomly selected the PPEP-1 homolog from the P. alvei strain DSM 29 (UniProt ID, K4ZRC1), from here on named PPEP-2, to study in more detail (Fig. 1). Analysis of the primary sequence of PPEP-2 showed 47% identity with PPEP-1. Moreover, most of the amino acid residues involved in the PPEP-1–substrate interaction (23) are conserved in PPEP-2 (arrowheads in Fig. 1, upper panel).
Figure 1.

Genomic organization of PPEP-2 and its substrate (VMSP) in P. alvei DSM 29. A homolog of C. difficile PPEP-1 was identified in the genome of P. alvei (PPEP-2). Primary sequence alignment of PPEP-2 and PPEP-1 showed an overall sequence identity of 47% (upper part, N-terminal signal secretion sequences were removed for simplicity; numbering was according to PPEP-2). The predicted secondary structure is shown at the top of the alignment. α, α-helix; β, β-sheet; T, β-turns/coils; η, 310-helices). Next to the gene encoding PPEP-2, a gene was identified encoding a protein with a predicted von Willebrand factor A (VWFA) domain, several Muc-binding protein (MucBP) domains, and three surface layer homology (SLH) domains. We call this protein VMSP, according to these predicted domains (VWFA, Mucbp, Surface-layer homology Protein). Between the last MucBP domain and the first SLH domain, two PPEP-2 cleavage sites are found (PLPPVP). Arrowheads refer to PPEP-1 contacts (23).
The gene encoding PPEP-1 in C. difficile (ppep-1/cd2830) is found adjacent to cd2831, the gene that encodes its substrate (21, 22). In CD2831, multiple PPEP-1 cleavage sites are present, located just above the site of attachment to the peptidoglycan layer. Interestingly, in the genomes of all PPEP-2 encoding the Paenibacillus species, an adjacent ORF is found encoding a homolog of a putative S-layer protein (Table S1 and Fig. 1) with 1–3 times the PLPPVP (or highly similar) sequence (Fig. S2A). This suggests that PLPPVP is the PPEP cleavage site in these proteins.
All putative PPEP substrate proteins in Paenibacillus contain a signal sequence and vary in length between 1091 amino acids (Paenibacillus popilliae ATCC14706) and 1739 amino acids (Paenibacillus phoceensis). We performed a conserved domain analysis using the InterPro Domain architecture tool (25) of the putative PPEP substrate proteins in Paenibacillus, and we observed that these have a clear modular structure (examples given in Fig. S2B). They contain a predicted von Willebrand factor A domain (VWFA, IPR002035/cd00198), several Muc-binding protein domains (MucBP, IPR009459/pfam06458), and three surface-layer-homology domains (SLH, IPR001119/pfam00395). We therefore named these large surface proteins with a tripartite organization, VMSP, according to these predicted domains (VWFA, Mucbp, Surface-layer homology Protein). The two putative PPEP-2 cleavage sites, PLPPVP, in the VMSP of P. alvei DSM 29 are located between the last mucin binding domain and the first SLH domain (Fig. 1).
PPEP-2 is a secreted Pro-Pro endopeptidase cleaving between two prolines in a PLPPVP motif
To test whether endogenous PPEP-2 is secreted, we first analyzed conditioned culture medium of growing P. alvei cells by SDS-PAGE, followed by LC-MS/MS analysis of a tryptic digest from proteins migrating at the expected molecular weight of PPEP-2 (Fig. S3, upper panel). Among other proteins, PPEP-2 was indeed identified by multiple tryptic peptides, and an overall sequence coverage of 54% was found. Importantly, relaxing the search to semi-tryptic specificity, N-terminal peptides of endogenous PPEP-2 were identified (e.g. 28QEQSILDKLVVLPSGEYNHSEAAAMK53, Fig. S3, lower panel), matching the predicted mature protein following removal of the signal peptide.
To analyze the activity and specificity of PPEP-2, we produced recombinant PPEP-2 and tested the activity against the synthetic peptide YPSSKPLPPVPPVQPLPPVPKLETS from the P. alvei DSM 29 VMSP (residues 1098–1122, Fig. 1). Following incubation with PPEP-2, three product peptides were found, corresponding to the predicted cleavages between the adjacent prolines within the PLPPVP motif (Fig. 2A). Importantly, no cleavage was observed between the two prolines within the PVPPVQ motif, demonstrating the specificity of PPEP-2 for amino acids surrounding the scissile bond. To substantiate these findings, we also tested PPEP-2 against a peptide library that was previously used to define the specificity of PPEP-1. This confirmed the preference of PPEP-2 for a Pro at the P3 and Leu at the P2 position (Fig. S4).
Figure 2.

PPEP-2 is a Pro-Pro endopeptidase with a high specificity for the P2 and P3 positions of the cleavage site compared with PPEP-1. A, MALDI-ToF–MS spectrum of the cleavage products formed after a 1-h incubation of a synthetic VMSP-derived peptide (YPSSKPLPPVPPVQPLPPVPKLETS, amino acids 1098–1122) containing two PPEP-2–specific cleavage sites, with PPEP-2. B, comparison of substrate specificities of PPEP-2 (left panel) and PPEP-1 (right panel). The progress curves show the increase of fluorescence during a 1-h incubation upon the protease-mediated cleavages (50 μm FRET substrate peptide and 200 ng of enzyme). Black, cleavage of a FRET peptide containing PLPPVP; red, cleavage of a FRET peptide containing VNPPVP.
Next, the real-time kinetics of the reaction of PPEP-2 toward a FRET peptide containing the PLPPVP motif were determined, which showed similar kinetics as for the reaction of PPEP-1 with its optimal substrate peptide VNPPVP (Fig. 2B). We also tested the PLPPVP peptide with PPEP-1 and the VNPPVP peptide with PPEP-2, but in both cases poor cleavage was observed (Fig. 2B). Hence, PPEP-1 and PPEP-2 show a clear difference in the specificity for the P2 and P3 positions.
To demonstrate the presence of active PPEP-2 in the cell culture medium of growing P. alvei cells, we incubated a peptide containing the optimal PLPPVP motif with conditioned medium of a P. alvei DSM 29 culture. MALDI-ToF MS analysis demonstrated that this peptide was cleaved between the two prolines (Fig. S5A). To evaluate the presence of the PPEP-2 substrate VMSP in the culture medium, we also analyzed the high-molecular weight proteins from P. alvei conditioned medium (Fig. S3) by LC-MS/MS. Indeed, among other proteins, three tryptic peptides from VMSP were found (Fig. S5B, in red).
Overall, the above data show that P. alvei PPEP-2 is a genuine, secreted Pro-Pro endopeptidase with a different specificity for the P2 and P3 positions of the cleavage site compared with PPEP-1.
Atomic structure of PPEP-2
To obtain a more detailed view of the enzyme, we crystallized P. alvei PPEP-2 and solved the crystal structure at a resolution of 1.75 Å. Data collection and refinement statistics are presented in Table 1. One asymmetric unit (ASU) of PPEP-2 contains four protein chains. Of those, chains A (Fig. 3) and B are almost identical (r.m.s.d. Cα 0.3 Å). The overall fold of PPEP-2 is very similar to that of PPEP-1 (r.m.s.d. Cα 1.28 Å); therefore, we kept the domain annotations (Fig. 3) like those used previously (23). As the crystallization condition contained a 20-fold excess of cadmium over zinc, substitution of the active-site ion could be expected. Careful examination of the calculated electron density maps and occupancy refinement of equivalently placed ions suggests an approximate 50:50 occupancy of the two ions for all chains in the ASU. Comparison with the previously published PPEP-1 holoenzyme structure suggests that this substitution has no effect on the position of the coordinating residues. Therefore, for the remainder of the discussion, we present this as only a zinc for the sake of clarity.
Table 1.
