

Abstract
Formation of the vasculature by angiogenesis is critical for proper development, but angiogenesis also contributes to the pathogenesis of various disorders, including cancer and cardiovascular diseases. Vascular endothelial zinc finger 1 (Vezf1), is a Krüppel-like zinc finger protein that plays a vital role in vascular development. However, the mechanism by which Vezf1 regulates this process is not fully understood. Here, we show that Vezf1−/− mouse embryonic stem cells (ESC) have significantly increased expression of a stem cell factor, Cbp/p300-interacting transactivator 2 (Cited2). Compared with WT ESCs, Vezf1−/− ESCs inefficiently differentiated into endothelial cells (ECs), which exhibited defects in the tube-formation assay. These defects were due to reduced activation of EC-specific genes concomitant with lower enrichment of histone 3 acetylation at Lys27 (H3K27) at their promoters. We hypothesized that overexpression of Cited2 in Vezf1−/− cells sequesters P300/CBP away from the promoters of proangiogenic genes and thereby contributes to defective angiogenesis in these cells. This idea was supported by the observation that shRNA-mediated depletion of Cited2 significantly reduces the angiogenic defects in the Vezf1−/− ECs. In contrast to previous studies that have focused on the role of Vezf1 as a transcriptional activator of proangiogenic genes, our findings have revealed a role for Vezf1 in modulating the expression of the antiangiogenic factor Cited2. Vezf1 previously has been characterized as an insulator protein, and our results now provide insights into the mechanism, indicating that Vezf1 can block inappropriate, nonspecific interactions of promoters with cis-located enhancers, preventing aberrant promoter activation.
Keywords: embryonic stem cell, endothelial cell, cell differentiation, E1A-binding protein p300 (P300), gene regulation, chromatin immunoprecipitation (ChiP), DNA methylation, histone acetylation, vascular endothelial growth factor (VEGF), angiogenesis, Cited2, H3K27 acetylation, Vezf1
Introduction
Development of a proper vascular system is indispensable for embryogenesis. Accurate spatial and temporal control of gene expression is required in endothelial cells (ECs),4 which are committed to the formation of the vasculature (1, 2). Angiogenesis involves migration, growth, and differentiation of ECs and takes place during development as well as in adulthood. Angiogenesis is regulated by an interplay between pro- and anti-angiogenic factors (3). Hif-1α is a major pro-angiogenic factor, which interacts with p300/CBP and activates the expression of a number of pro-angiogenic genes including VEGF, thus initiating new vessel formation. Treatment of cultured ECs with VEGF-A165 induces Hif-1α expression, suggesting a bidirectional stimulatory relationship between VEGF and Hif-1α (4). In vivo, Hif-1α can be induced by a variety of factors including hypoxic conditions, (5–7), and certain cytokines and growth factors under normoxic conditions (8–12). Among many known factors, Vezf1 (DB1/Bgp1) and Cited2 (Mrg1/p35srj) play important roles in regulation of angiogenesis during development and in adulthood.
Cited2 (Cbp/p300-interacting transactivator with Glu/Asp-rich carboxyl-terminal domain 2) also named Mrg1/p35srj is a ubiquitously expressed essential transcriptional regulator that binds strongly to the histone acetyltransferases p300 and CBP (cAMP-responsive element-binding protein). Cited2 plays a critical role in heart development, neurulation, and maintenance of fetal and adult hematopoietic stem cells. It is expressed throughout early embryogenesis and in embryonic stem cells (ESCs) (13–17, 19–21). Several studies have demonstrated that by competing with Hif-1α to bind CBP/P300, Cited2 prevents the activation of pro-angiogenic genes such as VEGF, and inhibits angiogenesis (22–24). For example, over-expression of Cited2 suppresses VEGF promoter activity, and siRNA knockdown of Cited2 increases VEGF promoter activity (25). The expression levels of Hif-1α responsive genes including VEGF is increased in Cited2−/− embryos (14). Another study showed that Cited2 is a negative regulator of fracture healing, and its expression is inversely related to the expression of genes involved in extracellular matrix remodeling and angiogenesis, such as matrix metalloprotease, VEGF, and Hif-1α (26). These studies suggest that the proper up-regulation of pro-angiogenic genes requires the levels of Cited2 to be tightly controlled especially at the onset of angiogenesis. Because its aberrant high expression in a specific developmental window can inhibit Hif-1α activity, this regulation can potentially be critical for angiogenesis in both embryonic stages and adulthood.
Mammalian Vezf1 is an essential transcription factor, which is expressed in the anterior-most mesoderm at E7.5 during development. Its expression is later restricted in the vascular endothelium, an observation that revealed its role in regulation of angiogenesis. Vezf1 null mice die at approximately E9.5 (27). Vezf1−/− ESCs grow slower and make smaller embryoid bodies, which have defects in vascularization and cease to grow a few days post-differentiation (28, 29). Vezf1 is expressed in both adult and embryonic ECs. Blocking the activity of Vezf1 by small molecule inhibitor Vec6 inhibits wound healing suggesting its role in postnatal angiogenesis (30). Vezf1 contains six Cys2/His2-type zinc finger motifs and binds poly(dG) or poly(dC) sequences (31, 32). It carries a glutamine-rich stretch and a proline-rich region that are characteristic of transcriptional activation or repression domains (33). It is proposed to act as a transcriptional activator of pro-angiogeneic genes including endothelin 1, microtubule turnover protein, stathmin/OP18, and metallothionein 1 (MT1) (34–36). However, no change in the expression of pro-angiogenic genes was seen in Vezf1−/− embryos (28). Other studies suggested an indirect role of Vezf1 by interacting with RhoB that promotes expression of RhoB-regulated pro-angiogenesis genes (30, 37). Therefore, the mechanism by which Vezf1 regulates angiogenesis is unclear.
