

ABSTRACT
During entry, the nonenveloped polyomavirus (PyV) simian virus 40 (SV40) traffics from the cell surface to the endoplasmic reticulum (ER), where it penetrates the ER membrane to reach the cytosol; the virus is then transported into the nucleus to cause infection. Although a coherent understanding of SV40's host entry is emerging, how the virus is ejected from the ER into the cytosol remains mysterious. Our previous analyses revealed that the cytosolic Hsc70-SGTA-Hsp105 complex binds to SV40 and extracts it from the ER into the cytosol. We now report that the nucleotide exchange factor (NEF) Bag2 stimulates SV40 release from Hsc70, thereby enabling successful virus arrival at the cytosol, which leads to infection. Hsp105, another NEF of Hsc70, displays a function overlapping that of Bag2, underscoring the importance of this release reaction. Our findings identify a new component of an extraction machinery essential during membrane penetration of a nonenveloped virus and provide further mechanistic insights into this process.
IMPORTANCE How a nonenveloped virus penetrates a biological membrane to cause infection is a mystery. For the nonenveloped polyomavirus SV40, transport across the ER membrane to reach the cytosol is an essential virus infection step. Here, we identify a novel component of a cytosolic Hsc70-dependent chaperone complex called Bag2 that extracts SV40 from the ER into the cytosol. Bag2 does this by triggering SV40 release from Hsc70, thus ensuring that the virus reaches the cytosol en route for productive infection.
KEYWORDS: endoplasmic reticulum, membrane transport, nonenveloped virus, protein chaperone, simian virus 40
INTRODUCTION
Enveloped and nonenveloped viruses must devise strategies to penetrate a host membrane in order to cause infection. However, this process is likely intrinsically different between the two. For an enveloped virus, encasement in a lipid bilayer allows membrane fusion between viral and host membranes (1). In contrast, a nonenveloped virus, which lacks a surrounding lipid bilayer, must mount an alternative strategy for membrane penetration (1, 2). Although this process is poorly understood, key strategies are becoming clearer. To prime the membrane transport process, a nonenveloped virus undergoes conformational changes induced by host factors (proteases, chaperones, and reductases) or the environment (low pH) at the membrane penetration site. These conformational changes can generate a hydrophobic viral particle that enables the virus to bind to and integrate into the limiting membrane. To complete membrane penetration, recent studies suggest a new strategy in which host factors are in fact exploited to “pull” a nonenveloped virus across the membrane (3, 4), but this last step of nonenveloped virus membrane transport remains enigmatic.
The nonenveloped polyomaviruses (PyVs) are highly prevalent and cause significant diseases in immunocompromised individuals (5, 6). Due to its structural and genetic similarities to human PyV, studies on the simian PyV simian virus 40 (SV40) have provided significant insights into the cellular infection pathway of its human counterparts. Structurally, SV40 is composed of 72 VP1 pentamers that encase the viral DNA genome (7, 8, 9). To maintain stability, these pentamers are linked by disulfide bonds, as well as noncovalent interactions established when the VP1 C-terminal arm extends from one pentamer to an adjacent pentamer (7, 8). Each VP1 pentamer harbors a hydrophobic VP2 or VP3 protein (7, 8, 10). SV40 infection is initiated when the virus binds to the ganglioside GM1 receptor at the plasma membrane (11–13). Upon endocytosis, the virus is delivered in a retrograde manner to endosomal compartments (14) and then to the endoplasmic reticulum (ER), where it penetrates the ER membrane in order to reach the cytosol (14–17). From the cytosol, the virus continues into the nucleus, where replication and transcription of the viral genome occur (18, 19).
In the ER, SV40 and other PyV family members experience conformational changes resulting from interactions with ER-resident chaperones that unfold the VP1 capsid and redox enzymes that reduce and isomerize the capsid disulfide bonds (20–24). These events render the virus hydrophobic by exposing underlying VP2 and VP3, allowing insertion of the virus into the ER membrane (25–27). Our laboratory previously identified the fact that ER membrane J proteins (B12, B14, and C18) act as docking sites to recruit a cytosolic chaperone complex that functions to extract SV40 from the ER into the cytosol (3, 4, 28). To date, the established components of this protein complex are Hsc70, SGTA, and Hsp105 (3, 4, 28).
In one model describing the extraction process, Hsc70 binds to the membrane-embedded SV40 when the ER membrane J proteins convert ATP-Hsc70 to ADP-Hsc70, which displays a high substrate-binding affinity. Once ADP-Hsc70 has reverted to ATP-Hsc70 via the action of a nucleotide exchange factor (NEF), ATP-Hsc70 releases SV40 due to its low affinity for the substrate. Iterative cycles of binding to and release from Hsc70 eject the virus into the cytosol. In the context of this reaction, SGTA regulates Hsc70's ability to engage SV40 (28) and binds directly to SV40 (3), while Hsp105 promotes SV40 disassembly, which likely facilitates the virus extraction process (4).
Here, we report that the Bag2 NEF is a novel component of the cytosolic virus extraction machinery. Using both cell-based and in vitro assays performed under loss-of-function conditions, our findings reveal that Bag2 is necessary for SV40 extraction from the ER into the cytosol because it acts as a NEF to trigger SV40 release from Hsc70, thereby enabling the virus to successfully reach the cytosol. These findings underscore the importance of host NEF activity during SV40 infection and reveal additional insights into the molecular mechanism of nonenveloped virus membrane penetration.
RESULTS
Bag2 binds to ER membrane J proteins.
Through a previously reported unbiased RNA interference (RNAi) screen, the ER membrane J proteins DNAJB12 (B12), DNAJB14 (B14), and DNAJC18 (C18) were found to be essential in promoting ER-to-cytosol membrane transport of SV40 (29). Using a biochemical protein-protein interaction strategy, the components of a cytosolic protein complex composed of Hsc70, SGTA, and Hsp105 were subsequently identified as binding partners of these three ER membrane J proteins that function to extract SV40 from the ER into the cytosol (3, 4, 28, 30). Whether additional cytosolic components play a role in this virus membrane transport process is unknown.
In this context, we previously reported that when 3×FLAG-tagged B12 (3×FLAG-B12) stably expressed in a 293T cell line was immunoprecipitated and the precipitated material was subjected to mass spectrometry analysis, peptides corresponding to the cytosolic NEF Bag2 were identified (31) (Fig. 1A). Likewise, when 3×FLAG-C18 stably expressed in a 293T cell line was immunoprecipitated and the precipitated sample was analyzed by mass spectrometry, Bag2-derived peptides were also found (Fig. 1A). A similar analysis for 3×FLAG-B14 was not performed. These findings raise the possibility that Bag2 represents a novel binding partner of B12/C18. To test this, S-tagged B12 (B12-S), B14 (B14-S), C18 (C18-S), and the control green fluorescent protein (GFP) (GFP-S) were transiently transfected in 293T cells; the S-tagged protein was affinity purified (AP); and the AP sample was subjected to SDS-PAGE, followed by immunoblotting. We found that endogenous Bag2 coprecipitated with B12-S, B14-S, and C18-S but minimally with GFP-S (Fig. 1B, top row), suggesting that Bag2 interacts with the ER membrane J proteins. Moreover, endogenous Bag2 can also bind to endogenous B12 (Fig. 1C, top row), indicating that this interaction is not an overexpression artifact. These results demonstrate that the ER membrane J proteins interact with Bag2.