Crystallographic data collection and refinement statistics
SAD is single-wavelength anomalous dispersion; CC is correlation coefficient. Data-merging statistics were output by the program Phenix (56). Data for the highest resolution shell are shown in parentheses.
| PPEP-2–SAD | PPEP-2–HiRes | |
|---|---|---|
| Data reduction | ||
| Space group | P 43 | P 43 |
| a, b, c (Å) | 85.1 85.1 113.5 | 84.9, 84.9, 113.3 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.8 | 0.9786 |
| Resolution range (Å) | 34.57–2.57 (2.7–2.57) | 36–1.75 (1.8–1.75) |
| Total reflections | 172,173 (10190) | 603,119 (64260) |
| Unique reflections | 25,492 (2560) | 80,685 (8079) |
| Multiplicity | 6.8 (4.0) | 7.5 (7.9) |
| Completeness (%) | 98.84 (99.65) | 99.8 (99.8) |
| Mean I/σ(I) | 18.17 (2.62) | 14.9 (2.0) |
| Wilson B-factor (Å2) | 52.5 | 27.5 |
| Rmerge | 0.077 (0.45) | 0.080 (0.949) |
| Rmeas | 0.083 (0.52) | 0.086 (1.02) |
| Rpim | 0.031 (0.25) | 0.031 (0.362) |
| CC½ | 0.998 (0.827) | 0.998 (0.633) |
| Refinement statistics | ||
| Reflections used in refinement | 80,563 (8079) | |
| Reflections used for Rfree | 2130 (206) | |
| Rwork | 0.180 (0.270) | |
| Rfree | 0.209 (0.312) | |
| CCwork | 0.953 (0.743) | |
| CCfree | 0.955 (0.714) | |
| No. of non-hydrogen atoms | 6313 | |
| Protein | 5627 | |
| Ligands | 84 | |
| Solvent | 602 | |
| Protein residues | 728 | |
| r.m.s.d. bonds (Å) | 0.014 | |
| r.m.s.d. angles (°) | 1.58 | |
| Ramachandran favored (%) | 98.19 | |
| Ramachandran allowed (%) | 1.81 | |
| Ramachandran outliers (%) | 0.00 | |
| Rotamer outliers (%) | 0.51 | |
| Molprobity Clashscore | 0.09 | |
| Average B-factor (Å) | ||
| Overall | 33.59 | |
| Protein | 32.21 | |
| Ligands | 64.78 | |
| Solvent | 42.13 | |
Figure 3.
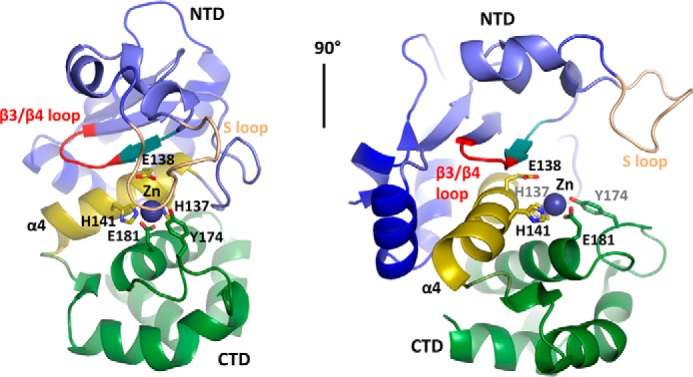
Atomic structure of PPEP-2. Atomic structure of chain A is in cartoon representation. Definition of the domains is the same as for PPEP-1: N-terminal domain (NTD), blue; active site (helix α4), yellow; C-terminal domain (CTD), green. Zinc-coordinating residues and residues involved in catalysis are shown as sticks. Zinc ion is shown as a sphere. For more details on the crystal unit, see Table 1 and the supporting information.
The α4-helix separates the α/β N-terminal (NTD) and all-α C-terminal (CTD) domains that represent two lobes forming the zinc-containing active site along the α4-helix. The long and flexible substrate loop (S-loop) overhangs from the NTD and covers the active site. Within the latter, the single zinc atom is coordinated by Glu-181 of α6, Tyr-174 of the α5/α6 loop, and His-137 and His-141 of the conserved metalloprotease HEXXH motif in α4, in which Glu-138 provides the catalytic base (26). Overall, the NTD is composed of a twisted four-stranded β-sheet and three α-helices (α1–α3) backing the β-sheet. As reported earlier, the edge strand β3 and the S-loop are involved in substrate recognition and binding (23). In this study, we have found that the S-loop might have a greater flexibility than previously anticipated, adopting at least several distinctly different conformations (Fig. S6). The CTD is mainly formed by four α-helices (α5–α8) and provides Tyr-174 to the catalytic site, which was proposed to act as electrophile during the catalysis (27, 28).
The other two copies in the ASU (chains C and D), although similar to each other, differ from chains A and B by a large conformational rearrangement of the S-loop, which now has a much more open conformation (Fig. S6). The flexible nature of this loop is also reflected in its partial disordering in chain D, where residues 96–112 are missing from the electron density map.
Contacts between individual PPEP-2 molecules within the crystal are rather extensive. Correspondingly, PISA analysis (29) of the crystal packing suggested a dimer or a tetramer as possible oligomeric states of the protein. To gain further insight into the state of PPEP-2 in solution and to compare it with that of PPEP-1, we performed small-angle X-ray scattering (SAXS) analysis. Given the self-association propensity of PPEP-2 observed in the course of purification, we carried out the SAXS experiments with size-exclusion chromatography separation (SEC-SAXS) to ensure that possible aggregation does not interfere with the SAXS analysis. We found that at a loading concentration as high as 25 mg/ml, PPEP-2 is predominantly presented by monomers with a mass of 20.5 kDa (Table S2), in good agreement with its calculated mass (21.1 kDa). Similar behavior was observed for PPEP-1 (Table S2; calculated mass monomer 21.6 kDa), which was also presented by monomers (19.4 kDa). Despite the overall similarity of the SAXS curves (Fig. 4A), PPEP-1 and PPEP-2 differed by small changes in the regions 0.18–0.26 and 0.35–0.45 Å−1. The best fit for the PPEP-2 crystal structure could be obtained for chain B (χ2 = 1.04; Fig. 4B), indicating that the crystal structure of the monomer fully describes its structure in solution. In addition, a very good match was found between the PPEP-2 crystal structure (chain B) and the ab initio molecular shape calculated by DAMMIN (29) directly from the SAXS curve (Fig. 4C, NSD = 1.45).
Figure 4.
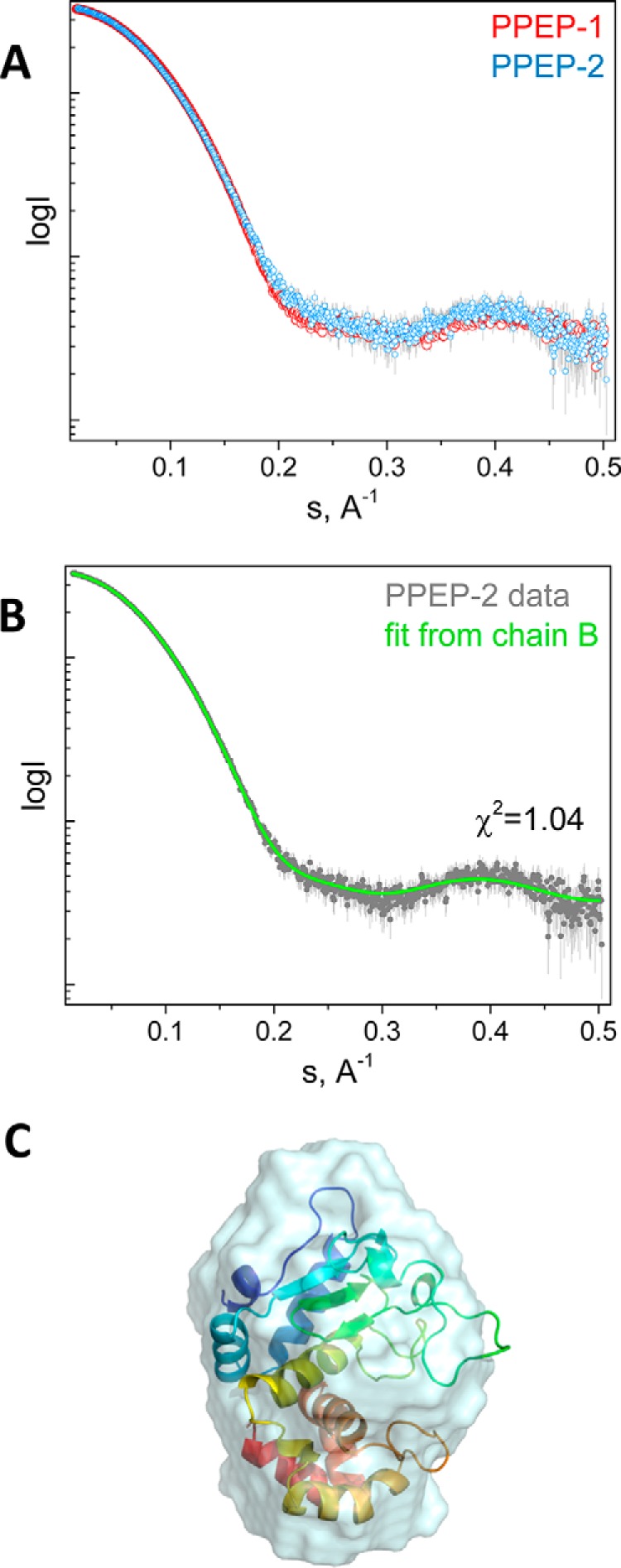
Analysis of PPEP-1 and PPEP-2 using SAXS. A, overlay of the SAXS profiles obtained for PPEP-1 and PPEP-2. B, fit from the best-fitting chain B of the PPEP-2 crystal structure to the PPEP-2 experimental curve. C, superposition of the PPEP-2 crystal structure chain B and the ab initio envelope obtained using DAMMIN (NSD = 1.45) (29).