To study the specific role of Vezf1 in endothelial development and angiogenesis, we examined the differentiation of WT and Vezf1−/− ESCs to ECs by treatment with VEGF-A165 and tested their angiogenic potential by a in vitro tube-formation assay. Our findings suggest that Vezf1 controls activation of angiogenesis in ECs by restricting Cited2 expression to basal levels, which allows Hif-1α–mediated activation of the pro-angiogenic genes. We observed a strong increase in the expression of Cited2 in Vezf1−/− ESCs compared with WT cells. In addition, the elevated expression of Cited2 in Vezf1−/− ESCs affected their efficiency of differentiation into ECs and attenuated the ability of Vezf1−/− ECs to form vascular structures in a tube-formation assay. Concomitant with this, there was reduced activation of endothelial/pro-angiogenic genes in differentiating Vezf1−/− ECs. Based on our data showing reduced levels of histone H3K27Ac at the promoters of angiogenesis-specific genes, we propose that high levels of Cited2 sequester the histone acetyltransferase p300 away from angiogenesis-specific gene promoters, thus reducing their activation and gene expression. Together these observations substantiate the critical role of Vezf1 in controlling the expression of developmental regulators such as Cited2. Given that the expression of Cited2 in ESCs is not completely turned off, we suggest the role of Vezf1 in fine-tuning Cited2 expression in ESCs. Our previous work using genome-wide ChIP-SEQ showed that binding sites for Vezf1 are mostly present in CpG-rich regions (38). We also showed that Vezf1 binds to the chicken β-globin insulator suggesting a role in regulating enhancer-mediated control of gene expression (32, 38). Based on these studies, we speculate that the insulator function of Vezf1 blocks inappropriate interactions of the Cited2 promoter with nearby enhancer/s, thus modulating the magnitude and spatiotemporal regulation of its expression.
Results
Cited2 expression is high in Vezf1−/− ESCs
To elucidate the mechanism of Vezf1, we had previously analyzed changes in gene expression of Vezf1−/− ESCs compared with WT ESCs using a microarray analysis (Fig. S1) (39). We found Cited2 among the top 20 genes that were up-regulated in Vezf1−/− ESCs by more than 5-fold. To confirm this observation, we measured the gene expression of Cited2 quantitatively in WT and Vezf1−/− ESCs by qRT-PCR and protein levels by Western blotting. The data show a 4–5-fold higher transcript and protein levels of Cited2 in Vezf1−/− ESCs compared with WT ESCs (Fig. 1, A and B). We found these observations consistent with our previously published ChIP-SEQ of Ser2 phosphorylated RNA Pol II (elongating form of RNA Pol II) in WT and Vezf1−/− ESCs (38). Data analysis of elongating RNA Pol II showed more than a 2-fold higher enrichment in the Cited2 gene body in Vezf1−/− ESCs compared with the WT ESCs (Fig. 1C). Based on the known function of Cited2 as an anti-angiogenic factor (22–24), we hypothesized that a presence of high Cited2 level in Vezf1−/− cells could interrupt or delay the differentiation of ECs, or reduce their angiogenic potential.
Figure 1.

A, gene expression analysis of Cited2 by RT-qPCR in WT and Vezf1−/− ESCs showed a 4–5-fold higher expression of Cited2 in Vezf1−/− ESCs. Gene expression was normalized to Gapdh and represented relative to gene expression in WT ESC, set to 1. B, 50 μg of total cell extract were loaded from the WT and Vezf1−/− ESCs followed by Western blot analysis using anti-Cited2 antibody and anti-GAPDH antibody for loading control. C, screen shot from UCSC genome browser showing the occupancy of Ser2-phosphorylated Pol II at the Cited2 locus in WT and Vezf1−/− ESCs. These data were obtained from the previously published publically available ChIP-SEQ data. WT, wildtype ESCs; Vezf1−/−, Vezf1-knockout ESCs.
Vezf1−/− ESCs are defective in EC differentiation
Cited2 expression is critical for pluripotency and differentiation of ESCs; therefore, we tested if overexpression of Cited2 affected the pluripotency of Vezf1−/− ESCs. We quantified the expression of the pioneer transcription factor, Oct4, in Vezf1−/− ESCs compared with that in the WT ESCs and observed no significant difference in its expression (Fig. 2A). Additionally, both WT and Vezf1−/− ESCs showed positive alkaline phosphate staining suggesting that high expression of Cited2 had little if any effect on the pluripotency of Vezf1−/− ESCs (Fig. 2B). Previous studies showed a reduced growth of Vezf1−/− EBs and defects in their vascular structures, but reported little or no difference in the endothelial differentiation in 3D cultures (27). We performed in vitro differentiation of WT and Vezf1−/− ESCs to ECs on gelatinized plates in the presence of 10 ng/μl of VEGF-A165. The differentiation of ESCs to ECs was monitored by microscopy. During differentiation, the WT ESCs showed an expected loss of ∼5–10% of cells, and differentiated efficiently into ECs. Comparatively, during the first 3 days of differentiation, over 80% Vezf1−/− cells died, leading to a reduced efficiency of EC derivation from the Vezf1−/− ESCs. This was confirmed by positive alkaline phosphatase staining of the surviving Vezf1−/− cells at 10 days post-differentiation (Fig. 2C).
Figure 2.

A, gene expression analysis by RT-qPCR of Oct4 in WT and Vezf1−/− ESCs. B, alkaline phosphatase (AP) staining for pluripotency in WT and Vezf1−/− ESCs; the presence of dark blue stain indicates positive for pluripotency. C, WT and Vezf1−/− ESCs were differentiated using 10 ng/μl of VEGF-A165 for 5, 6, 7, and 10 days and visualized using brightfield microscopy at ×10 magnification. Unlike the WT cells, the Vezf1−/− cells were unable to differentiate and most of the cells died. The field view is the representation of proliferation during differentiation and cell number. The panel for D10 shows alkaline phosphatase staining. A strong signal of Vezf1−/− cells indicates presence of undifferentiated stem cells. WT, wildtype ESCs; Vezf1−/−, Vezf1 knockout ESCs; UD, undifferentiated; D4–D10, days post-differentiation.
Given that the expression of the EC lineage is driven by VEGF-A in an autoregulatory loop (9, 40), we tested if increasing the concentration of VEGF-A165 in the medium could improve EC differentiation and survival of Vezf1−/− cells. EC differentiation was monitored by measurement of endothelial-specific gene expression including VEGF-A, Flk1, Flt1, CD31, and Tie2 (41). WT and Vezf1−/− ESCs were differentiated using 20, 40, and 60 ng/μl of VEGF-A165. Increasing the VEGF-A165 to 20 ng/μl stimulated Vezf1−/− cells to differentiate to ECs with a higher efficiency (Fig. 3A). In both WT and Vezf1−/− ESCs, differentiation induced repression of the pluripotency gene, Oct4, and activation of endothelial-specific genes, VEGF-A, Flk1, CD31, and Tie2, albeit at lower levels in the Vezf1−/− cells (Fig. 3B). Reduced activation of CD31 and Tie2 in Vezf1−/− cells suggested a partial defect in EC differentiation. WT and Vezf1−/− ECs day (D) 6 and D8 post-differentiation treated with 20, 40, and 60 ng/μl of VEGF-A165 were collected for gene expression analysis by qRT-PCR. The data showed that at all three concentrations of VEGF-A165, the expression of VEGF-A and its receptor, Flk1, was significantly higher in WT ECs compared with that in the Vezf1−/− ECs, indicating that gene expression is not further rescued by higher doses of VEGF-A165 (Fig. 4). Previous studies have shown that Hif-1α expression can also be activated by treatment of ECs with VEGF-A165 thus showing that VEGF regulates the expression of its own transcription factor (4). In WT and Vezf1−/− ECs, we checked the expression of Hif-1α and Flt1, which is the angiogenesis-specific VEGF receptor. Although no difference in the expression of Hif-1α was observed between WT and Vezf1−/− ECs, similar to Flk1 and VEGF-A, Flt1 expression was also comparatively lower in Vezf1−/− ECs at 20, 40, and 60 ng/μl of VEGF-A165 in both D6 (Fig. 4, A–D) and D8 (Fig. 4, E–H) post-differentiation. These data show that in Vezf1−/− ECs, reduced expression of angiogenic genes including VEGF is not due to lower Hif-1α expression, but potentially due to its reduced activity.
