

Proteins can be conjugated covalently by ubiquitin or ubiquitin-like proteins, such as SUMO and ISG15. ISG15 is highly induced in viral infection, and ISG15 conjugation, termed ISGylation, plays important regulatory roles in viral growth. Although ISGylation has been shown to negatively affect many viruses, including human cytomegalovirus (HCMV), viral countermeasures that might modulate ISGylation are not well understood. In the present study, we show that the transmembrane protein encoded by HCMV UL50 inhibits ISGylation by causing proteasomal degradation of UBE1L, an E1-activating enzyme for ISGylation. This pUL50 activity requires membrane targeting. In support of this finding, RNF170, an ER-associated ubiquitin E3 ligase, interacts with pUL50 and promotes UL50-mediated UBE1L ubiquitination and degradation. Our results provide the first evidence, to our knowledge, that viruses can regulate ISGylation by directly targeting the ISGylation E1 enzyme.
KEYWORDS: cytomegalovirus, UL50, ISGylation, UBE1L
ABSTRACT
Interferon-stimulated gene 15 (ISG15) encodes a ubiquitin-like protein that can be conjugated to proteins via an enzymatic cascade involving the E1, E2, and E3 enzymes. ISG15 expression and protein ISGylation modulate viral infection; however, the viral mechanisms regulating the function of ISG15 and ISGylation are not well understood. We recently showed that ISGylation suppresses the growth of human cytomegalovirus (HCMV) at multiple steps of the virus life cycle and that the virus-encoded pUL26 protein inhibits protein ISGylation. In this study, we demonstrate that the HCMV UL50-encoded transmembrane protein, a component of the nuclear egress complex, also inhibits ISGylation. pUL50 interacted with UBE1L, an E1-activating enzyme for ISGylation, and (to a lesser extent) with ISG15, as did pUL26. However, unlike pUL26, pUL50 caused proteasomal degradation of UBE1L. The UBE1L level induced in human fibroblast cells by interferon beta treatment or virus infection was reduced by pUL50 expression. This activity of pUL50 involved the transmembrane (TM) domain within its C-terminal region, although pUL50 could interact with UBE1L in a manner independent of the TM domain. Consistently, colocalization of pUL50 with UBE1L was observed in cells treated with a proteasome inhibitor. Furthermore, we found that RNF170, an endoplasmic reticulum (ER)-associated ubiquitin E3 ligase, interacted with pUL50 and promoted pUL50-mediated UBE1L degradation via ubiquitination. Our results demonstrate a novel role for the pUL50 transmembrane protein of HCMV in the regulation of protein ISGylation.
IMPORTANCE Proteins can be conjugated covalently by ubiquitin or ubiquitin-like proteins, such as SUMO and ISG15. ISG15 is highly induced in viral infection, and ISG15 conjugation, termed ISGylation, plays important regulatory roles in viral growth. Although ISGylation has been shown to negatively affect many viruses, including human cytomegalovirus (HCMV), viral countermeasures that might modulate ISGylation are not well understood. In the present study, we show that the transmembrane protein encoded by HCMV UL50 inhibits ISGylation by causing proteasomal degradation of UBE1L, an E1-activating enzyme for ISGylation. This pUL50 activity requires membrane targeting. In support of this finding, RNF170, an ER-associated ubiquitin E3 ligase, interacts with pUL50 and promotes UL50-mediated UBE1L ubiquitination and degradation. Our results provide the first evidence, to our knowledge, that viruses can regulate ISGylation by directly targeting the ISGylation E1 enzyme.
INTRODUCTION
Interferon-stimulated gene 15 (ISG15) encodes a ubiquitin-like protein that is covalently conjugated to lysine residues of a substrate protein through an enzymatic cascade involving E1, E2, and E3 enzymes, similar to the ubiquitin conjugation pathway (1, 2). UBE1L is the E1-activating enzyme for ISG15, and UbcH8, a ubiquitin E2-conjugating enzyme, also functions as the ISG15 E2-conjugating enzyme. E3 ligases for ISGylation in human cells include the HERC domain- and RCC1-like domain-containing protein 5 (Herc5), estrogen-responsive finger protein (EFP), and a human homolog of the Drosophila ariadne gene product (HHARI). ISG15, UBE1L, UbcH8, and Herc5 are all interferon (IFN) inducible. ISG15 can be removed reversibly from substrates by an ISG15-specific protease, USP18 (also known as UBP43), which is also IFN inducible (reviewed in reference 3). USP18 also acts as a negative regulator of the innate immune response, independent of its ISG15-deconjugating (deISGylating) activity, by association with a subunit of the type I IFN receptor, IFNAR2 (4, 5).
ISG15 or ISGylation represses the growth of diverse viruses, including influenza virus (types A and B), human immunodeficiency virus, avian sarcoma/leukosis virus, hepatitis C virus, Japanese encephalitis virus, Sindbis virus, vesicular stomatitis virus, dengue virus, West Nile virus, Ebola virus, porcine reproductive and respiratory syndrome virus, Chikungunya virus, herpes simplex virus 1 (HSV-1), murine gammaherpesvirus 68, and vaccinia virus (VACV) (6, 7; reviewed in reference 8). Recently, the antiviral activities of ISG15 or ISGylation were also demonstrated for Kaposi's sarcoma-associated herpesvirus (KSHV) (9), respiratory syncytial virus (10), and human cytomegalovirus (HCMV) (11, 12). In contrast to the antiviral activities of ISG15, free ISG15 has also been shown to negatively regulate the type I IFN response in humans by promoting sustained expression of USP18, which negatively regulates IFN signaling, suggesting an ISGylation-independent role for ISG15 in IFN pathway regulation (13–16).
Studies on the antiviral mechanisms of ISG15 have demonstrated that ISGylation regulates various steps of the virus life cycle. ISGylation can directly inhibit the functions of viral proteins. ISGylation of the influenza A virus (IAV) NS1 protein inhibits its binding to importin-α, blocking NS1 nuclear import (17), while ISGylation of the influenza B virus (IBV) NP protein inhibits its oligomerization, inhibiting the formation of the viral ribonucleoprotein complex (18). ISGylation of HCMV pUL26 regulates its ubiquitination and inhibits its activity to suppress tumor necrosis factor alpha (TNF-α)-mediated NF-κB activation (12). ISGylation can also affect viral growth by regulating cellular proteins. ISGylation of Nedd4 inhibits release of Ebola virus VP40 virus-like particles (19), while ISGylation of CHMP5 inhibits release of retroviruses (20, 21), and probably HCMV (12). ISGylation also affects microRNA (miRNA) functions (22) and the formation of autophagic clusters, inhibiting the growth of KSHV and HSV-1, respectively (23). Furthermore, ISGylation affects the early steps of the virus life cycle, such as virus entry of norovirus and viral gene expression in HCMV (12, 24).
Viral countermeasures against the antiviral effects of ISG15 and/or ISGylation have been demonstrated for some viruses. The IBV NS1 protein binds to ISG15 and suppresses the levels of ISGylation as it prevents the E1 enzyme from conjugating ISG15 (25). Furthermore, binding of NS1 to ISGylated NP protein appears to block the dominant negative effect of ISGylated NP on viral RNA synthesis (18). In VACV infection, the VACV E3 protein can bind to ISG15 and inhibit its antiviral activity (26). During HCMV infection, pUL26 binds to ISG15, UBE1L, and Herc5, and these interactions appear to be responsible for inhibiting ISGylation (12). Viruses also encode deISGylation enzymes. Nairoviruses and arteriviruses encode an ovarian tumor (OTU) domain-containing protease (27), while severe acute respiratory syndrome coronavirus (SARS-CoV) encodes papain-like protease (PLpro) enzymes that cleave ISG15 from its target proteins (28). However, viral mechanisms that regulate ISGylation enzymes are less well known.