Structural comparison of PPEP-1 and PPEP-2 provides insights into their specificity
In an attempt to explain the different specificity of PPEP-1 and PPEP-2, we have superposed our crystal structure of PPEP-2 with the structure of PPEP-1 in complex with its substrate (Fig. 5A) (PDB entry 5A0X (23)). We focused on the N-terminal half of the substrate recognition site, as this is where the optimal substrates for the two enzymes differ. This N-terminal half of the substrate motif aligns with the β3 strand and the immediately following β3/β4 loop. To this end, substrate binding to PPEP-1 involves a formation of two main-chain–main-chain H-bonds to residue Gly-117 as well as two H-bonds to residue Ser-119 (to the main-chain amine and the side-chain OH group, respectively) (Fig. 5A).
Figure 5.
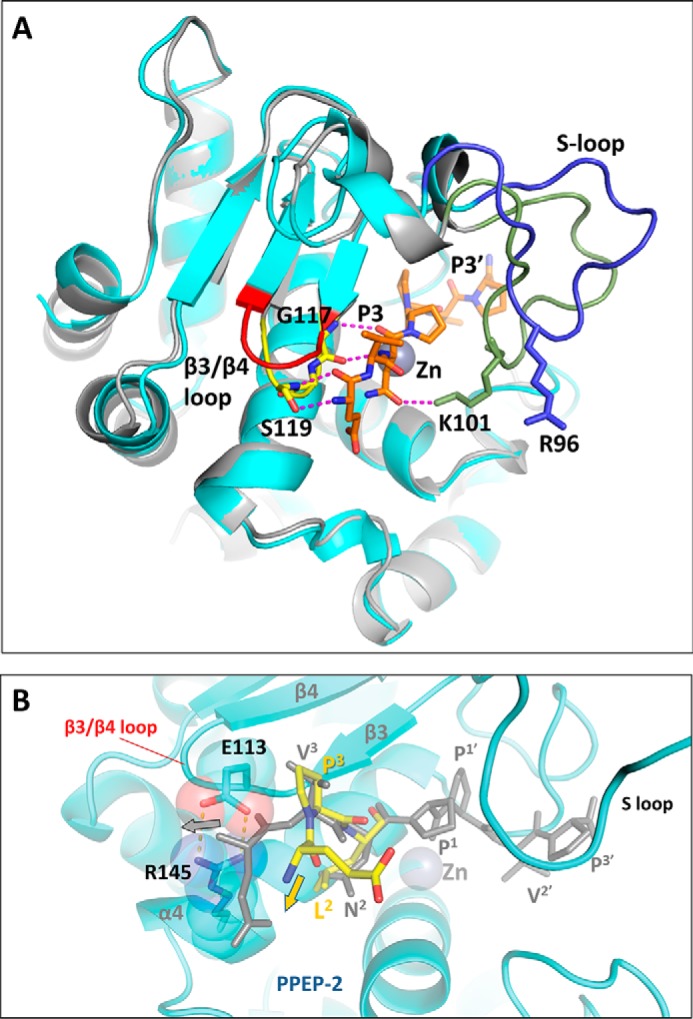
Structural comparison of substrate recognition in PPEP-1 and PPEP-2. A, superposition of the substrate-bound PPEP-1 structure (PDB code 5A0X, gray) with PPEP-2 (this work, chain B, cyan). The substrate peptide bound to PPEP-1 is shown in orange. The β3/β4 loop is shown in yellow for PPEP-1 and in red for PPEP-2. The S-loop is shown in green for PPEP-1 and in blue for PPEP-2. Hydrogen bonds between the peptide and PPEP-1 are shown as magenta dashed lines. B, close-up of the substrate peptide bound to PPEP-2 (yellow, model). For clarity, only the nonprime moiety of the modeled peptide is shown. A proline at the P3 position, as present in the optimal PPEP-2 substrate peptide, produces an additional kink in the polypeptide. As a result, the upstream polypeptide chain deviates away from the salt bridge formed by residues Glu-113 and Arg-145. For comparison, the substrate peptide bound to PPEP-1 (gray, crystal structure), with a Val at the P3 position, is overlaid. With PPEP-2, this conformation results in a sterical clash with the salt bridge.
Importantly, within a 4 Å distance to the N-terminal half of the bound substrate, the structural differences between PPEP-1 and PPEP-2 are essentially localized to the β3/β4 loop. The sequence of this loop is completely different in PPEP-1 (117GGST120) compared with PPEP-2 (112SERV115). In the latter, this loop moves somewhat upwards and away from the α4-helix (Fig. 5A). Importantly, the loop in PPEP-2 is stabilized by a salt bridge between residues Glu-113 and Arg-145 located on the α4-helix (Fig. 5B). This salt bridge is missing in the PPEP-1 structure. Beyond the β3/β4 loop, there is a difference in only one other substrate-interacting residue; in PPEP-1, the asparagine in the P2 position makes an H-bond with residue Lys-101, whereas the equivalent residue in PPEP-2, Arg-96, is located away from the substrate due to a distinct conformation of the S-loop (Fig. 5A).
Hence, the substrate recognition site in PPEP-2 is highly similar to that of PPEP-1, except for the differences outlined above. Thanks to this high-structural similarity, we could readily model the binding of the optimal peptide PLPPVP to PPEP-2 (Fig. 5B). This modeling strongly suggests that the Glu-113–Arg-145 salt bridge observed in PPEP-2 but not in PPEP-1 would create a steric hindrance for the binding of the VNPPVP peptide. In contrast, the presence of the Pro residue in position P3 of the PPEP-2 substrate peptide (PLPPVP) produces a kink in the peptide chain, directing the latter away from the salt bridge (Fig. 5B). Of note, this residue pair is conserved in many members of the Paenibacillus genus (Fig. S1).
Modifying the specificity of PPEP-2
Overall, our data demonstrate that the cleavage specificity of PPEP-1 and PPEP-2 differs in positions P3 and P2 relative to the cleavage site (Fig. 2B). Moreover, based on our structural analysis, we predict that the β3/β4 loop (GGST in PPEP-1 and SERV in PPEP-2) is important for the specificity (Fig. 5). To gain further insight into the role of this loop, we decided to exchange the four amino acids between PPEP-1 and PPEP-2. Recombinant PPEP-2 with the PPEP-1 β3/β4 loop, PPEP-2GGST, was first screened against our peptide library, which showed that it has the highest activity against a peptide containing VLPPVP instead of PLPPVP (data not shown). To study this further, we compared the steady state kinetics of the reaction of the PLPPVP and VLPPVP peptides with both PPEP-2 and PPEP-2GGST. First of all, the lowest Km and highest kcat values (70 μm and 8 s−1, respectively) were found for PPEP-2 with its optimal substrate peptide PLPPVP, whereas much lower efficiency was observed when the Pro at the P3 position was replaced by a Val (Table 2 and Fig. S7). In line with our prediction on the role of the β3/β4 loop, there was a clear shift in the selectivity of PPEP-2 when this loop was replaced by that of PPEP-1 (Table 2 and Fig. S7).
Table 2.