Figure 3.

A and B, WT and Vezf1−/− ESCs were differentiated using 20 ng/μl of VEGF-A165. A, differentiating WT and Vezf1−/− ECs were visualized using brightfield microscopy. Whereas most of the WT cells undergo distinct morphological changes, only a fraction acquire a similar morphology in Vezf1−/− cells. B, gene expression by RT-qPCR plotted as a relative change to the expression in UD where UD was set to 1. Endothelial specific genes, Flk1, VEGF-A, CD31, and Tie2 show an expected increase in expression in differentiating WT ECs. Differentiating Vezf1−/− ECs, however, show significantly low expression of all tested endothelial-specific genes. A decrease in Oct4 expression is observed in both WT and Vezf1−/− ECs indicating loss of pluripotency. WT, wildtype; Vezf1−/−, Vezf1 knockout; UD, undifferentiated; D2–D9, days post-differentiation.
Figure 4.

A–D, gene expression analysis in WT and Vezf1−/− D6 ECs, differentiated using 20, 40, and 60 ng/μl of VEGF-A165. E–H, gene expression analysis in WT and Vezf1−/− D8 ECs differentiated using 20, 40 and 60 ng/μl of VEGF-A165. The expression of all genes is increased in D8 compared with D6 post-differentiation. Higher doses of VEGF-A165 have no further effect on expression of VEGF-A, Flk1, Hif-1α, and Flt1 in both WT or in Vezf1−/− cells. The data represents average and S.D. of 3 to 4 replicates. WT, wildtype; Vezf1−/−, Vezf1 knockout; UD, 20, 40, 60 ng/μl of VEGF-A165 used for differentiation.
Vezf1−/− ECs are defective in forming vascular networks in 3D cultures
We next tested the angiogenic potential of Vezf1−/− ECs by an in vitro tube-formation assay. We differentiated WT and Vezf1−/− ESCs to ECs for 8 days in the presence of VEGF-A165 at 20 ng/μl. The ECs were collected and placed in Matrigel to form vascular networks or tubes in 3D culture. The mouse endothelial cell line, MSS31, was used as a positive control. Whereas WT ECs formed distinct tubes within 4–6 h in Matrigel, Vezf1−/− ECs showed significant defects in tube formation as indicated by the shorter tube length (Fig. 5, A and B).
Figure 5.
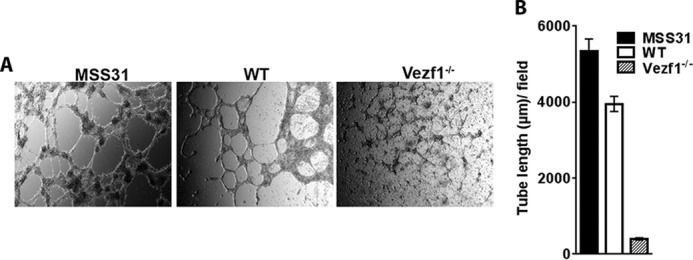
A, differentiated WT and Vezf1−/− EC plated in VEGF-supplemented Matrigel were incubated at 37 °C for 5–15 h. The formation of tube structures is visualized by brightfield microscopy. Mouse endothelial cell line MSS31 is used as a positive control. The images were taken at ×10 magnification at 12 h for MSS31, and 6 h for WT and Vezf1−/−. B, measurement of tube length using ImageJ software. Compared with MSS31 and WT ECs, Vezf1−/− ECs were unable to make tube-like structures in Matrigel. WT, wildtype; Vezf1−/−, Vezf1 knockout.
Taken together, these data show that Vezf1−/− ESCs have reduced competence to differentiate into ECs and to form vascular structures in Matrigel. Given that Flt1 receptor function is required for tubulogenesis (42), the inability of Vezf1−/− ECs to form tubes in Matrigel could be the consequence of strongly reduced expression of Flt1 in these cells. Because no change was observed in Hif-1α gene expression, we predict that the reduced expression of EC-specific genes is due to high Cited2 expression in Vezf1−/− ESCs and ECs.
Induced repression of Cited2 partially rescues EC differentiation and vascular defects in Vezf1−/− ECs
We next tested the hypothesis that the defective vasculature formation by Vezf1−/− ECs is due to anomalous high expression of Cited2. We therefore asked if depletion of Cited2 in Vezf1−/− ESCs could rescue their ability to differentiate and make vascular networks. Vezf1−/− ESCs were transfected with Cited2 shRNA, to generate stable transgenic ESCs lines, Vezf1−/sh. Of the nine transgenic lines, some showed more than 10-fold reduction in Cited2 expression when compared with WT ESCs. Because Cited2 is known to be important for pluripotency (43), we chose to use the Vezf1−/sh cell line (7-2), in which Cited2 expression is reduced to levels similar to that of WT ESC (Fig. 6, A and B). The Vezf1−/sh ESCs were differentiated to ECs using 20, 40, and 60 ng/μl of VEGF-A165. Compared with Vezf1−/− ESCs, Vezf1−/sh ESCs showed better survival and higher efficiency of EC differentiation at 20 ng/μl of VEGF-A165, which was similar to the WT cells (Fig. 6C). The Vezf1−/sh-derived ECs were collected on day 6 post-differentiation and RNA was extracted for gene-expression analysis. The data show that expression of VEGF-A was largely rescued, whereas Flt1 and Flk1 were partially rescued when compared with their expression in WT and Vezf1−/− ECs (Fig. 6D). Next, we tested if the derived ECs form vascular structures by a tube-formation assay. WT, Vezf1−/−, and Vezf1−/sh-derived ECs ((7-2) and (3-3)) were collected after 10 days and used to perform an in vitro tube-formation assay. Vezf1−/sh (3-3) ESCs have ∼7-fold reduced expression of Cited2 compared with WT ESCs. This cell line was therefore used to test the effect of Cited2 deficiency on EC differentiation and tube formation. The repression of Cited2 in Vezf1−/sh ECs largely rescued the defective tube formation on Matrigel, which was more prominent in Vezf1−/sh (7-2) compared with the (3-3) cell line (Fig. 7, A and B). This observation supports the known role of Cited2 in pluripotency and differentiation of ESCs and emphasizes the importance of the appropriate levels of developmental transcription factors for proper differentiation. These data directly support our hypothesis that an aberrant high expression of Cited2 prevents the activation of EC-specific gene expression potentially by sequestering p300 from the promoters of angiogenesis-specific genes.
