

During an attempt to explore the diversity of deltacoronaviruses among mammals and birds in Dubai, four novel deltacoronaviruses were detected in fecal samples from eight birds of four different species: FalCoV UAE-HKU27 from a falcon, HouCoV UAE-HKU28 from a houbara bustard, PiCoV UAE-HKU29 from a pigeon, and QuaCoV UAE-HKU30 from five quails. Genome analysis revealed evidence of recent interspecies transmission between falcons and their prey, houbara bustards and pigeons, possibly along the food chain, as well as avian-to-swine transmission. Recombination, which is known to occur frequently in some coronaviruses, was also common among these deltacoronaviruses and occurred predominantly at the spike region. Such recombination, involving the receptor binding protein, may contribute to the emergence of new viruses capable of infecting new hosts. Birds in the Middle East are hosts for diverse deltacoronaviruses with potential for interspecies transmission.
KEYWORDS: Middle East, coronavirus, deltacoronavirus, falcon, houbara bustard, interspecies jumping, pigeon, quail
ABSTRACT
The emergence of Middle East respiratory syndrome showed once again that coronaviruses (CoVs) in animals are potential source for epidemics in humans. To explore the diversity of deltacoronaviruses in animals in the Middle East, we tested fecal samples from 1,356 mammals and birds in Dubai, The United Arab Emirates. Four novel deltacoronaviruses were detected from eight birds of four species by reverse transcription-PCR (RT-PCR): FalCoV UAE-HKU27 from a falcon, HouCoV UAE-HKU28 from a houbara bustard, PiCoV UAE-HKU29 from a pigeon, and QuaCoV UAE-HKU30 from five quails. Complete genome sequencing showed that FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 belong to the same CoV species, suggesting recent interspecies transmission between falcons and their prey, houbara bustards and pigeons, possibly along the food chain. Western blotting detected specific anti-FalCoV UAE-HKU27 antibodies in 33 (75%) of 44 falcon serum samples, supporting genuine infection in falcons after virus acquisition. QuaCoV UAE-HKU30 belongs to the same CoV species as porcine coronavirus HKU15 (PorCoV HKU15) and sparrow coronavirus HKU17 (SpCoV HKU17), discovered previously from swine and tree sparrows, respectively, supporting avian-to-swine transmission. Recombination involving the spike protein is common among deltacoronaviruses, which may facilitate cross-species transmission. FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 originated from recombination between white-eye coronavirus HKU16 (WECoV HKU16) and magpie robin coronavirus HKU18 (MRCoV HKU18), QuaCoV UAE-HKU30 from recombination between PorCoV HKU15/SpCoV HKU17 and munia coronavirus HKU13 (MunCoV HKU13), and PorCoV HKU15 from recombination between SpCoV HKU17 and bulbul coronavirus HKU11 (BuCoV HKU11). Birds in the Middle East are hosts for diverse deltacoronaviruses with potential for interspecies transmission.
IMPORTANCE During an attempt to explore the diversity of deltacoronaviruses among mammals and birds in Dubai, four novel deltacoronaviruses were detected in fecal samples from eight birds of four different species: FalCoV UAE-HKU27 from a falcon, HouCoV UAE-HKU28 from a houbara bustard, PiCoV UAE-HKU29 from a pigeon, and QuaCoV UAE-HKU30 from five quails. Genome analysis revealed evidence of recent interspecies transmission between falcons and their prey, houbara bustards and pigeons, possibly along the food chain, as well as avian-to-swine transmission. Recombination, which is known to occur frequently in some coronaviruses, was also common among these deltacoronaviruses and occurred predominantly at the spike region. Such recombination, involving the receptor binding protein, may contribute to the emergence of new viruses capable of infecting new hosts. Birds in the Middle East are hosts for diverse deltacoronaviruses with potential for interspecies transmission.
INTRODUCTION
Coronaviruses (CoVs) infect humans and a wide variety of animals, causing respiratory, enteric, hepatic, and neurological diseases with various severities. Based on genotypic and serological characterization, CoVs were traditionally divided into three distinct groups (1–3). In 2008, the Coronavirus Study Group of the International Committee for Taxonomy of Viruses (ICTV) replaced the traditional groups 1, 2, and 3 CoVs with three genera, Alphacoronavirus, Betacoronavirus, and Gammacoronavirus (4). As a result of the unique mechanism of viral replication, CoVs have a high frequency of recombination (1). Their tendency for recombination and the inherently high mutation rates typical of RNA viruses may allow rapid adaptation to new hosts and ecological niches (5–9).
The severe acute respiratory syndrome (SARS) epidemic and the discovery of SARS coronavirus (SARS-CoV)-like viruses from palm civets in China have boosted interest in the discovery of novel CoVs in both humans and animals (10–15). In 2004, a novel human CoV (HCoV) of the genus Alphacoronavirus, human coronavirus NL63 (HCoV NL63), was reported independently by two groups (16, 17). In 2005, we also described the discovery of another novel HCoV, human coronavirus HKU1 (HCoV HKU1), in the genus Betacoronavirus (18–20). Regarding animal CoVs, we and others have described the discovery of SARS-CoV-like viruses in horseshoe bats in Hong Kong and other provinces of China (21, 22). Based on these findings, we expanded molecular surveillance studies to examine the diversity of CoVs in bats of southern China, during which at least nine other novel CoVs were discovered, including two novel lineages in Betacoronavirus, lineages C and D (23–25). Other novel CoVs in bats and other animals have also been discovered by various research groups, which has broadened our knowledge on the diversity and evolution of CoVs (7, 26–29).
Birds are the reservoir of major emerging viruses, most notably, avian influenza viruses (30). Due to their flocking behavior and abilities to fly over long distances, birds have the potential to disseminate emerging viruses efficiently among themselves, to other animals, and to humans. Yet, the number of known CoVs in birds is relatively small compared to that in bats. In 2009, we described the discovery of three novel CoVs in three families of birds, named bulbul coronavirus HKU11 (BuCoV HKU11), thrush coronavirus HKU12 (ThCoV HKU12), and munia coronavirus HKU13 (MunCoV HKU13) (31). These three CoVs formed a unique group of CoVs, which were subsequently classified as a novel genus of CoV, Deltacoronavirus (4). Recently, we have further discovered seven additional deltacoronaviruses: one from pigs, named porcine coronavirus HKU15 (PorCoV HKU15), and six from birds, named white-eye coronavirus HKU16 (WECoV HKU16), sparrow coronavirus HKU17 (SpCoV HKU17), magpie robin coronavirus HKU18 (MRCoV HKU18), night-heron coronavirus HKU19 (NHCoV HKU19), wigeon coronavirus HKU20 (WiCoV HKU20), and common-moorhen coronavirus HKU21 (CMCoV HKU21) (32). Subsequently, PorCoV HKU15 was found to be present widely in pigs in Asia and North America, and it has been associated with fatal outbreaks in pig farms (33–42). The findings supported that deltacoronaviruses have the potential for avian-to-mammalian transmission and emergence in mammals. We hypothesize that there are other, previously unrecognized deltacoronaviruses present in geographical locations other than Hong Kong, which may also have the potential for emergence. To test this hypothesis, we carried out a molecular epidemiology study in 1,356 mammals and birds in Dubai, The United Arab Emirates (UAE), in the Middle East. Based on the results of comparative genome and phylogenetic analyses, we propose four novel CoVs in Deltacoronavirus. The results of seroepidemiological studies are also discussed.
RESULTS
Animal surveillance and identification of four novel deltacoronaviruses.
A total of 1,356 fecal samples from 1,164 mammals and 192 birds were obtained (Table 1). Reverse transcription-PCR (RT-PCR) for a 440-bp fragment in the RNA-dependent RNA polymerase (RdRp) genes of CoVs was positive in specimens from eight birds of four species. Sequencing results suggested the presence of four novel deltacoronaviruses: the first (falcon CoV UAE-HKU27 [FalCoV UAE-HKU27]) from one falcon (family Falconidae), the second (houbara CoV UAE-HKU28 [HouCoV UAE-HKU28]) from one houbara bustard (family Otididae), the third (pigeon CoV UAE-HKU29 [PiCoV UAE-HKU29]) from one pigeon (family Columbidae), and the fourth (quail CoV UAE-HKU30 [QuaCoV UAE-HKU30]) from five quails (family Phasianidae) (Table 1; Fig. 1). None of the 1,164 mammals tested were positive.
TABLE 1.
Animals screened in the present study and their associated coronaviruses
| Animal | No. of specimens tested | No. (%) of specimens positive for CoV | CoV (n) |
|---|---|---|---|
| Birds | 192 | 8 (4.16) | |
| Black swan | 1 | 0 | |
| Crowned crane | 1 | 0 | |
| Eclectus parrot | 1 | 0 | |
| Falcon | 34 | 1 (2.94) | Falcon CoV UAE-HKU27 (1) |
| Flamingo | 3 | 0 | |
| Gray parrot | 1 | 0 | |
| Heuglin's bustard | 10 | 0 | |
| Houbara bustard | 36 | 1 (2.77) | Houbara CoV UAE-HKU28 (1) |
| Kori bustard | 4 | 0 | |
| Myna | 1 | 0 | |
| Ostrich | 4 | 0 | |
| Peacock | 1 | 0 | |
| Pigeon | 18 | 1 (7.14) | Pigeon CoV UAE-HKU29 (1) |
| Rhea | 1 | 0 | |
| Thick-knee | 1 | 0 | |
| Stone curlew | 22 | 0 | |
| Partridge | 8 | 0 | |
| Chicken | 19 | 0 | |
| Duck | 2 | 0 | |
| Guineafowl | 6 | 0 | |
| Pheasant | 7 | 0 | |
| Quail | 10 | 5 (50) | Quail CoV UAE-HKU30 (5) |
| Sand grouse | 1 | 0 | |
| Mammals | 1,164 | 0 | |
| Antelope | 90 | 0 | |
| Cat | 59 | 0 | |
| Cattle | 3 | 0 | |
| Camel | 754 | 0 | |
| Dog | 145 | 0 | |
| Goat | 34 | 0 | |
| Horse | 44 | 0 | |
| Lion | 7 | 0 | |
| Monkey | 6 | 0 | |
| Rabbit | 9 | 0 | |
| Rodent | 13 | 0 |
FIG 1.