Kinetic parameters of PPEP-2 and PPEP-2GGST–mediated cleavage of substrate peptides
The selectivity factor was calculated by dividing the kcat/Km for PPEP-2 by that for PPEP-2GGST. A minimum of three experiments was used for the determination of the Michaelis-Menten constants. See also Fig. S7.
| Substrate | PPEP-2 |
PPEP-2GGST |
Selectivity factor | ||||
|---|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | ||
| μm | s−1 | m−1·s−1 | μm | s−1 | m−1·s−1 | ||
| PLPPVP | 70 ± 14 | 8 ± 0.5 | 114,286 | 330 ± 65 | 2 ± 0.2 | 6061 | 19 |
| VLPPVP | 330 ± 70 | 3 ± 0.3 | 9091 | 128 ± 36 | 3 ± 0.3 | 23,438 | 0.4 |
Hence, notwithstanding the role of other structural elements, these data support the role of the β3/β4 loop in the specificity of PPEP-1 and PPEP-2. Of note, the reciprocal exchange of the PPEP-2 β3/β4 loop into PPEP-1 resulted in a soluble recombinant enzyme (PPEP-1SERV), which nonetheless was unable to cleave either VNPPVP or PLPPVP (data not shown), indicating that such a mutation may be incompatible with the conformation required for substrate binding and hydrolysis.
Discussion
Based on the gene organization of ppep-1/cd2830 and that of its substrate (cd2831) in C. difficile 630 (21), we hypothesized that Paenibacillus PPEPs would cleave between two prolines within the PLPPVP motif found in VMSP proteins encoded by the adjacent gene. Our biochemical characterization of recombinant PPEP-2 from P. alvei demonstrated that this prediction was indeed correct. The VMSP from P. alvei, containing two PPEP-2 cleavage sites, was identified in culture medium of P. alvei DSM 29-growing cells, indicating release by endogenous PPEP-2.
Our data show that PPEP-1 and PPEP-2 display a very high level of specificity with respect to the P2 and P3 position of the cleavage site. Despite these differences in specificity, PPEP-1 and PPEP-2 appear to be functionally similar because their substrates are proteins with domains involved in adhesion. The PPEP-1 substrate CD2831 is a collagen-binding protein (20), and the other C. difficile substrate protein, CD3246, probably mediates covalent attachment to a host protein through its reactive thioester bond (30). The two PPEP-1 substrates are covalently attached to the primary cell wall polymer, peptidoglycan, via a sortase-mediated reaction (31, 32). The PPEP-2 substrate, VMSP, in contrast, has three consecutive SLH domains at its C terminus. Secreted proteins containing these repeats are tethered to the envelope of Gram-positive bacteria through noncovalent interactions with secondary cell wall polymers (33–35). SLH-containing proteins often contain other domains that can be involved in biofilm formation (36), binding to specific ligands on the surface of the host cells (37), or have enzymatic activity (38). The N terminus of the PPEP-2 substrate VMSP contains a VWFA domain. In many eukaryotes, the VWFA fold is found in surface proteins involved in interactions with the extracellular matrix (39). The closest homologs of the VWFA domain of P. alvei VMSP are the tips of pilins RgrA of Streptococcus pneumoniae and GBS104 of Streptococcus agalactiae. Both these pili form stalks to project the VWFA domain to bind to respiratory or epithelial cells (40, 41). Paenibacillus VMSPs also contain 2–12 repeating units of MucBP domains, each ∼60 amino acids long. The closest structural homologs to the P. alvei repeats are the B1 domains of Lactobacillus reuteri MUB, a mucus-binding protein, and the β-GF module in Staphylococcus aureus SraP, a surface-exposed protein that promotes S. aureus adhesion to host epithelial cells via a specific binding to carbohydrates (42, 43). Unlike what the name suggests, the region corresponding to the B1 domains of MUB is not responsible for the mucin-binding activity but requires an additional (B2) domain (42). This B2 domain is missing in Paenibacillus VMSP MucBP repeats. Future research is needed to show which elements of the extracellular matrix are targeted by P. alvei VMSP. We propose VMSP is the sole in vivo PPEP-2 substrate. One other protein (K4Z6T3, nitrous oxide-stimulated promotor) in the P. alvei DSM 29 proteome was found to contain the sequence PLPPVP, but this protein does not contain a signal sequence or cell-surface–anchoring motif. Hence, we find it highly unlikely to be a relevant in vivo PPEP-2 substrate.
Interestingly, the expression of ppep-1 and the genes encoding its substrates are inversely regulated by the small cyclic dinucleotide c-di-GMP (21, 44, 45), providing an elegant mechanism by which their expression can be controlled in vivo. The c-di-GMP can bind to specific riboswitches, secondary RNA structures, in target genes, thereby regulating their expression. Two types of c-di-GMP riboswitsches have been described: a type I, which is activated at low concentrations of c-di-GMP, and a type II, which is activated at high concentrations of c-di-GMP (46–48). In C. difficile, ppep-1 has a type I riboswitch, whereas the genes encoding its substrates are under the control of a type II c-di-GMP riboswitch. In P. alvei DSM 29, both ppep-2 and its substrate gene (vmsp) have a putative type I riboswitch indicating that c-di-GMP is also an important mediator in the control of PPEP-2 and its substrate in P. alvei.
Besides the functional similarity, PPEP-1 and PPEP-2 share structural similarity as well. Indeed, according to the SAXS data, both proteins form stable compact monomers in solution with common hydrodynamic properties. Analysis of the crystal structures of both proteases reveals highly similar folds with the main differences associated with the positions of the S-loop, which is believed to be involved in the catalytic mechanism. This loop adopts slightly different conformations in the PPEP-1 crystal (PDB codes 5A0P, 5A0R, 5A0S, and 5A0X) (23) and the NMR (PDB code 2N6J) (49) structures available, but the largest differences in its conformation were found in the PPEP-2 crystal structure presented in this work (Fig. S6). In one of the chains, the corresponding loop is absent from the electron density maps (chain D), which additionally supports the notion that it is highly mobile. Of the other two types of PPEP-2 chains found in the crystal structure, one has the S-loop position similar to that of PPEP-1 (chains A and B), and the other one has a significantly different position resembling its open state (chain C) (Fig. S6). Such a structural pliability of the S-loop may play a role in the enzymatic activity of PPEPs by controlling the availability of the active site to substrate peptides. This hypothesis needs further exploration.
Apart from the common specificity for hydrolyzing a Pro–Pro bond, our data underscore the importance of the β3/β4 loop toward the cleavage preferences of PPEP-1 and PPEP-2. In particular, the salt bridge between Glu-113 and Arg-145 in PPEP-2 is likely to define its preference for a Pro at the P3 position. Ultimately, the co-crystal structure of PPEP-2 with the optimal substrate peptide could reveal the precise contacts contributing to the differences in substrate specificity. This will contribute to our understanding of these proteases and could also help to design specific inhibitors for the more clinically relevant pathogen, C. difficile.
The high similarity between PPEP-1 and PPEP-2 suggests that these are related via a common ancestral gene. The absence of a PPEP-1/PPEP-2 homolog in many other bacterial species, however, makes it likely that a horizontal gene transfer event has been the mechanism for acquisition. Although C. difficile species are anaerobic bacteria found in the cecum and distal colon of humans and animals, the majority of bacteria belonging to Paenibacillus genus are found in soil, often associated with plant roots (24). Yet, Paenibacillus can also be cultured from human gut microbiota (50) and the gut of insects (P. popilliae). Regardless whether these paenibacilli are gut commensals or are opportunistic due to the herbivorous diet, interactions are likely to occur between the C. difficile and Paenibacillus. Our data support a model where, after the transfer, PPEP developed altered specificity with different substrates but retained a common function in regulating bacterial adhesion. Hence, other Pro-Pro endopeptidases may exist, also in other human pathogenic bacteria, but they may be more distantly related and therefore less straightforward to predict.
Experimental procedures
Chemicals
Peptides were synthesized at the Leiden University Medical Center facility, as described previously (51). Synthetic FRET peptides had the sequence KDabcylEX1X2X3X4X5X6DEEdans, with X1–X6 corresponding to the six amino acids covering the P3–P3′ positions of the cleavage site. FRET peptides are named based on these six amino acids throughout the study. If not indicated otherwise, chemicals were from Sigma.