Figure 6.

A, gene expression analysis of Cited2 by RT-qPCR in Vezf1−/sh cell lines. Change in gene expression was plotted relative to that of WT ESCs, set to 1. Labels on the x axis represent various stable cell lines, of which the Vezf1−/sh cell line (7-2) has Cited2 expression reduced to the levels similar to WT ESCs. B, Western blot analysis using 50 μg of total cell extract from the WT, Vezf1−/−, and Vezf1−/sh (7-2) ESCs probed with anti-Cited2 antibody and anti-GAPDH. C, differentiation of WT, Vezf1−/−, and Vezf1−/sh (7-2) ESCs was induced using 20 ng/μl of VEGF-A165. D2–D6 are days post-differentiation. Compared with the differentiating WT cells, Vezf1−/sh (7-2) show similar morphology and cell number indicating at least a partial rescue of their ability to differentiate into ECs. D, gene expression analysis of Vezf1−/sh (7-2) ESCs, which were differentiated to ECs at 20, 40, and 60 ng/μl of VEGF-A165. Expression of pro-angiogeneic genes VEGF-A, Flk1, and Flt1 was measured. Compared with WT and Vezf1−/− cells, gene expression was largely rescued in Vezf1−/sh (7-2) ECs. The data represent the average and S.D. of 3 to 4 replicates. WT, wildtype ESCs; Vezf1−/−, Vezf1 knockout ESCs; Vezf1−/sh, stable transgenic Vezf1−/− ESCs expressing Cited2-shRNA; 20, 40, 60, ng/μl of VEGF-A165 used for differentiation.
Figure 7.
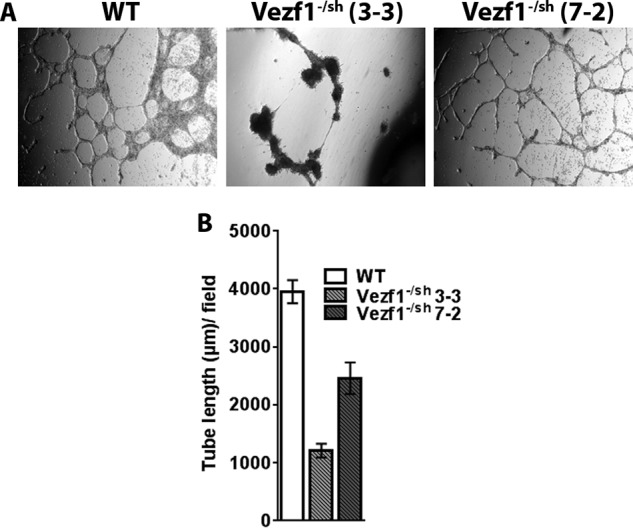
A, WT, Vezf1−/sh (7-2), and Vezf1−/sh (3-3) cells were differentiated for 10 days and used in a tube-formation assay. The Vezf1−/sh cell line (3-3) had about 7-fold lower Cited2 expression than WT. Compared with the WT cells, tube formation was rescued in Vezf1−/sh (7-2) ECs, which was absent in Vezf1−/sh (3-3) ECs. Images were taken at 12 h for Vezf1−/sh (3-3) and 6 h for WT and Vezf1−/sh (7-2). B, tube length was measured by ImageJ software and plotted.
P300 activity is regulated by Cited2 at the VEGF-A promoter
In response to VEGF signaling, P300 acetyltransferase interacts with Hif-1α, which targets it to the HBS (HIF-1–binding element) of the promoters of angiogenesis-specific genes where it acetylates histone H3 at Lys27. To test the impact of Cited2 expression on the activity of P300 histone acetyltransferase at VEGF and Flk-1 promoters, we performed a chromatin immunoprecipitation (ChIP) assay using anti-histone H3K27Ace antibody. We observed an expected increase in the fold-enrichment of H3K27Ace at VEGF and Flk-1 promoters in WT-differentiated ECs compared with the undifferentiated ESCs. However, this increase was markedly reduced in the differentiated Vezf1−/− ECs particularly at the VEGF promoter, which is a direct target of Hif-1α. As a control, we used the Oct4 promoter where H3K27 acetylation is decreased post-differentiation in both the WT and Vezf1−/− ESCs (Fig. 8A). Although Flk-1 has an HBS in its promoter, some studies indicate that it is targeted by both Hif-1α and HIF-1β (40). These data support our hypothesis that reduced P300 activity at angiogenesis-specific gene promoters inhibits their complete activation during Vezf1−/− EC differentiation.
Figure 8.

A, ChIP-qPCR of H3K27Ace showed an increase in the fold-enrichment of H3K27Ace at VEGF-A and Flk1 promoters in WT ECs that were reduced in Vezf1−/− ECs. At the Oct4 promoter, post-differentiation, deacetylation of H3K27 is accompanied by the loss of gene expression. Therefore, it serves as a negative control. B, DNA methylation was analyzed at Cited2 CpGi using bisulfite sequencing in undifferentiated ESCs. The average percent methylation of 27 CpG sites in the CpGi was plotted. C, gene expression analysis of Cited2 in WT and Dnmt3b−/− ESCs. There was no significant difference in Cited2 expression levels. D, ChIP-qPCR using anti-Vezf1 antibody showed an increase in the fold-enrichment at Cited2 promoter in WT ESCs. Fold-enrichment below 1 in Vezf1−/− ESCs indicates absence of binding, and was used as negative control. The data in each bar graph represents average and S.D. of 3 to 4 replicates. E, model showing the effect of Cited2 on Hif-1α-mediated regulation of pro-angiogenic genes. Vezf1 regulates the expression of Cited2 at basal levels in ESCs before the induction of endothelial differentiation. This allows the interaction of Hif-1α and P300 that activate the pro-angiogenic genes. In the absence of Vezf1, high expression of Cited2 sequesters P300 away from Hif-1α, thus inhibiting the activation of pro-angiogenic genes. WT, wildtype; Vezf1−/−, Vezf1 knockout; Dnmt3b−/−, Dnmt3b knockout; UD, undifferentiated; D7, days post-differentiation; P1 and P2, two primer pairs in Cited2 promoter used in ChIP-qPCR.