FIG 1.

Bag2 binds to ER membrane J proteins. (A) Total numbers of peptides corresponding to B12, C18, and Bag2 identified by mass spectrometry using 3×FLAG-B12- or 3×FLAG-C18-immunoprecipitated material from HEK 293T cells. (B) HEK 293T cells were transfected with either GFP-S, B12-S, B14-S, or C18-S. The cells were lysed, and the resulting cell extracts were subjected to affinity purification and SDS-PAGE, followed by immunoblotting using the indicated antibodies. Input represents 8% of the total sample used for affinity purification. (C) HEK 293T cell extracts were incubated with an anti-B12 or a control IgG antibody. The immunoprecipitated (IP) materials were subjected to SDS-PAGE, followed by immunoblotting using the indicated antibodies. (D) As for panel C, except that HEK 293T cells were transfected with GFP-FLAG, B12-FLAG, or QPD B12-FLAG, and the cell extracts were incubated with FLAG antibody-conjugated agarose beads. The asterisk indicates an unidentified protein that cross-reacted with the Hsc70 antibody. (E) As for panel C, except that HEK 293T cells were transfected with GFP-FLAG, WT Bag2*-FLAG, or I160A Bag2*-FLAG, and the cell extracts were incubated with FLAG antibody-conjugated agarose beads.
A J protein binds to and stimulates the ATPase activity of Hsc70 to generate ADP-Hsc70, while a NEF associates with Hsc70, converting ADP-Hsc70 to ATP-Hsc70. In this scenario, Hsc70 bridges the interaction between a J protein and a NEF. Accordingly, we asked if B12's interaction with Bag2 depends on Hsc70. To test this, we transfected 293T cells with either a FLAG-tagged B12 mutant that cannot bind to Hsc70 (QPD B12-FLAG), wild-type B12 (WT B12-FLAG), or the control GFP (GFP-FLAG) construct. When the FLAG-tagged proteins were immunoprecipitated, endogenous Hsc70 was found in the WT B12-FLAG- but not the QPD B12-FLAG-precipitated sample (Fig. 1D, second row from top), indicating that WT B12, but not QPD B12, binds to Hsc70, as previously reported (31). Importantly, endogenous Bag2 was observed in the WT B12-FLAG-precipitated but minimally in the QPD B12-FLAG-precipitated material (Fig. 1D, top row). These results suggest that B12's interaction with Bag2 depends on Hsc70.
To corroborate these data, we performed the reverse experiment in which a Bag2 mutant previously shown to lack Hsc70 binding (I160A Bag2*) (32) (the asterisk in the Bag2 constructs indicates that the backbone Bag2 sequence was modified, rendering it resistant to a Bag2-specific small interfering RNA [siRNA] [see Materials and Methods]) was tested for its ability to associate with B12. Indeed, endogenous B12 and Hsc70 were pulled down when transfected FLAG-tagged WT Bag2 (WT Bag2*-FLAG), but not I160A Bag2 (I160A Bag2*-FLAG), was precipitated (Fig. 1E, top and second panels). Collectively, these findings demonstrate that the B12-Bag2 interaction is mediated by Hsc70.
Bag2 promotes SV40 infection.
As B12 and Hsc70 are both necessary host factors for SV40 entry, we asked if Bag2 plays a role in SV40 infection. Successful SV40 infection can be monitored by expression of the virus-encoded large T antigen in the host nucleus. Accordingly, simian CV-1 cells (used classically to study SV40 infection) were transfected with either the control siRNA (scrambled), an siRNA targeting Bag2, Hsp105 (a positive control), or both Bag2 and Hsp105 siRNAs. The levels of Bag2 and Hsp105 proteins under the knockdown conditions are shown in Fig. 2A. When these cells were incubated with SV40 and analyzed by immunofluorescence microscopy, we found that SV40 infection robustly decreased in Bag2-depleted cells (Fig. 2B), similar to the extent observed when Hsp105 was knocked down, as previously reported (4). Simultaneous depletion of Bag2 and Hsp105 potently impaired virus infection, although it did not appear to exacerbate the single-knockdown phenotype. This finding suggests that neither NEF is sufficient to support SV40 infection.
FIG 2.

Bag2 promotes SV40 infection. (A) siRNA knockdown of Bag2 and Hsp105. Cell extracts derived from CV-1 cells transfected with the indicated siRNA(s) were subjected to SDS-PAGE and immunoblotting with the indicated antibodies. The asterisk indicates an unidentified protein that cross-reacted with the actin antibody. (B) CV-1 cells transfected with the indicated siRNAs were infected with SV40; 24 hpi, the cells were permeabilized, fixed, and stained for large T antigen. At least 300 cells were counted per condition over three biological replicates. The graph represents means and SD. Student's two-tailed t test was used to determine statistical significance. (C) As for panel B, except cells were transfected with the indicated constructs 24 h prior to infection with SV40. The cells were then fixed, permeabilized, and stained for large T antigen and FLAG. Only cells expressing the FLAG construct were counted. At least 100 cells were counted per condition over three biological replicates. The graph represents means and SD. Student's two-tailed t test was used to determine statistical significance.
To ensure that the block in SV40 infection in Bag2 siRNA-treated cells is specifically a result of depleting Bag2 and is not due to unintended off-target effects, we performed a knockdown-rescue experiment. To this end, cells transfected with either scrambled or Bag2 siRNA were cotransfected with either GFP-FLAG, WT Bag2*-FLAG, or I160A Bag2*-FLAG. The cells were incubated with SV40, and large T antigen expression was assessed in FLAG-expressing cells. Using this approach, we found that expressing WT, but not I160A, Bag2 largely restored SV40 infection in Bag2-depleted cells (Fig. 2C), indicating that the decrease in SV40 infection resulting from the Bag2 siRNA is due to the loss of Bag2. These data demonstrate that Bag2 promotes SV40 infection, and this is dependent on Bag2's ability to interact with Hsc70.
Bag2 is important for ER-to-cytosol transport of SV40.