Culturing of P. alvei cells and preparation of conditioned medium
P. alvei DSM 29 (ATCC 6344) was kindly provided by Prof. Dr. Schäffer, Dept. für NanoBiotechnologie, Universität für Bodenkultur, Wien, Austria. First, P. alvei cells were streaked on MHE agar plates (Biomerieux, France) from a −80 °C glycerol stock and incubated overnight at 37 °C. Then, a single colony from the plate was inoculated into 20 ml of liquid media (brain-heart infusion broth (Mediaproducts, BV)) and grown for 24 h while shaking at 250 rpm at 37 °C. Twenty ml of a culture was then centrifuged at 14,000 × g for 5 min. The supernatant was collected, filtered through a 0.2-μm filter (Whatman FT 30), and subsequently concentrated to 0.2–1 ml on an Amicon Ultra-15 centrifuge filter unit (Millipore, Germany) with 10-kDa cutoff. The concentrates were kept at −20 °C until further analysis.
DNA isolation, PCR, and Sanger sequencing
P. alvei genomic DNA was isolated using the Qiagen QIAamp DNA mini kit following the manufacturer's instructions.
The PPEP-2 sequence, missing the predicted signal peptide and corresponding to residues Gln-28 to Asn-217 of the immature protein (UniProt ID: K4ZRC1), was amplified from the P. alvei DSM 29 genomic DNA using Phusion DNA polymerase (New England Biolabs) with the primers 5′-GCAACAGATTGGTGGTGGACAGGAGCAGTCCATTTTGG and 5′-TCGAGGAGAGTTTAGATTAGTTGGCAAACAGCTTAGCCAT-3′. Using the In-Fusion system (Takara), the purified PCR product was cloned into the SUMO fusion vector pETRUK, an in-house T7-based expression plasmid derived from pETHSUL (52) containing a modified SUMO sequence with an altered pI. Following transformation and amplification of the recombinant plasmid in NEB5-α (New England Biolabs), the sequence of a single clone, termed pETRUK-PPEP-2, was validated by Sanger sequencing. The resulting construct encodes an additional non-native glycine between the end of the SUMO sequence and the starting Gln-28 of PPEP-2. This was introduced to aid cleavage and solubility of the purified fusion product.
The recombinant PPEP-2GGST, where residues 112–115 of PPEP-2 (SERV) were replaced by residues 91–94 of PPEP-1 (GGST), was prepared by oligo-directed mutagenesis. The pETRUK-PPEP-2 plasmid was amplified using the primers5′-GTGggtggaagtacaGTTGCAGTTCGCATTGGATATAG-3′ and 5′-AACtgtacttccaccCACTCCGGGAACATCATCCCA-3′. The resultant PCR was treated with DpnI and then transformed into NEB5-α cells. Following amplification and purification of the plasmid DNA, the integrity of the mutant construct was confirmed by Sanger sequencing.
The fragment between two genes c1_07810 and c1_07820 of P. alvei DSM 29 was amplified using Q5 polymerase using the fragment-specific primers (forward, 5′-CATTGCTGACGACGACACTT-3′; reverse, 5′-TTCTCCAACCTTACCGCTTG-3′) and analyzed by Sanger sequencing.
Production and purification of recombinant PPEPs
The pETRUK-PPEP-2 plasmid was transformed into the Escherichia coli expression strain RosettaTM 2(DE3)pLysS (Novagen). A 2-ml preculture, grown in Luria broth for 8 h, was used to inoculate 2 liters of ZYP-5052 auto-induction medium supplemented with an additional 1 mm ZnCl2 (53). This culture was grown overnight for ∼16 h at 24 °C and then shifted to 18 °C for a further 24 h. The cells were harvested by centrifugation (10,000 × g for 15 min at 4 °C), and the cell pellet was frozen in portions of 5 g (wet weight) and stored at −80 °C prior to protein purification.
Cells were disrupted using an ultrasonic disintegrator in 50 ml of a lysis buffer (20 mm HEPES-NaOH (pH 8.0), 250 mm NaCl, 10 mm MgCl2, 20 μg/ml lysozyme, 6 units/ml cryonase (Cold-Active Nuclease, Takara) and 0.05% Triton X-100). Insoluble material was pelleted by centrifugation (20,000 × g for 30 min at 4 °C). The clarified supernatant was applied on two tandem coupled 5-ml HiTrapSP HP (GE Healthcare) ion-exchange columns equilibrated in cation-exchange buffer A (20 mm HEPES-NaOH (pH 8.0), 250 mm NaCl). After washing of unbound proteins with the same buffer, a linear gradient (0–35% in 10 column volumes) of cation-exchange buffer B (20 mm HEPES-NaOH (pH 8.0), 1 m NaCl) was applied. The fractions containing the highest amounts of PPEP-2 were combined, and the protein concentration was determined photometrically at A280. To cleave off the SUMO tag, the combined sample of PPEP-2 was mixed with SUMO hydrolase (200:1 molar ratio) and incubated for 2 h at 21 °C. Then, the sample was dialyzed against 6 liters of buffer (10 mm Tris-HCl (pH 7.0), 25 mm (NH4)2SO4, 10% (w/v) glycerol) with three changes after 2, 10, and 3 h. Precipitated protein was removed by centrifugation (20,000 × g for 30 min at 4 °C), and the supernatant was applied on two tandem coupled 5-ml HiTrapQ HP (GE Healthcare) ion-exchange columns equilibrated in anion-exchange buffer A (10 mm Tris-HCl (pH 7.0)). A linear gradient (0–20% in 25 column volumes) of anion-exchange buffer B (10 mm Tris-HCl (pH 7.0), 1 m NaCl) was applied. Fractions containing the highest purity of PPEP-2 were combined, and the sample was concentrated on Amicon Ultracentrifugal filters (3 kDa cutoff) until the concentration was ∼35 μg/μl. Aliquots of 1.7 mg of protein were snap frozen in liquid nitrogen and stored at −80 °C. Recombinant PPEP-1 used for the enzymatic assays was produced and purified as described previously (21). Recombinant PPEP-1 used for the SAXS experiments was prepared as described above for PPEP-2, following amplification from the template pET16b-10×His-PPEP-1 (21) using the primers 5′-GCGAACAGATTGGTGGTGGAGATAGTACTACTATACAACAAAATA-3′ and 5′-TCGAGGAGAGTTTAGATTATTTAGCTAAATTTTGCAAAA-3′ and cloning into pETRUK vector.
MALDI-ToF and MALDI-FTICR MS analysis
For MALDI-MS analysis, samples were desalted and purified in one step with 0.6-μl C18 ZipTip pipette tips (Millipore, Germany). First, the tips were equilibrated with 50% acetonitrile, 0.1% formic acid (v/v) solution and washed twice with 0.1% TFA. One μl of the sample was aspirated and directly dispensed. The bound peptides were eluted directly on a stainless steel target plate with 2,5-dihydroxybenzoic acid matrix dissolved in 50% acetonitrile, 0.1% TFA (v/v) at the concentration of 10 mg/ml. The eluted sample was allowed to dry at room temperature. MALDI-ToF MS and MALDI-FT-ICR-MS analyses were performed as described previously (54, 55).
Enzyme activity assays
Five μl of 10 μm standard peptide YPSSKPLPPVPPVQPLPPVPKLETS in PBS buffer were mixed with 2 μl of 100 ng/μl PPEP-2 in the same buffer and incubated overnight at 37 °C.
For a standard FRET assay, 50 μm FRET substrate peptide was incubated with 100 ng of enzyme or 5 μl of the P. alvei conditioned medium, in a total volume of 100 μl (in PBS buffer) in a 96-well black plate (Cellstar, Greiner). Cleavage of the fluorescent FRET substrate peptides was measured in a 96-well plate reader (Mithras LB940, Berthold Technologies, Germany) at 355 nm excitation and at 485 nm emission wavelengths for 60 min with an interval of 1 min at 37 °C by following the increase in fluorescence as a result of the FRET–pair decoupling (21).