Transcriptional regulation of Cited2 expression by Vezf1
Vezf1−/− ESCs show a genome-wide loss of DNA methylation at several CpG islands (CpGi's) flanking tissue-specific genes and a significant decrease in the expression of the DNA methyltransferase, Dnmt3b (39). Cited2 is encoded by a relatively small gene, which has a large CpGi at its promoter and exon 1 (Fig. 1C). We therefore asked if the increase in Cited2 expression in Vezf1−/− ESCs could be due to loss of DNA methylation at its CpGi. To test the potential role of Dnmt3b in regulation of Cited2 CpGi DNA methylation, genomic DNA from Dnmt3b−/− ESCs, Vezf1−/− ESCs, and WT ESCs was extracted to quantify DNA methylation using bisulfite sequencing. Our data show very low CpG methylation at Cited2 CpGi in WT ESCs that did not change in Vezf1−/− and Dnmt3b−/− ESCs (Fig. 8B). Cited2 gene expression also showed no change in Dnmt3b−/− compared with that in WT ESCs (Fig. 8C). These data confirm that expression of Cited2 is not regulated by Dnmt3b or changes in DNA methylation at its CpGi and support the direct role of Vezf1 in regulating Cited2 expression. Therefore, we investigated the binding of Vezf1 near the Cited2 promoter by ChIP assay using custom-made rabbit polyclonal anti-Vezf1 antibody, which was previously characterized and used in ChIP studies in ESCs (12, 34). We observed a high relative enrichment of Vezf1 at the Cited2 promoter in WT ESCs (Fig. 8D). These data suggest a direct regulation of Cited2 expression by Vezf1 through its binding at the promoter-associated CpGi.
Taken together our in vitro differentiation experiments show that the aberrant high expression of Cited2 in Vezf1−/− ESCs suppresses their angiogenic potential by sequestering P300/CBP away from the pioneer transcription factor Hif-1α. This abbreviates the promoter activation of the downstream pro-angiogenic genes (Fig. 8E). These findings suggest that Vezf1 regulates angiogenesis by fine-tuning the level of anti-angiogenic factor Cited2.
Discussion
The transcription factor Vezf1 is highly expressed in vascular endothelium and its role in vascular development has been observed by several earlier studies (28, 34, 36, 44, 45). For example, recent studies have shown that a small molecule inhibitor of Vezf1, Vec6, prevents wound healing and angiogenesis (30). Although the function of Vezf1 has been suggested through its role as a transcriptional activator of some genes that are known to promote angiogenesis, previous studies were performed using semi-quantitative RT-PCR to measure the expression of pro-angiogenic genes in Vezf1−/− embryos. These data showed no change in the expression of pro-angiogenic factors compared with WT embryos (28). Unlike the previous study (28), we used quantitative RT-PCR to measure gene expression changes and our data show about 2–3-fold lower expression of several pro-angiogenic genes, including CD31, Tie2, VEGF-A, and its receptors Flk-1 and Flt-1 in the in vitro derived Vezf1−/− ECs. The defective EC differentiation of Vezf1−/− ESCs was also supported by impaired morphological changes associated with EC differentiation. We further show that the expression of some of these genes can be largely rescued by down-regulating Cited2, which is aberrantly overexpressed in Vezf1−/− ESCs. Our data support the previously suggested role of Vezf1 in angiogenesis, however, through a different mechanism. In contrast to its previously predicted role as a transcriptional activator, our data show that Vezf1 restricts the expression of the anti-angiogenic gene Cited2 to basal levels, ensuring a balanced gene expression during angiogenesis. Our data also emphasize that small but quantifiable changes in gene expression of developmental transcription factors and regulators can have profound effect on cell differentiation.
Cited2 (Mrg1/p35srj) belongs to a family of transactivators that lack direct DNA binding but contain glutamic acid/aspartic acid (ED)-rich tail, which interacts with P300/CBP acetyltransferase. Whereas, on one hand, Cited2 competes with Hif-1α to interact with P300/CBP, Cited2 promoter has HIF1-binding sites and its expression is up-regulated in hypoxia by Hif-1α. Therefore, Cited2 participates in a negative-feedback loop with Hif-1α in which Cited2 accumulates during hypoxia. During restoration of normaxia, it will inhibit Hif-1α activity and prevent hypervascularization. An anomalous high expression of Cited2 at the onset of angiogenesis could interfere with Hif-1α-mediated activation of pro-angiogenic genes, consequently the expression of Cited2 must be tightly controlled. We propose that in undifferentiated ESCs, Vezf1 modulates the expression of Cited2, thus enabling Hif-1α–mediated gene activation during angiogenesis. This mechanism is supported by our data showing that overexpression of Cited2 causes defective tube formation by Vezf1−/− ECs when there is no difference in Hif-1α expression between the WT and Vezf1−/− cells. Cited2 is expressed throughout early embryogenesis and its expression in ESCs is critical for pluripotency, and appropriate differentiation (13, 43). Our data showing absence of rescue in a Vezf1−/sh (3-3) cell line, which has Cited2 expression significantly lower than WT ESCs, supports the role of the basal expression of Cited2 in maintenance of pluripotency and differentiation potential of ESCs. Vezf1-mediated regulation of Cited2 expression is also potentially relevant during adult angiogenesis and wound healing where Vezf1 could down-regulate or maintain low Cited2 expression in the ECs. This prediction is supported by our observation from a published microarray analysis of Vezf1-silenced BVEC's, listing Cited2 among the up-regulated genes (30). This study also showed that loss of Vezf1 causes inhibition of wound healing and blood vessel formation (30).
Based on the previously characterized role of Vezf1 as an insulator binding protein, it is highly plausible that Vezf1 insulates its target promoters from interaction with nonspecific enhancers, and in the case of Cited2 from the enhancers in the downstream gene, β-taxilin (Txlnb). The insulator function of Vezf1 is supported by our published ChIP-SEQ studies showing that a significant number of Vezf1-binding sites are adjacent to insulator protein, CTCF. Vezf1 shows widespread binding at CpGi's present in the regulatory elements of genes including promoters, enhancers, and insulators (32, 38). By using in vitro EC differentiation as a developmental model system, it will be important to explore the regulatory potential of Vezf1-mediated insulator function in modulating gene expression during development.