HCMV belongs to the betaherpesvirus subfamily and typically causes latent and persistent infection. However, reactivation of virus in immunocompromised individuals, such as AIDS patients and transplantation recipients, and primary infections of newborns can result in severe disease (29). During lytic infection, HCMV gene expression occurs in a three-step sequential fashion, with immediate early (IE), early (E), and late (L) gene expression. HCMV expresses proteins that are able to counteract IFN production and subsequent ISG activation. IE2 and pp65 inhibit IFN production (30–32), whereas IE1 directly interacts with STAT2 and PML to suppress the IFN response (33–37). HCMV infection reduces levels of JAK1 and p48, two components of the type I IFN signaling pathway (38, 39). The stability and phosphorylation of STAT proteins are modulated during HCMV infection (40, 41). ISG15 expression and levels of ISGylation are upregulated immediately after HCMV infection via signaling pathways, including one mediated by cGAS/STING (11). However, HCMV appears to be armed with countermeasures, such as pUL26, which inhibits ISGylation at later stages of infection (12).
During HCMV infection, the core of the viral nuclear egress complex (NEC) is formed by two conserved viral proteins: pUL50 and pUL53. The integral membrane protein pUL50 is associated with the inner nuclear membrane (INM) by its transmembrane (TM) domain, whereas the nuclear phosphoprotein pUL53 carries a classical nuclear localization signal and accumulates in the nuclear rim upon interaction with the INM-associated pUL50 protein. Once the core NEC is formed, further NEC proteins, such as p32/gC1qR, emerin, and protein kinases, such as the viral protein kinase pUL97, cellular protein kinase C isoform a (PKCa), and cellular cyclin-dependent kinase 1 (CDK1), are recruited (42–46) in order to phosphorylate nuclear lamins, resulting in lamina disassembly (47–49). The recently solved crystal structure of the pUL50-pUL53 complex suggests that pUL50-pUL53 heterodimers assemble to form hexameric ring-like structures, acting as a scaffold for binding of further NEC proteins (50). The association of nuclear capsids with the NEC proteins has been demonstrated (51).
In this study, we demonstrate that the HCMV-encoded transmembrane protein pUL50 acts as a negative regulator of protein ISGylation by interacting with UBE1L and inducing its proteasomal degradation. We also show that this activity of pUL50 requires its transmembrane domain. Furthermore, we demonstrate that RNF170, an endoplasmic reticulum (ER)-associated ubiquitin E3 ligase, interacts with pUL50 and is involved in pUL50-mediated UBE1L degradation.
RESULTS
HCMV pUL50 interacts with UBE1L.
To identify HCMV proteins that regulate the ISG15 pathway, we performed yeast two-hybrid interaction assays and identified the transmembrane protein pUL50, encoded by HCMV UL50, as a potential UBE1L-binding protein. We further investigated the interaction of pUL50 with components of the ISG15 pathway. When we performed coimmunoprecipitation (co-IP) assays by using 293T cells cotransfected with UL50 and UBE1L, UbcH8, Herc5, or ISG15AA (a conjugation-inactive form), pUL50 interacted strongly with UBE1L and to a lesser extent with ISG15AA, while showing weak or no binding to UbcH8 and Herc5 (Fig. 1A). An isoform of pUL50 of about 26 kDa (denoted UL50-p26), which appears to start from an internal methionine, was also expressed from the UL50 gene. To further investigate the interaction of pUL50 with UBE1L and ISG15 during HCMV infection, we used a recombinant HCMV strain that expresses pUL50 with a C-terminal hemagglutinin (HA) tag. Although this recombinant virus showed reduced growth compared to that of its parental virus, which might be due to limited stability of the tagged protein (52), the pUL50-HA protein expressed from the recombinant virus was localized normally at the nuclear rim and interacted with all known protein ligands (53). In HF cells infected with this UL50-HA virus, immunoprecipitation of pUL50-HA with anti-HA antibody coprecipitated UBE1L and ISG15, while immunoprecipitation with anti-Flag antibody (as a control) did not (Fig. 1B and C). The results of these co-IP assays confirmed the specific interaction of pUL50 with UBE1L and ISG15 in both transfected and virus-infected cells.
FIG 1.

Interaction of pUL50 with UBE1L and ISG15. (A) 293T cells in six-well plates were cotransfected with plasmids encoding pUL50-HA (0.5 μg) and expressing Myc-tagged UBE1L (1 μg), UbcH8 (0.5 μg), Herc5 (1 μg), or ISG15AA (1 μg), as indicated. Forty-eight hours after transfection, cell lysates were prepared and immunoprecipitated (IP) with anti-Myc antibody, followed by immunoblotting (IB) with anti-HA antibody. The expression level of each protein in total cell lysate was also determined by immunoblotting. A small isoform of pUL50, of approximately 26 kDa (UL50-p26), was also detected. The amounts of pUL50-HA proteins coimmunoprecipitated relative to the input amounts of proteins were quantitated, and the relative binding strengths are shown in the graph. (B) HF cells in 150-mm dishes were infected with recombinant HCMV (AD169) containing the UL50-HA gene at a multiplicity of infection (MOI) of 3. Seventy-two hours after infection, cell lysates were prepared and immunoprecipitated with anti-HA or anti-Flag (as a negative control) antibody, followed by immunoblotting with anti-UBE1L. (C) Similar immunoprecipitation was performed 48 h after infection, followed by immunoblotting with anti-ISG15 antibody. The expression level of each protein in total cell lysate was also determined by immunoblotting. Levels of β-actin are shown as a loading control.
pUL50 inhibits ISGylation and causes proteasomal degradation of UBE1L.
We investigated whether pUL50, like HCMV pUL26 (12), regulates protein ISGylation by using cotransfection/ISGylation assays, in which ISGylated proteins are detected in cells cotransfected with plasmids encoding ISG15GG (an active form), UBE1L, UbcH8, and Herc5. We found that pUL50 expression downregulated ISGylation in this assay in a dose-dependent manner (Fig. 2).
FIG 2.
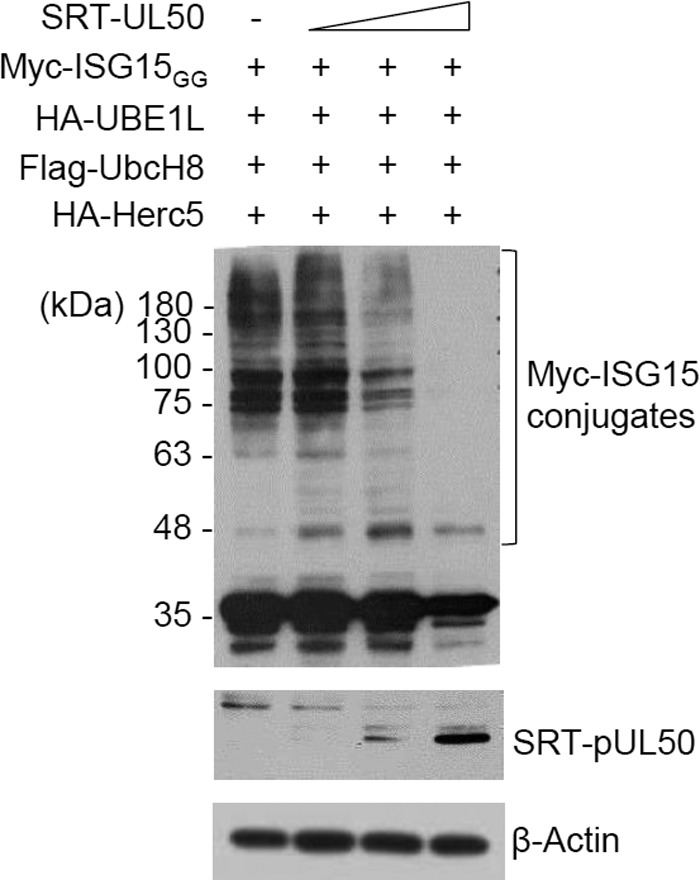
Effect of pUL50 expression on levels of ISGylation. 293T cells in six-well plates were cotransfected with plasmids (0.25 μg) expressing Myc-ISG15GG, HA-UBE1L, Flag-UbcH8, and HA-Herc5 and increasing amounts (0.2, 0.5, and 1 μg) of a plasmid expressing SRT-pUL50, as indicated. Forty-eight hours after transfection, cell lysates were prepared and subjected to immunoblotting with anti-Myc or anti-SRT antibody. Levels of β-actin are shown as a loading control.
