

Abstract
Cardiolipin (CL) is a signature phospholipid of the mitochondria required for the formation of mitochondrial respiratory chain (MRC) supercomplexes. The destabilization of MRC supercomplexes is the proximal cause of the pathology associated with the depletion of CL in patients with Barth syndrome. Thus, promoting supercomplex formation could ameliorate mitochondrial dysfunction associated with CL depletion. However, to date, physiologically relevant small-molecule regulators of supercomplex formation have not been identified. Here, we report that ethanolamine (Etn) supplementation rescues the MRC defects by promoting supercomplex assembly in a yeast model of Barth syndrome. We discovered this novel role of Etn while testing the hypothesis that elevating mitochondrial phosphatidylethanolamine (PE), a phospholipid suggested to overlap in function with CL, could compensate for CL deficiency. We found that the Etn supplementation rescues the respiratory growth of CL-deficient Saccharomyces cerevisiae cells in a dose-dependent manner but independently of its incorporation into PE. The rescue was specifically dependent on Etn but not choline or serine, the other phospholipid precursors. Etn improved mitochondrial function by restoring the expression of MRC proteins and promoting supercomplex assembly in CL-deficient cells. Consistent with this mechanism, overexpression of Cox4, the MRC complex IV subunit, was sufficient to promote supercomplex formation in CL-deficient cells. Taken together, our work identifies a novel role of a ubiquitous metabolite, Etn, in attenuating mitochondrial dysfunction caused by CL deficiency.
Keywords: mitochondria, cardiolipin, mitochondrial respiratory chain complex, cytochrome c oxidase (Complex IV), phospholipid, Barth syndrome, ethanolamine, respiratory supercomplexes
Introduction
Mitochondrial membrane phospholipid composition determines the function and formation of the mitochondrial respiratory chain (MRC).4 Previous work on cardiolipin (CL), a signature phospholipid of mitochondrial membranes, has identified its critical roles in electron transport and energy transformation reactions (1, 2). Both in vivo and in vitro experiments have demonstrated that CL exerts its effect on the MRC by facilitating supercomplex formation (3–5). MRC supercomplexes consist of supramolecular assemblies of the MRC complex III and complex IV in the yeast Saccharomyces cerevisiae (6) and complexes I, III, and IV in higher eukaryotes (7). These respiratory supercomplexes are proposed to stabilize individual MRC complexes, minimize the generation of reactive oxygen species, and enhance catalytic efficiency by substrate channeling (8, 9). The recent cryo-EM–based determination of high-resolution structures of the respiratory supercomplexes (10–12) have firmly established their existence and suggested the dynamic nature of their assembly. However, apart from CL, no other metabolite has been known to regulate their formation.
The biomedical relevance of CL in MRC function and supercomplex assembly can be gleaned from the finding that its depletion results in a life-threatening disorder in humans called Barth syndrome (BTHS). BTHS is a severely debilitating X-linked genetic disorder characterized by cardiomyopathy, skeletal muscle myopathy, neutropenia, growth-delay, and exercise intolerance (13). BTHS is caused by loss-of-function mutations in the evolutionarily conserved tafazzin (TAZ) gene (14), which encodes a mitochondrial transacylase that remodels CL (15, 16). Studies from BTHS patient cells as well as from a number of model systems, including the yeast S. cerevisiae taz1Δ cells, have shown that mitochondrial dysfunction caused by perturbation in CL remodeling is the primary cause of BTHS pathology (17–22). The downstream consequences of this altered mitochondrial phospholipid composition include the destabilization of MRC supercomplexes and increased oxidative stress (23–25).
In addition to its well-characterized role in MRC supercomplex formation, CL exerts its effect on mitochondrial bioenergetics at multiple levels. For example, CL is required for the expression of both mitochondrial and nuclear DNA-encoded MRC subunits (26, 27) and optimal MRC complex IV activity (4). The loss of CL also results in decreased mitochondrial membrane potential and protein import (28). Finally, the loss of CL perturbs efficient coupling of electron transport with ATP synthesis (29, 30). Like CL, phosphatidylethanolamine (PE) is also required for optimal mitochondrial bioenergetics by preserving the catalytic activities of the MRC complexes (31–33), whereas PC has been shown to be redundant as far as MRC function and assembly is concerned (31, 34, 35). Both PE and CL are synthesized in situ, unlike other mitochondrial phospholipids (Fig. 1). In addition to the mitochondrial pathway, PE can be biosynthesized either by the nonmitochondrial ethanolamine–Kennedy pathway or by a lyso-PE–requiring pathway (Fig. 1).
Figure 1.

Biochemical pathways for PE, PC, and CL biosynthesis in the yeast S. cerevisiae. CL biosynthesis occurs exclusively in the mitochondria, where Crd1 synthesizes nascent CLp from PG. The resulting CLp is deacylated by the phospholipase Cld1 to produce monolysocardiolipin (MLCL), which is then reacylated by the transacylase Taz1 to form mature cardiolipin (CLm). Mutations in the human homologue of TAZ1 result in Barth syndrome. CL biosynthesis depends on the import of PA from endoplasmic reticulum, which is converted to CDP-DAG by Tam41. Pgs1 catalyzes conversion of CDP-DAG to PGP, which is then dephosphorylated to PG by Gep4. PE biosynthesis in yeast can occur by the following: 1) Psd1-catalyzed decarboxylation of PS in the mitochondria; 2) incorporation of Etn via the cytosolic/endoplasmic reticulum Kennedy pathway enzymes Eki1, Ect1, and Ept1, respectively; and 3) the acylation of lyso-PE by Ale1. The Kennedy pathway enzymes Cki1, Pct1, and Cpt1 can utilize choline to biosynthesize PC, a bilayer-forming phospholipid that is imported into mitochondria. CLp, precursor cardiolipin; CLm, mature cardiolipin; PG, phosphatidylglycerol; PGP, phosphatidylglycerol phosphate; CDP, cytidine diphosphate; DAG, diacylglycerol.
Because of their common site of biosynthesis, reciprocal regulation of their levels, and the propensity to form nonbilayer structures, PE and CL have been proposed to have overlapping functions (36–43). These findings formed the basis of our hypothesis that MRC defects due to disruptions in CL biosynthesis can be rescued by elevating mitochondrial PE levels. We have recently shown that exogenous Etn supplementation can increase mitochondrial PE levels (31), allowing us to test the proposed hypothesis. In this study, we used an Etn supplementation strategy in yeast models of CL deficiency to demonstrate that Etn can rescue respiratory chain defects of CL-depleted strains. Surprisingly, Etn-mediated rescue was independent of its incorporation into PE. Thus, our work identifies a novel role of Etn as a regulator of MRC biogenesis.
Results
Ethanolamine supplementation rescues the respiratory growth of CL-deficient cells
Previous reports have shown that CL deficiency in yeast cells is accompanied by an increase in PE levels (19, 42, 43), suggesting a compensatory response to the lack of CL. Therefore, we asked whether further elevation of mitochondrial PE levels in CL-deficient taz1Δ and crd1Δ cells could compensate for the loss of CL and rescue the respiratory growth defect (20). To elevate mitochondrial PE levels, we utilized our recently described Etn supplementation strategy (31) and showed that Etn rescues the respiratory growth of taz1Δ and crd1Δ cells in a dose-dependent manner, with an EC50 of 0.5 and 1.04 mm, respectively (Fig. 2, A–C). Whereas Etn fully rescued the respiratory growth of CL-depleted taz1Δ cells, it only partially rescued the growth of crd1Δ cells, which are completely deficient in CL (Fig. 2, A and C). Notably, the rescue was also observed in BY4742 genetic background (Fig. S1).
Figure 2.

Ethanolamine supplementation rescues respiratory growth of CL-deficient yeast cells. A, 10-fold serial dilutions of WT, taz1Δ, and crd1Δ cells were seeded on to the SC glucose, SC ethanol, and SC ethanol + 2 mm Etn media, and images were captured after 2 days of growth on SC glucose, 5 days on SC ethanol ± 2 mm Etn at 30 °C (upper panel), and 7 days on SC ethanol ± 2 mm Etn at 37 °C (lower panel). Data are representative of at least three independent experiments. B, growth of taz1Δ and crd1Δ cells in SC ethanol with the indicated concentrations of Etn (0–5 mm) was monitored by measuring absorbance at 600 nm at 72 or 48 h for taz1Δ cells and crd1Δ cells, respectively, and EC50 values for Etn were calculated using GraphPad Prism. Data are expressed as mean ± S.D. (n = 3). For growth comparison, WT cells were cultured in SC ethanol medium without Etn supplementation for the indicated temperatures and time periods. C, growth of BY4741 WT, taz1Δ, and crd1Δ cells in SC glucose, SC ethanol, and SC ethanol + 2 mm Etn media at 30 °C (upper panel) and 37 °C (lower panel) was monitored by measuring absorbance at 600 nm. Data are representative of at least three independent measurements. D, mitochondrial phospholipid composition of WT cells grown in SC ethanol and taz1Δ and crd1Δ cells grown in SC ethanol ± Etn. Data are expressed as mean ± S.D. (n = 3); **, p < 0.005; *, p < 0.05; NS, not significant. PI, phosphatidylinositol; PG, phosphatidylglycerol.
