

ABSTRACT
In response to virus-induced shutoff host protein synthesis, dynamic aggregates containing mRNA, RNA-binding proteins and translation factors termed stress granules (SGs) often accumulate within the cytoplasm. SGs typically form following phosphorylation and inactivation of the eukaryotic translation initiation factor 2α (eIF2α), a substrate of the double-stranded RNA (dsRNA)-activated kinase protein kinase R (PKR). The detection of innate immune sensors and effectors like PKR at SGs suggests a role in pathogen nucleic acid sensing. However, the functional importance of SGs in host innate responses is unclear and has primarily been examined in response to infection with select RNA viruses. During infection with the DNA virus herpes simplex virus 1 (HSV-1), the virus-encoded virion host shutoff (VHS) endoribonuclease is required to restrict interferon production, PKR activation, and SG formation, although the relationship between these activities remains incompletely understood. Here, we show that in cells infected with a VHS-deficient HSV-1 (ΔVHS) dsRNA accumulated and localized to SGs. Surprisingly, formation of dsRNA and its concentration at SGs was not required for beta interferon mRNA induction, indicating that suppression of type I interferon induction by VHS does not stem from its control of dsRNA accumulation. Instead, STING signaling downstream of cGMP-AMP synthase (cGAS)-dependent DNA sensing is required for beta interferon induction. In contrast, significantly less PKR activation is observed when SG assembly is disrupted by ISRIB, an inhibitor of phosphorylated eIF2α-mediated translation repression, or depleting SG scaffolding proteins G3BP1 or TIA1. This demonstrates that PKR activation is intimately linked to SG formation and that SGs form important hubs to potentiate PKR activation during infection.
IMPORTANCE Formation of cytoplasmic stress granules that are enriched for innate immune sensors and effectors is suppressed during many viral infections. It is unclear, however, to what extent this is a side effect of viral efforts to maintain protein synthesis or intentional disruption of a hub for innate immune sensing. In this study, we utilize a herpes simplex virus 1 mutant lacking the RNA nuclease VHS which upon infection induces SGs, PKR activation, and beta interferon to address this question. We show that dsRNA is localized to SGs and that SGs can function to promote PKR activation in the context of a DNA virus infection, but we find no evidence to support their importance for interferon induction during HSV-1 infection.
KEYWORDS: stress granules, PKR, DNA sensing, antiviral immunity, VHS, HSV-1 replication, stress granules, virion host shutoff
INTRODUCTION
Stress granules (SGs) are cytoplasmic aggregates of messenger ribonucleoprotein complexes (mRNPs) stalled at translation initiation that transiently form during a number of cell stress conditions. Enriched at SGs are RNA binding proteins, translation initiation factors, and small ribosomal proteins bound to these mRNAs and which have been shown to dynamically exchange between the granules and the cytosol (reviewed in reference 1). Typically, SG formation is dependent on phosphorylation and inactivation of eukaryotic translation initiation factor 2α (eIF2α) accomplished in mammalian cells by one of four kinases that act as stress sensors: heme-regulated inhibitor (EIF2AK1), double-stranded RNA activated protein kinase R (PKR; EIF2AK2), PKR-like endoplasmic reticulum kinase (PERK; EIF2AK3), and general control nondepressible 2 (EIF2AK4). The formation of SGs has been suggested to help preserve mRNAs and allow reversible arrest and recovery of translation upon cell stress, but the contribution of SG formation to stress recovery is contentious and difficult to dissect from the translational inhibition which causes their formation (2). In the context of viral infection, however, SGs may play additional roles (3).
Several cell intrinsic, antiviral innate immune pathways exist in parallel to detect and respond to viral infection upon recognition of pathogen-associated molecular patterns (PAMPs), such as cytoplasmic double-stranded DNA (dsDNA) or double-stranded RNA (dsRNA). In recent years, several sensors and components of these pathways have been found localized to SGs and the possibility of SGs providing dynamic platforms for PAMP recognition raised. RIG-I like receptor MDA5 (IFIH1), which senses long dsRNA, and RIG-I (DDX58), which senses 5′-triphosphate single-stranded RNA and shorter dsRNA in the cytosol (4–6), have been shown to localize to SGs (7, 8). These sensors interact with MAVS localized at the mitochondrial membrane, which in turn activates TBK1 (TANK-binding kinase 1) and a signaling cascade that results in production of type I (α and β) interferon (IFN) (9). Paracrine IFN sensing in neighboring cells activates the JAK/STAT pathway to mediate the upregulation of a suite of antiviral IFN-stimulated genes (ISGs); the mechanisms of action of only a few ISGs are fully understood (10).