We next examined the effect of pUL50 on the expression levels of individual components of the ISG15 pathway by using cotransfection assays. pUL50 expression effectively reduced UBE1L in a dose-dependent manner, although high-level expression of pUL50 also reduced Herc5 and, to a lesser extent, ISG15. Treatment of cells with MG132, a proteasome inhibitor, largely restored the loss of UBE1L, Herc5, and ISG15 (Fig. 3A, B, and D). pUL50 alone did not downregulate UbcH8 expression, although MG132 treatment appeared to affect the UbcH8 level (Fig. 3C). As a specificity control, pUL26, which inhibits ISGylation, did not reduce the level of UBE1L (Fig. 3E). These results demonstrate that pUL50 expression causes degradation of UBE1L and (to a lesser extent) Herc5 and ISG15 through proteasomal activity. Consistently, the reduction of UBE1L expression by pUL50 was not seen at the transcriptional level (Fig. 1F). Since UBE1L appeared to be regulated directly by pUL50 through a relatively strong interaction, we focused on UBE1L regulation by pUL50 expression.
FIG 3.

Effect of pUL50 expression on levels of ISGylation enzymes. (A to E) 293T cells in six-well plates were cotransfected with a plasmid (1 μg) expressing HA-UBE1L (A and E), HA-Herc5 (B), Flag-UbcH8 (C), or Myc-ISG15AA (D) and increasing amounts (0.04, 0.2, and 1 μg) of a plasmid expressing pUL50-HA (A to D) or SRT-pUL26 (E), as indicated. Twenty-four hours after transfection, cells were left untreated or treated with MG132 (20 μM) for 24 h, and immunoblotting was performed as indicated. Levels of β-actin are shown as a loading control. The amounts of the UA-UBE1L, HA-Herc5, Flag-UbcH8, and Myc-ISG15AA proteins relative to those of β-actin are indicated below the blots. (F) 293T cells in six-well plates (two separate sets) were cotransfected with a plasmid expressing HA-UBE1L and increasing amounts of a plasmid expressing pUL50-HA, as for panel A. Forty-eight hours after transfection, cell lysates from one set were prepared for immunoblotting, while mRNAs from the other set were prepared for RT-PCR analysis. Immunoblotting was performed as described for panel A. The results of RT-PCR, showing the levels of UBE1L and β-actin mRNAs, are also shown.
As an IFN-inducible gene, UBE1L was highly induced in HF cells infected with UV-inactivated HCMV (UV-HCMV), but this induction was mitigated with intact HCMV infection due to the expression of viral gene products that counteract IFN-β production and activation of ISGs (Fig. 4A). We investigated whether pUL50 expression affects levels of UBE1L in HF cells induced by IFN-β treatment or virus infection. Control HF cells and cells expressing pUL50-HA were produced by use of retroviral vectors. After induction of UBE1L by IFN-β treatment, cells were treated with cycloheximide (CHX), and the level of UBE1L was monitored. The half-life of IFN-β-induced UBE1L became shorter in pUL50-HA-expressing cells than in control cells (compare the UBE1L levels 4 h after CHX treatment in Fig. 4B, lanes 3 and 8). The half-life of pUL50-HA was shorter than that of UBE1L, and the reduction of pUL50-HA expression from 8 h after CHX treatment restored UBE1L levels (Fig. 4B, lanes 9 and 10). We also found that the expression of pUL50-HA in HF cells by recombinant adenoviral vectors reduced the UBE1L level induced by UV-HCMV infection (Fig. 4C). Collectively, these results show that pUL50 expression downregulates the expression of endogenous UBE1L in HF cells.
FIG 4.

Effect of pUL50 expression on UBE1L level induced by IFN-β treatment or viral infection. (A) HF cells were mock infected or infected with UV-HCMV or HCMV (Toledo strain) at an MOI of 3 for 24 or 48 h. The levels of the UBE1L, IE1/IE2, and pUL50 proteins were determined by immunoblotting. Bands indicating pUL50 and its 26-kDa isoform (UL50-p26) are indicated with arrowheads. Levels of β-actin are shown as a loading control. (B) Control and pUL50-HA-expressing HF cells produced by retroviral vectors (MIN) were left untreated or treated with IFN-β (1,000 U/ml) for 48 h and then incubated with or without cycloheximide (CHX; 100 μg/ml) for the indicated times. The levels of UBE1L, pUL50-HA, and β-actin were detected by immunoblotting. The relative levels of UBE1L (normalized to levels of β-actin) are shown in the graph. (C) HF cells were mock infected or infected with recombinant adenoviruses (Ad-lacZ or Ad-UL50-HA) at an MOI of 5 for 24 h and then superinfected with UV-HCMV (Toledo) for 24 h. The levels of UBE1L, pUL50-HA, and β-actin were determined by immunoblotting.
pUL50 requires the TM domain in its C-terminal region for ISGylation inhibition.
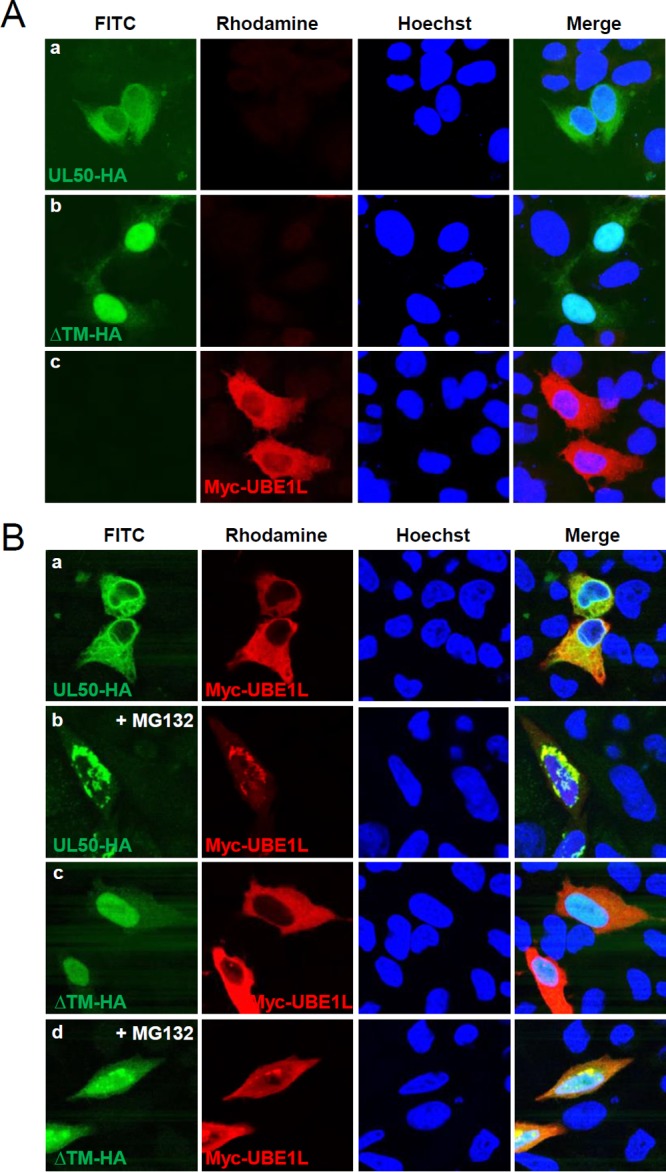
pUL50 consists of the conserved N-terminal globular domain, including the pUL53-binding region, and a C-terminal region containing the transmembrane (TM) domain (50, 54). To identify the region of pUL50 required for ISGylation inhibition, several N- or C-terminally truncated UL50 mutants were produced and used for cotransfection/ISGylation assays. The results suggested that the UL50 constructs containing the C-terminal region encompassing the TM domain effectively inhibited ISGylation. To address the role of the TM domain in ISGylation inhibition, two TM domain-deleted UL50 mutant proteins, ΔTM and 1-358, were produced and used in cotransfection/ISGylation assays. Unlike intact pUL50, both the ΔTM and 1-358 mutants did not inhibit ISGylation or cause the loss of UBE1 and Herc5 (Fig. 5A and B). Both the ΔTM and 1-358 mutants still interacted with UBE1L in co-IP assays (Fig. 5C). Unlike intact pUL50, both mutants failed to accumulate in the nuclear rim of HF cells as expected, suggesting that pUL50 binds to UBE1L independent of its membrane targeting (Fig. 5D).