For the determination of the steady-state kinetics, reactions were performed using 0 (control), 25, 50, 75, 100, 150, 250, 400, and 800 μm of the substrate peptides. To achieve confident detection of fluorescent readout for recombinant PPEP-2 and PPEP-2GGST, the amount of enzyme in a reaction mix varied depending on enzyme–substrate combination: 100 ng for PPEP-2 with the PLPPVP peptide; 2 μg of PPEP-2 with VLPPVP peptide, and 200 ng of PPEP-2GGST for both peptides. The enzymatic reaction was initiated by adding enzyme into the reaction mix. The samples were incubated at 37 °C, and at 3-min intervals 15 μl of the reaction was added to an equal volume of 1% TFA to stop the reaction. Then, 20 μl of the stopped reaction mix was diluted 5 times with 100 mm phosphate buffer (pH 7.5), and fluorescence was measured as described above. For the 400 and 800 μm samples, an extra dilution of 4× was used. All dilutions were made to prevent inner filter-quenching effects. Initial velocities were derived from the slope of each individual 12-min reaction. Conversion of the initial velocities from unit·min−1 into nanomole·s−1 was done by the measurement of fluorescence derived from completely cleaved substrate peptides at each concentration mentioned above. Then, the converted values were fitted to the Michaelis-Menten equation in GraphPad Prism 6, where Km and kcat values were calculated.
In-gel tryptic digestion and identification of proteins by LC-MS/MS analysis
Briefly, gel bands of interest were excised from the SDS-polyacrylamide gel and subjected to an in-gel digestion procedure according to the standard protocol of washing with 25 mm NH4HCO3, reduction with 10 mm DTT, followed by alkylation with 55 mm iodoacetamide and overnight trypsin digestion at 37 °C.
Tryptic digests were analyzed by nano-LC-MS using an Easy-nLC 1000 (ThermoFisher Scientific, Germany) coupled online to a Q Exactive hybrid quadrupole Orbitrap mass spectrometer (ThermoFisher Scientific, Germany). Peptides were trapped on an in-house–made trap column (C18, 10 × 15 μm, 3-μm particles, Reprosil Acu, Dr. Maisch) equilibrated with 0.1% formic acid and separated on an in-house–made analytical reverse–phase column (C18, 7 μm × 30 cm with a taper tip, 3-μm particles, Reprosil Acu, Dr. Maisch) using a linear gradient from 0 to 30% phase B in 120 min, where phase B consisted of 0.1% formic acid in acetonitrile at a flow rate of 250 nl/min.
Data-dependent analysis in the m/z 300–1400 range was performed where the 10 most intense ions in full MS mode were subjected to the fragmentation with higher-energy collisional dissociation at 27 V (only 2+, 3+, and 4+ ions were analyzed: AGC target 3,000,000 ions; maximum injection time 20 ms; dynamic exclusion of 10 s; resolution 70,000). Next, the obtained raw data were converted into the MGF peak lists with Proteome Discoverer 1.4 software (ThermoFisher Scientific, Germany). The converted data were searched against the P. alvei DSM 29 database using Mascot 2.2 (Mascot Daemon 2.5.1) for protein identification with the following settings: trypsin cleavage specificity with one missed cleavage allowed; carbamidomethylation of cysteine as a fixed modification; oxidation of methionine as a variable modification; MS tolerance 10 ppm; MS/MS tolerance 20 milli-mass units.
Crystallization
Sparse matrix crystallization screening with commercial precipitation solution kits using the hanging drop technique has yielded the initial crystallization conditions. The optimized PPEP-2 crystals were obtained upon mixing 2 μl of purified protein (12 mg/ml) with 2 μl of 2.2 m (NH4)2SO4, 0.2 m CdSO4, 0.01 m ZnCl2 (pH 9.0) adjusted with NH4OH. The mix was equilibrated at 20 °C against 1 ml of the same precipitant solution. Single well ordered crystals appeared after 5–7 days and were allowed to grow until the maximal size of 100 μm for another week. For data collection, the crystals were transferred in the same crystallization solution with 3.5 m (NH4)2SO4, incubated during 2 min, and flash-frozen in liquid nitrogen.
Structure solution and refinement
Structure determination was based on data collected from a single crystal on the PROXIMA 1 beamline at 100 K (Soleil Synchrotron, France). Data reduction statistics are provided in Table 1. Initial attempts to phase the structure using molecular replacement failed. A dataset collected using synchrotron radiation at 6.89 keV (1.8 Å) was used to perform a SAD phasing with phenix.autsol (56). The positions of 14 cadmium atoms per asymmetric unit could be found. A model of PPEP-2 was then manually placed into the RESOLVE solvent-flattened electron density map using Coot (57). Following the initial positioning of four chains in the asymmetric unit, the model was refined against a higher resolution dataset collected from the same crystal at 12.67 keV (0.9786 Å). The model was iteratively improved by rounds of manual rebuilding with Coot and refinement with phenix.refine (56). Coordinates and the structure factors for PPEP-2 have been deposited into the Protein Data Bank (PDB) under the accession code 6FPC.
Size-exclusion chromatography coupled with SAXS
Fifty microliters of either PPEP-2 or PPEP-1 (25 mg/ml) were injected on a size-exclusion column (Biosec3-100, 3 μm, 300 Å, 4.6 × 300 mm, Agilent) equilibrated with a SEC-SAXS Buffer (20 mm HEPES (pH 8.0), 300 mm KCl, 5% glycerol). Proteins were separated at 0.3 ml/min at 15 °C. SEC-SAXS measurements were performed at the SWING beamline at Synchrotron Soleil (Gif-sur-Yvette, France) (58) by exposing the eluting sample to the X-ray beam for 750 ms with a gap time of 250 ms between frames. For buffer subtraction, 180 SAXS frames were collected after a sample injection but prior to the column void volume. For both samples, 255 frames around the elution peak were collected. Radial averaging of the collected frames, buffer averaging, and subsequent subtraction from the sample data were performed using the Foxtrot application (SWING beamline). Further analysis was performed using the HPLC-SAXS module within the UltraScan Solution Modeler software package (58).
The overall hydrodynamic parameters were assessed by using the ATSAS 2.8.2 software (59). Calculation of the theoretical scattering profile and fitting to the experimental SAXS data were performed using Crysol (60) for which the PPEP-2 structure, chain B, was supplemented with four amino acids at the N terminus (GQEQ) and the Asn-217 residue at the C terminus that were missing from the crystallographic model. Ab initio molecular shape was calculated using 10 independent DAMMIN (29) runs, and the best-fitting model (χ2 = 1.12) was presented overlaid with the crystal structure of PPEP-2 (chain B).
Bioinformatics
PPEP-1 (PDB code 5A0X, chain A) and PPEP-2 (chain B, this work) structures were superimposed using the TM-align algorithm (61). Thereafter, the target peptide bound to PPEP-1 in the 5A0X (chain A) structure was used as a template to model the target peptide of PPEP-2 using the Small Molecule Discovery Suite, Schrödinger, LLC. To this end, the target PPEP-1 peptide was mutated in the P2 and P3 positions to produce the PPEP-2 peptide and thereafter minimized by running the Glide algorithm. Invariant residues in positions P1–P3′ were fixed.
Author contributions
O. I. K., H. C. v. L., and P. J. H. conceptualization; O. I. K., T. M. S., S. D. W., H. C. v. L., N. N. S., S. V. S., and P. J. H. data curation; O. I. K., T. M. S., S. D. W., and P. J. H. formal analysis; O. I. K., T. M. S., S. D. W., and P. J. H. investigation; O. I. K., H. C. v. L., and P. J. H. writing-original draft; O. I. K., S. D. W., H. C. v. L., N. N. S., S. V. S., and P. J. H. writing-review and editing; T. M. S., H. C. v. L., and N. N. S. visualization; S. D. W., N. N. S., and S. V. S. validation; S. D. W., J .C., J. W. D., and P. A. v. V. methodology; J. C., J. W. D., and P. A. v. V. resources; S. V. S. and P. J. H. supervision; P. J. H. project administration.
Supplementary Material
Acknowledgments
We thank Prof. Christina Schäffer (Universität für Bodenkultur Wien, Austria) for providing the P. alvei cells. We further greatly appreciate the support from the beamline scientists at the PROXIMA 1 and SWING beamlines (Soleil Synchrotron, France) in facilitating collection of X-ray scattering data. We also thank Steff De Graef (Laboratory for Biocrystallography, KU Leuven) for help with the X-ray data collection.
This work was supported in part by funds from the Department of Pharmaceutical and Pharmacological Sciences of KU Leuven. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Tables S1–S2.