Interfering with SG formation during infection with two RNA viruses, influenza A and Newcastle disease virus, has been shown to impair IFN induction downstream of RIG-I, inferring a functional role for SGs in the support of PAMP sensing (7, 11). Investigations of encephalomyocarditis (EMCV) infection in which IFN is induced via MDA5 provide contradictory results, however, with different groups reporting that IFN-β levels were either reduced or unaffected by SG disruption (8, 12). The dsRNA-activated kinase PKR is frequently the eIF2α-kinase responsible for translational inhibition and SG formation during virus infection, and its activation has the effect of globally limiting infected-cell protein synthesis. PKR also accumulates at SGs under certain stress conditions and has been suggested to be further activated by this localization (13, 14). Forcing SG assembly by overexpression of the RNA binding protein Ras GTPase-activating protein-binding protein 1 (G3BP1) resulted in PKR activation (15), though whether a correlation between SG assembly and PKR activation occurs during normal SG formation and influences the antiviral activity of PKR during infection is unclear. A further defense mechanism that impairs protein synthesis is activated when oligoadenylate synthetase (OAS) enzymes encounter dsRNA. Their subsequent activation leads to the production of 2′-5′-oligoadenylate, which in turn activates the cellular endoribonuclease RNase L, which cleaves both mRNAs and rRNA (16). OAS2 and RNase L have each been shown to localize to SGs (7, 14), though the functional consequences of this localization are completely unexplored.
Many RNA and DNA viruses express proteins that are able to prevent SG assembly (reviewed in references 3 and 17). While some also benefit the virus by maintaining protein synthesis, for example, the influenza virus protein NS1 which prevents both PKR activation and SG formation (18), others disrupt stress granule scaffolding proteins directly, arguing that SGs themselves are intrinsically antiviral. The essential SG component G3BP1 is the most common target; it is cleaved by poliovirus (19), bound and inhibited by chikungunya virus nsP3 protein (20), and a dengue virus-encoded RNA (21) and sequestered and repurposed in vaccinia virus infection (22). An alternate strategy is adopted by herpes simplex virus 1 (HSV-1), which employs the virion host shutoff (VHS) endoribonuclease (23, 24). Infection with a mutant virus lacking VHS (ΔVHS) results in PKR activation and PKR-dependent SG formation in multiple cell types (23–26). Destabilization of host mRNAs by VHS may also directly contribute to its disruption of SG formation. Studies with HSV-2 found that whereas wild type (WT)-infected cells were refractory to SG induction by external stimuli, those infected with a mutant lacking a related VHS endonuclease were not (27).