FIG 5.

Requirement of the TM domain for ISGylation inhibition by pUL50. (A) 293T cells in six-well plates were cotransfected with plasmids (0.25 μg) expressing Myc-ISG15GG, HA-UBE1L, Flag-UbcH8, and HA-Herc5 and a plasmid (1 μg) expressing pUL50-HA (wild type [Wt] or the ΔTM or 1-358 mutant), as indicated. Forty-eight hours after transfection, immunoblotting was performed with anti-Myc or anti-HA antibody. Levels of β-actin are shown as a loading control. The positions of the wild-type pUL50 protein and its 26-kDa isoform (UL50-p26) are indicated. (B) Structures of the UL50 constructs used for panel A. The α-helix, globular domain, and transmembrane (TM) regions and the UL53 binding region are indicated. The relatively conserved regions (CR1 and CR2) in other herpesvirus homologs are also indicated. The numbers indicate amino acid positions. (C) 293T cells in six-well plates were cotransfected with plasmids encoding pUL50-HA (wild type or mutant) (1 μg) and Myc-UBE1L (0.5 μg), as indicated. Forty-eight hours after transfection, cell lysates were prepared and immunoprecipitated with anti-HA antibody, followed by immunoblotting with anti-Myc antibody. The expression level of each protein in total cell lysate was also determined by immunoblotting with anti-UBE1L or anti-HA antibody. The position of endogenous (endo.) UBE1L is indicated. Levels of β-actin are shown as a loading control. (D) HF cells in chamber slides were transfected with UL50 plasmids. Forty-eight hours after transfection, cells were fixed with 4% paraformaldehyde and stained with anti-HA antibody. Hoechst dye was used to stain cell nuclei. Representative confocal laser scanning microscopic images are shown.
The activity of pUL50 in binding to and degrading UBE1L was further investigated by indirect immunofluorescence assay (IFA). Unlike wild-type pUL50, the TM domain-deleted UL50 proteins did not accumulate at the nuclear rim and were largely nuclear (Fig. 6A, panels a and b). UBE1L has been shown to be localized largely in the cytoplasm (55). Consistently, UBE1L was mainly distributed in the cytoplasm of HeLa cells (Fig. 6A, panel c). When UBE1L was coexpressed with wild-type pUL50, the pUL50 localization was not apparently altered by UBE1L expression, and UBE1L did not accumulate at high levels at the nuclear rim where pUL50 concentrated (Fig. 6B, panel a); however, when cotransfected cells were treated with MG132, UBE1L and pUL50 were colocalized largely in the nuclear periphery and within the nucleus (Fig. 6B, panel b). When cells were cotransfected with UBE1L and the ΔTM UL50 mutant, the localization of the ΔTM UL50 protein was not affected by UBE1L; however, when cotransfected cells were treated with MG132, the proteins were only partly colocalized within the nucleus (Fig. 6B, panels c and d). Our finding that UBE1L was effectively colocalized with wild-type pUL50 in the nuclear periphery when proteasomal activity was inhibited supports the idea that pUL50 expression causes UBE1L degradation in a manner dependent on its membrane targeting through the TM domain.
FIG 6.
Colocalization of UL50 proteins and UBE1L in MG132-treated cells. (A) HeLa cells in chamber slides were singly transfected with a plasmid containing the UL50-HA gene (wild type or ΔTM) or Myc-UBE1L. Forty-eight hours after transfection, double-label IFA was performed with anti-HA and anti-Myc antibodies. Hoechst dye was used to stain cell nuclei. (B) HeLa cells were cotransfected with plasmids expressing the Myc-UBE1L and UL50-HA proteins (Wt or ΔTM mutant), as indicated. Forty-eight hours after transfection, cells were left untreated or treated with MG132 (5 μM) for 16 h, and double-label IFA was performed as described for panel A. Representative confocal microscopic images are shown.
The ubiquitin E3 ligase RNF170 is involved in pUL50-mediated UBE1L degradation.
To understand the mechanism by which pUL50 induces UBE1L degradation, we examined the possible role of several ubiquitin E3 ligases in UBE1L degradation. In a previous proteomics study of the NEC interactome (53), several peptides from ubiquitin E3 ligases, such as RNF170, were found to be associated with the NEC, although their significance was low. We found that expression of RNF170 variant 1 (RNF170v1) and variant 3 (RNF170v3) promoted pUL50-mediated UBE1L degradation in cotransfection assays, although expression of RNF170 proteins alone, without pUL50 coexpression, did not affect the UBE1L level (Fig. 7A and B). In co-IP assays, RNF170 proteins were found to interact with pUL50 (Fig. 7C). RNF170 proteins have been reported to be associated with the ER (56). RNF170 proteins were colocalized with pUL50 at sites including the nuclear rim in cotransfected HeLa cells (Fig. 8A) and in UL50-HA virus-infected HF cells (Fig. 8B), supporting the results of co-IP assays demonstrating that RNF170 is associated with pUL50.
FIG 7.

Involvement of RNF170 in pUL50-mediated UBE1L degradation. (A and B) 293T cells in six-well plates were cotransfected with plasmids expressing HA-UBE1L (0.5 μg) or pUL50-HA (0.25 μg) and a plasmid (2 μg) expressing RNF170v1-3×Flag (A) or RNF170v3-3×Flag or GFP-RNF170v3 (B), as indicated. Twenty-four hours after transfection, immunoblotting was performed as indicated. Levels of β-actin are shown as a loading control. The amounts of HA-UBE1L relative to those of β-actin are indicated under blots. (C) 293T cells were cotransfected with plasmids expressing pUL50-HA (0.5 μg) and RNF170v1-3×Flag or RNF170v3-3×Flag (2 μg), as indicated. Twenty-four hours after transfection, cell lysates were prepared and immunoprecipitated with anti-Flag antibody, followed by immunoblotting with anti-HA antibody. The expression level of each protein in total cell lysate was also determined by immunoblotting. The amounts of pUL50-HA proteins coimmunoprecipitated relative to the input amounts of proteins were quantitated, and relative binding strengths are indicated under the blot. A diagram comparing the structures of RNF170v1 and RNF170v3 is shown. RNF170v1 and RNF170v3 share the N-terminal 133 amino acids. RNF170v1 contains two more transmembrane domains (orange boxes) in its C-terminal region.
FIG 8.
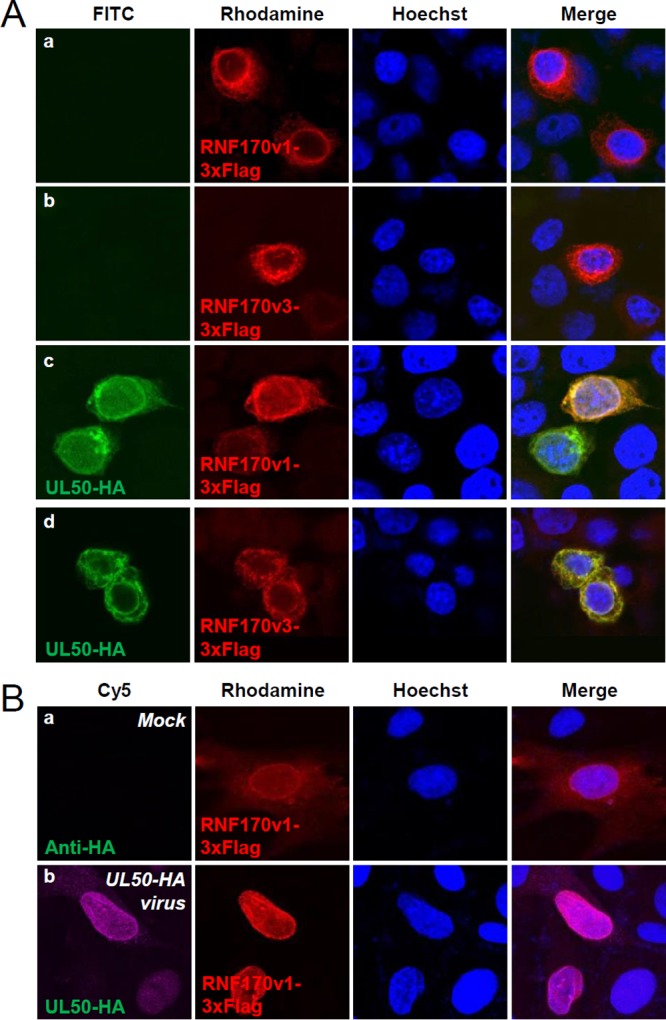
Colocalization of RNF170 with UL50 proteins. (A) HeLa cells in chamber slides were singly transfected with plasmids expressing RNF170v1-3×Flag (a) or RNF170v3-3×Flag (b) or cotransfected with plasmids containing the UL50-HA gene and expressing RNF170v1-3×Flag (c) or RNF170v3-3×Flag (d). Forty-eight hours after transfection, double-label IFA was performed with anti-HA and anti-Flag antibodies. Hoechst dye was used to stain cell nuclei. Representative confocal microscopic images are shown. (B) HF cells in chamber slides were transfected via electroporation with a plasmid expressing RNF170v1-3×Flag. Twenty-four hours after transfection, cells were mock infected (a) or infected with UL50-HA virus at an MOI of 3 (b) for 120 h. Double-label IFA was performed as described for panel A.