Experimental procedures
Embryonic stem cell culture
Undifferentiated WT, Dnmt3b−/−, and Vezf1−/− ESCs were cultured in Dulbecco's modified Eagle's medium containing 15% ESC qualified fetal bovine serum (Millipore), supplemented with nonessential amino acids, glutamine, 1000 units/ml of leukemia inhibitory factor (LIF) (ESGRO; Chemicon International), and 50 μm β-mercaptoethanol. Cells were cultured on 0.1% gelatin for one passage before switching to differentiation conditions.
Endothelial lineage differentiation
ESCs were plated at a density of 3 × 103 cells/cm2 on gelatinized plates in ESC medium with LIF and incubated overnight to attach. The next day the media was removed and cells were washed with PBS. Differentiation was induced by adding ESC medium without LIF and VEGF-A165 (R&D Systems) at 20–60 ng/μl. VEGF-A165 was supplemented to the culture every alternate day for 10 days to drive differentiation into endothelial lineage. Cell morphology was monitored using phase-contrast microscopy and pluripotency was monitored by alkaline phosphatase staining.
Tube-formation assay
The ECs on D10 post-differentiation were collected by trypsinization and counted using Bio-Rad Cell Counter. The tube-formation assay was performed by plating 2 × 105 ECs on a 24-well plate coated with VEGF supplemented Matrigel (BD Biosciences) according to the manufacturer's protocol (46, 47). The cells were incubated at 37 °C for 3–18 h. Tubing was scored using images from phase-contrast microscopy (29). The length of the tubes was measured by ImageJ software.
Transfection and generation of stable ESC lines
The lentivirus construct pLKO.1 carrying shRNA specific for Cited2 was purchased from Dharmacon. The recombinant lentivirus containing Cited2 shRNA was packaged using Vira Power (Fisher Thermo Scientific) in 293FT cells using the manufacturer's protocol. For shRNA-mediated depletion of Cited2, WT, and Vezf1−/− ESCs were transfected by lentivirus at a multiplicity of infection of 2 followed by selection of transgenic lines with stably integrated lentivirus construct using 3 μg/ml of puromycin.
Gene expression by quantitative RT-PCR and Western blotting
RNA from cells was purified using TRIzol (Invitrogen, 15596026) according to the manufacturer's protocol. Genomic DNA contamination was removed by DNase (Roche Applied Science, 04716728001) treatment at 37 °C overnight. Quantitative RT-PCR was performed for equal amounts of RNA by using Verso One-Step kit (Thermo Scientific, AB-4104A). The data were analyzed and gene expression was normalized to Gapdh expression. The change in expression is represented either as normalized gene expression or as relative gene expression that is changed relative to expression in the undifferentiated cells, set to 1. See Table S1 for primers used. Western blot analysis was performed using commercially available antibodies anti-Cited2 (ab108345, Abcam) and anti-Gapdh (sc47724, Santa Cruz) according to the manufacturers' recommendation.
DNA-methylation assay
DNA-methylation assay was performed by bisulfite sequencing (Bis-SEQ). Bisulfite sequencing was performed using the EpiTect Fast Bisulfite Conversion Kit (Qiagen, 59802). Genomic DNA was purified from WT, Vezf1−/−, and Dnmt3b−/− ESCs. 1 μg of genomic DNA was used and bisulfite-converted DNA was amplified using nested primers and Taq polymerase (New England Biolabs, M0267L). Products from the inner PCR were used to generate a library for high throughput sequencing using Wide-SEQ. The primers used are listed in Table S1.
ChIP
ChIP was performed using cross-linked chromatin from WT and Vezf1−/− ESCs using a previously published protocol (18). Briefly, cells were cross-linked for 5 min with 1% formaldehyde in buffer (0.1 m NaCl, 1 mm EDTA, 0.5 mm EGTA, and 50 mm HEPES, pH 8). Nuclei were isolated and chromatin was sheared to 0.5–1-kb fragments using a Covaris E210 device, according to the manufacturer's protocols. Antibodies (8 μg) were immobilized on Protein A/G magnetic beads (Life Technologies, 10002D and 10004D) by overnight incubation. The magnetic beads were washed to remove unconjugated antibody and mixed with 8 μg of sonicated chromatin. After an overnight incubation, magnetic beads were washed, and bound DNA was purified. DNA was quantified using PicoGreen (Life Technologies, P11495) in a NanoDrop 3300 fluorospectrometer. Quantitative PCR was then performed using equal amounts of IN (input) and IP (immunoprecipitated sample) DNA. Fold-enrichment was calculated as follows: Ct(IN)–Ct(IP) and the fold-change was calculated by using 2∧(Ct(IN)–Ct(IP)). The fold-enrichment of 1 or less indicates no binding. See Table S1 for primers used. The H3K27Ace antibody used is commercially available (39133, Active Motif) and anti-Vezf1 antibody is a previously characterized custom-made polyclonal antibody.
Author contributions
L. A., M. H., Q. Y., and H. G. formal analysis; L. A., M. H., and A. B. N. validation; L. A., M. H., Q. Y., and A. B. N. investigation; L. A. and Q. Y. methodology; L. A., M. H., and H. G. writing-review and editing; M. H. visualization; M. H. and H. G. project administration; Q. Y. and H. G. writing-original draft; H. G. conceptualization; H. G. supervision; H. G. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Sandra Rossie for reading the manuscript, Dr. Taiping Chen for providing Dnmt3b−/− ESCs, and Dr. Heidi Stuhlmann for Vezf1−/− ESCs.
This work was supported by American Heart Association (AHA) Scientist Development Award 17SDG33700153 (to H. G.) a Graduate Student Fellowship from the King Saud University (KSU) (to L. A.) and NCI, National Institutes of Health Grant P30 CA023168. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1 and Table S1.
- EC
- endothelial cell
- VEGF
- vascular endothelial growth factor
- Cited2
- Cbp/p300-interacting transactivator with Glu/Asp-rich carboxyl-terminal domain 2
- CBP
- cAMP-responsive element-binding protein
- ESC
- embryonic stem cell
- Vezf1
- vascular endothelial zinc finger 1
- LIF
- leukemia inhibitory factor
- IN
- input
- IP
- immunoprecipitated
- qPCR
- quantitative PCR
- Gapdh
- glyceraldehyde-3-phosphate dehydrogenase
- D
- days post-differentiation.