The atomic coordinates and structure factors (code 6FPC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PPEP
- Pro-Pro endopeptidase
- FTICR
- Fourier-transform ion cyclotron resonance
- ASU
- asymmetric unit
- SAXS
- small-angle X-ray scattering
- SEC
- size-exclusion chromatography
- PDB
- Protein Data Bank
- SLH
- surface-layer homology
- r.m.s.d.
- root mean square deviation
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- SUMO
- small ubiquitin-related modifier
- VWFA
- von Willebrand factor A
- MucBP
- mucin-binding protein
- NSD
- normalized spatial discrepancy.
References
- 1. Turk B. (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799 10.1038/nrd2092 [DOI] [PubMed] [Google Scholar]
- 2. Klein T., Eckhard U., Dufour A., Solis N., and Overall C. M. (2018) Proteolytic cleavage-mechanisms, function, and “Omic” approaches for a near-ubiquitous posttranslational modification. Chem. Rev. 118, 1137–1168 [DOI] [PubMed] [Google Scholar]
- 3. Vergnolle N. (2016) Protease inhibition as new therapeutic strategy for GI diseases. Gut 65, 1215–1224 10.1136/gutjnl-2015-309147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. López-Otín C., and Bond J. S. (2008) Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437 10.1074/jbc.R800035200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banerjee G., and Ray A. K. (2017) Impact of microbial proteases on biotechnological industries. Biotechnol. Genet. Eng. Rev. 33, 119–143 10.1080/02648725.2017.1408256 [DOI] [PubMed] [Google Scholar]
- 6. Dos Santos Aguilar J. G., and Sato H. H. (2018) Microbial proteases: production and application in obtaining protein hydrolysates. Food Res. Int. 103, 253–262 10.1016/j.foodres.2017.10.044 [DOI] [PubMed] [Google Scholar]
- 7. Kasana R. C., Salwan R., and Yadav S. K. (2011) Microbial proteases: detection, production, and genetic improvement. Crit. Rev. Microbiol. 37, 262–276 10.3109/1040841X.2011.577029 [DOI] [PubMed] [Google Scholar]
- 8. Cunningham D. F., and O'Connor B. (1997) Proline specific peptidases. Biochim. Biophys. 1343, 160–186 10.1016/S0167-4838(97)00134-9 [DOI] [PubMed] [Google Scholar]
- 9. Rawlings N. D., Barrett A. J., and Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350 10.1093/nar/gkr987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vanhoof G., Goossens F., De Meester I., Hendriks D., and Scharpé S. (1995) Proline motifs in peptides and their biological processing. FASEB J. 9, 736–744 10.1096/fasebj.9.9.7601338 [DOI] [PubMed] [Google Scholar]
- 11. Olsen J. V., Ong S. E., and Mann M. (2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614 10.1074/mcp.T400003-MCP200 [DOI] [PubMed] [Google Scholar]
- 12. Stepniak D., Spaenij-Dekking L., Mitea C., Moester M., de Ru A., Baak-Pablo R., van Veelen P., Edens L., and Koning F. (2006) Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G621–G629 10.1152/ajpgi.00034.2006 [DOI] [PubMed] [Google Scholar]
- 13. Tack G. J., van de Water J. M., Bruins M. J., Kooy-Winkelaar E. M., van Bergen J., Bonnet P., Vreugdenhil A. C., Korponay-Szabo I., Edens L., von Blomberg B. M., Schreurs M. W., Mulder C. J., and Koning F. (2013) Consumption of gluten with gluten-degrading enzyme by celiac patients: a pilot-study. World J. Gastroenterol. 19, 5837–5847 10.3748/wjg.v19.i35.5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rey M., Yang M., Lee L., Zhang Y., Sheff J. G., Sensen C. W., Mrazek H., Halada P., Man P., McCarville J. L., Verdu E. F., and Schriemer D. C. (2016) Addressing proteolytic efficiency in enzymatic degradation therapy for celiac disease. Sci. Rep. 6, 30980 10.1038/srep30980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schräder C. U., Lee L., Rey M., Sarpe V., Man P., Sharma S., Zabrouskov V., Larsen B., and Schriemer D. C. (2017) Neprosin, a selective prolyl endoprotease for bottom-up proteomics and histone mapping. Mol. Cell. Proteomics 16, 1162–1171 10.1074/mcp.M116.066803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Friebe S., van der Goot F. G., and Bürgi J. (2016) The ins and outs of anthrax toxin. Toxins 8, E69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marshall N. C., Finlay B. B., and Overall C. M. (2017) Sharpening host defenses during infection: proteases cut to the chase. Mol. Cell. Proteomics 16, S161–S171 10.1074/mcp.O116.066456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tonry J. H., McNichol B. A., Ramarao N., Chertow D. S., Kim K. S., Stibitz S., Schneewind O., Kashanchi F., Bailey C. L., Popov S., and Chung M.-C. (2012) Bacillus anthracis protease InhA regulates BslA-mediated adhesion in human endothelial cells. Cell. Microbiol. 14, 1219–1230 10.1111/j.1462-5822.2012.01791.x [DOI] [PubMed] [Google Scholar]
- 19. Cafardi V., Biagini M., Martinelli M., Leuzzi R., Rubino J. T., Cantini F., Norais N., Scarselli M., Serruto D., and Unnikrishnan M. (2013) Identification of a novel zinc metalloprotease through a global analysis of Clostridium difficile extracellular proteins. PLoS One 8, e81306 10.1371/journal.pone.0081306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hensbergen P. J., Klychnikov O. I., Bakker D., Dragan I., Kelly M. L., Minton N. P., Corver J., Kuijper E. J., Drijfhout J. W., and van Leeuwen H. C. (2015) Clostridium difficile secreted Pro-Pro endopeptidase PPEP-1 (ZMP1/CD2830) modulates adhesion through cleavage of the collagen binding protein CD2831. FEBS Lett. 589, 3952–3958 10.1016/j.febslet.2015.10.027 [DOI] [PubMed] [Google Scholar]
- 21. Hensbergen P. J., Klychnikov O. I., Bakker D., van Winden V. J., Ras N., Kemp A. C., Cordfunke R. A., Dragan I., Deelder A. M., Kuijper E. J., Corver J., Drijfhout J. W., and van Leeuwen H. C. (2014) A novel secreted metalloprotease (CD2830) from Clostridium difficile cleaves specific proline sequences in LPXTG cell surface proteins. Mol. Cell. Proteomics 13, 1231–1244 10.1074/mcp.M113.034728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corver J., Cordo' V., van Leeuwen H. C., Klychnikov O. I., and Hensbergen P. J. (2017) Covalent attachment and Pro-Pro endopeptidase (PPEP-1)-mediated release of Clostridium difficile cell surface proteins involved in adhesion. Mol. Microbiol. 105, 663–673 10.1111/mmi.13736 [DOI] [PubMed] [Google Scholar]
- 23. Schacherl M., Pichlo C., Neundorf I., and Baumann U. (2015) Structural basis of proline-proline peptide bond specificity of the metalloprotease Zmp1 implicated in motility of Clostridium difficile. Structure 23, 1632–1642 10.1016/j.str.2015.06.018 [DOI] [PubMed] [Google Scholar]
- 24. Grady E. N., MacDonald J., Liu L., Richman A., and Yuan Z. C. (2016) Current knowledge and perspectives of Paenibacillus: a review. Microb. Cell Fact. 15, 203 10.1186/s12934-016-0603-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Finn R. D., Attwood T. K., Babbitt P. C., Bateman A., Bork P., Bridge A. J., Chang H. Y., Dosztányi Z., El-Gebali S., Fraser M., Gough J., Haft D., Holliday G. L., Huang H., Huang X., et al. (2017) InterPro in 2017–beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 10.1093/nar/gkw1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matthews B. W. (1988) Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 21, 333–340 10.1021/ar00153a003 [DOI] [Google Scholar]
- 27. Grams F., Dive V., Yiotakis A., Yiallouros I., Vassiliou S., Zwilling R., Bode W., and Stöcker W. (1996) Structure of astacin with a transition-state analogue inhibitor. Nat. Struct. Biol. 3, 671–675 10.1038/nsb0896-671 [DOI] [PubMed] [Google Scholar]
- 28. Yiallouros I., Grosse Berkhoff E., and Stöcker W. (2000) The roles of Glu93 and Tyr149 in astacin-like zinc peptidases. FEBS Lett. 484, 224–228 10.1016/S0014-5793(00)02163-3 [DOI] [PubMed] [Google Scholar]