To confirm that Etn supplementation results in increased mitochondrial PE in taz1Δ and crd1Δ cells, we analyzed the mitochondrial phospholipid composition of cells grown with and without Etn supplementation. Consistent with the previous reports (19, 42, 43), mitochondrial phospholipid analysis of CL-deficient cells revealed that, when compared with WT cells, PE was already elevated by ∼25 and 50% in taz1Δ and crd1Δ cells, respectively (Fig. 2D). Addition of Etn to the growth media further increased mitochondrial PE levels in taz1Δ cells by 25% (Fig. 2D). The increase in PE was accompanied by a concomitant decrease in PC levels (Fig. 2D). However, in crd1Δ cells, neither PE nor PC was significantly altered upon Etn supplementation (Fig. 2D). These results indicate that exogenous supplementation of phospholipid precursor Etn leads to increased levels of PE in taz1Δ but not in crd1Δ mitochondria.
Ethanolamine-mediated rescue of CL deficiency is independent of its incorporation into PE
Although significant elevation of PE in taz1Δ cells upon Etn supplementation can explain the rescue of respiratory growth, the lack of a significant increase in PE upon Etn supplementation in crd1Δ cells suggests that PE may not be responsible for the observed rescue. To test this idea, we deleted the gene encoding the rate-limiting enzyme of the Kennedy pathway, ethanolaminephosphate cytidylyltransferase (Ect1), in taz1Δ and crd1Δ backgrounds. We analyzed the growth of ect1Δtaz1Δ and ect1Δcrd1Δ cells in respiratory media with and without Etn and observed that Etn supplementation rescued growth of the double mutants despite the absence of Ect1 (Fig. 3A and Fig. S2A). To confirm that the deletion of ECT1 abolished the increase in PE levels upon Etn supplementation, we measured the cellular PE levels in WT and individual single and double mutants with and without Etn supplementation. As expected in the ect1Δ genetic background, the cellular PE did not increase with Etn supplementation (Fig. 3B and Fig. S2B).
Figure 3.

Ethanolamine-mediated rescue of respiratory growth of CL-deficient cells is independent of PE biosynthesis. A, 10-fold serial dilutions of WT, ect1Δ, taz1Δ, and ect1Δtaz1Δ cells were seeded on to the indicated media, and images were captured after 2 days of growth on SC glucose and 7 days on SC ethanol ± 2 mm Etn at 37 °C. Data are representative of at least three independent experiments. B, total cellular PE levels of the indicated strains grown in SC ethanol ± 2 mm Etn. Data are expressed as mean ± S.D. (n = 3); *, p < 0.05, NS, not significant. C, 10-fold serial dilutions of WT, eki1Δcki1Δ, taz1Δ, eki1Δcki1Δtaz1Δ. D, WT, hnm1Δ, taz1Δ, and hnm1Δtaz1Δ cells were seeded onto the indicated media, and images were captured after 2 days of growth on SC glucose and 7 days on SC ethanol ± 2 mm Etn at 37 °C. Data are representative of at least three independent experiments. E, box plots of relative intracellular Etn abundance from WT, hnm1Δ, taz1Δ, and hnm1Δtaz1Δ grown in SC ethanol ± Etn. Etn levels were measured by LC-MS and expressed as peak intensity in arbitrary units (A.U.). Box plots show individual data points, median, first, and third quartiles, and greatest values within 1.5 inter-quartile range. Data are representative of at least three independent experiments. Data are expressed as mean ± S.D. (n = 3). F, WT and hnm1Δ cells were labeled with 1 μCi of [14C]Etn for 24 h, and radioactive Etn counts were measured in cpm (CPM) and expressed as % of WT. Data are expressed as mean ± S.D. (n = 3); ***, p < 0.001. G, growth of taz1Δhnm1Δ cells in SC ethanol with indicated concentrations of Etn (0–10 mm) was monitored by measuring absorbance at 600 nm at 72 h, and EC50 values for Etn were calculated using GraphPad Prism. Data are expressed as mean ± S.D. (n = 3). H, WT cells, grown in presence or absence of 10 mm choline, were radiolabeled with 1 μCi of [14C]Etn for 24 h, and intracellular radioactivity was measured and expressed as % of WT intensity. Data are expressed as mean ± S.D. (n = 3); ***, p < 0.001. I, 10-fold serial dilutions of WT, taz1Δ, and crd1Δ cells were seeded onto the indicated media, and images were captured after 2 days of growth on SC glucose and 7 days on SC ethanol ± Cho at 37 °C. Data are representative of at least three independent experiments.
To independently test the role of PE in rescuing CL deficiency, we used lyso-PE supplementation, which activates an alternative pathway for mitochondrial PE elevation (44). Lyso-PE supplementation did not rescue the respiratory growth of either the taz1Δ or the crd1Δ cells (Fig. S3, A and B) but, as expected, did rescue respiratory growth of psd1Δ cells that are deficient in mitochondrial PE (Fig. S3C). Together, these results suggest that Etn-mediated rescue of CL-depleted cells is independent of PE biosynthesis and is mediated by either Etn itself or its downstream metabolites.
Kennedy pathway intermediates are not required for the ethanolamine-mediated rescue of CL-deficient cells
To test the requirement of the Kennedy pathway intermediates in Etn-mediated rescue of taz1Δ cells, we constructed a triple mutant eki1Δcki1Δtaz1Δ strain that cannot convert Etn to phosphoethanolamine (PEtn) (45). We found that Etn supplementation was able to rescue the respiratory growth of the triple mutant in a manner similar to taz1Δ cells (Fig. 3C). This result suggests that Etn itself, not PEtn or the downstream metabolites of the Kennedy pathway, mediates the respiratory growth rescue observed in CL-deficient cells. Furthermore, we found that Etn-mediated rescue was independent of the Etn/Cho transporter, Hnm1, because deletion of HNM1 failed to abrogate Etn-mediated rescue (Fig. 3D). To test whether Etn is able to enter cells lacking Hnm1, we measured Etn levels in hnm1Δ and hnm1Δtaz1Δ cells by LC-MS (LC-MS) as well as by using radiolabeled [14C]Etn. Both of these experiments demonstrated that Etn could indeed enter hnm1Δ cells, albeit at ∼5-fold lower levels (Fig. 3, E and F). Consistent with the lower efficiency of Etn uptake in hnm1Δ cells, the EC50 of Etn was found to be ∼4-fold higher in taz1Δhnm1Δ cells as compared with taz1Δ cells (Fig. 3G). In addition to Etn, Hnm1 is known to transport choline (Cho), which also regulates the abundance of Hnm1 (46). Therefore, co-supplementation of Etn with Cho is expected to decrease Etn uptake. Indeed, co-incubation of 10 mm choline with radiolabeled [14C]Etn decreased its uptake by ∼8-fold (Fig. 3H). Under similar experimental conditions, Etn-mediated rescue of taz1Δ and crd1Δ cells is abrogated indicating that cellular uptake of Etn is required to rescue the respiratory growth defects of CL-deficient cells (Fig. 3I). These results suggested that either Etn or Etn-containing metabolites are responsible for the rescue. To identify other intracellular Etn-containing metabolites that may confer the rescue phenotype, we utilized LC-MS to investigate downstream products of Etn that may rescue CL-deficient cells. We found Etn supplementation resulted in an increase in N-palmitoylethanolamine levels (Fig. S4), raising the possibility that N-palmitoylethanolamine may serve as the bioactive mediator. However, we did not observe a positive genetic interaction between ECT1 and TAZ1 (Fig. 3A), i.e. the growth rate of ect1Δtaz1Δ cells, which have higher levels of N-palmitoylethanolamine even without Etn supplementation (Fig. S4), was not higher than that of taz1Δ cells, thus ruling out this possibility. Taken together, these results suggest a specific role of Etn in ameliorating the respiratory growth defect in CL-depleted cells.