Phylogenetic analysis of amino acid sequences of the 371-bp fragments (excluding primer sequences) of RNA-dependent RNA polymerases (RdRps) of coronaviruses (CoVs) identified from birds from Dubai in the present study. The tree was reconstructed by the maximum-likelihood method using PhyML 3.0 with the substitution model general time reversible with gamma distributed rate variation and estimated proportion of invariable sites (GTR+G+I). Bootstrap values were calculated from 1,000 trees. The scale bar indicates the number of nucleotide substitutions per site. The eight newly identified coronaviruses are shown in bold. The viruses and their respective DDBJ/ENA/GenBank accession numbers are as follow: ALCCoV, Asian leopard cat coronavirus (EF584908); Badger SARS-CoV, SARS-related Chinese ferret badger CoV (AY545919); BCoV, bovine CoV (NC_003045); BdCoV HKU22, bottlenose dolphin CoV HKU22 (KF793826); BuCoV HKU11, bulbul CoV HKU11 (FJ376619); BWCoV SW1, Beluga whale CoV SW1 (NC_010646); Camel MERS-CoV, camel Middle East respiratory syndrome CoV (KT751244); ChRCoV HKU24, China Rattus CoV HKU24 (KM349742); Civet SARS-CoV, SARS-related palm civet CoV (AY304488); CMCoV HKU21, common-moorhen CoV HKU21 (NC_016996); DcCoV HKU23, dromedary camel CoV HKU23 (KF906251); FalCoV UAE-HKU27, falcon CoV UAE-HKU27; FIPV, feline infectious peritonitis virus (AY994055); GiCoV, giraffe CoV (EF424622); HouCoV UAE-HKU28, houbara CoV UAE-HKU28; HCoV 229E, human CoV 229E (NC_002645); HCoV HKU1, human CoV HKU1 (NC_006577); HCoV NL63, human CoV NL63 (NC_005831); HCoV OC43, human CoV OC43 (NC_005147); Human MERS-CoV, human Middle East respiratory syndrome CoV (JX869059); Human SARS-CoV, severe acute respiratory syndrome-related human CoV (NC_004718); IBV, infectious bronchitis virus (NC_001451); IBV-partridge, partridge coronavirus (AY646283); IBV-peafowl, peafowl coronavirus (AY641576); MHV, murine hepatitis virus (NC_001846); MRCoV HKU18, magpie robin CoV HKU18 (NC_016993); MunCoV HKU13, munia CoV HKU13 (FJ376622); NHCoV HKU19, night-heron CoV HKU19 (NC_016994); PEDV, porcine epidemic diarrhea virus (NC_003436); PHEV, porcine hemagglutinating encephalomyelitis virus (NC_007732); PiCoV UAE-HKU29, pigeon CoV UAE-HKU29; Pi-BatCoV HKU5, Pipistrellus bat CoV HKU5 (NC_009020); PorCoV HKU15, porcine CoV HKU15 (NC_016990); PRCV, porcine respiratory CoV (DQ811787); QuaCoV UAE-HKU30, quail CoV UAE-HKU30; RbCoV HKU14, rabbit CoV HKU14 (JN874559); Rh-BatCoV HKU2, Rhinolophus bat CoV HKU2 (EF203064); Ro-BatCoV HKU9, Rousettus bat CoV HKU9 (NC_009021); SACoV, sable antelope CoV (EF424621); SARSr-Rs-BatCoV HKU3, SARS-related Rhinolophus bat CoV HKU3 (DQ022305); Sc-BatCoV 512, Scotophilus bat CoV 512 (NC_009657); SpCoV HKU17, sparrow CoV HKU17 (NC_016992); TGEV, transmissible gastroenteritis virus (AJ271965); ThCoV HKU12, thrush CoV HKU12 (FJ376621); Ty-BatCoV HKU4, Tylonycteris bat CoV HKU4 (NC_009019); WECoV HKU16, white-eye CoV HKU16 (NC_016991); WiCoV HKU20, wigeon CoV HKU20 (NC_016995).
Genome organizations and coding potentials of the four novel deltacoronaviruses.
The complete genome sequences of one strain each of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 and two strains of QuaCoV UAE-HKU30 were obtained by assembly of the sequences of RT-PCR products from the RNA extracted from the corresponding individual specimens.
The genome sizes of the four novel CoVs ranged from 25,871 bases (QuaCoV UAE-HKU30 strain 1101F) to 26,162 bases (FalCoV UAE-HKU27 strain 988F), and their G+C contents ranged from 0.39 to 0.42 (Table 2). Their genome organizations are typical of CoVs, with the gene order (5′ to 3′) replicase ORF1ab, spike (S), envelope (E), membrane (M), and nucleocapsid (N) (Fig. 2 and Table 3). Both the 5′ and 3′ ends contain short untranslated regions. The replicase ORF1ab occupies 18.363 to 18.678 kb of the genomes (Table 3). This open reading frame (ORF) encodes a number of putative proteins, including nsp3 (which contains the putative papain-like protease [PLpro]), nsp5 (putative chymotrypsin-like protease [3CLpro]), nsp12 (putative RdRp), nsp13 (putative helicase), and other proteins of unknown functions. Overall, the cleavage sites for the nonstructural proteins in ORF1ab of FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and QuaCoV UAE-HKU30 were similar to those of other deltacoronaviruses, except for nsp3/nsp4 and nsp15/16 in FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 (Table 4). In fact, the amino acids downstream from the putative cleavage site at nsp3/nsp4 are quite variable across different deltacoronaviruses (Table 4). The amino acids downstream from the putative cleavage site at nsp15/nsp16 are AL instead of SL (Table 4).
TABLE 2.
Comparison of genomic features and amino acid identities of the four novel deltacoronaviruses, representative alpha-, beta-, and gammacoronaviruses, and other deltacoronaviruses
| Genus and CoV | Genome size (bases) | G+C content | Amino acid identity (%) |
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FalCoV UAE-HKU27 |
HouCoV UAE-HKU28 |
PiCoV UAE-HKU29 |
QuaCoV UAE-HKU30 |
|||||||||||||||||||
| 3CLpro | RdRp | Hel | S | N | 3CLpro | RdRp | Hel | S | N | 3CLpro | RdRp | Hel | S | N | 3CLpro | RdRp | Hel | S | N | |||
| Alphacoronavirus | ||||||||||||||||||||||
| HCoV 229E | 27,317 | 0.38 | 35.8 | 49.1 | 47.7 | 44.8 | 21.4 | 35.5 | 49.0 | 47.7 | 44.8 | 21.4 | 35.8 | 49.1 | 47.7 | 44.8 | 21.4 | 34.8 | 48.9 | 50.6 | 41.1 | 22.3 |
| HCoV NL63 | 27,553 | 0.34 | 36.7 | 48.9 | 47.5 | 39.3 | 19.8 | 36.7 | 48.8 | 47.5 | 39.5 | 20.5 | 37.0 | 48.9 | 47.5 | 39.4 | 20.5 | 36.0 | 48.8 | 49.6 | 37.7 | 21.5 |
| Betacoronavirus | ||||||||||||||||||||||
| Lineage A | ||||||||||||||||||||||
| βCoV | 31,028 | 0.37 | 36.5 | 51.9 | 47.2 | 24.9 | 22.7 | 36.5 | 52.0 | 47.2 | 24.9 | 22.5 | 38.1 | 52.1 | 47.2 | 25.0 | 22.5 | 37.2 | 51.4 | 48.6 | 25.4 | 23.4 |
| HCoV HKU1 | 29,926 | 0.32 | 38.1 | 51.6 | 47.2 | 25.5 | 20.1 | 37.8 | 51.6 | 47.2 | 25.8 | 20.1 | 36.9 | 51.7 | 47.2 | 25.8 | 20.1 | 38.1 | 51.2 | 48.3 | 25.0 | 21.3 |
| Lineage B | ||||||||||||||||||||||
| SARS-CoV | 29,751 | 0.41 | 35.8 | 50.2 | 50.2 | 25.9 | 24.2 | 35.5 | 50.2 | 50.0 | 24.5 | 24.0 | 35.8 | 50.3 | 50.0 | 24.5 | 24.0 | 34.8 | 51.1 | 51.9 | 25.0 | 25.3 |
| Lineage C | ||||||||||||||||||||||
| MERS-CoV | 30,119 | 0.41 | 36.7 | 50.7 | 48.9 | 25.8 | 24.2 | 36.4 | 50.7 | 48.9 | 25.0 | 23.5 | 36.7 | 50.8 | 48.9 | 25.0 | 23.5 | 35.8 | 51.4 | 50.2 | 25.3 | 23.1 |