The roles of pUL50 and RNF170 in UBE1L degradation through proteasomal activity were further investigated by performing cotransfection/ubiquitination assays. We found that pUL50 effectively increased UBE1L ubiquitination, supporting the finding that proteasome activity is required for UBE1L degradation by UL50 (Fig. 9, compare lanes 2 and 4). Furthermore, we found that RNF170 promoted the pUL50-mediated ubiquitination of UBE1L (Fig. 9, compare lanes 2 and 3 and lanes 4 and 5). Taken together, our results suggest that RNF170 is involved in pUL50-mediated UBE1L degradation through the ubiquitin-proteasome pathway.
FIG 9.
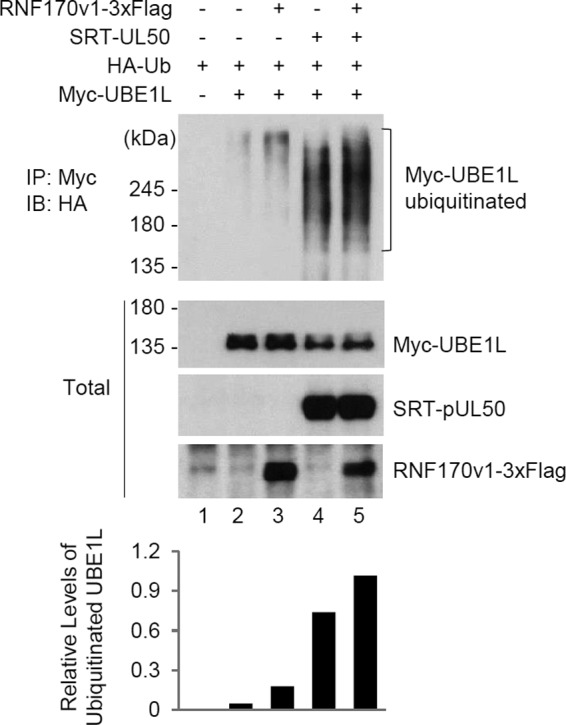
Effects of UL50 and RNF170 on UBE1L ubiquitination. 293T cells in six-well plates were cotransfected with plasmids expressing Myc-UBE1L (0.25 μg), HA-Ub (0.5 μg), SRT-pUL50 (0.5 μg), and RNF170v1-3×Flag (1.5 μg), as indicated. Twenty-four hours after transfection, total cell lysates were immunoprecipitated with anti-Myc antibody, followed by immunoblotting with anti-HA antibody to determine the ubiquitination level of UBE1L. The total protein levels of the Myc-UBE1L, SRT-pUL50, and RNF170v1-3×Flag proteins in cell lysates were also determined by immunoblotting with anti-Myc, anti-pUL50, and anti-Flag antibodies, respectively. The relative levels of ubiquitinated UBE1L are shown in the graph.
DISCUSSION
In this study, we demonstrated that the transmembrane protein pUL50 of HCMV interacts with UBE1L, an E1-activating enzyme for ISGylation, and causes its proteasomal degradation. pUL50 expression in HCMV-permissive cells reduced the UBE1L level that was induced by IFN-β treatment or UV-HCMV infection. Studies on the mechanism underlying this pUL50 activity revealed that although the C-terminal TM domain is not essential for UBE1L binding, it is required for UBE1L degradation. Moreover, we found that RNF170, an ER-associated ubiquitin E3 ligase, interacts with pUL50 and promotes pUL50-mediated UBE1L ubiquitination for degradation. Taken together, these results give rise to the idea that pUL50 recruits UBE1L to the ER membrane, where RNF170 facilitates UBE1L ubiquitination for proteasomal degradation. Given the role of the ISGylation pathway in host defense against virus infection (8), the findings of the present study reveal an unexpected role for pUL50, a component of the NEC that mediates the nuclear egress of the capsid, in counteracting the host immune response by downregulating protein ISGylation.
pUL50 is the second HCMV protein identified that inhibits ISGylation. We previously showed that pUL26 interacts with ISG15, UBE1L, and Herc5 and inhibits ISGylation, although the underlying mechanism is unclear (12). In the current study, we also demonstrated that pUL50 interacts with ISG15 and UBE1L. Although both pUL26 and pUL50 target ISG15 and UBE1L, they appear to use different mechanisms to inhibit ISGylation, since only pUL50 induced UBE1L degradation. Given that pUL50 is a membrane protein that targets the ER and eventually the INM to act as a regulator of the nuclear egress of the capsid, the requirement of the TM domain of pUL50 for UBE1L degradation suggests that the ER is the site of UBE1L degradation. In support of this model, pUL50 was colocalized with UBE1L at the nuclear periphery, which appeared to include the compressed ER membrane, in the presence of a proteasome inhibitor. Furthermore, an ER-associated ubiquitin E3 ligase was found to bind to pUL50 and promote UL50-mediated UBE1L degradation. Recently, HSV-1 pUL34, a homolog of HCMV pUL50, was shown to remodel the global ER architecture, including ER compression around the nuclear membrane during virus infection, which may lead to the recruitment of nuclear egress regulators to the nuclear membrane (57). Therefore, it is also an intriguing scenario that the pUL50-triggered remodeling of the ER architecture is responsible for the exposure of UBE1L to ER-associated ubiquitin E3 ligases, such as RNF170. This activity of pUL50 may also be related to the high level of pUL50-induced loss of Herc5, which is associated with polyribosomes, and (to a lesser extent) of ISG15, which weakly interacts with pUL50.
Analyses of the expression of pUL50 with a C-terminal HA tag from transfected or recombinant virus-infected cells revealed that a smaller isoform of pUL50, of about 26 kDa (referred to as UL50-p26 in this study), is expressed from the UL50 gene. This isoform appears to initiate from an internal methionine codon and encompasses the C-terminal half of pUL50, including the TM domain, but not the N-terminal globular domain necessary for pUL53 binding to form the NEC. We observed that this isoform was expressed earlier than pUL50 in virus-infected cells, but it became less abundant at the late phases of infection and was capable of moderately inhibiting ISGylation in cotransfection/ISGylation assays. However, whether UL50-p26 plays a regulatory role in viral infection needs further investigation.
Since Herc5, an E3 ligase for ISGylation, is associated with polyribosomes (58), abundantly expressed viral proteins may often be subjected to ISGylation in virus-infected cells. We previously showed that many viral proteins are ISGylated in cotransfection/ISGylation assays (12). The finding that pUL50 interacts with UBE1L and ISG15 prompted us to test whether pUL50 is a substrate for ISGylation. In cotransfection/ISGylation assays, pUL50 was indeed ISGylated within the central CR2 region of the N-terminal globular domain. Analysis of lysine-to-arginine mutants suggested that ISGylation occurred in several lysine residues in the CR2 region, making it difficult to further investigate the role of pUL50 ISGylation. It is conceivable that ISGylation of pUL50 may affect its targeting to the INM or interaction with pUL53. Determining whether ISGylation of pUL50 occurs during infection and affects its functions will require further study.