Respiratory growth rescue of CL-deficient cells by Etn analogues
To test the specificity of Etn-mediated rescue, we supplemented respiratory media with other water-soluble lipid precursors, including monomethylethanolamine (MME), dimethylethanolamine (DME), choline (Cho), serine (Ser), and propanolamine (Prn) (Fig. 4A). MME, DME, and Cho act as substrates for the biosynthesis of phosphatidylmonomethylethanolamine (PMME), phosphatidyldimethylethanolamine (PDME), and PC, respectively. Both PMME and PDME serve as intermediates of PC, the most abundant mitochondrial phospholipid. Serine is a precursor for phosphatidylserine (PS), a less abundant mitochondrial phospholipid, whereas Prn is an Etn analog that is a substrate for phosphatidylpropanolamine (PP), a non-natural phospholipid (47). We found that Prn, and to a lesser extent MME, supplementation rescued respiratory growth of taz1Δ and crd1Δ cells, whereas DME, Cho, and Ser supplementation did not restore the respiratory growth of taz1Δ or crd1Δ cells (Figs. 4, B and C, and Fig. S5). These results show that the rescue is specific to Etn and its structurally similar analogues MME and Prn.
Figure 4.

Respiratory growth of CL-deficient cells is rescued by ethanolamine analogues propanolamine and monomethylethanolamine. A, chemical structures of Etn, MME, DME, Cho, Ser, and Prn. B, growth of WT and taz1Δ cells at 37 °C; C, WT and crd1Δ cells at 30 °C in SC ethanol (SCE), with the indicated supplementations. Growth was monitored by measuring absorbance at 600 nm and presented as a heat map. Data are average of five independent measurements.
Ethanolamine supplementation rescues MRC supercomplex levels in taz1Δ cells
To determine the biochemical mechanism of Etn-mediated respiratory growth rescue of CL-depleted cells, we determined the supra-molecular assembly of MRC complexes in taz1Δ cells, which are known to have reduced levels of MRC supercomplexes (3, 4, 23). Etn supplementation in taz1Δ cells partially restored the formation of the large supercomplex (III2IV2) and reduced the amounts of the free complex III dimer (III2) (Fig. 5, A and B) and the complex IV monomer (Fig. 5, C and D). Consistent with the respiratory growth rescue, Prn supplementation resulted in PP biosynthesis (Fig. S6A) and restoration of MRC supercomplex levels in taz1Δ cells (Fig. S6B). To determine whether the Etn-mediated restoration of supercomplex levels in taz1Δ cells was independent of an increase in mitochondrial PE levels, we examined supercomplex formation in taz1Δect1Δ cells, which cannot synthesize PE via the Kennedy pathway. Similar to taz1Δ cells, Etn was able to promote the formation of the large supercomplex in taz1Δect1Δ cells (Fig. 5, E and F). These results identify Etn and Prn as novel regulators of respiratory supercomplex formation in CL-depleted taz1Δ cells.
Figure 5.
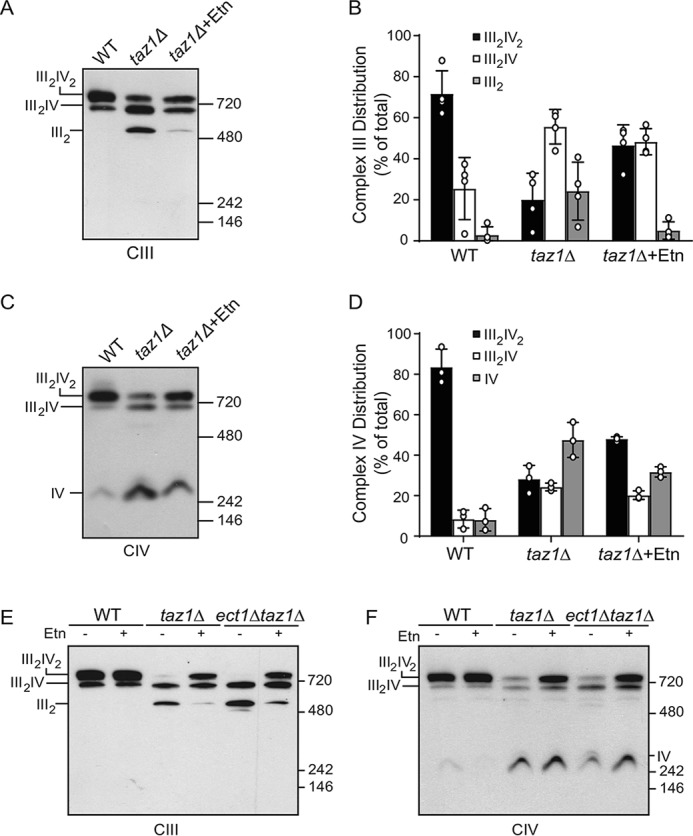
Ethanolamine supplementation partially restores supercomplex formation in taz1Δ cells. A, mitochondria from WT cells grown in SC ethanol and taz1Δ cells grown in SC ethanol ± 2 mm Etn were solubilized by 1% digitonin and subjected to BN-PAGE/Western blot analysis. Complexes containing MRC complex III were detected by anti-Rip1 antibody. B, relative abundance of MRC complex III containing complexes from A were quantified by densitometric analysis. Data are expressed as mean ± S.D. (n = 4). C, samples from A were probed with anti-Cox2 antibody to detect MRC complex IV containing supercomplexes and complex IV monomer. D, relative abundance of MRC complex IV containing complexes from C were quantified by densitometric analysis. Data are expressed as mean ± S.D. (n = 3). E, mitochondria from WT, taz1Δ, and ect1Δtaz1Δ cells grown in SC ethanol ± 2 mm Etn were solubilized by 1% digitonin and subjected to BN-PAGE/Western blot analysis and MRC complex III; F, complex IV was detected by probing with anti-Rip1 and anti-Cox2 antibodies, respectively. III2IV2, large supercomplex; III2IV, small supercomplex; III2, complex III dimer; IV, complex IV monomer.
Ethanolamine supplementation restores the expression of MRC subunits in crd1Δ cells
Previous reports have shown that a complete lack of CL in crd1Δ cells results in a more pronounced decrease in supercomplex formation and that CL is specifically and absolutely required for supercomplex reconstitution in vitro (5). Consistent with these reports, we found that the supercomplex formation in crd1Δ cells was not restored with Etn supplementation (Fig. 6, A–D). However, Etn supplementation did increase the levels of the complex IV monomer (Fig. 6, C and D), which were due to increased expression of MRC subunits of complex IV (Fig. 6E). Additionally, Etn supplementation also restored the levels of MRC complex III subunits (Fig. 6E). Consistent with an increase in MRC complex IV levels, the MRC complex IV activity was restored in crd1Δ mitochondria (Fig. 6F). These results suggest that critical levels of CL are required for supercomplex formation and that the observed partial rescue of respiratory growth of crd1Δ cells is due to the restoration of complex IV activity.
Figure 6.

Ethanolamine supplementation restores MRC complex IV levels and activity in crd1Δ cells. A, mitochondria from WT cells grown in SC ethanol and crd1Δ cells grown in SC ethanol ± 2 mm Etn were solubilized by 1% digitonin and subjected to BN-PAGE/Western blot analysis. MRC complex III containing complexes were detected by anti-Rip1 antibody. B, relative abundance of MRC complex III containing complexes from A were quantified by densitometric analysis. Data are expressed as mean ± S.D. (n = 3). C, samples from A were probed with anti-Cox2 antibody to detect MRC complex IV containing supercomplexes and complex IV monomer. D, relative abundance of MRC complex IV containing complexes from C were quantified by densitometric analysis. Data are expressed as mean ± S.D. (n = 3). E, MRC subunits of complexes II (Sdh1), III (Cor1 and Qcr2), and IV (Cox2 and Cox4) were analyzed by SDS-PAGE/Western blotting. Mitochondrial proteins aconitase and porin were used as a loading control. Data are representative of at least three independent experiments. F, digitonin-solubilized MRC complexes from WT and crd1Δ cells were separated by CN-PAGE, followed by in-gel activity staining for complex IV. In-gel activity of complex IV was quantified by densitometric analysis, and relative activity was plotted. Data were normalized to WT cells and expressed as mean ± S.D. (n = 3); *, p < 0.05.
Cox4 overexpression rescues MRC supercomplex levels in CL-deficient cells
To determine whether increased complex IV biogenesis upon Etn supplementation is sufficient to restore supercomplex formation in CL-deficient cells, we overexpressed the Cox4 subunit of MRC complex IV in taz1Δ and crd1Δ cells (Fig. 7A). Indeed, overexpression of Cox4 was sufficient to restore MRC supercomplexes in CL-deficient cells (Fig. 7, B and C). These results are consistent with the idea that Etn stimulates supercomplex formation by increasing the expression of MRC complex IV subunits.
Figure 7.