| Ty-BatCoV HKU4 | 30,286 | 0.38 | 36.0 | 51.2 | 48.9 | 25.1 | 24.2 | 36.3 | 51.2 | 48.9 | 25.3 | 24.1 | 36.6 | 51.3 | 48.9 | 25.3 | 24.1 | 35.7 | 50.6 | 49.7 | 24.0 | 23.8 |
| Lineage D | ||||||||||||||||||||||
| Ro-Bat-CoV HKU9 | 29,114 | 0.41 | 38.0 | 52.5 | 49.2 | 26.7 | 24.3 | 38.0 | 52.4 | 49.0 | 26.6 | 24.5 | 38.3 | 52.5 | 49.0 | 26.6 | 24.5 | 37.4 | 51.7 | 51.1 | 26.9 | 24.2 |
| Gammacoronavirus | ||||||||||||||||||||||
| IBV | 27,608 | 0.38 | 42.9 | 54.8 | 53.4 | 27.9 | 28.2 | 42.9 | 54.9 | 53.4 | 28.5 | 28.4 | 43.3 | 55.0 | 53.4 | 28.6 | 28.4 | 44.6 | 54.6 | 56.1 | 29.5 | 29.0 |
| Deltacoronavirus | ||||||||||||||||||||||
| BuCoV HKU11 | 26,476 | 0.39 | 85.0 | 88.8 | 94.5 | 45.2 | 71.5 | 84.4 | 89.1 | 94.7 | 45.0 | 72.1 | 85.0 | 88.9 | 94.7 | 45.2 | 72.1 | 81.1 | 87.5 | 89.4 | 68.1 | 73.4 |
| ThCoV HKU12 | 26,396 | 0.38 | 84.7 | 86.9 | 94.5 | 46.3 | 78.6 | 84.7 | 87.0 | 94.4 | 47.0 | 79.1 | 85.3 | 86.8 | 94.4 | 46.8 | 79.1 | 82.4 | 87.2 | 89.9 | 47.9 | 79.9 |
| MunCoV HKU13 | 26,552 | 0.43 | 77.9 | 87.6 | 88.3 | 46.2 | 74.1 | 77.5 | 87.6 | 88.1 | 46.5 | 74.4 | 77.9 | 87.4 | 88.1 | 46.4 | 74.4 | 84.0 | 89.9 | 96.1 | 73.0 | 77.4 |
| PorCoV HKU15 | 25,421 | 0.43 | 80.8 | 87.4 | 88.1 | 45.7 | 75.4 | 80.1 | 87.5 | 88.0 | 46.0 | 75.7 | 80.8 | 87.3 | 88.0 | 45.9 | 75.7 | 91.2 | 95.2 | 98.5 | 71.8 | 90.9 |
| WECoV HKU16 | 26,027 | 0.40 | 86.6 | 91.9 | 96.8 | 47.1 | 82.7 | 86.3 | 91.9 | 97.0 | 47.7 | 83.6 | 87.0 | 91.7 | 97.0 | 47.8 | 83.6 | 78.5 | 86.7 | 88.4 | 62.2 | 75.5 |
| SpCoV HKU17 | 26,067 | 0.45 | 80.8 | 87.5 | 88.3 | 67.3 | 75.9 | 80.1 | 87.6 | 88.1 | 67.9 | 76.8 | 80.8 | 87.4 | 88.1 | 67.7 | 76.8 | 93.8 | 95.8 | 98.7 | 44.7 | 91.8 |
| MRCoV HKU18 | 26,674 | 0.47 | 78.8 | 87.1 | 88.3 | 68.6 | 73.4 | 78.5 | 87.1 | 88.1 | 68.2 | 73.6 | 78.8 | 86.9 | 88.1 | 68.2 | 73.6 | 85.7 | 90.3 | 96.3 | 45.0 | 78.2 |
| NHCoV HKU19 | 26,064 | 0.38 | 58.3 | 72.3 | 75.9 | 45.6 | 50.4 | 57.6 | 72.3 | 76.2 | 46.0 | 50.7 | 57.9 | 72.3 | 76.2 | 46.2 | 50.7 | 53.7 | 72.3 | 78.4 | 41.8 | 51.6 |
| WiCoV HKU20 | 26,211 | 0.39 | 58.3 | 72.1 | 73.8 | 45.6 | 49.3 | 57.7 | 72.0 | 73.8 | 45.7 | 50.8 | 58.3 | 71.9 | 73.8 | 45.7 | 50.8 | 59.0 | 71.3 | 75.4 | 43.0 | 51.4 |
| CMCoV HKU21 | 26,212 | 0.35 | 76.2 | 84.8 | 89.1 | 47.1 | 64.7 | 75.9 | 84.7 | 89.2 | 46.5 | 65.3 | 76.5 | 84.5 | 89.2 | 46.5 | 65.3 | 71.7 | 83.5 | 84.6 | 51.5 | 61.6 |
| FalCoV UAE-HKU27 | 26,155 | 0.39 | - | - | - | - | - | 99.3 | 98.9 | 99.5 | 94.5 | 100 | 99.3 | 98.7 | 99.5 | 94.5 | 99.1 | 81.8 | 86.3 | 88.3 | 45.8 | 74.2 |
| HouCoV UAE-HKU28 | 26,155 | 0.39 | 98.7 | 98.9 | 99.5 | 94.5 | 99.1 | - | - | - | - | - | 99.3 | 99.8 | 100 | 99.8 | 100 | 81.1 | 86.4 | 88.1 | 46.0 | 74.8 |
| PiCoV UAE-HKU29 | 26,162 | 0.39 | 99.3 | 98.7 | 99.5 | 94.5 | 99.1 | 98.7 | 99.8 | 100 | 99.8 | 99.1 | - | - | - | - | - | 81.8 | 86.2 | 88.1 | 46.1 | 74.8 |
| QuaCoV UAE-HKU30 | 25,871 | 0.42 | 80.8 | 86.3 | 88.3 | 45.8 | 74.8 | 80.1 | 86.4 | 88.1 | 46.0 | 74.8 | 80.8 | 86.2 | 88.1 | 46.1 | 74.8 | - | - | - | - | - |
FIG 2.

Genome organization of members of Deltacoronavirus. Open reading frames downstream of spike (S) gene are magnified to show the differences among the genomes of the 10 CoVs. Papain-like protease (PLpro), chymotrypsin-like protease (3CLpro), and RNA-dependent RNA polymerase (RdRp) genes are represented by green boxes. S, envelope (E), membrane (M), and nucleocapsid (N) genes are represented by orange boxes. Putative accessory proteins are represented by blue boxes. The novel coronaviruses discovered in this study are shown in bold. Abbreviations for the viruses are the same as those in Fig. 1.
TABLE 3.
Coding potentials and putative transcription regulatory sequences of novel deltacoronavirus genomes
| CoV | ORF | Location (nt) | Frame(s) | Length |
TRS location (nt) | TRS sequence (distance [nt] to AUG) | |
|---|---|---|---|---|---|---|---|
| aa | nt | ||||||
| FalCoV UAE-HKU27 | 1ab | 595–19271 | +1, +3 | 6,226 | 18,678 | 71 | ACACCA(523)AUG |
| S | 19253–22855 | +2 | 1,201 | 3,603 | 19112 | ACACCA(140)AUG | |
| E | 22849–23097 | +1 | 83 | 249 | 22828 | ACACCU(20)AUG | |
| M | 23090–23746 | +2 | 219 | 657 | 23072 | ACACCA(17)AUG | |
| NS6 | 23746–24027 | +1 | 94 | 282 | 23726 | ACGCCA(19)AUG | |
| N | 24051–25085 | +3 | 345 | 1,035 | 24042 | ACACCA(8)AUG | |
| NS7a | 24079–24735 | +1 | 219 | 657 | 24042 | ACACCA(36)AUG | |
| NS7b | 25066–25272 | +1 | 69 | 207 | 25042 | ACAACG(18)AUG | |
| NS7c | 25259–25636 | +2 | 126 | 378 | 25244 | AGACCU(14)AUG | |
| NS7d | 25629–25826 | +3 | 66 | 198 | 25618 | AUACCA(10)AUG | |
| HouCoV UAE-HKU28 | 1ab | 588–19264 | +3, +2 | 6,226 | 18,678 | 70 | ACACCA(517)AUG |
| S | 19246–22848 | +1 | 1,201 | 3,603 | 19105 | ACACCA(140)AUG | |
| E | 22842–23090 | +3 | 83 | 249 | 22821 | ACACCU(20)AUG | |
| M | 23083–23739 | +1 | 219 | 657 | 23065 | ACACCA(17)AUG | |
| NS6 | 23739–24020 | +3 | 94 | 282 | 23719 | ACGCCA(19)AUG | |
| N | 24044–25078 | +2 | 345 | 1,035 | 24035 | ACACCA(8)AUG | |
| NS7a | 24072–24728 | +3 | 219 | 657 | 24035 | ACACCA(36)AUG | |
| Ns7b | 25059–25265 | +3 | 69 | 207 | 25040 | ACAACG(18)AUG | |
| NS7c | 25252–25629 | +1 | 126 | 378 | 25237 | AGACCU(14)AUG | |
| Ns7d | 25622–25819 | +2 | 66 | 198 | 25611 | AUACCA(10)AUG | |
| PiCoV UAE-HKU29 | 1ab | 588–19264 | +3, +2 | 6,226 | 18,678 | 70 | ACACCA(517)AUG |
| S | 19246–22848 | +1 | 1,201 | 3,603 | 19105 | ACACCA(140)AUG | |
| E | 22842–23090 | +3 | 83 | 249 | 22821 | ACACCU(20)AUG | |
| M | 23083–23739 | +1 | 219 | 657 | 23065 | ACACCA(17)AUG | |
| NS6 | 23739–24020 | +3 | 94 | 282 | 23719 | ACGCCA(19)AUG | |
| N | 24044–25078 | +2 | 345 | 1,035 | 24035 | ACACCA(8)AUG | |
| NS7a | 24072–24728 | +3 | 219 | 657 | 24035 | ACACCA(36)AUG | |
| Ns7b | 25059–25265 | +3 | 69 | 207 | 25040 | ACAACG(18)AUG | |
| NS7c | 25252–25629 | +1 | 126 | 378 | 25237 | AGACCU(14)AUG | |
| Ns7d | 25622–25819 | +2 | 66 | 198 | 25611 | AUACCA(10)AUG | |
| QuaCoV UAE-HKU30 | 1ab | 522–19312 | +3, +2 | 6,121 | 18,363 | 62 | ACACCA(459)AUG |
| S | 19294–22770 | +1 | 1,159 | 3,477 | 19153 | ACACCA(140)AUG | |
| E | 22764–23015 | +3 | 84 | 252 | 22740 | CAACCA(23)AUG | |
| M | 23008–23661 | +1 | 218 | 654 | 22987 | ACACCA(20)AUG | |
| NS6 | 23661–23942 | +3 | 94 | 282 | 23614 | ACACCA(46)AUG | |
| N | 23967–24992 | +3 | 342 | 1,026 | 23959 | ACACCA(7)AUG | |
| NS7a | 24061–24663 | +1 | 201 | 603 | 23978 | GCTCCA(82)AUG | |
| NS7b | 25003–25419 | +1 | 139 | 417 | 24998 | ACACCA(4)AUG | |
| NS7c | 25358–25579 | +2 | 74 | 222 | 25347 | ACACCA(10)AUG | |
TABLE 4.