ISGylation appears to modulate both early and late events of HCMV infection. In our previous study, ISGylation enhancement by overexpression of E1, E2, and E3 enzymes and ISG15 reduced the activation of viral immediate early, early, and late promoters and the release of virions, while ISGylation reduction by short hairpin RNA (shRNA)-mediated Herc5 knockdown increased both viral gene expression and virion release (12). ISGylation of endosomal proteins was recently shown to promote the fusion of multivesicular bodies with lysosomes or autophagosomes and to inhibit exosome secretion and virus spread (59, 60). Considering that pUL50, which is expressed at a delayed early time after infection, targets the ER, is associated with ER-resident E3 ligase, and causes UBE1L degradation, it is tempting to study a regulatory role for pUL50 in these vesicle-related events.
MATERIALS AND METHODS
Cell culture, transfection, and reagents.
Human foreskin fibroblasts (HF cells), human embryonic kidney (HEK) 293T and 293A cells, and HeLa cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). Nonessential amino acids (NEAA) were also added to the medium for HEK293A cells. DNA transfection of 293T, 293A, and HeLa cells was performed using the polyethylenimine (PEI) version of the cationic polymer procedure (61). Electroporation of HF cells was conducted using a Microporator MP-100 instrument (Digital Bio) as described previously (33). Interferon beta (IFN-β) was purchased from Chemicon. Cycloheximide (CHX) and the proteasome inhibitor MG132 were purchased from Sigma.
Viruses.
Recombinant HCMV (Toledo strain) was produced by transfecting a Toledo bacmid gifted from Hua Zhu (UMDNJ-New Jersey Medical School) into HF cells (62). The IE1-deleted CR208 virus (Towne strain) (63), which was provided by Edward Mocarski (Emory University School of Medicine), was grown in IE1-expressing HF cells (34). HCMV (AD169 strain)-GFP UL50-HA (52) was grown in HF cells. UV-inactivated viruses were produced by irradiating the virus stock with UV light three times at 0.72 J/cm2, using a model CL-1000 cross-linker (UVP).
Expression plasmids.
Mammalian expression plasmids for Myc-UBE1L (pMK86), Myc-UbcH8 (pMK92), Myc-Herc5 (pMK89), and SRT-UL50 (pMK55) were produced in the pcDNA6 vector (Life Technologies) by using Gateway technology. A plasmid (pCS3-MT) expressing Myc-ISG15GG (an active form of ISG15) (pYJ12) and plasmids (pSG5) expressing Flag-UbcH8 (pYJ23) and HA-Herc5 (pYJ29) were previously described (12). pcDNA3.1-UL50-HA was previously described (42). The following mutant versions of pcDNA3.1-UL50-HA were produced using Stratagene QuikChange site-directed mutagenesis: the 1-358 (pMK132), Δ359-381 (ΔTM) (pMK183), and M199V (pMK113) mutants. RNF170 variant 1 (RNF170v1) and variant 3 (RNR170v3) were PCR amplified from the cDNA and from a plasmid expressing GFP-RNF170 (a gift from Ho Chul Kang [Ajou University]), respectively, and subcloned into a pcDNA3.1 derivative containing 3×Flag, which was provided by Cheol-Sang Hwang (Pohang University of Science and Technology), to create plasmids expressing C-terminally Flag-tagged RNF170 proteins. The plasmid expressing HA-ubiquitin (HA-Ub) was previously described (62).
Adenoviral and retroviral vectors.
Adenoviral vectors expressing pUL50-HA (pMK181) were produced in pAD/CMV/V5-DEST (Invitrogen) by using Gateway technology. Recombinant adenoviruses were grown in 293A cells after transfection with adenoviral vector DNAs that were linearized by use of the PacI restriction enzyme. Retroviral vectors expressing pUL50-HA (pMK85) were produced on the background of pMIN (64) by using Gateway technology. To grow recombinant retroviruses, 293T cells were transfected with retroviral vectors and packaging plasmids pHIT60 (expressing the Gag and Pol proteins of murine leukemia virus) and pMD-G (expressing the envelope G protein of vesicular stomatitis virus). At 48 h, the culture supernatants were collected and used to transduce HF cells in the presence of Polybrene (7.5 μg/ml). The transduced cells were selected with G418 (0.4 mg/ml) and maintained in medium containing G418 (0.1 mg/ml).
Reverse transcription-PCR (RT-PCR).
Total RNAs were isolated from virus-infected cells by use of Tri reagent (Molecular Research Center, Inc.) and a MaXtract High Density tube (Qiagen). cDNAs were synthesized using the random hexamer/oligo(dT) primers in a QuantiTect reverse transcriptase kit (Qiagen). PCR was performed using the following primers: UBE1L forward, 5′-AGGTGGCCAAGAACTTGGTT-3′; UBE1L reverse, 5′-CACCACCTGGAAGTCCAACA-3′; β-actin forward, 5′-AGCGGGAAATCGTGCGTG-3′; and β-actin reverse, 5′-CAGGGTACATGGTGGTGCC-3′. The relative DNA levels in agarose gel images were quantitated using ImageJ (NIH).
Antibodies.
Anti-HA rat monoclonal antibody (MAb) 3F10 and anti-Myc mouse MAb 9E10, which were conjugated to horseradish peroxidase (HRP) or labeled with fluorescein isothiocyanate (FITC), were purchased from Roche. Mouse MAb 810R, detecting IE1 and IE2, was purchased from Chemicon. Anti-β-actin and anti-Flag mouse MAbs were purchased from Sigma. A rabbit polyclonal antibody (PAb) to UBE1L was obtained from Abcam. A mouse MAb against the SRT epitope has been described previously (65). An HRP-conjugated anti-green fluorescent protein (anti-GFP) mouse MAb was purchased from Santa Cruz. A rabbit anti-peptide PAb for pUL50 was raised against a mixture of the synthetic peptides N′-C-RTAGKRSSRTAS-C′ (pUL50 residues 205 to 216), N′-C-DVGGSARRPLEE-C′ (residues 297 to 318), and N′-C-RARSGPSRPQSG-C′ (residues 346 to 357).
Immunoblot analysis.
Cells were washed with phosphate-buffered saline (PBS), and total cell lysates were prepared by boiling the cell pellets in sodium dodecyl sulfate (SDS) loading buffer. Equal amounts of the clarified cell extracts were separated in an SDS-polyacrylamide gel and electroblotted onto nitrocellulose membranes. The blots were blocked with PBS plus 0.1% Tween 20 (PBST) containing 5% nonfat dry milk by incubation for 1 h at room temperature. After being washed with PBST three times, the blots were incubated with the appropriate antibodies in PBST for 1 h at room temperature. Antibody dilutions were 1:2,500 for anti-Flag, 1:10,000 for anti-SRT, 1:1,000 for anti-UL50, 1:10,000 for anti-UBE1L, and 1:1,000 for anti-ISG15 antibodies. After three 5-min washes with PBST, the blots were incubated with HRP-conjugated goat anti-mouse IgG or anti-rabbit IgG (Amersham) at a 1:5,000 dilution for 1 h at room temperature. HRP-conjugated anti-HA, anti-Myc, and anti-GFP antibodies were used at a 1:5,000 dilution. The blots were then washed three times with PBST, and the protein bands were visualized by use of an enhanced chemiluminescence system (Amersham). The relative protein levels in immunoblots were quantitated using ImageJ (NIH).
Indirect immunofluorescence assay (IFA).
Cells in chamber slides were fixed in 4% paraformaldehyde for 5 min and permeabilized in PBS containing 0.2% Triton X-100 for 20 min at 4°C. The cells were then incubated with appropriate primary antibodies (anti-HA at a 1:200 dilution; anti-Myc and anti-Flag at a 1:2,000 dilution) in PBS for 1 h at 37°C, followed by incubation with appropriate secondary antibodies at a 1:100 dilution for 1 h at 37°C. FITC-conjugated anti-HA antibody was also used at a 1:100 dilution. Mounting solution containing Hoechst dye and an antifade reagent (Molecular Probes) was used. For double labeling, two different antibodies were incubated together. Slides were examined and photographed by use of a Carl Zeiss LSM710Meta confocal microscope system.