Cox4 overexpression rescues mitochondrial respiratory chain supercomplex assembly in cardiolipin-deficient cells. A, mitochondria from WT, taz1Δ, and crd1Δ cells, with or without Cox4 overexpression, were subjected to SDS-PAGE/Western blotting. B, mitochondria from WT, taz1Δ, and crd1Δ cells, with or without Cox4 overexpression, were solubilized by 1% digitonin and subjected to BN-PAGE/Western blot analysis. MRC complex III containing complexes were detected by anti-Rip1. C, MRC complex IV containing complexes were detected by anti-Cox2 antibodies, respectively. Data are representative of three independent measurements.
Ethanolamine supplementation reduces protein carbonylation in CL-deficient cells
One of the hallmarks of CL depletion and BTHS pathology across multiple model systems, including the yeast BTHS model taz1Δ, is increased oxidative stress (25, 48, 49). To determine whether Etn supplementation could alleviate oxidative stress in CL-deficient cells, we measured cellular protein carbonylation, a sensitive indicator of oxidative stress (25). Consistent with a previous report (25), we observed that taz1Δ and crd1Δ cells had increased cellular protein carbonylation indicative of enhanced oxidative stress. Etn supplementation reduced protein carbonylation in both taz1Δ and crd1Δ cells (Fig. 8A). To determine whether Etn-mediated reduction of protein carbonylation was independent of PE biosynthesis, we measured protein carbonylation in the double mutants taz1Δect1Δ and crd1Δect1Δ and found that Etn was able to reduce carbonylation (Fig. 8B). Together, these results demonstrate that Etn supplementation reduces oxidative stress in taz1Δ and crd1Δ cells.
Figure 8.
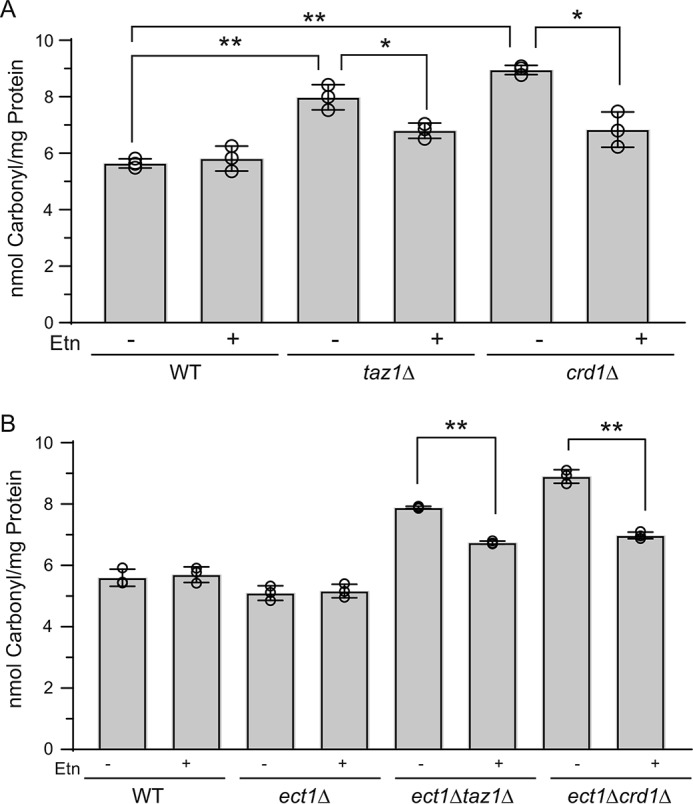
Ethanolamine supplementation reduces protein carbonylation in CL-deficient cells. WT, taz1Δ, and crd1Δ (A) or WT, ect1Δ, ect1Δtaz1Δ, and ect1Δcrd1Δ cells (B) were grown in SC ethanol ± 2 mm Etn to early stationary phase; protein was extracted, and protein carbonylation was measured as described under “Experimental procedures.” Data are expressed as mean ± S.D. (n = 3); **, p < 0.005; *, p < 0.05.
Ethanolamine supplementation does not rescue respiratory growth defects of cells lacking MRC assembly factors
Next, we wanted to determine whether Etn-mediated rescue is specific to CL deficiency or whether any MRC assembly defect could be rescued. To test this possibility, we performed rescue experiments on a number of MRC assembly factors, including the recently discovered mitochondrial proteins, Rcf1 and Rcf2, which are proposed to promote supercomplex formation (50–52). Consistent with the previous report (51), rcf1Δ cells exhibited more pronounced growth defect compared with rcf2Δ cells in respiratory media (Fig. 9A). However, Etn supplementation did not rescue the respiratory growth of either rcf1Δ or rcf2Δ cells (Fig. 9A). In addition, Etn supplementation could not rescue the respiratory growth of sdh2Δ, bcs1Δ, shy1Δ, and atp12Δ cells, which are mutants defective in MRC complexes II–V, respectively (Fig. 9, B and C). Together, these results suggest that Etn-mediated rescue of mitochondrial defects is specific to CL deficiency.
Figure 9.

Ethanolamine-mediated rescue is specific to CL deficiency. A, growth of WT, rcf1Δ, and rcf2Δ cells in SC glucose and SC ethanol ± 2 mm Etn media was monitored at 30 °C (upper panel) and 37 °C (lower panel) by measuring absorbance at 600 nm. Data are representative of at least two independent measurements. B, 10-fold serial dilutions of WT, taz1Δ, sdh2Δ, bcs1Δ, shy1Δ, and atp12Δ cells were seeded onto the indicated media, and images were captured after 2 days of growth on SC glucose and 5 days on SC ethanol ± 2 mm Etn at 30 °C. Data are representative of at least three independent trials. C, mitochondria from YP galactose grown WT, sdh2Δ, bcs1Δ, and shy1Δ cells were subjected to Western blot analysis. Mitochondrial respiratory chain complexes II, III, and IV subunits were probed using anti-Sdh2, anti-Rip1, and anti-Cox2 antibodies, respectively. Porin was used as a loading control.
Discussion
We report a novel role for Etn in ameliorating mitochondrial dysfunction caused by perturbations in mitochondrial membrane phospholipid composition due to CL deficiency. We serendipitously discovered this new role of Etn while testing the hypothesis that elevating mitochondrial PE by exogenous Etn supplementation could rescue mitochondrial defects caused by CL deficiency. We found that Etn ameliorates mitochondrial dysfunction in CL-deficient cells but, surprisingly, without increasing PE levels. Specifically, Etn exerted its protective effect by increasing the expression of MRC complex III and IV subunits and thereby restoring MRC supercomplex formation in CL-depleted cells.
Whereas the phospholipid precursors choline and inositol are known to influence cellular physiology (53), very little is known about the role of Etn outside of its incorporation into PE. Previous work has shown that Etn and PEtn can directly modulate mitochondrial function (54, 55). In fact, pharmacological elevation of PEtn by meclizine, an anti-nausea drug, has been shown to protect the heart, brain, and the kidney from the ischemia–reperfusion injury by altering mitochondrial bioenergetics (56, 57). Therefore, we asked whether Etn mediated its effect by incorporation into PEtn. We addressed this question by constructing a triple knockout strain, taz1Δeki1Δcki1Δ, which cannot phosphorylate ethanolamine. Our results show that Etn supplementation was still able to rescue the respiratory-deficient growth of the triple knockout cells, indicating that PEtn is unlikely to contribute to the observed rescue (Fig. 3C). The protective effect of Etn on CL-deficient cells was specific because other phospholipid precursors, including Ser and Cho, were unable to rescue the respiratory growth defect of CL-deficient cells (Fig. 4). We determined that the amine group of ethanolamine is critical for its biological activity because addition of the methyl groups on the nitrogen of the amine group progressively diminished its efficacy, as exemplified by the lack of rescue with DME and Cho and only a partial rescue with MME (Fig. 4). Consistent with this observation, Prn, a structural analogue of Etn containing the primary amine group, was also able to rescue the respiratory growth of CL-deficient cells with the same efficacy as that of Etn.
Because the Etn-mediated rescue was independent of the Etn–Kennedy pathway, we considered the possibility that Etn can be incorporated into some other metabolite, which in turn can exert its protective effect. Therefore, we utilized a LC-MS system to discover Etn-containing molecules and found N-palmitoylethanolamine levels to be significantly increased upon Etn supplementation (Fig. S4). This observation raised the possibility that the protective effect of Etn may, in fact, be mediated by N-palmitoylethanolamine, which has been shown to increase upon oxidative stress in yeast (58). However, we noticed that there were higher basal levels of N-palmitoylethanolamine in ect1Δtaz1Δ cells even without Etn supplementation (Fig. S4), and these cells still exhibit respiratory growth defect (Fig. 3A), thus ruling out any possible role of N-palmitoylethanolamine in mediating the protective effect. Interestingly, higher basal levels of N-palmitoylethanolamine in ect1Δtaz1Δ cells suggest that blocking the Etn–Kennedy pathway reroutes endogenous Etn to N-palmitoylethanolamine.