References
- 1. Carmeliet P. (2000) Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389–395 10.1038/74651 [DOI] [PubMed] [Google Scholar]
- 2. Risau W. (1997) Mechanisms of angiogenesis. Nature 386, 671–674 10.1038/386671a0 [DOI] [PubMed] [Google Scholar]
- 3. Marcelo K. L., Goldie L. C., and Hirschi K. K. (2013) Regulation of endothelial cell differentiation and specification. Circ. Res. 112, 1272–1287 10.1161/CIRCRESAHA.113.300506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deudero J. J., Caramelo C., Castellanos M. C., Neria F., Fernández-Sánchez R., Calabia O., Peñnate S., and González-Pacheco F. R. (2008) Induction of hypoxia-inducible factor 1α gene expression by vascular endothelial growth factor. J. Biol. Chem. 283, 11435–11444 10.1074/jbc.M703875200 [DOI] [PubMed] [Google Scholar]
- 5. Imanirad P., and Dzierzak E. (2013) Hypoxia and HIFs in regulating the development of the hematopoietic system. Blood Cells Mol. Dis. 51, 256–263 10.1016/j.bcmd.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Semenza G. L. (1999) Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15, 551–578 10.1146/annurev.cellbio.15.1.551 [DOI] [PubMed] [Google Scholar]
- 7. Semenza G. L. (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 7, 345–350 10.1016/S1471-4914(01)02090-1 [DOI] [PubMed] [Google Scholar]
- 8. Zelzer E., Levy Y., Kahana C., Shilo B. Z., Rubinstein M., and Cohen B. (1998) Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 17, 5085–5094 10.1093/emboj/17.17.5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hellwig-Bürgel T., Rutkowski K., Metzen E., Fandrey J., and Jelkmann W. (1999) Interleukin-1β and tumor necrosis factor-α stimulate DNA binding of hypoxia-inducible factor-1. Blood 94, 1561–1567 [PubMed] [Google Scholar]
- 10. Görlach A., Diebold I., Schini-Kerth V. B., Berchner-Pfannschmidt U., Roth U., Brandes R. P., Kietzmann T., and Busse R. (2001) Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ. Res. 89, 47–54 10.1161/hh1301.092678 [DOI] [PubMed] [Google Scholar]
- 11. Spinella F., Rosanò L., Di Castro V., Natali P. G., and Bagnato A. (2002) Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1α in ovarian carcinoma cells. J. Biol. Chem. 277, 27850–27855 10.1074/jbc.M202421200 [DOI] [PubMed] [Google Scholar]
- 12. Laughner E., Taghavi P., Chiles K., Mahon P. C., and Semenza G. L. (2001) HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 21, 3995–4004 10.1128/MCB.21.12.3995-4004.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pacheco-Leyva I., Matias A. C., Oliveira D. V., Santos J. M., Nascimento R., Guerreiro E., Michell A. C., van De Vrugt A. M., Machado-Oliveira G., Ferreira G., Domian I., and Bragança J. (2016) CITED2 cooperates with ISL1 and promotes cardiac differentiation of mouse embryonic stem cells. Stem Cell Rep. 7, 1037–1049 10.1016/j.stemcr.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yin Z., Haynie J., Yang X., Han B., Kiatchoosakun S., Restivo J., Yuan S., Prabhakar N. R., Herrup K., Conlon R. A., Hoit B. D., Watanabe M., and Yang Y.-C. (2002) The essential role of Cited2, a negative regulator for HIF-1α, in heart development and neurulation. Proc. Natl. Acad. Sci. U.S.A. 99, 10488–10493 10.1073/pnas.162371799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bamforth S. D., Bragança J., Eloranta J. J., Murdoch J. N., Marques F. I., Kranc K. R., Farza H., Henderson D. J., Hurst H. C., and Bhattacharya S. (2001) Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 29, 469–474 10.1038/ng768 [DOI] [PubMed] [Google Scholar]
- 16. Bamforth S. D., Bragança J., Farthing C. R., Schneider J. E., Broadbent C., Michell A. C., Clarke K., Neubauer S., Norris D., Brown N. A., Anderson R. H., and Bhattacharya S. (2004) Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 36, 1189–1196 10.1038/ng1446 [DOI] [PubMed] [Google Scholar]
- 17. Qu X., Lam E., Doughman Y. Q., Chen Y., Chou Y. T., Lam M., Turakhia M., Dunwoodie S. L., Watanabe M., Xu B., Duncan S. A., and Yang Y. C. (2007) Cited2, a coactivator of HNF4α, is essential for liver development. EMBO J. 26, 4445–4456 10.1038/sj.emboj.7601883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petell C. J., Alabdi L., He M., San Miguel P., Rose R., and Gowher H. (2016) An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids Res. 44, 7605–7617 10.1093/nar/gkw426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Du J., and Yang Y. C. (2013) Cited2 in hematopoietic stem cell function. Curr. Opin. Hematol. 20, 301–307 10.1097/MOH.0b013e3283606022 [DOI] [PubMed] [Google Scholar]
- 20. Du J., and Yang Y. C. (2012) HIF-1 and its antagonist Cited2: regulators of HSC quiescence. Cell Cycle 11, 2413–2414 10.4161/cc.20803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kranc K. R., Schepers H., Rodrigues N. P., Bamforth S., Villadsen E., Ferry H., Bouriez-Jones T., Sigvardsson M., Bhattacharya S., Jacobsen S. E., and Enver T. (2009) Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell 5, 659–665 10.1016/j.stem.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bragança J., Eloranta J. J., Bamforth S. D., Ibbitt J. C., Hurst H. C., and Bhattacharya S. (2003) Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding protein, and CITED2. J. Biol. Chem. 278, 16021–16029 10.1074/jbc.M208144200 [DOI] [PubMed] [Google Scholar]
- 23. Fox S. B., Bragança J., Turley H., Campo L., Han C., Gatter K. C., Bhattacharya S., and Harris A. L. (2004) CITED4 inhibits hypoxia-activated transcription in cancer cells, and its cytoplasmic location in breast cancer is associated with elevated expression of tumor cell hypoxia-inducible factor 1α. Cancer Res. 64, 6075–6081 10.1158/0008-5472.CAN-04-0708 [DOI] [PubMed] [Google Scholar]
- 24. Freedman S. J., Sun Z. Y., Kung A. L., France D. S., Wagner G., and Eck M. J. (2003) Structural basis for negative regulation of hypoxia-inducible factor-1α by CITED2. Nat. Struct. Biol. 10, 504–512 10.1038/nsb936 [DOI] [PubMed] [Google Scholar]
- 25. Agrawal A., Gajghate S., Smith H., Anderson D. G., Albert T. J., Shapiro I. M., and Risbud M. V. (2008) Cited2 modulates hypoxia-inducible factor-dependent expression of vascular endothelial growth factor in nucleus pulposus cells of the rat intervertebral disc. Arthritis Rheum. 58, 3798–3808 10.1002/art.24073 [DOI] [PubMed] [Google Scholar]