As Bag2 is recruited to the ER membrane J proteins via Hsc70 (Fig. 1), we envision that Bag 2 is strategically localized to promote cytosol entry of SV40 from the ER membrane. To test this, we employed a cell-based, semipermeabilization assay that detects SV40 arrival in the cytosol from the ER. This assay, developed in our laboratory, is routinely used to characterize ER-to-cytosol membrane transport of PyV family members (33). In the assay, cells are treated with the gentle detergent digitonin, which permeabilizes the plasma membrane while maintaining the membrane integrity of intracellular organelles. Centrifugation of the semipermeabilized cells generates two fractions, a supernatant fraction that contains cytosolic proteins and virus that successfully reached the cytosol from the ER (cytosol), and a pellet fraction that harbors membranes, including the ER and virus that is trapped in the ER (membrane). Using this method, cytosolic Hsp90 largely partitioned to the cytosol fraction (Fig. 3A, compare 2nd row to 7th row), while ER-resident PDI remained in the membrane fraction (Fig. 3A, compare 8th row to 3rd row), verifying the integrity of the fractionation protocol. Importantly, under these conditions, we found that the level of SV40 VP1 in the cytosol decreased markedly when Bag2 was knocked down (Fig. 3A, top row; the VP1 band intensity in the cytosol fraction is quantified in panel B), similar to the effect observed when Hsp105 is depleted, as previously reported (4). Using a biochemical extraction approach designed to isolate ER-localized SV40 from the membrane fraction (see Materials and Methods), we found that knockdown of neither Bag2 nor Hsp105 significantly affected the level of ER-localized SV40 (Fig. 3A, 9th row), indicating that these cytosolic factors do not facilitate ER arrival of the virus from the cell surface. Hence, Bag2 plays an important role in promoting cytosol entry of SV40 from the ER, consistent with its function in supporting SV40 infection (Fig. 2).
FIG 3.

Bag2 is important for ER-to-cytosol transport of SV40. (A) CV-1 cells transfected with the indicated siRNAs were infected with SV40 and subjected to semipermeabilization. The resulting cytosol-, membrane-, and ER-localized fractions were subjected to SDS-PAGE and immunoblotting with the indicated antibodies. The amount loaded for the cytosol was 50% of the total cytosol fraction, whereas the amount loaded for the membrane was 20% of the total membrane fraction. Hsp90 and PDI acted as both loading and fractionation controls. (B) The VP1 band intensity in the cytosol fraction from panel A was quantified with Image J software (National Institutes of Health), normalized relative to the Hsp90 loading control bands, and graphed as a percentage of the VP1 band intensity in the scrambled-siRNA-treated sample. The graph represents the means and SD from at least 3 biological replicates. Student's two-tailed t test was used to determine statistical significance.
Depletion of Bag2 traps SV40 in the ER-to-cytosol membrane penetration site.
As an independent approach to evaluate whether Bag2 promotes extraction of SV40 from the ER into the cytosol, we used a microscopy-based strategy. We and others previously demonstrated that upon reaching the ER, SV40 reorganizes selective ER membrane proteins (including the membrane J proteins and the BAP31 ER membrane protein) to distinct punctate structures in the ER harboring the virus, called foci (3, 4, 17, 30). Our findings further suggest that these virus-induced foci act as cytosol entry sites (3, 4, 30). We therefore reasoned that if Bag2 extracts SV40 from the ER into the cytosol, depletion of Bag2 should trap the virus in the foci, consequently augmenting this virus-containing structure. To test this, CV-1 cells transfected with either the scrambled or Bag2 siRNA were infected with SV40 and stained for SV40 VP1 and BAP31. Indeed, we found that depletion of Bag2 increased the number of cells containing at least one BAP31+ focus (Fig. 4A, compare bottom and top rows); the relative size of VP1 harbored in the BAP31+ focus also increased when Bag2 was depleted (Fig. 4B). These findings are consistent with the idea that Bag2 plays an important role in extracting SV40 from the ER into the cytosol.
FIG 4.

Depletion of Bag2 traps SV40 in the ER-to-cytosol membrane penetration site. (A) CV-1 cells were transfected with the indicated siRNAs for 48 h, followed by 16 h of SV40 infection. The cells were fixed, permeabilized, stained using anti-BAP31 and anti-VP1 antibodies, and imaged by fluorescence microscopy. The enlarged images (3× zoom) correspond to the boxed areas in the merged images. The arrowheads indicate BAP31 foci. (B) The size of the VP1 focus was quantified based on the measured area (in pixels) using Image J software. The graph represents the means and SD of at least 30 cells counted in at least three biological replicates. Student's two-tailed t test was used to determine statistical significance. (C) As for panel A, except cells were transfected with the indicated constructs 24 h prior to infection with SV40. The cells were then fixed, permeabilized, and stained using anti-BAP31, anti-FLAG, or anti-S antibodies. Only cells expressing the indicated FLAG- or S-tagged constructs were counted. The data plotted are the numbers of cells with at least one BAP31 focus, graphed as a percentage of the scrambled siRNA-treated sample. The graph represents the means and SD of at least 100 cells counted in at least three biological replicates. Student's two-tailed t test was used to determine statistical significance. (D) Representative images of the data in panel C. The arrowheads indicate BAP31 foci. The asterisks indicate cells expressing the indicated constructs.
We further asked whether reexpression of WT Bag2, I160A Bag2, or HspBP1, a cytosolic NEF previously shown to be dispensable during SV40 infection (4), in Bag2-depleted cells affected focus formation. For this, CV-1 cells transfected with either the scrambled or Bag2 siRNA were cotransfected with GFP-FLAG, WT Bag2*-FLAG, I160A Bag2*-FLAG, or HspBP1-S. The cells were then infected with SV40 and stained for BAP31. Only cells expressing the FLAG-tagged (or S-tagged) protein were assessed. Again, we found that depleting Bag2 increased the number of cells containing at least one BAP31+ focus (the extent of focus formation is quantified in Fig. 4C; representative images are shown in panel D). Importantly, add-back of WT Bag2*-FLAG, but not I160A Bag2*-FLAG or HspBP1-S, in the Bag2-depleted cells reduced the number of cells harboring at least one BAP31+ focus to control levels (the extent of focus formation is quantified in Fig. 4C; see representative images in Fig. 4D). These results indicate that restoring Bag2 expression in Bag2-depleted cells can promote cytosol entry of the virus from the ER, thereby decreasing the focus structures. Thus, our microscopy-based analyses are in agreement with data from the semipermeabilization assay, strengthening the hypothesis that Bag2 plays a decisive role in extracting SV40 from the ER into the cytosol.
Bag2 triggers release of SV40 from Hsc70.
Our data indicate that Bag2 binds to B12 via Hsc70. Because Bag2 functions as a NEF to induce substrate release from Hsc70 (32, 34), we hypothesized that Bag2 induces release of SV40 from Hsc70 to promote cytosol entry. To test this, we transfected (CV-1-derived) COS-7 cells expressing Hsc70-S with scrambled or Bag2 siRNA and precipitated Hsc70-S. (COS-7 cells were used because they support higher transfection efficiency than CV-1 cells). Importantly, under the Bag2 knockdown condition, Hsc70-S precipitated significantly more VP1 than cells treated with scrambled siRNA (Fig. 5A, top row; the VP1 band intensity is quantified in panel B), indicating that SV40 is trapped on Hsc70 in the absence of Bag2. This result is consistent with the hypothesis that Bag2 stimulates SV40 release from Hsc70.