Putative cleavage sites at the junctions between nonstructural proteins in FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and QuaCoV UAE-HKU30 compared with those in other deltacoronaviruses
| nsp pair | Cleavage site in: |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PorCoV HKU15 | WECoV HKU16 | SpCoV HKU17 | MRCoV HKU18 | NHCoV HKU19 | WiCoV HKU20 | CMCoV HKU21 | FalCoV UAE-HKU27 | HouCoV UAE-HKU28 | PiCoV UAE-HKU29 | QuaCoV UAE-HKU30 | |
| nsp2/nsp3 | AG/SD | AG/SD | AG/SD | AG/AD | VG/GL | DG/VY | AG/VS | AG/SD | AG/SD | AG/SD | AG/SD |
| nsp3/nsp4 | AG/AP | AG/AR | AG/AP | AG/AM | TG/GN | GG/SK | AG/KF | AG/RK | AG/RK | AG/RK | AG/AP |
| nsp4/nsp5 | LQ/AG | LQ/AG | LQ/AG | LQ/AG | VQ/AG | VQ/SG | VQ/AG | LQ/AG | LQ/AG | LQ/AG | LQ/AG |
| nsp5/nsp6 | LQ/SG | LQ/SN | LQ/SG | LQ/SG | LQ/GT | LQ/AN | LQ/AS | LQ/SG | LQ/SG | LQ/SG | LQ/SG |
| nsp6/nsp7 | VQ/NK | VQ/NK | VQ/NK | VQ/NK | VQ/NK | VQ/NR | VQ/NR | VQ/NR | VQ/NR | VQ/NR | VQ/NK |
| nsp7/nsp8 | VQ/AV | VQ/AV | VQ/AV | VQ/AV | LQ/VV | LQ/VV | LQ/VV | LQ/AV | LQ/AV | LQ/AV | VQ/AV |
| nsp8/nsp9 | LQ/NN | LQ/NN | LQ/NN | LQ/NN | LQ/NN | CQ/NN | LQ/NN | LQ/NN | LQ/NN | LQ/NN | LQ/NN |
| nsp9/nsp10 | LQ/AS | LQ/AN | LQ/AN | LQ/AN | LQ/SS | LQ/AN | LQ/AT | LQ/AN | LQ/AN | LQ/AN | LQ/AN |
| nsp10/nsp11 | LQ/NS | LQ/GS | LQ/NS | LQ/NS | LQ/LG | LQ/SN | LQ/NT | LQ/GS | LQ/GS | LQ/GS | LQ/NS |
| nsp12/nsp13 | LQ/AS | LQ/AS | LQ/AS | LQ/AS | LQ/AT | LQ/AT | LQ/AS | LQ/AS | LQ/AS | LQ/AS | LQ/AS |
| nsp13/nsp14 | LQ/SS | LQ/SS | LQ/SS | LQ/AG | VQ/SL | VQ/AE | VQ/CS | LQ/SS | LQ/SS | LQ/SS | LQ/SG |
| nsp14/nsp15 | LQ/NL | LQ/NL | LQ/NL | LQ/NL | LQ/TL | LQ/TL | LQ/TI | LQ/NL | LQ/NL | LQ/NL | LQ/NL |
| nsp15/nsp16 | LQ/SL | VQ/SL | LQ/SL | LQ/SL | VQ/AL | LQ/SL | VQ/SL | VQ/AL | VQ/AL | VQ/AL | LQ/SL |
The four novel CoVs display similar genome organizations and differ only in the number of ORFs downstream of N (Fig. 2). Their transcription regulatory sequences (TRSs) conform to the consensus motif 5′-ACACCA-3′ (Table 3), which is unique to deltacoronaviruses. Interestingly, similar to the case for other deltacoronaviruses, the perfect TRSs of S in the genomes of the four novel CoVs were separated from the corresponding AUG by a large number of bases (140) (Table 3). This is in contrast to the relatively small number of bases between the TRS for S and the corresponding AUG in alphacoronaviruses, betacoronaviruses, and gammacoronaviruses (range, 0 bases in HCoV NL63, Rhinolophus bat coronavirus HKU2 [Rh-BatCoV HKU2], HCoV HKU1, bovine coronavirus [BCoV], human coronavirus OC43 [HCoV OC43], mouse hepatitis virus [MHV], porcine hemagglutinating encephalomyelitis virus, SARS-CoV, and SARS-related Rhinolophus bat coronavirus HKU3 [SARSr-Rh-BatCoV HKU3] to 52 bases in infectious bronchitis virus [IBV]). Similar to the case for other deltacoronaviruses, the genomes of the four novel CoVs encode putative PLpro, which is homologous to PL2pro of alphacoronaviruses and betacoronavirus subgroup A and to PLpro of betacoronavirus subgroups B, C, and D and gammacoronaviruses. In addition, one ORF (NS6) is found between M and N in the genomes of the four novel CoVs. In all the four novel CoVs, one ORF (NS7a) overlapping with N is present. For FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29, three ORFs (NS7b, -7c, and -7d), and for QuaCoV UAE-HKU30, two ORFs (NS7b and -7c), are present downstream of N. A BLAST search revealed no amino acid similarities between these putative nonstructural proteins and other known proteins, and no functional domain was identified by PFAM and InterProScan, except that NS7a of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 was homologous to NS7a of WECoV HKU16, NS7b was homologous to NS7b of CMCoV HKU21, and NS7c was homologous to NS7a of ThCoV HKU12 and that NS7a, NS7b, and NS7c of QuaCoV UAE-HKU30 were homologous to NS7a, NS7b, and NS7c of SpCoV HKU17, respectively. Transmembrane helices, predicted by TMHMM, were found only in NS7c of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 at positions 4 to 23 and 30 to 52 among all the putative accessory proteins downstream from the N genes of FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and QuaCoV UAE-HKU30. Each of the genomes of FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and QuaCoV UAE-HKU30 contains a stem-loop II motif (s2m) (residues 25924 to 25965, 25924 to 25965, 25932 to 25973, and 26649 to 26690, respectively), a conserved RNA element downstream of N and upstream of the poly(A) tail, similar to those in IBV, TCoV, SARSr-Rh-BatCoV, and SARS-CoV as well as other deltacoronaviruses.
Comparisons of the amino acid identities of the seven conserved replicase domains for species demarcation (ADRP, nsp5 [3CLpro], nsp12 [RdRp], nsp13 [Hel], nsp14 [ExoN], nsp15 [NendoU], and nsp16 [O-MT]) (4) among the four novel CoVs are shown in Tables 5 and 6. In all seven domains, the amino acid sequences of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 showed more than 90% identity to each other, but their overall amino acid sequences showed only 84.4% identity to those of WECoV HKU16, indicating that these three CoVs should be subspecies of a novel CoV species (Table 5). For QuaCoV UAE-HKU30, its overall amino acid sequences showed more than 90% identity to those of PorCoV HKU15 and SpCoV HKU17, indicating that QuaCoV UAE-HKU30, PorCoV HKU15, and SpCoV HKU17 should be subspecies of the same CoV species (Table 6).
TABLE 5.