To understand the biochemical basis by which Etn restored the respiratory growth of CL-depleted cells, we examined the formation and function of MRC supercomplexes. Previous reports have shown that CL deficiency results in diminished MRC supercomplexes, leading to respiratory defects in both the yeast and mammalian models of BTHS (23, 24). Remarkably, exogenous supplementation of Etn was able to rescue MRC supercomplex levels in the CL-depleted mitochondrial membranes of taz1Δ cells (Fig. 5). The observed Etn-mediated rescue occurred despite MLCL accumulation (Fig. 2D), which has been reported to be the primary cause of mitochondrial dysfunction (20, 21). Thus, our finding suggests that detrimental effects of MLCL accumulation can be overcome.
Previous studies have shown that the loss of CL and its precursor phosphatidylglycerol causes an inhibition of translation of MRC subunits, specifically decreasing the levels of complex III and IV subunits (26, 27). Thus Etn-mediated rescue of complexes III and IV subunit levels provides a biochemical mechanism for the rescue of respiratory growth of CL-deficient cells (Fig. 6E). We further demonstrated that overexpression of the complex IV subunit is sufficient to restore supercomplex formation in CL-deficient cells (Fig. 7), a finding consistent with the recent report showing that supercomplex formation is the function of MRC complex IV abundance (59). Based on these observations, we propose a model where the primary effect of Etn is to restore the expression of MRC complex III and IV subunits (Figs. 6, E and F, and 7), which in turn rescue complex IV activity and promote supercomplex formation in CL-deficient cells. Although the molecular mechanism by which Etn increases the expression of MRC subunits remains elusive at this time, a number of important conclusions can be drawn from our study. First, Etn mediates its effect on mitochondria either directly or by incorporation into a yet unidentified metabolite that is neither PE nor N-palmitoylethanolamine. Second, the protective effect of Etn is specific to CL deficiency because Etn supplementation fails to rescue mitochondrial dysfunction in cells lacking MRC assembly factors. Third, Etn is able to overcome the accumulation of MLCL, which has been shown to be the primary cause of mitochondrial dysfunction in yeast models of BTHS (20, 21). Fourth, increased cytosolic Etn levels in Etn-supplemented hnm1Δ cells suggest existence of alternative Etn import machinery. Finally, exogenously supplemented Etn or endogenous Etn, which builds up in cells with a disruption in the Etn–Kennedy pathway, is rerouted to N-acylethanolamine biosynthesis.
In a recent review, Henry et al. (53) predicted novel roles of lipid precursors in cellular physiology and suggested the suitability of the yeast S. cerevisiae model system to uncover such processes. Our work, demonstrating a novel role of the soluble lipid precursor Etn in ameliorating mitochondrial dysfunction caused by CL deficiency in yeast model of BTHS, provides strong support for their prediction.
Experimental procedures
Yeast strains, growth medium composition, and culture conditions
S. cerevisiae strains used in this study are listed in Table 1. These strains were confirmed by PCR as well as by replica plating on dropout plates. Single, double, and triple knockout yeast strains were constructed by one-step gene disruption using hygromycin or clonNat (nourseothricin) resistance cassettes amplified from pFA6a-hphNT1 or pFA6a-natNT2 plasmids, respectively (60). The primers used for one-step gene disruption are listed in Table 2. For growth in liquid media, strains were pre-cultured in YPD medium (1% yeast extract, 2% peptone, and 2% dextrose) and inoculated into SC media (0.2% dropout mix containing amino acid and other supplements as described previously (61), 0.17% yeast nitrogen base without amino acids and ammonium sulfate, and 0.5% ammonium sulfate) containing either 2% glucose, 2% lactate, pH 5.5, or 2% ethanol and grown to late logarithmic phase. Solid media were prepared by the addition of 2% agar to the media described above. Yeast strains were inoculated into liquid SC glucose, SC lactate, and SC ethanol at a starting A600 of 0.1, and growth was monitored for up to 1 day (SC glucose), 2 days (SC lactate), or 3 days (SC ethanol), respectively. For growth on solid media, 10-fold serial dilutions of overnight pre-cultures were seeded on SC glucose or SC ethanol plates and incubated at 30 or 37 °C for 2 days (SC glucose) and 5 or 7 days (SC ethanol), respectively. For Etn supplementation experiments, 2 mm Etn was added to SC growth medium. For media containing lyso-PE, 1% (v/v) Tergitol Nonidet P-40 was included, and lyso-PE was added to the final concentration of 0.5 mm from a sterile 25 mm stock solution in 10% (v/v) Tergitol Nonidet P-40 (44).
Table 1.
S. cerevisiae strains used in this study
| Yeast strains | Genotype | Source |
|---|---|---|
| BY4742 WT | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0 | M. L. Greenberg |
| BY4742 taz1Δ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, taz1Δ::hphNT1 | This study |
| VGY1 | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, crd1Δ::URA3 | M. L. Greenberg |
| BY4741 WT | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | M. L. Greenberg |
| BY4741 taz1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, taz1Δ::kanMX4 | Open Biosystems |
| BY4741 crd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, crd1Δ::kanMX4 | Open Biosystems |
| BY4741 psd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, psd1Δ::kanMX4 | Open Biosystems |
| BY4741 ect1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, ect1Δ::hphNT1 | This study |
| BY4741 taz1Δect1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, taz1Δ::kanMX4, ect1Δ::hphNT1 | This study |
| BY4741 crd1Δect1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, crd1Δ::kanMX4, ect1Δ::hphNT1 | This study |
| BY4741 eki1Δcki1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, eki1Δ::natNT2, cki1Δ::hphNT1 | This study |
| BY4741 eki1Δcki1Δtaz1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, taz1Δ::kanMX4, eki1Δ::natNT2, cki1Δ::hphNT1 | This study |
| BY4741 hnm1Δtaz1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, hnm1Δ::kanMX4, taz1Δ::hphNT1 | This study |
| BY4741 hnm1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, hnm1Δ::kanMX4 | Open Biosystems |
| BY4741 sdh2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sdh2Δ::kanMX4 | Open Biosystems |
| BY4741 bcs1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, bcs1Δ::kanMX4 | Open Biosystems |
| BY4741 shy1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, shy1Δ::kanMX4 | Open Biosystems |
| BY4741 atp12Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, atp12Δ::kanMX4 | Open Biosystems |
| BY4741 rcf1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rcf1Δ::kanMX4 | Open Biosystems |
| BY4741 rcf2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rcf2Δ::kanMX4 | Open Biosystems |
Table 2.
Primers used in this study
The boldface letters represent sequences that are complementary to the flanking sequences of the hygromycin and clonNat (nourseothricin) resistance cassettes of pFA6a-hphNT1 or pFA6a-natNT2 plasmids, respectively.
| Name | Sequence (5′ to 3′) |
|---|---|
| COX4F | CCCGTTCTCGAGCAGCTGACTGTCCACCAATAAGATC |
| COX4R | CCCGGTGGATCCCGAAGCAATACCGGTAAGCTGCTTA |
| TAZ1 S1 | CATTTTCAAAAAAAAAAAAAGTAAAGTTTTCCCTATCAAATGCGTACGCTGCAGGTCGAC |
| TAZ1 S2 | TGAAATTTAAGCAATTAAATTCGTGTAAATACTAGCATGTAATCGATGAATTCGAGCTCG |
| ECT1 S1 | AATGCTTTACAGGATCGGGACTTGAAATATACTGACTGGATGCGTACGCTGCAGGTCGAC |
| ECT1 S2 | CCATTTAATTTACGTTCGAAGAAGTTTTCAACATTTGTTTAATCGATGAATTCGAGCTCG |
| EKI1 S1 | TAGCAGAAATTAACAGATACAGATCTGCAATTTGGCATAATGCGTACGCTGCAGGTCGAC |
| EKI1 S2 | ATCGCAGTGAATAGAAAAATACTTGATTGTGTATACAGCTTATCGATGAATTCGAGCTCG |
| CKI1 S1 | TACACACACATAGATACGCACGTAAAATTAGAGCAAAAGATGCGTACGCTGCAGGTCGAC |
| CKI1 S2 | TTATTTCCTTGGCCTTTGTTGAAGGAATTCGTATACGTATTATCGATGAATTCGAGCTCG |
Plasmids
Yeast COX4 gene was cloned into a multicopy plasmid (pRS426) under control of the native promoter using primers listed in Table 2.