FIG 5.
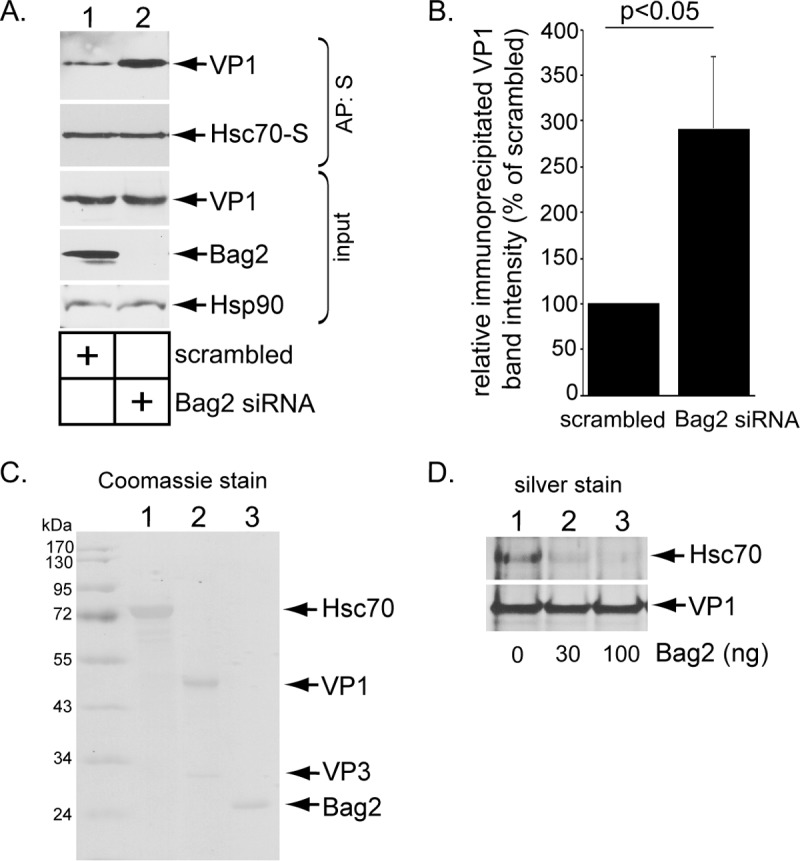
Bag2 releases SV40 from Hsc70. (A) COS-7 cells were treated with the indicated siRNAs for 24 h, followed by transfection of the Hsc70-S construct. After 24 h, the cells were infected with SV40 for 16 h, and the resulting cell extracts were subjected to affinity purification, SDS-PAGE, and immunoblotting with the indicated antibodies. (B) The VP1 band intensity from panel A was quantified with Image J software (National Institutes of Health), normalized relative to the affinity-purified Hsc70-S level, and graphed as a percentage of the VP1 band intensity in the scrambled-siRNA-treated sample. The graph represents the means and SD from three biological replicates. Student's two-tailed t test was used to determine statistical significance. (C) Coomassie staining of commercially available Hsc70 and Bag2, along with purified SV40. Note that the level of VP2 in the SV40 preparation is below the threshold of detection. (D) After SV40 was incubated with Hsc70 (in the presence of 1 mM DTT), precipitation of SV40 VP1 coprecipitated Hsc70. The SV40-Hsc70 complex was incubated with a control buffer containing only ATP, or the indicated amount of Bag2 with ATP, followed by reimmunoprecipitation of SV40 VP1. The reimmunoprecipitated material was subjected to SDS-PAGE followed by silver staining.
To strengthen this finding, we reconstituted the virus release reaction using purified components, including Hsc70, SV40, and Bag2 (Fig. 5C). In the ER, SV40 undergoes disulfide bond reduction, priming it for insertion into the ER membrane (22, 23). The reductant dithiothreitol (DTT) was therefore added to purified SV40 to partially mimic this reduced viral conformation. The reduced SV40 was incubated with Hsc70, followed by pulldown of SV40 in order to isolate the SV40-Hsc70 complex. Importantly, when this complex was subsequently incubated with increasing concentrations of Bag2 in the presence of ATP and the SV40 was reisolated, the level of bound Hsc70 decreased compared to addition of a control buffer containing only ATP (Fig. 5D, top row, compare lanes 2 and 3 to lane 1). These findings demonstrate that Bag2 can promote disengagement of SV40 from Hsc70 in vitro, in agreement with the premise that Bag2 induces the release of SV40 from Hsc70 in the intact cell.
Bag2 and Hsp105 play overlapping roles.
Our results show that depleting either Bag2 or Hsp105 inhibits SV40 infection (Fig. 2B). Because Hsp105 is also a NEF of Hsc70 (35–37), we asked if Bag2 and Hsp105 perform overlapping roles. To test this, we transfected cells with scrambled, Bag2, or Hsp105 siRNA and cotransfected the cells with either GFP-FLAG, WT Bag2*-FLAG, or Hsp105*-FLAG (Hsp105*-FLAG is a construct that is resistant to the Hsp105 siRNA). Only FLAG-expressing cells were assessed for large T antigen expression in order to evaluate SV40 infection. We found that add-back of Bag2 largely restored the decrease in SV40 infection in Bag2-depleted cells (Fig. 6A, compare columns 3 and 1, and 2C), while add-back of Hsp105 rescued the block in SV40 infection in Hsp105-depleted cells (Fig. 6A, compare 6th and 1st columns from left), which is consistent with our previous report (4). Strikingly, expression of Hsp105 fully restored SV40 infection in Bag2-depleted cells (Fig. 6A, compare 4th and 1st columns from left), and likewise, expression of Bag2 rescued SV40 infection in Hsp105-depleted cells (Fig. 6A, compare 7th and 1st columns from left). These results indicate that Bag2 and Hsp105 display overlapping functions.
FIG 6.

Bag2 and Hsp105 play overlapping roles. (A) CV-1 cells were transfected with the indicated siRNAs for 24 h, followed by DNA transfection of the indicated FLAG constructs. After 24 additional hours, the cells were infected with SV40 for 24 h, fixed, permeabilized, and stained for FLAG and large T antigen. Only cells expressing the indicated FLAG constructs were counted. At least 100 cells were counted per condition over three biological replicates. The graph represents means and SD. Student's two-tailed t test was used to determine statistical significance. (B) As for panel A, except cells were infected with SV40 for 16 h prior to being fixed, permeabilized, and stained for FLAG and BAP31. Only cells expressing the indicated FLAG constructs were counted. The graph represents the means and SD of at least 100 cells counted in at least three biological replicates. (C) Representative images of the data in panel B. The arrowheads indicate BAP31 foci. The asterisks indicate cells expressing the indicated constructs. (D) HEK 293T cells were transfected with the indicated FLAG-tagged constructs and lysed, and the resulting cell extract was subjected to immunoprecipitation, SDS-PAGE, and immunoblotting with the indicated antibodies.