Comparison of amino acid identities of the seven conserved replicase domains for species demarcation among FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and WECoV-HKU16
| CoV | Domain | Amino acid identity (%) |
|||
|---|---|---|---|---|---|
| WECoV HKU16 | FalCoV UAE-HKU27 | HouCoV UAE-HKU28 | PiCoV UAE-HKU29 | ||
| WECoV HKU16 | ADRP | 68.4 | 68.7 | 68.8 | |
| 3CLpro | 86.6 | 86.3 | 87.0 | ||
| RdRp | 91.9 | 91.9 | 91.7 | ||
| Hel | 96.8 | 97.0 | 97.0 | ||
| ExoN | 93.6 | 93.8 | 93.8 | ||
| NendoU | 86.2 | 85.6 | 85.6 | ||
| O-MT | 89.2 | 89.2 | 88.9 | ||
| Concatenated | 84.4 | 84.4 | 84.4 | ||
| FalCoV UAE-HKU27 | ADRP | 98.6 | 98.6 | ||
| 3CLpro | 98.7 | 98.7 | |||
| RdRp | 98.9 | 98.7 | |||
| Hel | 99.5 | 99.5 | |||
| ExoN | 99.8 | 99.8 | |||
| NendoU | 99.1 | 99.1 | |||
| O-MT | 100 | 99.6 | |||
| Concatenated | 99.1 | 99.1 | |||
| HouCoV UAE-HKU28 | ADRP | 99.9 | |||
| 3CLpro | 99.3 | ||||
| RdRp | 99.8 | ||||
| Hel | 100 | ||||
| ExoN | 100 | ||||
| NendoU | 100 | ||||
| O-MT | 99.6 | ||||
| Concatenated | 99.9 | ||||
TABLE 6.
Comparison of amino acid identities the seven conserved replicase domains for species demarcation among QuaCoV UAE-HKU30, PorCoV-HKU15, and SpCoV-HKU17
| CoV | Domain | Amino acid identity (%) |
||
|---|---|---|---|---|
| PorCoV HKU15 | SpCoV HKU17 | QuaCoV UAE-HKU30 | ||
| PorCoV HKU15 | ADRP | 89.3 | 83.0 | |
| 3CLpro | 97.1 | 91.2 | ||
| RdRp | 97.8 | 95.2 | ||
| Hel | 99.2 | 98.5 | ||
| ExoN | 97.3 | 96.1 | ||
| NendoU | 96.3 | 91.1 | ||
| O-MT | 97.5 | 96.4 | ||
| Concatenated | 95.0 | 91.3 | ||
| SpCoV HKU17 | ADRP | 83.0 | ||
| 3CLpro | 93.8 | |||
| RdRp | 95.8 | |||
| Hel | 98.7 | |||
| ExoN | 97.1 | |||
| NendoU | 92.0 | |||
| O-MT | 95.7 | |||
| Concatenated | 91.8 | |||
Phylogenetic analyses.
The phylogenetic trees reconstructed using the nucleotide sequences of 3CLpro, RdRp, Hel, S, and N of the four novel CoVs and other deltacoronaviruses are shown in Fig. 3, and the corresponding pairwise amino acid identities are shown in Table 2. In all five trees, FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 were clustered together (Fig. 3). In the 3CLpro, RdRp, Hel, and N trees, FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 were clustered with WECoV HKU16, whereas QuaCoV UAE-HKU30 was clustered with PorCoV HKU15 and SpCoV HKU17 (Fig. 3). However, in the S tree, FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 were most closely related to MRCoV HKU18, whereas QuaCoV UAE-HKU30 was most closely related to MunCoV HKU13 (Fig. 3).
FIG 3.

Phylogenetic analyses of chymotrypsin-like protease (3CLpro), RNA-dependent RNA polymerase (RdRp), helicase (Hel), spike (S) protein, and nucleocapsid (N) protein of falcon CoV-HKU27, houbara CoV-HKU28, pigeon CoV-HKU29, and quail CoV-HKU30. The trees were reconstructed by the maximum-likelihood method using PhyML 3.0 with the following substitution models: Le and Gascuel (LG) with gamma distributed rate variation (G) (3CLpro); LG with G, estimated proportion of invariable sites (I), and empirical frequencies (F) (RdRp); LG+G+F (Hel and N); and Whelan and Goldman (WAG)+G+I+F (S). Bootstrap values were calculated from 1,000 trees, and 314, 944, 599, 1,561, and 392 amino acid positions in 3CLpro, RdRp, Hel, S, and N, respectively, were included in the analyses. The scale bars indicate the number of amino acid substitutions per site. Viruses characterized in this study are in bold. Abbreviations for the viruses are the same as those in Fig. 1.
Recombination analysis.
For the FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 cluster and QuaCoV UAE-HKU30, bootscan analysis showed possible recombination sites in the S gene (positions 20300 to 24300 for the FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 cluster [Fig. 4A] and positions 19900 to 23300 for QuaCoV UAE-HKU30 [Fig. 4B]).
FIG 4.

Detection of possible recombination by bootscan analysis. Bootscanning was conducted with Simplot version 3.5.1 (F84 model; window size, 1,000 bp; step, 200 bp). (A) Falcon CoV UAE-HKU27 (FalCoV UAE-HKU27) was used as the query sequence and compared with the genome sequences of white-eye coronavirus HKU16 (WECoV HKU16), magpie robin coronavirus HKU18 (MRCoV HKU18), and ThCoV HKU12 thrush coronavirus HKU12 (ThCoV HKU12). (B) Quail CoV UAE-HKU30 (QuaCoV UAE-HKU30) was used as the query sequence and compared with the genome sequences of sparrow coronavirus HKU17 (SpCoV HKU17), munia coronavirus HKU13 (MunCoV HKU13), and ThCoV HKU12.
Estimation of divergence dates.
Using the Bayesian Skyline under a relaxed-clock model with an uncorrelated log-normal distribution, the mean evolutionary rate of CoVs was estimated as 1.027 × 10−4 nucleotide substitutions per site per year for the RdRp gene. Molecular clock analysis using the RdRp gene showed that the mean time to the most recent common ancestor (tMRCA) of Deltacoronavirus was estimated to be April 82 (95% highest posterior density [HPD], 822 BC to 1824), that of HouCoV UAE-HKU28/PiCoV UAE-HKU29 to be March 2011 (95% HPD, October 2009 to February 2013), that of FalCoV UAE-HKU27/HouCoV UAE-HKU28/PiCoV UAE-HKU29 to be June 1981 (95% HPD, January 1964 to June 2010), and that of QuaCoV UAE-HKU30 to be February 1954 (95% HPD, October 1921 to September 2007) (Fig. 5).
FIG 5.

Estimation of the mean time to the most recent common ancestor (tMRCA) for Deltacoronavirus. The time-scaled phylogeny was summarized from all Markov chain Monte Carlo (MCMC) phylogenies of the RNA-dependent RNA polymerase (RdRp) gene data set analyzed under the relaxed-clock model with an uncorrelated log-normal distribution in BEAST version 1.7.4. Viruses characterized in this study are in bold. Abbreviations for the viruses are the same as those in Fig. 1.
Western blot analysis.
Prominent immunoreactive bands were visible for 33 (75%) of 44 falcon serum samples. The band size observed (42 kDa) was consistent with the expected size of 41.1 kDa for the full-length His6-tagged recombinant N protein (Fig. 6).
FIG 6.
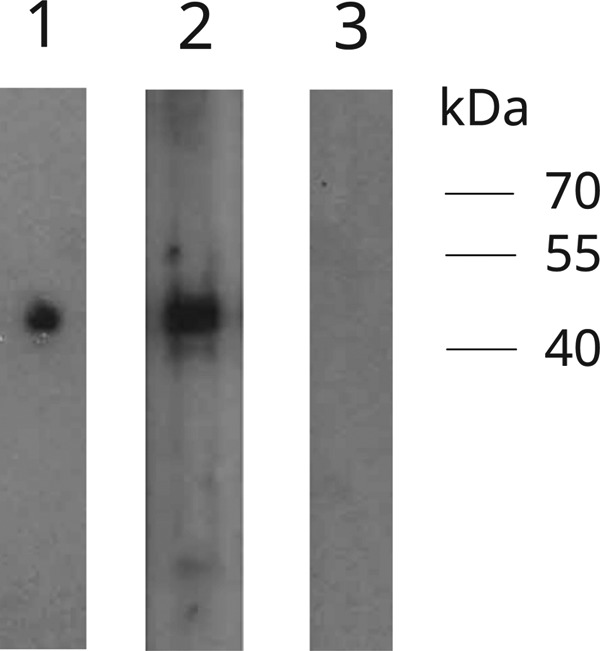
Western blot analysis of falcon CoV UAE-HKU27 (FalCoV UAE-HKU27) using nucleocapsid (N) protein expressed in Escherichia coli. Lane 1, positive control; lane 2, falcon serum sample (FS7) strongly positive for antibodies against FalCoV UAE-HKU27 N protein; lane 3: falcon serum sample (FS5) negative for antibodies against FalCoV UAE-HKU27 N protein.