Co-IP assays.
Cell lysates were prepared by sonication in 1 ml co-IP buffer (50 mM Tris-Cl [pH 7.4], 50 mM NaF, 5 mM sodium phosphate, 150 mM NaCl, 0.1% NP-40, protease inhibitors [Sigma]) by use of a Vibra-Cell microtip probe (Sonics and Materials) for 10 s (pulse on, l s; pulse off, 3 s). Cell lysates were incubated with appropriate antibodies (10 μg) for 16 h at 4°C. Next, 30 μl of a 50% slurry of protein A- and G-Sepharose (Amersham) was added, and the mixture was incubated for 2 h at 4°C to allow adsorption. The mixture was then pelleted and washed seven times with co-IP buffer. The beads were resuspended and boiled for 5 min in loading buffer. Each sample was analyzed by SDS-PAGE and immunoblotting with appropriate antibodies.
UBE1L ubiquitination assays.
293T cells were cotransfected with plasmids expressing target (Myc-UBE1L) or effector proteins and a plasmid expressing HA-Ub, and ubiquitination assays were performed as described previously (62). In brief, 24 h after transfection, cells were harvested and cell pellets were resuspended with 2% SDS lysis buffer containing protease inhibitors (Sigma) and boiled for 10 min. Cell lysates were diluted 10-fold with co-IP buffer and sonicated using a Vibra-Cell microtip probe. The clarified cell lysates were immunoprecipitated with anti-Myc antibody and then immunoblotted with anti-HA antibody as described above.
ACKNOWLEDGMENTS
We thank Dong-Er Zhang, Chin Ha Chung, Ho Chul Kang, and Cheol-Sang Hwang for providing plasmids and antibodies. We also thank Edward Mocarski and Hua Zhu for mutant and recombinant viruses.
This work was supported by grants from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (grant HI14C2114); the National Research Foundation of Korea (NRF), funded by the Ministry of Education (grant 2016R1A2B4011848); and the Deutsche Forschungsgemeinschaft (grant DFG MA1289/11-1).
REFERENCES
- 1.Haas AL, Ahrens P, Bright PM, Ankel H. 1987. Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J Biol Chem 262:11315–11323. [PubMed] [Google Scholar]
- 2.Loeb KR, Haas AL. 1992. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J Biol Chem 267:7806–7813. [PubMed] [Google Scholar]
- 3.Skaug B, Chen ZJ. 2010. Emerging role of ISG15 in antiviral immunity. Cell 143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arimoto KI, Lochte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, Wilmes S, Fan JB, Heinisch JJ, Li Z, Yan M, Pellegrini S, Colland F, Piehler J, Zhang DE. 2017. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol 24:279–289. doi: 10.1038/nsmb.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. 2006. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J 25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingues P, Bamford CG, Boutell C, McLauchlan J. 2015. Inhibition of hepatitis C virus RNA replication by ISG15 does not require its conjugation to protein substrates by the HERC5 E3 ligase. J Gen Virol 96:3236–3242. doi: 10.1099/jgv.0.000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. 2012. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol 86:3839–3850. doi: 10.1128/JVI.06466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morales DJ, Lenschow DJ. 2013. The antiviral activities of ISG15. J Mol Biol 425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs SR, Stopford CM, West JA, Bennett CL, Giffin L, Damania B. 2015. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 1 interacts with a member of the interferon-stimulated gene 15 pathway. J Virol 89:11572–11583. doi: 10.1128/JVI.01482-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez-Sanz R, Mata M, Bermejo-Martin J, Alvarez A, Cortijo J, Melero JA, Martinez I. 2016. ISG15 is upregulated in respiratory syncytial virus infection and reduces virus growth through protein ISGylation. J Virol 90:3428–3438. doi: 10.1128/JVI.02695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bianco C, Mohr I. 2017. Restriction of human cytomegalovirus replication by ISG15, a host effector regulated by cGAS-STING double-stranded-DNA sensing. J Virol 91:e02483-16. doi: 10.1128/JVI.02483-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YJ, Kim ET, Kim YE, Lee MK, Kwon KM, Kim KI, Stamminger T, Ahn JH. 2016. Consecutive inhibition of ISG15 expression and ISGylation by cytomegalovirus regulators. PLoS Pathog 12:e1005850. doi: 10.1371/journal.ppat.1005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL. 2012. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, Lequin MH, Kros JM, Stam W, Hermann M, Willemsen R, Brouwer RW, Van IWF, Martin-Fernandez M, de Coo I, Dudink J, de Vries FA, Bertoli Avella A, Prinz M, Crow YJ, Verheijen FW, Pellegrini S, Bogunovic D, Mancini GM. 2016. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 213:1163–1174. doi: 10.1084/jem.20151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, Duehr J, Sanal O, Tezcan I, Mansouri N, Tabarsi P, Mansouri D, Francois-Newton V, Daussy CF, Rodriguez MR, Lenschow DJ, Freiberg AN, Tortorella D, Piehler J, Lee B, Garcia-Sastre A, Pellegrini S, Bogunovic D. 2016. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun 7:11496. doi: 10.1038/ncomms11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu JY, Wang QK, Yalnizoglu D, Radoshevich L, Uze G, Gros P, Rozenberg F, Zhang SY, Jouanguy E, Bustamante J, Garcia-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S. 2015. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao C, Hsiang TY, Kuo RL, Krug RM. 2010. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci U S A 107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao C, Sridharan H, Chen R, Baker DP, Wang S, Krug RM. 2016. Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat Commun 7:12754. doi: 10.1038/ncomms12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okumura A, Pitha PM, Harty RN. 2008. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci U S A 105:3974–3979. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuang Z, Seo EJ, Leis J. 2011. Mechanism of inhibition of retrovirus release from cells by interferon-induced gene ISG15. J Virol 85:7153–7161. doi: 10.1128/JVI.02610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pincetic A, Kuang Z, Seo EJ, Leis J. 2010. The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J Virol 84:4725–4736. doi: 10.1128/JVI.02478-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai L, Bai L, Lin Z, Qiao J, Yang L, Flemington EK, Zabaleta J, Qin Z. 2016. Transcriptomic analysis of KSHV-infected primary oral fibroblasts: the role of interferon-induced genes in the latency of oncogenic virus. Oncotarget 7:47052–47060. doi: 10.18632/oncotarget.9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katzenell S, Leib DA. 2016. Herpes simplex virus and interferon signaling induce novel autophagic clusters in sensory neurons. J Virol 90:4706–4719. doi: 10.1128/JVI.02908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez MR, Monte K, Thackray LB, Lenschow DJ. 2014. ISG15 functions as an interferon-mediated antiviral effector early in the murine norovirus life cycle. J Virol 88:9277–9286. doi: 10.1128/JVI.01422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan W, Krug RM. 2001. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J 20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerra S, Caceres A, Knobeloch KP, Horak I, Esteban M. 2008. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog 4:e1000096. doi: 10.1371/journal.ppat.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frias-Staheli N, Giannakopoulos NV, Kikkert M, Taylor SL, Bridgen A, Paragas J, Richt JA, Rowland RR, Schmaljohn CS, Lenschow DJ, Snijder EJ, Garcia-Sastre A, Virgin HW IV. 2007. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng X, Agnihothram S, Mielech AM, Nichols DB, Wilson MW, St John SE, Larsen SD, Mesecar AD, Lenschow DJ, Baric RS, Baker SC. 2014. A chimeric virus-mouse model system for evaluating the function and inhibition of papain-like proteases of emerging coronaviruses. J Virol 88:11825–11833. doi: 10.1128/JVI.01749-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mocarski ES, Shenk T, Griffiths PD, Pass RF. 2013. Cytomegaloviruses, p 1960–2014. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 30.Abate DA, Watanabe S, Mocarski ES. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol 78:10995–11006. doi: 10.1128/JVI.78.20.10995-11006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci U S A 100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor RT, Bresnahan WA. 2005. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced beta interferon production. J Virol 79:3873–3877. doi: 10.1128/JVI.79.6.3873-3877.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huh YH, Kim YE, Kim ET, Park JJ, Song MJ, Zhu H, Hayward GS, Ahn JH. 2008. Binding STAT2 by the acidic domain of human cytomegalovirus IE1 promotes viral growth and is negatively regulated by SUMO. J Virol 82:10444–10454. doi: 10.1128/JVI.00833-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YE, Ahn JH. 2015. Positive role of promyelocytic leukemia protein in type I interferon response and its regulation by human cytomegalovirus. PLoS Pathog 11:e1004785. doi: 10.1371/journal.ppat.1004785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krauss S, Kaps J, Czech N, Paulus C, Nevels M. 2009. Physical requirements and functional consequences of complex formation between the cytomegalovirus IE1 protein and human STAT2. J Virol 83:12854–12870. doi: 10.1128/JVI.01164-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulus C, Krauss S, Nevels M. 2006. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci U S A 103:3840–3845. doi: 10.1073/pnas.0600007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scherer M, Otto V, Stump JD, Klingl S, Muller R, Reuter N, Muller YA, Sticht H, Stamminger T. 2016. Characterization of recombinant human cytomegaloviruses encoding IE1 mutants L174P and 1-382 reveals that viral targeting of PML bodies perturbs both intrinsic and innate immune responses. J Virol 90:1190–1205. doi: 10.1128/JVI.01973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller DM, Rahill BM, Boss JM, Lairmore MD, Durbin JE, Waldman JW, Sedmak DD. 1998. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J Exp Med 187:675–683. doi: 10.1084/jem.187.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD. 1999. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol 162:6107–6113. [PubMed] [Google Scholar]
- 40.Knoblach T, Grandel B, Seiler J, Nevels M, Paulus C. 2011. Human cytomegalovirus IE1 protein elicits a type II interferon-like host cell response that depends on activated STAT1 but not interferon-gamma. PLoS Pathog 7:e1002016. doi: 10.1371/journal.ppat.1002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le VT, Trilling M, Wilborn M, Hengel H, Zimmermann A. 2008. Human cytomegalovirus interferes with signal transducer and activator of transcription (STAT) 2 protein stability and tyrosine phosphorylation. J Gen Virol 89:2416–2426. doi: 10.1099/vir.0.2008/001669-0. [DOI] [PubMed] [Google Scholar]
- 42.Milbradt J, Auerochs S, Marschall M. 2007. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J Gen Virol 88:2642–2650. doi: 10.1099/vir.0.82924-0. [DOI] [PubMed] [Google Scholar]
- 43.Milbradt J, Auerochs S, Sticht H, Marschall M. 2009. Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J Gen Virol 90:579–590. doi: 10.1099/vir.0.005231-0. [DOI] [PubMed] [Google Scholar]
- 44.Sharma M, Coen DM. 2014. Comparison of effects of inhibitors of viral and cellular protein kinases on human cytomegalovirus disruption of nuclear lamina and nuclear egress. J Virol 88:10982–10985. doi: 10.1128/JVI.01391-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sonntag E, Hamilton ST, Bahsi H, Wagner S, Jonjic S, Rawlinson WD, Marschall M, Milbradt J. 2016. Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J Gen Virol 97:1676–1685. doi: 10.1099/jgv.0.000495. [DOI] [PubMed] [Google Scholar]
- 46.Sonntag E, Milbradt J, Svrlanska A, Strojan H, Hage S, Kraut A, Hesse AM, Amin B, Sonnewald U, Coute Y, Marschall M. 2017. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J Gen Virol 98:2569–2581. doi: 10.1099/jgv.0.000931. [DOI] [PubMed] [Google Scholar]
- 47.Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog 5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milbradt J, Hutterer C, Bahsi H, Wagner S, Sonntag E, Horn AH, Kaufer BB, Mori Y, Sticht H, Fossen T, Marschall M. 2016. The prolyl isomerase Pin1 promotes the herpesvirus-induced phosphorylation-dependent disassembly of the nuclear lamina required for nucleocytoplasmic egress. PLoS Pathog 12:e1005825. doi: 10.1371/journal.ppat.1005825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. 2010. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J Biol Chem 285:13979–13989. doi: 10.1074/jbc.M109.063628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walzer SA, Egerer-Sieber C, Sticht H, Sevvana M, Hohl K, Milbradt J, Muller YA, Marschall M. 2015. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J Biol Chem 290:27452–27458. doi: 10.1074/jbc.C115.686527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milbradt J, Sonntag E, Wagner S, Strojan H, Wangen C, Lenac Rovis T, Lisnic B, Jonjic S, Sticht H, Britt WJ, Schlotzer-Schrehardt U, Marschall M. 2018. Human cytomegalovirus nuclear capsids associate with the core nuclear egress complex and the viral protein kinase pUL97. Viruses 10:E35. doi: 10.3390/v10010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmeiser C, Borst E, Sticht H, Marschall M, Milbradt J. 2013. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J Gen Virol 94:2056–2069. doi: 10.1099/vir.0.052571-0. [DOI] [PubMed] [Google Scholar]
- 53.Milbradt J, Kraut A, Hutterer C, Sonntag E, Schmeiser C, Ferro M, Wagner S, Lenac T, Claus C, Pinkert S, Hamilton ST, Rawlinson WD, Sticht H, Coute Y, Marschall M. 2014. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol Cell Proteomics 13:2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, Marschall M. 2012. Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J Biol Chem 287:24004–24016. doi: 10.1074/jbc.M111.331207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaughlin PM, Helfrich W, Kok K, Mulder M, Hu SW, Brinker MG, Ruiters MH, de Leij LF, Buys CH. 2000. The ubiquitin-activating enzyme E1-like protein in lung cancer cell lines. Int J Cancer 85:871–876. doi:. [DOI] [PubMed] [Google Scholar]
- 56.Lu JP, Wang Y, Sliter DA, Pearce MM, Wojcikiewicz RJ. 2011. RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J Biol Chem 286:24426–24433. doi: 10.1074/jbc.M111.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maeda F, Arii J, Hirohata Y, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2017. Herpes simplex virus 1 UL34 protein regulates the global architecture of the endoplasmic reticulum in infected cells. J Virol 91:e00271-17. doi: 10.1128/JVI.00271-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durfee LA, Lyon N, Seo K, Huibregtse JM. 2010. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol Cell 38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villarroya-Beltri C, Baixauli F, Mittelbrunn M, Fernandez-Delgado I, Torralba D, Moreno-Gonzalo O, Baldanta S, Enrich C, Guerra S, Sanchez-Madrid F. 2016. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat Commun 7:13588. doi: 10.1038/ncomms13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villarroya-Beltri C, Guerra S, Sanchez-Madrid F. 2017. ISGylation—a key to lock the cell gates for preventing the spread of threats. J Cell Sci 130:2961–2969. doi: 10.1242/jcs.205468. [DOI] [PubMed] [Google Scholar]
- 61.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A 92:7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwon KM, Oh SE, Kim YE, Han TH, Ahn JH. 2017. Cooperative inhibition of RIP1-mediated NF-kappaB signaling by cytomegalovirus-encoded deubiquitinase and inactive homolog of cellular ribonucleotide reductase large subunit. PLoS Pathog 13:e1006423. doi: 10.1371/journal.ppat.1006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greaves RF, Mocarski ES. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus IE1 mutant. J Virol 72:366–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu SS, Kim JM, Kim S. 2000. High efficiency retroviral vectors that contain no viral coding sequences. Gene Ther 7:797–804. [DOI] [PubMed] [Google Scholar]
- 65.Lee JM, Kang HJ, Lee HR, Choi CY, Jang WJ, Ahn JH. 2003. PIAS1 enhances SUMO-1 modification and the transactivation activity of the major immediate-early IE2 protein of human cytomegalovirus. FEBS Lett 555:322–328. doi: 10.1016/S0014-5793(03)01268-7. [DOI] [PubMed] [Google Scholar]