As both Bag2 (this study) and Hsp105 (4) promote the arrival of SV40 in the cytosol from the ER, we assessed if Bag2 and Hsp105 play overlapping roles at this step. To this end, cells transfected with scrambled or Bag2 siRNA were cotransfected with GFP-FLAG, WT Bag2*-FLAG, or Hsp105*-FLAG. The cells were then infected with SV40 and stained for BAP31 and VP1 to detect formation of virus-induced focus structures that represent the virus' cytosol entry sites (Fig. 4A). We found that the enhanced focus formation observed in Bag2-depleted cells could be decreased to control levels by add-back of either WT Bag2*-FLAG or Hsp105*-FLAG (the extent of focus formation is quantified in Fig. 6B, with representative images in panel C), suggesting that SV40 trapped in the focus structure due to the absence of Bag2 can be released into the cytosol (thereby decreasing the focus structure) when Bag2 or Hsp105 is added back. These results corroborate the infection data, suggesting that the two cytosolic NEFs have overlapping functions.
Although Bag2 and Hsp105 must bind to Hsc70 to induce substrate release, whether they engage the same Hsc70 complex is unknown. We postulate that if Bag2 and Hsp105 in fact interact with the same Hsc70 complex, precipitation of Bag2 should correspondingly pull down Hsp105, and vice versa. To test this, Hsp105*-FLAG, WT Bag2*-FLAG, and the control GFP-FLAG were immunoprecipitated, and the precipitated material was subjected to immunoblotting. We found that precipitation of Hsp105*-FLAG (but not GFP-FLAG) pulled down endogenous (and transfected) Hsp105 and Hsc70, but not endogenous Bag2 (Fig. 6D, top three rows, lanes 2). Similarly, precipitation of WT Bag2*-FLAG coprecipitated endogenous (and transfected) Bag2 and Hsc70, but not endogenous Hsp105 (Fig. 6D, top three rows, lanes 3). These results suggest that Bag2 and Hsp105 may form separate Hsc70 complexes.
DISCUSSION
Host membrane penetration is essential for successful virus infection. For a nonenveloped virus, this process remains enigmatic. In the case of the nonenveloped PyV SV40, penetration of the virus across the ER membrane to reach the cytosol represents a decisive infection step. However, the molecular basis by which host factors are exploited to extract the virus from the ER into the cytosol has yet to be fully clarified. We previously demonstrated that Hsc70, SGTA, and Hsp105 form a cytosolic chaperone-cochaperone complex that promotes cytosol entry of the virus from the ER (3, 4, 28); whether Hsp70 or additional cytosolic components participate in this virus extraction process is unknown. Here, we pinpoint the cytosolic NEF Bag2 as a novel cellular component that promotes SV40 infection by supporting viral ER-to-cytosol membrane transport. Our results indicate that Bag2 associates with Hsc70 and ER membrane J proteins to generate a protein complex that is likely distinct from the Hsp105-Hsc70-J protein complex (Fig. 7). Importantly, during SV40 entry, Bag2 acts as a NEF to stimulate virus release from Hsc70 in order to support cytosol entry of the virus (Fig. 7, inset).
FIG 7.

The Hsc70-SGTA-Bag2 complex docks on an ER membrane J protein and promotes SV40 cytosol entry. In our model, Hsc70-containing protein complexes dock on an ER membrane J protein, such as B12. In association with its cochaperone, SGTA, an Hsc70 molecule recruits Bag2 or Hsp105, forming either the Hsc70-SGTA-Bag2 or Hsc70-SGTA-Hsp105 cytosolic extraction complex. During the final step of SV40's ER-to-cytosol membrane transport, a single membrane-embedded viral particle recruits many copies of either the Hsc70-SGTA-Bag2 or Hsc70-SGTA-Hsp105 protein complex. Iterative cycles of SV40 binding to and release from Hsc70 in turn provide the energy to extract the virus into the cytosol. (Inset) Importantly, Bag2 functions to trigger SV40 release from Hsc70 in this cycle. Bag2 does this by promoting ADP release from Hsc70, generating ATP-Hsc70, which displays low affinity for its substrate, such as SV40. “Focus” refers to the SV40 cytosol entry site in the ER membrane, where the cytosolic extraction machinery is recruited to the ER membrane J protein.
Our previous mass spectrometry data raised the possibility that Bag2 interacts with the ER membrane J protein B12 (31); a similar mass spectrometry approach also suggested that C18 engages Bag2. By immunoprecipitation and affinity purification, we confirmed that Bag2 indeed binds to B12 and C18, as well as the J protein B14, demonstrating that Bag2 is a bona fide binding partner of the J proteins. Using established Hsc70 binding-defective mutants of either Bag2 or B12, our results further indicate that the Bag2-B12 interaction occurs via an Hsc70 intermediate, revealing that this interaction is coupled through the Hsc70 chaperone.
Through knockdown-rescue experiments, we found that Bag2 executes an important Hsc70-dependent function to promote SV40 infection. We then took advantage of two independent strategies—a semipermeabilized fractionation method and a microscopy-based focus-tracking assay—to demonstrate that Bag2 promotes virus extraction from the ER to the cytosol to support virus infection. Hence, by strategic positioning at the ER membrane (via engaging the Hsc70-J protein complex), Bag2 triggers cytosol entry of SV40 from the ER. Whether Bag2 impacts ER-to-cytosol membrane transport of endogenous misfolded substrates is a critical question that needs to be addressed by future experiments. In this context, it is worth noting that ER-to-cytosol membrane transport is central to a major ER-associated degradation pathway designed to remove misfolded ER-resident proteins to the cytosol for degradation by the proteasome (38). In fact, Hsc70, the J proteins, and Bag2 have already been implicated in proteasomal degradation of misfolded substrates (39–42).
Mechanistically, we demonstrated via cell-based and in vitro approaches that Bag2 triggers SV40 release from Hsc70, thereby promoting arrival of SV40 in the cytosol. This result is in line with Bag2's established NEF activity, which normally converts ADP-Hsc70 to ATP-Hsc70 to trigger substrate release (32). Our data thus support the hypothesis that release of SV40 from Hsc70 is necessary for successful SV40 arrival in the cytosol.