DISCUSSION
Similar to the case for birds in Hong Kong, birds in the Middle East are also hosts for a diversity of deltacoronaviruses. In 2009, we reported the discovery of three novel avian CoVs that were phylogenetically distinct from infectious bronchitis virus (31). These three CoVs were found in fecal samples of bulbuls, thrushes, and munias in Hong Kong (31). In 2011, the ICTV approved the classification of these three avian CoVs as a novel genus, Deltacoronavirus, in the Coronaviridae family (4). In 2012, in a large epidemiological study, we discovered seven additional deltacoronaviruses (32). Six of them were from fecal samples from Japanese white eyes, tree sparrows, original magpie robins, black-crowned night herons, Eurasian wigeons, and common moorhens, respectively, and one was from fecal samples from pigs in Hong Kong (32). In the last few years, PorCoV HKU15 (now officially named Coronavirus HKU15) has been widely detected in fecal samples from pigs in Canada, China, Laos, Mexico, South Korea, Thailand, Vietnam, and the United States (33–41). Recently, we have also found PorCoV HKU15 in respiratory samples from pigs, which may have implications about the possible routes and sites of infections (42). In this study, four additional novel deltacoronaviruses were detected in fecal samples from falcons, houbara bustards, pigeons, and quails in Dubai. Similar to the case for other deltacoronaviruses, FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and QuaCoV UAE-HKU30 also have large genome sizes of 25,871 to 26,162 bases, owing to the presence of multiple ORFs downstream from the N gene. Continuous surveillance studies on birds in the Middle East and other regions will help to better understand the viral and host diversity of deltacoronaviruses and their potential for emergence in mammals.
Recent interspecies jumping events were observed in the deltacoronaviruses from falcons, houbara bustards, and pigeons, which were likely a result of predator-and-prey relationships along the food chain. According to the ICTV definition for demarcation of CoV species, where CoVs that share an overall amino acid identity of more than 90% in their seven conserved replicase domains (ADRP, nsp5 [3CLpro], nsp12 [RdRp], nsp13 [Hel], nsp14 [ExoN], nsp15 [NendoU], and nsp16 [O-MT]) should be regarded as the same species, FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 should be subspecies of a novel CoV species. Notably, the S proteins of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29, which are responsible for CoV receptor binding, also shared 94.5 to 99.8% amino acid identity, suggesting that the viruses have not evolved much yet to adapt to the corresponding avian host after jumping from one species to another. In fact, molecular clock analysis estimated that the tMRCA of HouCoV UAE-HKU28 and PiCoV UAE-HKU29 was just around 7 years ago and that of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 was just around 38 years ago (Fig. 5). Nevertheless, it should be noted that results of molecular clock estimation are only speculative for RNA viruses, which are known for episodic evolution and adaptation to different environments (43). It is interesting to note that falcons, houbara bustards, and pigeons are three radically different types of birds with unique behaviors and habitats. Falcons (order Falconiformes, family Falconidae) are medium-size birds of prey traditionally used for hunting wild quarry in the Arabian region. Houbara bustards (order Otidiformes, family Otididae) are large birds that are geographically restricted to arid habitats. Pigeons (order Columbiformes, family Columbidae) are relatively smaller and are globally distributed. Yet, these birds are ecologically closely related, because falcons are known predators of pigeons and houbara bustards and are also fed with the meat of these birds (44, 45). Moreover, falcons are trained by Arabian falconers to hunt houbara bustards because Arabs and other Asians, such as Pakistanis, believe that their meat possesses aphrodisiac qualities and the meat is sold at high prices, despite banning of such controversial practices in some countries. Therefore, interspecies transmission of this CoV species is likely a result of the predator-and-prey relationship. Serological testing confirmed the presence of specific antibodies against FalCoV UAE-HKU27 in 75% of field falcon sera collected in Dubai. Such a relatively high seroprevalence is similar to those observed for other coronaviruses found in other animals, such as Middle East respiratory syndrome coronavirus (MERS-CoV) and dromedary CoV UAE-HKU23 in dromedaries and rabbit CoV HKU14 in rabbits (7, 46), supporting widespread genuine infection among the falcon population and excluding the possibility of remnant viruses in falcon fecal samples resulting from ingestion of pigeons and houbara bustards. The detection of viruses from only one pigeon was not surprising, as viral shedding is often transient during the acute infection phase. Further molecular studies will reveal whether these three closely related deltacoronaviruses share a receptor in these three phylogenetically distant birds, where viral adaptation to an evolutionarily conserved host-cell receptor might help offer facile interspecies transmissibility (47).
Recombination involving the S protein is likely a common phenomenon among deltacoronaviruses, which may facilitate interspecies transmission and adaptation to new animal hosts. Phylogenetic analysis showed that the CoV species comprising FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 was most closely related to WECoV HKU16 in the 3CLpro, RdRp, Hel, and N genes but was only distantly related to WECoV HKU16 and most closely related to MRCoV HKU18 in the S gene, suggesting recombination events around the S gene region as demonstrated by bootscan analysis (Fig. 4). Regarding QuaCoV UAE-HKU30, PorCoV HKU15, and SpCoV HKU17, according to the ICTV definition for demarcation of CoV species, these three CoVs should be classified as subspecies of the same CoV species. However, in contrast to FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29, which are clustered together in all phylogenetic trees, QuaCoV UAE-HKU30, PorCoV HKU15, and SpCoV HKU17 showed phylogenetic positions shifting in different phylogenetic trees. QuaCoV UAE-HKU30 was clustered with PorCoV HKU15 and SpCoV HKU17 in the 3CLpro, RdRp, Hel, and N genes, whereas it was most closely related to MunCoV-HKU13 in the S gene (Fig. 3). Recombination around the S gene region was also demonstrated by bootscan analysis (Fig. 4). Similarly, PorCoV HKU15 and/or Asian leopard cat CoV (ALCCoV) was most closely related to SpCoV HKU17 in the 3CLpro, RdRp, Hel, and N genes but was only distantly related to SpCoV HKU17 and most closely related to BuCoV HKU11 in the S gene. This suggests that these mammalian deltacoronaviruses may have arisen from recombination events between SpCoV HKU17 and BuCoV HKU11 or related viruses. Recombination is not uncommon in other CoVs and was found to be responsible for the emergence of SARS-CoV (48–51) and generation of new genotypes or strains of other CoVs, including human CoV HKU1, human CoV OC43, and feline CoV type II strains (5, 6, 52). The present results suggest that recombination is also common among deltacoronaviruses, with the S gene being a frequent recombination site. Further studies may reveal if this may be an important mechanism for overcoming the mammalian species barrier through more efficient receptor binding to swine or other mammalian cells in PorCoV HKU15 and related viruses.
MATERIALS AND METHODS
Ethics statement.
Collection of animal fecal samples and field falcon sera in this study was approved by the Animal Ethic Committee of the Central Veterinary Research Laboratory and Ministry of Climate Change and Environment, UAE, according to Ministerial Decree no. 384 of 2008 on the executive by-law of the Federal Law no. 16 of 2007 concerning animal welfare. All the experimental procedures were performed in accordance with the International Guiding Principles for Biomedical Research Involving Animals regarding the care and use of animals.
Animal surveillance and sample collection.
All animal fecal samples were leftover specimens submitted to the Central Veterinary Research Laboratory in Dubai, UAE, for pathogen screening over a 24-month period (January 2013 to December 2014). A total of 1,356 fecal samples from 1,164 mammals and 192 birds were tested (Table 1). Serum samples from falcons in the field were collected in Dubai over a 4-month period (November 2015 to February 2016). All samples were taken from the caudal tibial vein (vena metatarsalis plantaris superficialis) under isoflurane anesthesia in serum tubes, centrifuged, and stored at −20°C until use.
RNA extraction.
Viral RNA was extracted from the fecal samples using the RNeasy minikit (Qiagen, Germany). The RNA was eluted in 50 μl of RNase-free water and was used as the template for RT-PCR.
RT-PCR of RdRp gene of CoVs using deltacoronavirus-conserved primers and DNA sequencing.
Initial CoV screening was performed by amplifying a 440-bp fragment of the RdRp gene of CoVs using deltacoronavirus-conserved primers (LPW16472 [5′-GTGGVTGTMTTAATGCACAGTC-3′] and LPW16473 [5′-TACTGYCTGTTRGTCATRGTG-3′]), which we published previously (32). Reverse transcription was performed using the SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). The PCR mixture (25 μl) contained cDNA, PCR buffer (10 mM Tris-HCl [pH 8.3], 50 mM KCl, 3 mM MgCl2, and 0.01% gelatin), 200 μM each deoxynucleoside triphosphate (dNTP), and 1.0 U Taq polymerase (Applied Biosystems, Foster City, CA). The mixtures were amplified by 60 cycles of 94°C for 1 min, 48°C for 1 min, and 72°C for 1 min and a final extension at 72°C for 10 min in an automated thermal cycler (Applied Biosystems). Standard precautions were taken to avoid PCR contamination, and no false-positive result was observed in negative controls.
The PCR products were gel purified using the QIAquick gel extraction kit (Qiagen). Both strands of the PCR products were sequenced twice with the ABI Prism 3130xl genetic analyzer (Applied Biosystems), using the two PCR primers. The sequences of the PCR products were compared with known sequences of the RdRp genes of CoVs in the DDBJ/ENA/GenBank sequence databases. A phylogenetic tree was reconstructed using 371-bp fragments of the RdRp gene with the maximum-likelihood (ML) method using the substitution model general time reversible with gamma distributed rate variation as well as estimated proportion of invariable sites (GTR+G+I) by PhyML.3.0.
Complete genome sequencing.