- 29. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 10.1016/S0006-3495(99)77443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walden M., Edwards J. M., Dziewulska A. M., Bergmann R., Saalbach G., Kan S. Y., Miller O. K., Weckener M., Jackson R. J., Shirran S. L., Botting C. H., Florence G. J., Rohde M., Banfield M. J., and Schwarz-Linek U. (2015) An internal thioester in a pathogen surface protein mediates covalent host binding. Elife 4, e66383 10.7554/eLife.06638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donahue E. H., Dawson L. F., Valiente E., Firth-Clark S., Major M. R., Littler E., Perrior T. R., and Wren B. W. (2014) Clostridium difficile has a single sortase, SrtB, that can be inhibited by small-molecule inhibitors. BMC Microbiol. 14, 219 10.1186/s12866-014-0219-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Leeuwen H. C., Klychnikov O. I., Menks M. A., Kuijper E. J., Drijfhout J. W., and Hensbergen P. J. (2014) Clostridium difficile sortase recognizes a (S/P)PXTG sequence motif and can accommodate diaminopimelic acid as a substrate for transpeptidation. FEBS Lett. 588, 4325–4333 10.1016/j.febslet.2014.09.041 [DOI] [PubMed] [Google Scholar]
- 33. Kern J., Wilton R., Zhang R., Binkowski T. A., Joachimiak A., and Schneewind O. (2011) Structure of surface layer homology (SLH) domains from Bacillus anthracis surface array protein. J. Biol. Chem. 286, 26042–26049 10.1074/jbc.M111.248070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fagan R. P., and Fairweather N. F. (2014) Biogenesis and functions of bacterial S-layers. Nat. Rev. Microbiol. 12, 211–222 10.1038/nrmicro3213 [DOI] [PubMed] [Google Scholar]
- 35. Kirk J. A., Banerji O., and Fagan R. P. (2017) Characteristics of the Clostridium difficile cell envelope and its importance in therapeutics. Microb. Biotechnol. 10, 76–90 10.1111/1751-7915.12372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Janesch B., Koerdt A., Messner P., and Schäffer C. (2013) The S-layer homology domain-containing protein SlhA from Paenibacillus alvei CCM 2051(T) is important for swarming and biofilm formation. PLoS ONE 8, e76566 10.1371/journal.pone.0076566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y., Wei Y., Yuan S., Tao H., Dong J., Zhang Z., Tian W., and Liu C. (2016) Bacillus anthracis S-layer protein BslA binds to extracellular matrix by interacting with laminin. BMC Microbiol. 16, 183 10.1186/s12866-016-0802-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Talamantes D., Biabini N., Dang H., Abdoun K., and Berlemont R. (2016) Natural diversity of cellulases, xylanases, and chitinases in bacteria. Biotechnol. Biofuels 9, 133 10.1186/s13068-016-0538-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Springer T. A. (2006) Complement and the multifaceted functions of VWA and integrin I domains. Structure 14, 1611–1616 10.1016/j.str.2006.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krishnan V., Dwivedi P., Kim B. J., Samal A., Macon K., Ma X., Mishra A., Doran K. S., Ton-That H., and Narayana S. V. (2013) Structure of Streptococcus agalactiae tip pilin GBS104: a model for GBS pili assembly and host interactions. Acta Crystallogr. D Biol. Crystallogr. 69, 1073–1089 10.1107/S0907444913004642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Izoré T., Contreras-Martel C., El Mortaji L., Manzano C., Terrasse R., Vernet T., Di Guilmi A. M., and Dessen A. (2010) Structural basis of host cell recognition by the pilus adhesin from Streptococcus pneumoniae. Structure 18, 106–115 10.1016/j.str.2009.10.019 [DOI] [PubMed] [Google Scholar]
- 42. MacKenzie D. A., Tailford L. E., Hemmings A. M., and Juge N. (2009) Crystal structure of a mucus-binding protein repeat reveals an unexpected functional immunoglobulin binding activity. J. Biol. Chem. 284, 32444–32453 10.1074/jbc.M109.040907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang Y. H., Jiang Y. L., Zhang J., Wang L., Bai X. H., Zhang S. J., Ren Y. M., Li N., Zhang Y. H., Zhang Z., Gong Q., Mei Y., Xue T., Zhang J. R., Chen Y., and Zhou C. Z. (2014) Structural insights into SraP-mediated Staphylococcus aureus adhesion to host cells. PLoS Pathog. 10, e1004169 10.1371/journal.ppat.1004169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peltier J., Shaw H. A., Couchman E. C., Dawson L. F., Yu L., Choudhary J. S., Kaever V., Wren B. W., and Fairweather N. F. (2015) Cyclic diGMP regulates production of sortase substrates of Clostridium difficile and their surface exposure through ZmpI protease-mediated cleavage. J. Biol. Chem. 290, 24453–24469 10.1074/jbc.M115.665091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soutourina O. A., Monot M., Boudry P., Saujet L., Pichon C., Sismeiro O., Semenova E., Severinov K., Le Bouguenec C., Coppée J.-Y., Dupuy B., and Martin-Verstraete I. (2013) Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PloS Genet. 9, e1003493 10.1371/journal.pgen.1003493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen A. G., Sudarsan N., and Breaker R. R. (2011) Mechanism for gene control by a natural allosteric group I ribozyme. RNA 17, 1967–1972 10.1261/rna.2757311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee E. R., Baker J. L., Weinberg Z., Sudarsan N., and Breaker R. R. (2010) An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 329, 845–848 10.1126/science.1190713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sudarsan N., Lee E. R., Weinberg Z., Moy R. H., Kim J. N., Link K. H., and Breaker R. R. (2008) Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321, 411–413 10.1126/science.1159519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rubino J. T., Martinelli M., Cantini F., Castagnetti A., Leuzzi R., Banci L., and Scarselli M. (2016) Structural characterization of zinc-bound Zmp1, a zinc-dependent metalloprotease secreted by Clostridium difficile. J. Biol. Inorg. Chem. 21, 185–196 10.1007/s00775-015-1319-6 [DOI] [PubMed] [Google Scholar]
- 50. Rajilić-Stojanović M., and de Vos W. M. (2014) The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 38, 996–1047 10.1111/1574-6976.12075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hiemstra H. S., Duinkerken G., Benckhuijsen W. E., Amons R., de Vries R. R., Roep B. O., and Drijfhout J. W. (1997) The identification of CD4+ T cell epitopes with dedicated synthetic peptide libraries. Proc. Natl. Acad. Sci. U.S.A. 94, 10313–10318 10.1073/pnas.94.19.10313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weeks S. D., Drinker M., and Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 53, 40–50 10.1016/j.pep.2006.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 54. Balog C. I., Mayboroda O. A., Wuhrer M., Hokke C. H., Deelder A. M., and Hensbergen P. J. (2010) Mass spectrometric identification of aberrantly glycosylated human apolipoprotein C-III peptides in urine from Schistosoma mansoni-infected individuals. Mol. Cell. Proteomics 9, 667–681 10.1074/mcp.M900537-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bondt A., Nicolardi S., Jansen B. C., Stavenhagen K., Blank D., Kammeijer G. S., Kozak R. P., Fernandes D. L., Hensbergen P. J., Hazes J. M., van der Burgt Y. E., Dolhain R. J., and Wuhrer M. (2016) Longitudinal monitoring of immunoglobulin A glycosylation during pregnancy by simultaneous MALDI-FTICR-MS analysis of N- and O-glycopeptides. Sci. Rep. 6, 27955 10.1038/srep27955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heirbaut M., Lermyte F., Martin E. M., Beelen S., Verschueren T., Sobott F., Strelkov S. V., and Weeks S. D. (2016) The preferential heterodimerization of human small heat shock proteins HSPB1 and HSPB6 is dictated by the N-terminal domain. Arch. Biochem. Biophys. 610, 41–50 10.1016/j.abb.2016.10.002 [DOI] [PubMed] [Google Scholar]
- 59. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Svergun D., Barberato C., and Koch M. H. (1995) CRYSOL-a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 61. Zhang Y., and Skolnick J. (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.