We previously found that Hsp105 is a crucial factor that stimulates cytosol entry of SV40 from the ER (4). Because Hsp105 can also act as a NEF of Hsc70 (35–37), we asked if Hsp105 and Bag2 displayed overlapping functions and found that they did. However, it is clear that neither NEF is sufficient to support SV40 infection or ER-to-cytosol transport of the virus. Why might ER-to-cytosol transport of SV40 require two distinct NEFs that display overlapping functions? One possibility is that a threshold level of NEFs beyond what is normally provided by Bag2 or Hsp105 alone is necessary for efficient ER-to-cytosol membrane extraction of SV40. Having both Bag2 and Hsp105 thus provides a sufficiently high level of NEF to carry out the substrate release reaction. However, the observation that the HspBP1 NEF is not involved in SV40 ER membrane penetration (4) suggests that only selective cytosolic NEFs that are membrane associated are strategically positioned to extract the virus from the ER into the cytosol. Another possibility is that the different cell types infected by SV40 may harbor only one of the two NEFs, and thus, having multiple NEFs capable of regulating the same SV40 entry step would ensure successful infection. The simultaneous use of two NEFs to cross the ER membrane is not unprecedented. In fact, cholera toxin requires both ER-resident NEFs, Sil1 and Grp170, to penetrate the ER membrane during the toxin's cellular-intoxication process (43).
Single knockdown of either Bag2 or Hsp105 robustly impaired SV40 infection, indicating that both Bag2 and Hsp105 play critical roles during SV40 infection. Double knockdown of Bag2 and Hsp105 did not cause a more severe block in infection than the single-knockdown condition, although this may reflect the sensitivity of the infection assay. Regardless, this finding suggests that Bag2 and Hsp105 operate within the same pathway during SV40 entry. Because each Hsc70 interacts with only one NEF (through its single ATPase domain) at any one point and Bag2 and Hsp105 engage different Hsc70 complexes (this study), the simplest model for these data is that an individual SV40 particle simultaneously recruits multiple Bag2-Hsc70 and Hsp105-Hsc70 protein complexes, as depicted in Fig. 7; the inclusion of SGTA in this model is based on our previous findings (3). We envision that iterative cycles of binding to and release from these Hsc70-dependent complexes provide the energy to extract SV40 from the ER into the cytosol. In sum, our work here identifies a novel member of a cytosolic protein complex that extracts a nonenveloped virus across a host membrane, which enables its cytosolic entry to cause infection, and provides further insights into this largely enigmatic process.
MATERIALS AND METHODS
Antibodies.
Endogenous Bag2, PDI, anti-VP1, and anti-S antibodies were purchased from Abcam (Cambridge, MA). Hsc70 and BAP31 antibodies were purchased from Invitrogen (Carlsbad, CA). Actin antibody was purchased from Cell Signaling (Danvers, MA). Anti-FLAG antibody was purchased from Sigma (St. Louis, MO). Monoclonal large T antigen Hsp90, and Hsp105 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-B12 antibody was purchased from Proteintech Group (Chicago, IL).
Reagents.
FLAG M2 antibody-conjugated beads, phenylmethylsulfonyl fluoride (PMSF), and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). S protein-conjugated beads and digitonin were purchased from EMD Millipore Chemicals (San Diego, CA). Opti-MEM and 0.25% trypsin were purchased from Invitrogen (Carlsbad, CA). Prolong Diamond antifade mount with DAPI (4′,6-diamidino-2-phenylindole) mounting reagent was purchased from Thermo Fisher (Carlsbad, CA). Phosphate-buffered saline (PBS) (1×) was purchased from Gibco (Carlsbad, CA). Purified Hsc70 was purchased from StressMarq Biosciences (Victoria, British Columbia, Canada), and purified Bag2 was purchased from Fitzgerald Industries International (Acton, MA). Magnetic protein G-conjugated Dynabeads were purchased from ThermoFisher (Carlsbad, CA).
Cells.
CV-1, COS-7, and HEK 293T (ATCC) cells were cultured in complete Dulbecco's modified Eagle medium (cDMEM) supplemented with 10% fetal bovine serum (FBS), 10 U/ml penicillin, and 10 μg/ml streptomycin from Gibco (Carlsbad, CA).
DNA plasmid and siRNA transfection.
To generate a Bag2 expression construct, Bag2 was cloned from a 293T cDNA library and amplified by PCR. PCR fragments were then inserted into a pcDNA3.1(−) backbone. To generate an siRNA-resistant Bag2 construct, overlapping PCR was performed using the following primers: forward, CTCCGTGGAGACTATCAGGAATCCCCAGCAGCAAGAATCCCT, and reverse, ATTCCTGATAGTCTCCACGGAGACTTCAACGGTGAGAGTTCT.
The amplified Bag2 fragment generates the following silent mutations (underlined): GTC TCC GTG GAG ACT ATC AGG AAT. The I160A Bag2 mutant was generated using point mutagenesis at residue I160, as previously reported (32). All the Bag2 constructs were inserted into the pcDNA3.1(−) backbone harboring a C-terminal FLAG tag and subjected to DNA sequencing to confirm the sequence. The Bag2 siRNA sequence is 5′-GUUGGCUUUAGCGUUGAUCUUCGCCUG-3′ (Life Technologies), and the scrambled siRNA sequence is that of the “all-star negative” siRNA from Qiagen. All DNA and siRNA transfections were incubated for at least 24 h.
Preparation of SV40.
Purified SV40 preparation using the OptiPrep gradient system (Sigma) has been described previously (33).
Immunoprecipitation.
For affinity purification of S-tagged J proteins, HEK 293T cells were seeded in 6-cm plates. The cells were transfected or not with B12-S, B14-S, C18-S, or GFP-S for 24 h in polyethyleneimine (PEI) and Opti-MEM and lysed in 1% Triton X-100 in physiological buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, and phenylmethylsulfonyl fluoride [PMSF]) at 4°C. Postlysis, the cells were centrifuged at 16,000 rpm at 4°C for 10 min. The supernatant was collected and rotated at 4°C for 2 h with S protein-conjugated beads. The beads were washed three times and boiled in 5× SDS sample buffer before being subjected to SDS-PAGE and immunoblotting. For immunoprecipitation of endogenous B12, cells were lysed, and the resulting lysate was incubated with an antibody against B12 (or a nonspecific IgG control antibody). The lysates were then incubated with protein A agarose beads for 2 h. The precipitated materials were processed as described above. For immunoprecipitation of FLAG-tagged proteins, HEK 293T cells were seeded in 6-cm plates and transfected with FLAG-tagged constructs in PEI and Opti-MEM for 24 h. The cells were lysed in 1% Triton X-100 in a physiological buffer at 4°C for 10 min and centrifuged at 16,000 rpm at 4°C for 10 min. The supernatant was collected and rotated at 4°C for 2 h with M2 FLAG-conjugated beads. The precipitated materials were processed as described above. To examine the interaction between SV40 and Hsc70 under Bag2 knockdown conditions, COS-7 cells were seeded in 6-cm plates and treated with either 50 nM Bag2 siRNA or a scrambled siRNA. After 24 h, the cells were transfected with Hsc70-S for 24 h. The cells were then infected with SV40 (multiplicity of infection [MOI], ∼20 to 30) for 16 h before being lysed in 1% Triton X-100 in a physiological buffer at 4°C for 10 min. The cells were then centrifuged at 16,000 rpm for 10 min at 4°C. The supernatant was collected and rotated with S protein-conjugated beads for 2 h at 4°C. After 2 h, the beads were washed three times in lysis buffer. The beads were then treated with 5× SDS sample buffer and subjected to SDS-PAGE and immunoblotting. The graph in Fig. 5B represents three biological replicates, and the VP1 band was quantified using Image J software (National Institutes of Health) and normalized to affinity-purified Hsc70-S.