One complete genome each of FalCoV UAE-HKU27, HouCoV UAE-HKU28, and PiCoV UAE-HKU29 and two complete genomes of QuaCoV UAE-HKU30 were amplified and sequenced using the RNAs extracted from the original fecal specimens as templates. The RNA was converted to cDNA by a combined random-priming and oligo(dT) priming strategy. The cDNA was amplified with degenerate primers designed by multiple alignments of the genomes of other CoVs with complete genomes available, using strategies described in our previous publications (7, 18, 29). Additional primers were designed from the results of the first and subsequent rounds of sequencing. The 5′ ends of the viral genomes were confirmed by rapid amplification of cDNA ends using the SMARTer 5′/3′ RACE kit (Clontech, Mountain View, CA). Sequences were assembled and manually edited to produce final sequences of the viral genomes.
Genome analysis.
The nucleotide sequences of the genomes and the deduced amino acid sequences of the ORFs were compared to those of other CoVs using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). Phylogenetic tree reconstruction was performed using the ML method with PhyML3.0. The best-fit substitution models were selected using PhyML with Smart Model Selection. Protein family analysis was performed using PFAM and InterProScan (53, 54). Prediction of transmembrane domains was performed using TMHMM (55).
Recombination analysis.
To detect possible recombination events, bootscan analysis was performed by using the nucleotide alignment of the genome sequences of the novel deltacoronaviruses with Simplot version 3.5.1, as previously described (56). The analysis was conducted using a sliding window of 1,000 nucleotides moving in 200-nucleotide steps with 1,000 bootstrap values. Possible recombination sites suggested by the bootscan analysis were confirmed through multiple-sequence alignments.
Estimation of divergence dates.
Divergence times for the genus Deltacoronavirus was calculated using a Bayesian Markov chain Monte Carlo (MCMC) approach as implemented in BEAST (version 1.7.4) as described previously (50, 57). RdRp gene sequence data were selected for analyses under the substitution model GTR+G+I using an unrelaxed log-normally distributed (Ucld) relaxed molecular clock and a Bayesian Skyline coalescent model. The MCMC run was 5 × 107 steps long, with sampling every 1,000 steps. Convergence was assessed on the basis of the effective sampling size after a 10% burn-in using Tracer version 1.6. tMRCA and the HPD regions at 95% were calculated. The trees were summarized in a target tree by the Tree Annotator program included in the BEAST package by choosing the tree with the maximum sum of posterior probabilities (maximum clade credibility) after a 10% burn-in.
Cloning and purification of His6-tagged recombinant N protein of FalCoV UAE-HKU27.
Cloning and purification of the His6-tagged recombinant N protein of FalCoV UAE-HKU27 was performed using a protocol we described previously (46, 58). To produce a plasmid for protein purification, primers LPW36358 (5′-GGAATTCCATATGATGAGCACTCCCACAGTCCCT-3′) and LPW36359 (5′-CCGCTCGAGATGCAGTTGAATCTCCATCCTG-3′) were used to amplify the gene encoding the N protein of FalCoV UAE-HKU27 by RT-PCR. The sequence coding for amino acid residues (aa) 1 to 344 of the N protein was amplified and cloned into the NdeI and XhoI sites of the expression vector pET-28b(+) (Merck, Germany) in frame and upstream of the histidine residue series. The recombinant N protein was expressed and purified using Ni-nitrilotriacetic acid (NTA) agarose (Qiagen) according to the manufacturer's instructions.
Western blot analysis.
Western blot analysis was performed according to our published protocol (46, 58). Briefly, 600 ng of purified His6-tagged recombinant N protein of FalCoV UAE-HKU27 was loaded into the well of a sodium dodecyl sulfate (SDS)-10% polyacrylamide gel and subsequently electrophoresed and then electroblotted onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The blot was cut into strips, and each separated strip was incubated with an individual serum sample, at a dilution of 1:1,000, obtained from falcons in Dubai. Antigen-antibody interaction was detected with an in-house-developed polyclonal guinea pig anti-falcon IgY antibody (59) at a dilution of 1:2,000, a horseradish peroxidase (HRP)-conjugated rabbit anti-guinea pig IgG antibody (Invitrogen) at a dilution of 1:4,000, and the WesternBright Quantum HRP substrate (Advansta, USA). Monoclonal anti-His6 antibody (clone HIS.H8; Invitrogen) at a concentration of 0.5 μg/ml, with HRP-conjugated goat anti-mouse IgG antibody (Invitrogen) at a dilution of 1:4,000 as the secondary antibody, was used as the positive control.
Accession number(s).
The nucleotide sequences of the five complete genomes of FalCoV UAE-HKU27, HouCoV UAE-HKU28, PiCoV UAE-HKU29, and QuaCoV UAE-HKU30 have been deposited in the DDBJ/ENA/GenBank sequence databases with the accession numbers LC364342 to LC364346.
ACKNOWLEDGMENTS
This work was partly supported by the Theme-based Research Scheme (project no. T11/707/15), University Grant Committee, Hong Kong, by the University Development Fund, The University of Hong Kong, Hong Kong, and by the Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Ministry of Education, China.
Patrick C. Y. Woo has provided scientific advisory/laboratory services for Gilead Sciences, Incorporated, for International Health Management Associates, Incorporated, for Merck Corporation, Incorporated, and for Pfizer, Incorporated. The other authors report no conflict of interest. These commercial companies had no role in study design, data collection, analysis, interpretation, or writing of the report. The authors alone are responsible for the content and the writing of the manuscript.
REFERENCES
- 1.Lai MMC, Cavanagh D. 1997. The molecular biology of coronaviruses. Adv Virus Res 48:1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziebuhr J. 2004. Molecular biology of severe acute respiratory syndrome coronavirus. Curr Opin Microbiol 7:412–419. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brian DA, Baric RS. 2005. Coronavirus genome structure and replication. Curr Top Microbiol Immunol 287:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2012. Family Coronaviridae, p 806–828. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy. Classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego, CA. [Google Scholar]
- 5.Herrewegh AA, Smeenk I, Horzinek MC, Rottier PJ, de Groot RJ. 1998. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J Virol 72:4508–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo PCY, Lau SKP, Yip CCY, Huang Y, Tsoi H-W, Chan K-H, Yuen K-Y. 2006. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol 80:7136–7145. doi: 10.1128/JVI.00509-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lau SKP, Woo PCY, Yip CCY, Fan RYY, Huang Y, Wang M, Guo R, Lam CSF, Tsang AKL, Lai KKY, Chan K-H, Che X-Y, Zheng B-J, Yuen K-Y. 2012. Isolation and characterization of a novel betacoronavirus subgroup a coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J Virol 86:5481–5496. doi: 10.1128/JVI.06927-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau SKP, Wernery R, Wong EYM, Joseph S, Tsang AKL, Patteril NAG, Elizabeth SK, Chan K-H, Muhammed R, Kinne J, Yuen K-Y, Wernery U, Woo PCY. 2016. Polyphyletic origin of MERS coronaviruses and isolation of a novel clade A strain from dromedary camels in the United Arab Emirates. Emerg Microbes Infect 5:e128. doi: 10.1038/emi.2016.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreno A, Franzo G, Massi P, Tosi G, Blanco A, Antilles N, Biarnes M, Majó N, Nofrarías M, Dolz R, Lelli D, Sozzi E, Lavazza A, Cecchinato M. 2017. A novel variant of the infectious bronchitis virus resulting from recombination events in Italy and Spain. Avian Pathol 46:28–35. doi: 10.1080/03079457.2016.1200011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 11.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. 2003. The genome sequence of the SARS-associated coronavirus. Science 300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 12.Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, Nicholls J, Yee WKS, Yan WW, Cheung MT, Cheng VCC, Chan KH, Tsang DNC, Yung RWH, Ng TK, Yuen KY, SARS Study Group. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 14.Woo PCY, Lau SKP, Tsoi H-W, Chan K-H, Wong BHL, Che X-Y, Tam VKP, Tam SCF, Cheng VCC, Hung IFN, Wong SSY, Zheng B-J, Guan Y, Yuen K-Y. 2004. Relative rates of non-pneumonic SARS coronavirus infection and SARS coronavirus pneumonia. Lancet 363:841–845. doi: 10.1016/S0140-6736(04)15729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng VCC, Lau SKP, Woo PCY, Yuen KY. 2007. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev 20:660–694. doi: 10.1128/CMR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH, Osterhaus AD. 2004. A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci U S A 101:6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, Berkhout B. 2004. Identification of a new human coronavirus. Nat Med 10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo PCY, Lau SKP, Chu C-M, Chan K-H, Tsoi H-W, Huang Y, Wong BHL, Poon RWS, Cai JJ, Luk W-K, Poon LLM, Wong SSY, Guan Y, Peiris JSM, Yuen K-Y. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woo PCY, Lau SKP, Tsoi H-W, Huang Y, Poon RWS, Chu C-M, Lee RA, Luk W-K, Wong GKM, Wong BHL, Cheng VCC, Tang BSF, Wu AKL, Yung RWH, Chen H, Guan Y, Chan K-H, Yuen K-Y. 2005. Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J Infect Dis 192:1898–1907. doi: 10.1086/497151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lau SKP, Woo PCY, Yip CCY, Tse H, Tsoi H-W, Cheng VCC, Lee P, Tang BSF, Cheung CHY, Lee RA, So L-Y, Lau Y-L, Chan K-H, Yuen K-Y. 2006. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol 44:2063–2071. doi: 10.1128/JCM.02614-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau SKP, Woo PCY, Li KSM, Huang Y, Tsoi H-W, Wong BHL, Wong SSY, Leung S-Y, Chan K-H, Yuen K-Y. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A 102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 23.Woo PCY, Lau SKP, Li KSM, Poon RWS, Wong BHL, Tsoi H-W, Yip BCK, Huang Y, Chan K-H, Yuen K-Y. 2006. Molecular diversity of coronaviruses in bats. Virology 351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo PCY, Wang M, Lau SKP, Xu H, Poon RWS, Guo R, Wong BHL, Gao K, Tsoi H-W, Huang Y, Li KSM, Lam CSF, Chan K-H, Zheng B-J, Yuen K-Y. 2007. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol 81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau SKP, Zhang L, Luk HKH, Xiong L, Peng X, Li KSM, He X, Zhao PS-H, Fan RYY, Wong ACP, Ahmed SS, Cai J-P, Chan JFW, Sun Y, Jin D, Chen H, Lau TCK, Kok RKH, Li W, Yuen K-Y, Woo PCY. 2018. Receptor usage of a novel bat lineage C betacoronavirus reveals evolution of MERS-related coronavirus spike proteins for human DPP4 binding. J Infect Dis doi: 10.1093/infdis/jiy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chu DKW, Peiris JSM, Chen H, Guan Y, Poon LLM. 2008. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J Gen Virol 89:1282–1287. doi: 10.1099/vir.0.83605-0. [DOI] [PubMed] [Google Scholar]
- 27.Mihindukulasuriya KA, Wu G, St Leger J, Nordhausen RW, Wang D. 2008. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J Virol 82:5084–5088. doi: 10.1128/JVI.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corman VM, Kallies R, Philipps H, Göpner G, Müller MA, Eckerle I, Brünink S, Drosten C, Drexler JF. 2014. Characterization of a novel betacoronavirus related to Middle East respiratory syndrome coronavirus in European hedgehogs. J Virol 88:717–724. doi: 10.1128/JVI.01600-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau SKP, Woo PCY, Li KSM, Tsang AKL, Fan RYY, Luk HKH, Cai J-P, Chan K-H, Zheng B-J, Wang M, Yuen K-Y. 2015. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A. J Virol 89:3076–3092. doi: 10.1128/JVI.02420-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li KS, Guan Y, Wang J, Smith GJD, Xu KM, Duan L, Rahardjo AP, Puthavathana P, Buranathai C, Nguyen TD, Estoepangestie ATS, Chaisingh A, Auewarakul P, Long HT, Hanh NTH, Webby RJ, Poon LLM, Chen H, Shortridge KF, Yuen KY, Webster RG, Peiris JSM. 2004. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- 31.Woo PCY, Lau SKP, Lam CSF, Lai KKY, Huang Y, Lee P, Luk GSM, Dyrting KC, Chan K-H, Yuen K-Y. 2009. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J Virol 83:908–917. doi: 10.1128/JVI.01977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo PCY, Lau SKP, Lam CSF, Lau CCY, Tsang AKL, Lau JHN, Bai R, Teng JLL, Tsang CCC, Wang M, Zheng B-J, Chan K-H, Yuen K-Y. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol 86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kochhar HS. 2014. Porcine epidemic diarrhea in Canada: an emerging disease case study. Can Vet J 55:1048–1049. [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S, Lee C. 2014. Outbreak-related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerg Infect Dis 20:1223–1226. doi: 10.3201/eid2007.140294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Byrum B, Zhang Y. 2014. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg Infect Dis 20:1227–1230. doi: 10.3201/eid2007.140296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong N, Fang L, Zeng S, Sun Q, Chen H, Xiao S. 2015. Porcine deltacoronavirus in mainland China. Emerg Infect Dis 21:2254–2255. doi: 10.3201/eid2112.150283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu H, Jung K, Vlasova AN, Chepngeno J, Lu Z, Wang Q, Saif LJ. 2015. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J Clin Microbiol 53:1537–1548. doi: 10.1128/JCM.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vui DT, Thanh TL, Tung N, Srijangwad A, Tripipat T, Chuanasa T, Nilubol D. 2015. Complete genome characterization of porcine epidemic diarrhea virus in Vietnam. Arch Virol 160:1931–1938. doi: 10.1007/s00705-015-2463-6. [DOI] [PubMed] [Google Scholar]
- 39.Janetanakit T, Lumyai M, Bunpapong N, Boonyapisitsopa S, Chaiyawong S, Nonthabenjawan N, Kesdaengsakonwut S, Amonsin A. 2016. Porcine deltacoronavirus, Thailand, 2015. Emerg Infect Dis 20:757–759. doi: 10.3201/eid2204.151852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorsirigool A, Saeng-chuto K, Temeeyasen G, Madapong A, Tripipat T, Wegner M, Tuntituvanont A, Intrakamhaeng M, Nilubol D. 2016. The first detection and full-length genome sequence of porcine deltacoronavirus isolated in Lao PDR. Arch Virol 161:2909–2911. doi: 10.1007/s00705-016-2983-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trujillo-Ortega ME, Beltran-Figueroa R, Garcia-Hernandez ME, Juarez-Ramirez M, Sotomayor-Gonzalez A, Hernandez-Villegas EN, Becerra-Hernandez JF, Sarmiento-Silva RE. 2016. Isolation and characterization of porcine epidemic diarrhea virus associated with the 2014 disease outbreak in Mexico: case report. BMC Vet Res 12:132. doi: 10.1186/s12917-016-0763-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woo PCY, Lau SKP, Tsang C-C, Lau CCY, Wong P-C, Chow FWN, Fong JYH, Yuen K-Y. 2017. Coronavirus HKU15 in respiratory tract of pigs and first discovery of coronavirus quasispecies in 5′-untranslated region. Emerg Microbes Infect 6:e53. doi: 10.1038/emi.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lauring AS, Frydman J, Andino R. 2013. The role of mutational robustness in RNA virus evolution. Nat Rev Microbiol 11:327–336. doi: 10.1038/nrmicro3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palleroni A, Miller CT, Hauser M, Marler P. 2005. Predation: prey plumage adaptation against falcon attack. Nature 434:973–974. [DOI] [PubMed] [Google Scholar]
- 45.Kane SA, Zamani M. 2014. Falcons pursue prey using visual motion cues: new perspectives from animal-borne cameras. J Exp Biol 217:225–234. doi: 10.1242/jeb.092403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woo PCY, Lau SKP, Wernery U, Wong EYM, Tsang AKL, Johnson B, Yip CCY, Lau CCY, Sivakumar S, Cai J-P, Fan RYY, Chan K-H, Mareena R, Yuen K-Y. 2014. Novel betacoronavirus in dromedaries of the Middle East, 2013. Emerg Infect Dis 20:560–572. doi: 10.3201/eid2004.131769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallagher T, Perlman S. 2013. Broad reception for coronavirus. Nature 495:176–177. doi: 10.1038/495176a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ, Luo CM, Tan B, Wang N, Zhu Y, Crameri G, Zhang SY, Wang LF, Daszak P, Shi ZL. 2013. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu B, Ge X, Wang LF, Shi Z. 2015. Bat origin of human coronaviruses. Virol J 12:221. doi: 10.1186/s12985-015-0422-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lau SKP, Feng Y, Chen H, Luk HKH, Yang W-H, Li KSM, Zhang Y-Z, Huang Y, Song Z-Z, Chow W-N, Fan RYY, Ahmed SS, Yeung HC, Lam CSF, Cai J-P, Wong SSY, Chan JFW, Yuen K-Y, Zhang H-L, Woo PCY. 2015. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J Virol 89:10532–10547. doi: 10.1128/JVI.01048-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu B, Zeng LP, Yang XL, Ge XY, Zhang W, Li B, Xie JZ, Shen XR, Zhang YZ, Wang N, Luo DS, Zheng XS, Wang MN, Daszak P, Wang LF, Cui J, Shi ZL. 2017. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog 13:e1006698. doi: 10.1371/journal.ppat.1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lau SKP, Lee P, Tsang AKL, Yip CCY, Tse H, Lee RA, So L-Y, Lau Y-L, Chan K-H, Woo PCY, Yuen K-Y. 2011. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol 85:11325–11337. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sonnhammer EL, von Heijne G, Krogh A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6:175–182. [PubMed] [Google Scholar]
- 56.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lau SKP, Wong ACP, Lau TCK, Woo PCY. 2017. Molecular evolution of MERS coronavirus: dromedaries as a recent intermediate host or long-time animal reservoir? Int J Mol Sci 18:2138. doi: 10.3390/ijms18102138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woo PCY, Lau SKP, Lam CSF, Tsang AKL, Hui S-W, Fan RYY, Martelli P, Yuen K-Y. 2014. Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. J Virol 88:1318–1331. doi: 10.1128/JVI.02351-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Wernery U, Tsang C-C, Hebel C, Damerau A, Kinne J, Cai J-P, Küspert H, Chan K-F, Joseph M, Xue S, Raghavan R, Tang JYM, Syriac G, Lau SKP, Jose S, Woo PCY. 2018. Serodiagnosis of aspergillosis in falcons (Falco spp.) by an Afmp1p-based enzyme-linked immunosorbent assay. Mycoses doi: 10.1111/myc.12776. [DOI] [PubMed] [Google Scholar]