- 26. Lee J. Y., Taub P. J., Wang L., Clark A., Zhu L. L., Maharam E. R., Leong D. J., Ramcharan M., Li Z., Liu Z., Ma Y. Z., Sun L., Zaidi M., Majeska R. J., and Sun H. B. (2009) Identification of CITED2 as a negative regulator of fracture healing. Biochem. Biophys. Res. Commun. 387, 641–645 10.1016/j.bbrc.2009.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiong J. W., Leahy A., Lee H. H., and Stuhlmann H. (1999) Vezf1: a Zn finger transcription factor restricted to endothelial cells and their precursors. Dev. Biol. 206, 123–141 10.1006/dbio.1998.9144 [DOI] [PubMed] [Google Scholar]
- 28. Kuhnert F., Campagnolo L., Xiong J. W., Lemons D., Fitch M. J., Zou Z., Kiosses W. B., Gardner H., and Stuhlmann H. (2005) Dosage-dependent requirement for mouse Vezf1 in vascular system development. Dev. Biol. 283, 140–156 10.1016/j.ydbio.2005.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou Z., Ocaya P. A., Sun H., Kuhnert F., and Stuhlmann H. (2010) Targeted Vezf1-null mutation impairs vascular structure formation during embryonic stem cell differentiation. Arterioscler. Thromb. Vasc. Biol. 30, 1378–1388 10.1161/ATVBAHA.109.200428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerald D., Adini I., Shechter S., Perruzzi C., Varnau J., Hopkins B., Kazerounian S., Kurschat P., Blachon S., Khedkar S., Bagchi M., Sherris D., Prendergast G. C., Klagsbrun M., Stuhlmann H., Rigby A. C., Nagy J. A., and Benjamin L. E. (2013) RhoB controls coordination of adult angiogenesis and lymphangiogenesis following injury by regulating VEZF1-mediated transcription. Nat. Commun. 4, 2824 10.1038/ncomms3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clark S. P., Lewis C. D., and Felsenfeld G. (1990) Properties of BGP1, a poly(dG)-binding protein from chicken erythrocytes. Nucleic Acids Res. 18, 5119–5126 10.1093/nar/18.17.5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dickson J., Gowher H., Strogantsev R., Gaszner M., Hair A., Felsenfeld G., and West A. G. (2010) VEZF1 elements mediate protection from DNA methylation. PLoS Genet. 6, e1000804 10.1371/journal.pgen.1000804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koyano-Nakagawa N., Nishida J., Baldwin D., Arai K., and Yokota T. (1994) Molecular cloning of a novel human cDNA encoding a zinc finger protein that binds to the interleukin-3 promoter. Mol. Cell. Biol. 14, 5099–5107 10.1128/MCB.14.8.5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miyashita H., and Sato Y. (2005) Metallothionein 1 is a downstream target of vascular endothelial zinc finger 1 (VEZF1) in endothelial cells and participates in the regulation of angiogenesis. Endothelium 12, 163–170 10.1080/10623320500227101 [DOI] [PubMed] [Google Scholar]
- 35. Miyashita H., Kanemura M., Yamazaki T., Abe M., and Sato Y. (2004) Vascular endothelial zinc finger 1 is involved in the regulation of angiogenesis: possible contribution of stathmin/OP18 as a downstream target gene. Arterioscler. Thromb. Vasc. Biol. 24, 878–884 10.1161/01.ATV.0000126373.52450.32 [DOI] [PubMed] [Google Scholar]
- 36. Aitsebaomo J., Kingsley-Kallesen M. L., Wu Y., Quertermous T., and Patterson C. (2001) Vezf1/DB1 is an endothelial cell-specific transcription factor that regulates expression of the endothelin-1 promoter. J. Biol. Chem. 276, 39197–39205 10.1074/jbc.M105166200 [DOI] [PubMed] [Google Scholar]
- 37. Lebowitz P. F., and Prendergast G. C. (1998) Functional interaction between RhoB and the transcription factor DB1. Cell Adhes. Commun. 6, 277–287 10.3109/15419069809010787 [DOI] [PubMed] [Google Scholar]
- 38. Gowher H., Brick K., Camerini-Otero R. D., and Felsenfeld G. (2012) Vezf1 protein binding sites genome-wide are associated with pausing of elongating RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 109, 2370–2375 10.1073/pnas.1121538109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gowher H., Stuhlmann H., and Felsenfeld G. (2008) Vezf1 regulates genomic DNA methylation through its effects on expression of DNA methyltransferase Dnmt3b. Genes Dev. 22, 2075–2084 10.1101/gad.1658408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu C.-J., Wang L.-Y., Chodosh L. A., Keith B., and Simon M. C. (2003) Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol. Cell. Biol. 23, 9361–9374 10.1128/MCB.23.24.9361-9374.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamaguchi T. P., Dumont D. J., Conlon R. A., Breitman M. L., and Rossant J. (1993) flk-1, an flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development 118, 489–498 [DOI] [PubMed] [Google Scholar]
- 42. Yang S., Xin X., Zlot C., Ingle G., Fuh G., Li B., Moffat B., de Vos A. M., and Gerritsen M. E. (2001) Vascular endothelial cell growth factor-driven endothelial tube formation is mediated by vascular endothelial cell growth factor receptor-2, a kinase insert domain-containing receptor. Arterioscler. Thromb. Vasc. Biol. 21, 1934–1940 10.1161/hq1201.099432 [DOI] [PubMed] [Google Scholar]
- 43. Kranc K. R., Oliveira D. V., Armesilla-Diaz A., Pacheco-Leyva I., Catarina Matias A., Luisa Escapa A., Subramani C., Wheadon H., Trindade M., Nichols J., Kaji K., Enver T., and Bragança J. (2015) Acute loss of Cited2 impairs Nanog expression and decreases self-renewal of mouse embryonic stem cells. Stem Cells 33, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bruderer M., Alini M., and Stoddart M. J. (2013) Role of HOXA9 and VEZF1 in endothelial biology. J. Vasc. Res. 50, 265–278 10.1159/000353287 [DOI] [PubMed] [Google Scholar]
- 45. Yang Z., and Li J. C. (2008) Stimulation of endothelin-1 gene expression by insulin via phosphoinositide-3 kinase-glycogen synthase kinase-3β signaling in endothelial cells. Life Sci. 82, 512–518 10.1016/j.lfs.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 46. Valster A., Tran N. L., Nakada M., Berens M. E., Chan A. Y., and Symons M. (2005) Cell migration and invasion assays. Methods 37, 208–215 10.1016/j.ymeth.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 47. Eccles S. A., Box C., and Court W. (2005) Cell migration/invasion assays and their application in cancer drug discovery. Biotechnol. Annu. Rev. 11, 391–421 10.1016/S1387-2656(05)11013-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.