Mitochondrial isolation
Isolation of mitochondria was performed as described previously (62). Mitochondria were isolated from yeast cells grown to late logarithmic phase and were subsequently used for SDS-PAGE Western blot analysis as well as in-gel activity assays. For obtaining gradient-purified mitochondrial fractions, crude mitochondria were loaded onto a sucrose step gradient (60, 32, 23 and 15%) and centrifuged at 134,000 × g for 1 h. The intact mitochondria re-covered from the gradient interface (60 and 32%) were washed in isotonic buffer, pelleted at 10,000 × g, and subsequently used for BN-PAGE/Western blot analysis and mitochondrial phospholipid quantification. Protein concentrations were determined by the BCA assay (ThermoFisher Scientific).
Mitochondrial and cellular phospholipid measurements
For the quantification of mitochondrial phospholipids, lipids were extracted from gradient-purified mitochondria (1.5 mg of protein) using the Folch method (63), and individual phospholipids were separated by two-dimensional TLC using the following solvent systems: chloroform/methanol/ammonium hydroxide (65:35:5) in the first dimension followed by chloroform/acetic acid/methanol/water (75:25:5:2.2) in the second dimension (47). Phospholipids were visualized with iodine vapor, scraped into acid-washed glass tubes, and Pi was quantified (64). For the quantification of cellular PE, phospholipids were extracted from yeast cells (0.5 g wet weight) using the Folch method (63); Pi was quantified, and 30 nmol of phospholipids were separated into individual phospholipid class on HPTLC Silica Gel 60 plates (EMD Millipore 1.11764.0001) by one-dimensional TLC using chloroform/methanol/ammonium hydroxide (50:50:3) (65). Phospholipids were visualized with copper sulfate charring, and bands were quantified using ImageJ (65).
SDS and blue native–PAGE
SDS-PAGE was performed on mitochondrial samples solubilized in lysis buffer (150 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitor mixture (Roche Diagnostics). Protein extracts were separated on NuPAGE 4–12% BisTris gels (Life Technologies, Inc.) and transferred to PVDF membranes using a Trans-Blot transfer cell (Bio-Rad). Membranes were blocked in 5% fatty acid–free BSA dissolved in TBS with 0.1% Tween 20 and probed with the indicated antibodies. BN-PAGE was performed to separate native MRC complexes as described previously (66). Briefly, yeast gradient–purified mitochondria were solubilized in buffer containing 1% digitonin (6 g of detergent/g of mitochondrial protein) and incubated for 15 min at 4 °C. Following a clarifying spin at 20,000 × g (30 min, 4 °C), 50× G-250 sample additive was added to the supernatant, and 20 μg of protein was loaded on a 3–12% gradient native PAGE BisTris gel (Life Technologies, Inc.). Western blotting was performed using a Mini-PROTEAN Tetra cell (Bio-Rad). The membrane was blocked in 5% nonfat milk in TBS with 0.1% Tween 20 and probed with antibodies as indicated. Primary antibodies used for yeast proteins were as follows: Cox2, 1:50,000 (Abcam 110271); Cox4, 1:5000 (Abcam 110272); Sdh1, 1:10,000 (from Dr. Dennis Winge); Sdh2, 1:5000 (from Dr. Dennis Winge); Rip1, 1:100,000 (from Dr. Vincenzo Zara); Cor1/QCR2, 1:50,000 (from Dr. Vincenzo Zara); Aco1, 1:2000 (from Dr. Chris Meisinger); porin, 1:100,000 (Abcam 110326). Secondary antibodies (1:5000) were incubated for 1 h at room temperature, and membranes were developed using Western Lightning Plus-ECL (PerkinElmer Life Sciences). Quantification was performed using the gel analysis method in ImageJ.
In-gel activity measurements
In-gel activity measurements for mitochondrial respiratory chain complexes were performed as described previously (67). Clear native-PAGE (CN-PAGE) was used to avoid interference of Coomassie Blue with activity measurements. Briefly, mitochondria solubilized in 1% digitonin were resolved on a 4–16% gradient native PAGE BisTris gel (Life Technologies, Inc.) with the addition of 0.05% n-dodecyl β-d-maltoside and 0.05% sodium deoxycholate in the cathode buffer. Gels were loaded with 90 μg of protein and incubated in MRC complex IV activity staining solutions as reported previously (67). Equal loading was determined by Coomassie Blue stain, and total protein and band intensity quantification was determined using the gel analysis method in ImageJ.
Steady-state labeling of yeast cells with [14C]ethanolamine
The yeast strains were grown in the presence of [14C]ethanolamine (1 μCi) for 24 h at 30 °C with shaking. 1 ml of ∼2.5 A600 cells were centrifuged at 3000 × g for 5 min, followed by washing four times with ice-cold water. The [14C]ethanolamine incorporation was measured using a scintillation counter by resuspending the cell pellets into the scintillation mixture. [14C]Etn incorporation values in hnm1Δ cells were normalized with WT values.
Quantification of protein carbonyl content
The protein carbonyl content was measured by determining the amount of 2,4-dinitrophenylhydrazone (DNP) formed upon reaction with 2,4-dinitrophenyl hydrazine (DNPH), as described previously (25, 68). Yeast cells were grown at 30 °C to the early stationary phase in SC ethanol medium, and cell extracts were used for subsequent protein carbonylation measurements. Nucleic acids were removed from the cell extracts with 1.0% streptomycin sulfate, and the resulting protein samples were incubated with 10 mm DNPH in 2 m HCl at room temperature for 60 min in the dark. Proteins were precipitated by addition of TCA to a final concentration of 10%, and the pellets were washed with ethanol/ethyl acetate (1:1) to remove the free DNPH. The final protein pellets were dissolved in 6 m guanidine hydrochloride solution containing 20 mm potassium phosphate, pH 2.4. The carbonyl content was calculated from the absorbance maximum of DNP measured at 370 nm.
Mass spectrometry
Ethanolamine and N-palmitoylethanolamine levels were determined in yeast cells using LC-MS (LC-MS) method, as described previously (55). Yeast cells cultured in 5 ml of SC ethanol medium with and without 2 mm Etn supplementation were pelleted, washed, and frozen. Cell pellets were resuspended in an adjusted volume of ice-cold 80:20 methanol/water with 150 μl of solvent per 10 mg of cells that were transferred into a 2.0-ml impact-resistant tube containing 300 mg of 1-mm zirconium beads. Samples underwent three 15-s homogenization cycles at 6400 Hz in a Precellys 24® tissue homogenizer. To ensure complete cell lysis, samples were thereafter sonicated for 2 min and vortexed for 30 s. Samples were then placed in a −20 °C freezer for 30 min to allow for complete protein precipitation. Samples were thereafter vortexed again for 30 s and centrifuged at 14,000 × g for 10 min at 4 °C, and supernatants were transferred to LC-MS vials containing 200-μl glass inserts. An injection volume of 2.0 μl was used so that ∼134 μg of yeast cells were injected for all samples. LC-MS/MS-based metabolomics analysis was performed using a ThermoFisher Scientific QExactive Orbitrap mass spectrometer coupled to a ThermoFisher Scientific Vanquish UPLC system. Chromatographic separation of polar metabolites was achieved using a Millipore (Sequant) Zic-pHILIC 2.1 × 150-mm, 5-μm column maintained at 25 °C. Compounds were eluted via a 19-min linear gradient starting from 90:10 acetonitrile, 20 mm ammonium bicarbonate to 45:55 acetonitrile, 20 mm ammonium bicarbonate. Chromatographic separation of nonpolar metabolites was achieved using an Agilent Eclipse Plus C18 RRHT 1.8 um 2.1 × 50-mm column maintained at 50 °C with a 13-min linear gradient beginning with 5:95 water with 0.2% acetic acid, acetonitrile:isopropanol (1:1) with 0.2% acetic acid and ending with 100% acetonitrile:isopropanol (1:1) with 0.2% acetic acid. A ThermoFisher Scientific Q-Exactive Orbitrap mass spectrometer was operated in positive and negative ion modes using a heated electrospray ionization source at 35,000 resolution for polar metabolites and 70,000 resolution for non-polar, 100-ms ion trap time for MS1 and 17,500 resolution, and 50-ms ion trap time for MS2 collection. For polar compounds, data were collected over a mass range of m/z 59–885, using a sheath gas flow rate of 40 units, auxiliary gas flow rate of 20 units, sweep gas flow rate of 2 units, spray voltage of 3.5 and 2.5 kV for positive- and negative-ion modes, respectively, capillary inlet temperature of 275 °C, auxillary gas heater temperature of 350 °C, and an S-lens RF level of 45. For MS2 collection, MS1 ions were isolated using a 1.0 m/z window and fragmented using a stepped normalized collision energy of 15, 30, and 45. For non-polar compounds, the mass range was m/z 120–1800, with an auxiliary gas flow rate of 10 units, spray voltage of 3.55 kV for negative ion mode, and capillary inlet temperature of 265 °C with all other parameters kept the same. Fragmented ions were placed on dynamic exclusion for 30 s before another round of fragmentation. Collected data were imported into the mzMine 2.26 software suite for analysis.