SV40 infection.
CV-1 cells (3 × 105) were seeded and transfected with 50 nM Bag2 siRNA, 25 nM Hsp105 siRNA, or 50 nM scrambled siRNA (along with Opti-MEM and RNAiMax) onto glass coverslips in 6-well plates. The cells were incubated at 37°C for 48 h. For knockdown-rescue experiments, 1.5 × 105 CV-1 cells were seeded and transfected using siRNA as described above. After 24 h of siRNA transfection, the cells were washed and transfected with the indicated FLAG-tagged constructs using Fugene and Opti-MEM, totaling 48 h of siRNA knockdown before infection. After 48 h, cells were infected with purified SV40 (MOI, ∼0.5 to 1.5) for 24 h. After 24 h, the cells were fixed in 1% paraformaldehyde (PFA) for 15 min, permeabilized in 0.2% Triton X-100 for 5 min, and incubated with rabbit anti-FLAG and mouse anti-large T antigen antibodies for 1 h at 25°C. The cells were then washed and incubated with anti-rabbit (Alexa Fluor 488) and anti-mouse (Alexa Fluor 594) antibodies in the dark at 25°C for 30 min. The cells were washed, dried, and mounted on slides using Prolong Diamond antifade mount with DAPI (Invitrogen). For evaluating SV40 infection in cells without DNA transfection, at least 1,000 cells were counted per condition. To assess SV40 infection in cells transfected with DNA, at least 100 cells were counted per condition. The graphs in Fig. 2B and C and 6A represent the mean and standard deviation (SD) of at least 3 biological replicates, with paired Student two-tailed t tests used to determine the P values.
ER-to-cytosol transport assay.
The protocol for the ER-to-cytosol transport assay has been described previously (33). Briefly, CV-1 cells were seeded in 6-cm plates and transfected with 50 nM Bag2 siRNA, 50 nM scrambled siRNA, or 25 nM Hsp105 siRNA for 24 h. After 24 h, the cells were infected with purified SV40 (MOI, ∼1 to 5) for 16 h. The cells were then treated with 0.1% digitonin and centrifuged. The resulting supernatant fraction represented the cytosol fraction, while the pellet fraction represented the membrane fraction. When the membrane fraction was treated with 1% Triton X-100 and the extracted material was isolated, the fraction then contained ER-localized SV40. The VP1 band intensity was quantified using Image J software (National Institutes of Health) and normalized to Hsp90 loading control bands.
Focus formation assay.
The focus formation assay method has been described previously (3, 4, 30). Briefly, control or knocked-down cells were fixed 16 h postinfection (hpi) (MOI, ∼1 to 5) and stained with anti-rat BAP31 and anti-mouse VP1 antibodies for 1 h at 25°C. The cells were then washed and incubated with anti-mouse (Alexa Fluor 488) and anti-rat (Alexa Fluor 594) antibodies in the dark at 25°C for 30 min. For the knockdown-rescue focus formation assay, cells were transfected with siRNA as described above for 24 h, followed by DNA transfection of the indicated constructs for 24 h. The cells were fixed as described above and stained with anti-rat BAP31 and anti-rabbit FLAG and S antibodies. The cells were washed and incubated with anti-rat (Alexa Fluor 594) and anti-rabbit (Alexa Fluor 488) antibodies as described above. At least 100 cells were counted per condition, and 3 biological replicates were completed. The graphs in Fig. 4C and 6B represent the means and standard deviations of at least 3 biological replicates, with paired Student two-tailed t tests used to determine P values. Using Image J (National Institutes of Health), the VP1 focus size was measured by transforming images to 8 bits, adjusting the threshold, and processing the images as binary with watershed. The focus size was then quantified in pixels squared.
Bag2-dependent release of Hsc70 from SV40.
To isolate the Hsc70-SV40 complex, 1 μg of Hsc70, 250 ng of SV40, 1 mM MgCl2, 2 mM ATP, and 1 mM DTT were incubated at 37°C in 0.1% Triton X-100 in PBS for 30 min. Anti-VP1 antibody was added to the samples for 30 min at 25°C, followed by addition of magnetic protein G Dynabeads for 30 min at 25°C. The beads were then washed 2 times in PBS. The beads then harbored the Hsc70-SV40 complex. To induce release of Hsc70 from SV40, purified Bag2 was added to samples at the indicated concentrations in the presence of 1 mM MgCl2, 1 mM KCl, and 2 mM ATP at 25°C for 10 min. The beads were subsequently washed 2 times in PBS, boiled in 5× SDS running buffer for 10 min, and subjected to silver staining.
ACKNOWLEDGMENTS
We thank Chelsey Spriggs for critical reviews of the manuscript. We especially thank Takamasa Inoue for his insights throughout this project.
This work was funded by grants from the National Institutes of Health (RO1 AI064296 and GM113722) to B.T.