Author contributions
W. B. B., C. D. B., J. K. N., K. A. L., G. Z., U. P., M. J., and V. M. G. data curation; W. B. B., C. D. B., J. K. N., G. L. A., U. P., M. J., and V. M. G. formal analysis; W. B. B., C. D. B., J. K. N., G. L. A., K. A. L., G. Z., U. P., and M. J. validation; W. B. B., C. D. B., J. K. N., G. L. A., K. A. L., G. Z., and U. P. investigation; W. B. B., C. D. B., J. K. N., K. A. L., G. Z., and U. P. methodology; W. B. B., C. D. B., J. K. N., and V. M. G. writing-original draft; W. B. B., C. D. B., and V. M. G. writing-review and editing; U. P., M. J., and V. M. G. supervision; M. J. and V. M. G. resources; M. J. and V. M. G. funding acquisition; V. M. G. conceptualization; V. M. G. project administration.
Supplementary Material
Acknowledgments
We thank Miriam L. Greenberg (Wayne State University) for yeast strains and Dennis Winge (University of Utah), Vincenzo Zara (Università del Salento), and Chris Meisinger (University of Freiburg) for their generous gift of antibodies. We also thank members of the Gohil lab, including Ashley Adams and Donna Iadarola, for their valuable comments in the preparation of this manuscript.
This work was supported in part by Welch Foundation Grant A-1810, American Heart Association Award 16GRNT31020028, National Institutes of Health Grants R01GM111672 (to V. M. G.) and 1R01ES027595, 1R03HL133720, and 1S10OD020025 (to M. J.), and the University of California, San Diego, Frontiers of Innovation Scholars Program (to K. A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
- MRC
- mitochondrial respiratory chain
- BTHS
- Barth syndrome
- PE
- phosphatidylethanolamine
- MLCL
- monolyso-cardiolipin
- PC
- phosphatidylcholine
- PS
- phosphatidylserine
- PA
- phosphatidic acid
- Etn
- ethanolamine
- Cho
- choline
- Prn
- propanolamine
- PP
- phosphatidylpropanolamine
- CL
- cardiolipin
- DME
- dimethylethanolamine
- PMME
- phosphatidylmonomethylethanolamine
- MME
- monomethylethanolamine
- PDME
- phosphatidyldimethylethanolamine
- DNP
- dinitrophenylhydrazone
- DNPH
- 2,4-dinitrophenyl hydrazine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PEtn
- phosphoethanolamine
- Prn
- propanolamine
- BN-PAGE
- blue native–PAGE
- CN-PAGE
- clear native-PAGE.
References
- 1. Horvath S. E., and Daum G. (2013) Lipids of mitochondria. Prog. Lipid Res. 52, 590–614 10.1016/j.plipres.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 2. Raja V., and Greenberg M. L. (2014) The functions of cardiolipin in cellular metabolism–potential modifiers of the Barth syndrome phenotype. Chem. Phys. Lipids 179, 49–56 10.1016/j.chemphyslip.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang M., Mileykovskaya E., and Dowhan W. (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 10.1074/jbc.C200551200 [DOI] [PubMed] [Google Scholar]
- 4. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., and Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 10.1074/jbc.M308366200 [DOI] [PubMed] [Google Scholar]
- 5. Bazán S., Mileykovskaya E., Mallampalli V. K., Heacock P., Sparagna G. C., and Dowhan W. (2013) Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J. Biol. Chem. 288, 401–411 10.1074/jbc.M112.425876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schägger H., and Pfeiffer K. (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783 10.1093/emboj/19.8.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaban Y., Boekema E. J., and Dudkina N. V. (2014) Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilization. Biochim. Biophys. Acta 1837, 418–426 10.1016/j.bbabio.2013.10.004 [DOI] [PubMed] [Google Scholar]
- 8. Genova M. L., and Lenaz G. (2014) Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 1837, 427–443 10.1016/j.bbabio.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 9. Acin-Perez R., and Enriquez J. A. (2014) The function of the respiratory supercomplexes: the plasticity model. Biochim. Biophys. Acta 1837, 444–450 10.1016/j.bbabio.2013.12.009 [DOI] [PubMed] [Google Scholar]
- 10. Letts J. A., Fiedorczuk K., and Sazanov L. A. (2016) The architecture of respiratory supercomplexes. Nature 537, 644–648 10.1038/nature19774 [DOI] [PubMed] [Google Scholar]
- 11. Gu J., Wu M., Guo R., Yan K., Lei J., Gao N., and Yang M. (2016) The architecture of the mammalian respirasome. Nature 537, 639–643 10.1038/nature19359 [DOI] [PubMed] [Google Scholar]
- 12. Wu M., Gu J., Guo R., Huang Y., and Yang M. (2016) Structure of mammalian respiratory supercomplex I1III2IV1. Cell 167, 1598–1609.e10 10.1016/j.cell.2016.11.012 [DOI] [PubMed] [Google Scholar]
- 13. Barth P. G., Scholte H. R., Berden J. A., Van der Klei-Van Moorsel J. M., Luyt-Houwen I. E., Van't Veer-Korthof E. T., Van der Harten J. J., and Sobotka-Plojhar M. A. (1983) An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355 10.1016/0022-510X(83)90209-5 [DOI] [PubMed] [Google Scholar]
- 14. Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., and Toniolo D. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 10.1038/ng0496-385 [DOI] [PubMed] [Google Scholar]
- 15. Xu Y., Kelley R. I., Blanck T. J., and Schlame M. (2003) Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 278, 51380–51385 10.1074/jbc.M307382200 [DOI] [PubMed] [Google Scholar]
- 16. Xu Y., Malhotra A., Ren M., and Schlame M. (2006) The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224 10.1074/jbc.M606100200 [DOI] [PubMed] [Google Scholar]
- 17. Vreken P., Valianpour F., Nijtmans L. G., Grivell L. A., Plecko B., Wanders R. J., and Barth P. G. (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382 10.1006/bbrc.2000.3952 [DOI] [PubMed] [Google Scholar]
- 18. Schlame M., Kelley R. I., Feigenbaum A., Towbin J. A., Heerdt P. M., Schieble T., Wanders R. J., DiMauro S., and Blanck T. J. (2003) Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999 10.1016/j.jacc.2003.06.015 [DOI] [PubMed] [Google Scholar]
- 19. Gu Z., Valianpour F., Chen S., Vaz F. M., Hakkaart G. A., Wanders R. J., and Greenberg M. L. (2004) Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51, 149–158 [DOI] [PubMed] [Google Scholar]
- 20. Ye C., Lou W., Li Y., Chatzispyrou I. A., Hüttemann M., Lee I., Houtkooper R. H., Vaz F. M., Chen S., and Greenberg M. L. (2014) Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant: implications for Barth syndrome. J. Biol. Chem. 289, 3114–3125 10.1074/jbc.M113.529487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baile M. G., Sathappa M., Lu Y. W., Pryce E., Whited K., McCaffery J. M., Han X., Alder N. N., and Claypool S. M. (2014) Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 289, 1768–1778 10.1074/jbc.M113.525733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valianpour F., Mitsakos V., Schlemmer D., Towbin J. A., Taylor J. M., Ekert P. G., Thorburn D. R., Munnich A., Wanders R. J., Barth P. G., and Vaz F. M. (2005) Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 46, 1182–1195 10.1194/jlr.M500056-JLR200 [DOI] [PubMed] [Google Scholar]
- 23. Brandner K., Mick D. U., Frazier A. E., Taylor R. D., Meisinger C., and Rehling P. (2005) Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214 10.1091/mbc.e05-03-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKenzie M., Lazarou M., Thorburn D. R., and Ryan M. T. (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 10.1016/j.jmb.2006.06.057 [DOI] [PubMed] [Google Scholar]
- 25. Chen S., He Q., and Greenberg M. L. (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 68, 1061–1072 10.1111/j.1365-2958.2008.06216.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ostrander D. B., Zhang M., Mileykovskaya E., Rho M., and Dowhan W. (2001) Lack of mitochondrial anionic phospholipids causes an inhibition of translation of protein components of the electron transport chain. A yeast genetic model system for the study of anionic phospholipid function in mitochondria. J. Biol. Chem. 276, 25262–25272 10.1074/jbc.M103689200 [DOI] [PubMed] [Google Scholar]
- 27. Su X., and Dowhan W. (2006) Translational regulation of nuclear gene COX4 expression by mitochondrial content of phosphatidylglycerol and cardiolipin in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 743–753 10.1128/MCB.26.3.743-753.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., and Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 10.1074/jbc.M909868199 [DOI] [PubMed] [Google Scholar]
- 29. Koshkin V., and Greenberg M. L. (2000) Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 347, 687–691 10.1042/0264-6021:3470687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koshkin V., and Greenberg M. L. (2002) Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 364, 317–322 10.1042/bj3640317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker C. D., Basu Ball W., Pryce E. N., and Gohil V. M. (2016) Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol. Biol. Cell 27, 2161–2171 10.1091/mbc.e15-12-0865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Böttinger L., Horvath S. E., Kleinschroth T., Hunte C., Daum G., Pfanner N., and Becker T. (2012) Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 423, 677–686 10.1016/j.jmb.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tasseva G., Bai H. D., Davidescu M., Haromy A., Michelakis E., and Vance J. E. (2013) Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 288, 4158–4173 10.1074/jbc.M112.434183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schuler M. H., Di Bartolomeo F., Mårtensson C. U., Daum G., and Becker T. (2016) Phosphatidylcholine affects inner membrane protein translocases of mitochondria. J. Biol. Chem. 291, 18718–18729 10.1074/jbc.M116.722694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holthuis J. C., and Menon A. K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57 10.1038/nature13474 [DOI] [PubMed] [Google Scholar]
- 36. Gohil V. M., and Greenberg M. L. (2009) Mitochondrial membrane biogenesis: phospholipids and proteins go hand in hand. J. Cell Biol. 184, 469–472 10.1083/jcb.200901127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gohil V. M., Thompson M. N., and Greenberg M. L. (2005) Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J. Biol. Chem. 280, 35410–35416 10.1074/jbc.M505478200 [DOI] [PubMed] [Google Scholar]
- 38. Osman C., Haag M., Potting C., Rodenfels J., Dip P. V., Wieland F. T., Brügger B., Westermann B., and Langer T. (2009) The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 184, 583–596 10.1083/jcb.200810189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Joshi A. S., Thompson M. N., Fei N., Hüttemann M., and Greenberg M. L. (2012) Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287, 17589–17597 10.1074/jbc.M111.330167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Becker T., Horvath S. E., Böttinger L., Gebert N., Daum G., and Pfanner N. (2013) Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins. J. Biol. Chem. 288, 16451–16459 10.1074/jbc.M112.442392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rietveld A. G., Killian J. A., Dowhan W., and de Kruijff B. (1993) Polymorphic regulation of membrane phospholipid composition in Escherichia coli. J. Biol. Chem. 268, 12427–12433 [PubMed] [Google Scholar]
- 42. Zhong Q., Gohil V. M., Ma L., and Greenberg M. L. (2004) Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J. Biol. Chem. 279, 32294–32300 10.1074/jbc.M403275200 [DOI] [PubMed] [Google Scholar]
- 43. Tuller G., Hrastnik C., Achleitner G., Schiefthaler U., Klein F., and Daum G. (1998) YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae. FEBS Lett. 421, 15–18 10.1016/S0014-5793(97)01525-1 [DOI] [PubMed] [Google Scholar]
- 44. Riekhof W. R., and Voelker D. R. (2006) Uptake and utilization of lyso-phosphatidylethanolamine by Saccharomyces cerevisiae. J. Biol. Chem. 281, 36588–36596 10.1074/jbc.M608851200 [DOI] [PubMed] [Google Scholar]
- 45. Kim K., Kim K. H., Storey M. K., Voelker D. R., and Carman G. M. (1999) Isolation and characterization of the Saccharomyces cerevisiae EKI1 gene encoding ethanolamine kinase. J. Biol. Chem. 274, 14857–14866 10.1074/jbc.274.21.14857 [DOI] [PubMed] [Google Scholar]
- 46. Fernández-Murray J. P., Ngo M. H., and McMaster C. R. (2013) Choline transport activity regulates phosphatidylcholine synthesis through choline transporter Hnm1 stability. J. Biol. Chem. 288, 36106–36115 10.1074/jbc.M113.499855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Storey M. K., Clay K. L., Kutateladze T., Murphy R. C., Overduin M., and Voelker D. R. (2001) Phosphatidylethanolamine has an essential role in Saccharomyces cerevisiae that is independent of its ability to form hexagonal phase structures. J. Biol. Chem. 276, 48539–48548 10.1074/jbc.M109043200 [DOI] [PubMed] [Google Scholar]
- 48. Hsu P., Liu X., Zhang J., Wang H. G., Ye J. M., and Shi Y. (2015) Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652 10.1080/15548627.2015.1023984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang G., McCain M. L., Yang L., He A., Pasqualini F. S., Agarwal A., Yuan H., Jiang D., Zhang D., Zangi L., Geva J., Roberts A. E., Ma Q., Ding J., Chen J., et al. (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623 10.1038/nm.3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Strogolova V., Furness A., Robb-McGrath M., Garlich J., and Stuart R. A. (2012) Rcf1 and Rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol. Cell. Biol. 32, 1363–1373 10.1128/MCB.06369-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen Y. C., Taylor E. B., Dephoure N., Heo J. M., Tonhato A., Papandreou I., Nath N., Denko N. C., Gygi S. P., and Rutter J. (2012) Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 15, 348–360 10.1016/j.cmet.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vukotic M., Oeljeklaus S., Wiese S., Vögtle F. N., Meisinger C., Meyer H. E., Zieseniss A., Katschinski D. M., Jans D. C., Jakobs S., Warscheid B., Rehling P., and Deckers M. (2012) Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 15, 336–347 10.1016/j.cmet.2012.01.016 [DOI] [PubMed] [Google Scholar]
- 53. Henry S. A., Kohlwein S. D., and Carman G. M. (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190, 317–349 10.1534/genetics.111.130286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Modica-Napolitano J. S., and Renshaw P. F. (2004) Ethanolamine and phosphoethanolamine inhibit mitochondrial function in vitro: implications for mitochondrial dysfunction hypothesis in depression and bipolar disorder. Biol. Psychiatry 55, 273–277 10.1016/S0006-3223(03)00784-4 [DOI] [PubMed] [Google Scholar]
- 55. Gohil V. M., Zhu L., Baker C. D., Cracan V., Yaseen A., Jain M., Clish C. B., Brookes P. S., Bakovic M., and Mootha V. K. (2013) Meclizine inhibits mitochondrial respiration through direct targeting of cytosolic phosphoethanolamine metabolism. J. Biol. Chem. 288, 35387–35395 10.1074/jbc.M113.489237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gohil V. M., Sheth S. A., Nilsson R., Wojtovich A. P., Lee J. H., Perocchi F., Chen W., Clish C. B., Ayata C., Brookes P. S., and Mootha V. K. (2010) Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 28, 249–255 10.1038/nbt.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kishi S., Campanholle G., Gohil V. M., Perocchi F., Brooks C. R., Morizane R., Sabbisetti V., Ichimura T., Mootha V. K., and Bonventre J. V. (2015) Meclizine preconditioning protects the kidney against ischemia-reperfusion injury. EBioMedicine 2, 1090–1101 10.1016/j.ebiom.2015.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Merkel O., Schmid P. C., Paltauf F., and Schmid H. H. (2005) Presence and potential signaling function of N-acylethanolamines and their phospholipid precursors in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1734, 215–219 10.1016/j.bbalip.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 59. Cui T. Z., Conte A., Fox J. L., Zara V., and Winge D. R. (2014) Modulation of the respiratory supercomplexes in yeast: enhanced formation of cytochrome oxidase increases the stability and abundance of respiratory supercomplexes. J. Biol. Chem. 289, 6133–6141 10.1074/jbc.M113.523688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 10.1002/yea.1142 [DOI] [PubMed] [Google Scholar]
- 61. Amberg D. C., Burke D. J., and Strathern J. N. (2005) Methods in yeast genetics: a Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 62. Meisinger C., Pfanner N., and Truscott K. N. (2006) Isolation of yeast mitochondria. Methods Mol. Biol. 313, 33–39 [DOI] [PubMed] [Google Scholar]
- 63. Folch J., Lees M., and Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 64. Bartlett G. R. (1959) Phosphorus assay in column chromatography. J. Biol. Chem. 234, 466–468 [PubMed] [Google Scholar]
- 65. Connerth M., Tatsuta T., Haag M., Klecker T., Westermann B., and Langer T. (2012) Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818 10.1126/science.1225625 [DOI] [PubMed] [Google Scholar]
- 66. Wittig I., Braun H. P., and Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- 67. Wittig I., Karas M., and Schägger H. (2007) High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteomics 6, 1215–1225 10.1074/mcp.M700076-MCP200 [DOI] [PubMed] [Google Scholar]
- 68. Reznick A. Z., and Packer L. (1994) Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. 233, 357–363 10.1016/S0076-6879(94)33041-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.