REFERENCES
- 1.Poranen MM, Daugelavicius R, Bamford DH. 2002. Common principles in viral entry. Annu Rev Microbiol 56:521–538. doi: 10.1146/annurev.micro.56.012302.160643. [DOI] [PubMed] [Google Scholar]
- 2.Tsai B. 2007. Penetration of nonenveloped viruses into the cytoplasm. Annu Rev Cell Dev Biol 23:23–43. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed] [Google Scholar]
- 3.Walczak CP, Ravindran MS, Inoue T, Tsai B. 2014. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog 10:e1004007. doi: 10.1371/journal.ppat.1004007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravindran MS, Bagchi P, Inoue T, Tsai B. 2015. A non-enveloped virus hijacks host disaggregation machinery to translocate across the endoplasmic reticulum membrane. PLoS Pathog 11:e1005086. doi: 10.1371/journal.ppat.1005086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalianis T, Hirsch HH. 2013. Human polyomaviruses in disease and cancer. Virology 437:63–72. doi: 10.1016/j.virol.2012.12.015. [DOI] [PubMed] [Google Scholar]
- 6.DeCaprio JA, Garcea RL. 2013. A cornucopia of human polyomaviruses. Nat Rev Microbiol 11:264–276. doi: 10.1038/nrmicro2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liddington RC, Yan Y, Moulai J, Sahli R, Benjamin TL, Harrison SC. 1991. Structure of simian virus 40 at 3.8-A resolution. Nature 354:278–284. doi: 10.1038/354278a0. [DOI] [PubMed] [Google Scholar]
- 8.Stehle T, Yan Y, Benjamin TL, Harrison SC. 1994. Structure of murine polyomavirus complexed with an oligosaccharide receptor fragment. Nature 369:160–163. doi: 10.1038/369160a0. [DOI] [PubMed] [Google Scholar]
- 9.Stehle T, Gamblin SJ, Yan Y, Harrison SC. 1996. The structure of simian virus 40 refined at 3.1 A resolution. Structure 4:165–182. doi: 10.1016/S0969-2126(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 10.Chen XS, Stehle T, Harrison SC. 1998. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J 17:3233–3240. doi: 10.1093/emboj/17.12.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith AE, Lilie H, Helenius A. 2003. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett 555:199–203. doi: 10.1016/S0014-5793(03)01220-1. [DOI] [PubMed] [Google Scholar]
- 12.Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J 22:4346–4355. doi: 10.1093/emboj/cdg439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campanero-Rhodes MA, Smith A, Chai W, Sonnino S, Mauri L, Childs RA, Zhang Y, Ewers H, Helenius A, Imberty A, Feizi T. 2007. N-glycolyl GM1 ganglioside as a receptor for simian virus 40. J Virol 81:12846–12858. doi: 10.1128/JVI.01311-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J Virol 85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian M, Cai D, Verhey KJ, Tsai B. 2009. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog 5:e1000465. doi: 10.1371/journal.ppat.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kartenbeck J, Stukenbrok H, Helenius A. 1989. Endocytosis of simian virus 40 into the endoplasmic reticulum. J Cell Biol 109:2721–2729. doi: 10.1083/jcb.109.6.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A. 2011. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol 13:1305–1314. doi: 10.1038/ncb2339. [DOI] [PubMed] [Google Scholar]
- 18.Clever J, Yamada M, Kasamatsu H. 1991. Import of simian virus 40 virions through nuclear pore complexes. Proc Natl Acad Sci U S A 88:7333–7337. doi: 10.1073/pnas.88.16.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakanishi A, Clever J, Yamada M, Li PP, Kasamatsu H. 1996. Association with capsid proteins promotes nuclear targeting of simian virus 40 DNA. Proc Natl Acad Sci U S A 93:96–100. doi: 10.1073/pnas.93.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magnuson B, Rainey EK, Benjamin T, Baryshev M, Mkrtchian S, Tsai B. 2005. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol Cell 20:289–300. doi: 10.1016/j.molcel.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 21.Gilbert J, Ou W, Silver J, Benjamin T. 2006. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J Virol 80:10868–10870. doi: 10.1128/JVI.01117-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, Helenius A. 2007. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131:516–529. doi: 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 23.Walczak CP, Tsai B. 2011. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J Virol 85:2386–2396. doi: 10.1128/JVI.01855-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoue T, Dosey A, Herbstman JF, Ravindran MS, Skiniotis G, Tsai B. 2015. ERdj5 reductase cooperates with protein disulfide isomerase to promote simian virus 40 endoplasmic reticulum membrane translocation. J Virol 89:8897–8908. doi: 10.1128/JVI.00941-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daniels R, Rusan NM, Wadsworth P, Hebert DN. 2006. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol Cell 24:955–966. doi: 10.1016/j.molcel.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Rainey-Barger EK, Magnuson B, Tsai B. 2007. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J Virol 81:12996–13004. doi: 10.1128/JVI.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuksin D, Norkin LC. 2012. Disassembly of simian virus 40 during passage through the endoplasmic reticulum and in the cytoplasm. J Virol 86:1555–1562. doi: 10.1128/JVI.05753-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dupzyk A, Williams JM, Bagchi P, Inoue T, Tsai B. 2017. SGTA-dependent regulation of Hsc70 promotes cytosol entry of Simian Virus 40 from the endoplasmic reticulum. J Virol 12:e00232-. doi: 10.1128/JVI.00232-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodwin EC, Lipovsky A, Inoue T, Magaldi TG, Edwards AP, Van Goor KE, Paton AW, Paton JC, Atwood WJ, Tsai B, DiMaio D. 2011. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. mBio 2:e00101-. doi: 10.1128/mBio.00101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bagchi P, Walczak CP, Tsai B. 2015. The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration. J Virol 89:4058–4068. doi: 10.1128/JVI.03574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue T, Tsai B. 2017. Regulated Erlin-dependent release of the B12 transmembrane J-protein promotes ER membrane penetration of a non-enveloped virus. PLoS Pathog 13:e1006439. doi: 10.1371/journal.ppat.1006439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Z, Page RC, Gomes MM, Kohli E, Nix JC, Herr AB, Patterson C, Misra S. 2008. Structural basis of nucleotide exchange and client binding by the Hsp70 cochaperone Bag2. Nat Struct Mol Biol 15:1309–1317. doi: 10.1038/nsmb.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue T, Tsai B. 2011. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog 7:e1002037. doi: 10.1371/journal.ppat.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rauch JN, Gestwicki JE. 2014. Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J Biol Chem 289:1402–1414. doi: 10.1074/jbc.M113.521997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. 2006. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J 25:2519–2528. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raviol H, Bukau B, Mayer MP. 2006. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett 580:168–174. doi: 10.1016/j.febslet.2005.11.069. [DOI] [PubMed] [Google Scholar]
- 37.Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. 2013. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J Biol Chem 288:21399–21411. doi: 10.1074/jbc.M113.479253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berner N, Reutter KR, Wolf DH. 2 February 2018. Protein quality control of the endoplasmic reticulum and ubiquitin-proteasome-triggered degradation of aberrant proteins: yeast pioneers the path. Annu Rev Biochem. doi: 10.1146/annurev-biochem-062917-012749. [DOI] [PubMed] [Google Scholar]
- 39.Arndt V, Daniel C, Nastainczyk W, Alberti S, Hohfeld J. 2005. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol Biol Cell 16:5891–5900. doi: 10.1091/mbc.e05-07-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Paula CA, Santiago FE, de Oliveira AS, Oliveira FA, Almeida MC, Carrettiero DC. 2016. The co-chaperone BAG2 mediates cold-induced accumulation of phosphorylated tau in SH-SY5Y cells. Cell Mol Neurobiol 36:593–602. doi: 10.1007/s10571-015-0239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin L, Guo J, Zheng Q, Zhang H. 2016. BAG2 structure, function and involvement in disease. Cell Mol Biol Lett 21:18. doi: 10.1186/s11658-016-0020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schonbuhler B, Schmitt V, Huesmann H, Kern A, Gamerdinger M, Behl C. 2016. BAG2 interferes with CHIP-mediated ubiquitination of HSP72. Int J Mol Sci 18:E69. doi: 10.3390/ijms18010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams JM, Inoue T, Chen G, Tsai B. 2015. The nucleotide exchange factors Grp170 and Sil1 induce cholera toxin release from BiP to enable retrotranslocation. Mol Biol Cell 26:2181–2189. doi: 10.1091/mbc.e15-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]