In cell culture, HSV-1 ΔVHS replicates to near normal levels in single cycle infections but is more sensitive to type I IFN compared to WT (28) and severely attenuated in animal models of both lytic and latent infections (29). VHS degrades RNA with little specificity in vitro, cutting after cytidine and uridine residues (30); however, in infected cells it preferentially cleaves translating mRNAs (31), a propensity attributed to interactions with translation initiation factors eIF4H and eIF4A (32, 33). With a few exceptions, the majority of cellular mRNAs are targeted (30, 34), suppressing host antiviral responses and likely enabling viral mRNAs to better compete for limiting translation components. Viral mRNAs do not completely escape, and their accelerated degradation by VHS ensures sharp transitions between temporal classes of viral genes during infection (28). Accumulation of late viral gene products is impaired ΔVHS infection, and this translational defect has both PKR-dependent and -independent aspects (25). Failure to produce adequate levels of the late-expressed dsRNA binding protein Us11, which inhibits PKR activation (35), by ΔVHS likely contributes to PKR-dependent phenotypes (36). Expression of ICP34.5, which interacts with the protein phosphatase 1α catalytic subunit to dephosphorylate eIF2α (37), is insufficient to limit phosphorylated eIF2α and SG accumulation in ΔVHS-infected cells, although the underlying reason for this remains unknown (24, 28). Type I IFN is also induced during infection with a ΔVHS virus, and viral replication is partially rescued in IFN-αβγ receptor knockout mouse embryonic fibroblasts (28), suggesting that failure to suppress IFN and IFN-mediated responses is important for the in vivo attenuation of ΔVHS. Activation of PKR and detection of increased viral transcripts during ΔVHS infection (28) suggest that dsRNA is present and contributing to the IFN response, though cGMP-AMP synthase (cGAS)-mediated signaling downstream of cytosolic sensing of DNA may also regulate ΔVHS replication (38). Indeed, sensing of the dsDNA genome has been widely implicated in the innate immune response to HSV-1, where signaling to adaptor protein STING from sensor cGAS via the second messenger molecule 2′3′-cGAMP is thought to mediate type I IFN production (39–42). IFN-γ-inducible protein 16 (IFI16) has also been implicated as either an alternate/additional DNA sensor or in a role supporting transcriptional activation downstream of cGAS sensing (39, 43, 44). VHS is not the only HSV-1 protein implicated in controlling the IFN response. The activities of ICP0, VP16, ICP27, US3, VP24, US11, UL36, UL37, and UL42 (reviewed in reference 45), and recently UL46 (VP11/12) (46), each reportedly restrict IFN production mediated by cytosolic dsRNA or DNA sensing or downstream signaling events shared by both pathways. It remains unclear which viral functions are the most important and whether this varies by infected cell type.
Here, we dissect the interplay between SG formation, PKR activation, and type I IFN production, which are each suppressed by VHS. Surprisingly, while dsRNA accumulates and partially localizes to SGs during ΔVHS infection, neither dsRNA nor SG formation are required for the production of IFN-β mRNA or ISG-products, ruling out an important role for SGs in HSV-1-mediated IFN induction. Instead, we find that DNA sensing plays the major role in IFN-β induction in the absence of VHS and restricts the replication of ΔVHS. In contrast, SG formation is crucial for full activation of PKR, suggesting that SGs potentiate PKR activation in a feed-forward mechanism.
RESULTS
dsRNA is generated during ΔVHS HSV-1 infection and partially localizes to SGs.
To test the hypothesis that SGs might facilitate dsRNA sensing in HSV-1 infection, we first examined whether dsRNA is formed and its subcellular localization. Primary normal human dermal fibroblasts (NHDFs) were infected with ΔVHS or WT HSV-1, and the distribution of dsRNA and SGs was evaluated by indirect immunofluorescence (Fig. 1). Samples were imaged at 18 h postinfection (hpi) since SGs accumulate late in infection with ΔVHS HSV-1 (23, 24). Costaining for viral protein ICP4 and the SG component G3BP1 revealed G3BP1-positive SGs in a majority of ΔVHS-infected cells (Fig. 1A) in keeping with previous findings (24). We then examined dsRNA distribution using the monoclonal antibody J2 (47) (Fig. 1B). No staining for dsRNA was observed in WT F-strain HSV-1-infected cells beyond that of mock-infected cells; however, abundant dsRNA staining was detected in ΔVHS-infected cells. Specifically, cells with dsRNA staining also stained positively for SGs. PKR is necessary for SG formation during ΔVHS infection (25), and the J2 antibody detects dsRNA of 40 bp or longer (48), similar to the 30-bp minimal RNA duplex required to activate PKR (49). Therefore, our data are consistent with the idea that dsRNA is generated during infection in the absence of VHS, and this dsRNA activates PKR, resulting in SG formation. Closer examination of dsRNA distribution indicated that though not completely colocalized, some G3BP1-stained SGs also stained for dsRNA (Fig. 1B, zoom panel). This raises the possibility that SGs may serve as platforms for sensing of dsRNA by PKR and MDA5 or RIG-I.
FIG 1.
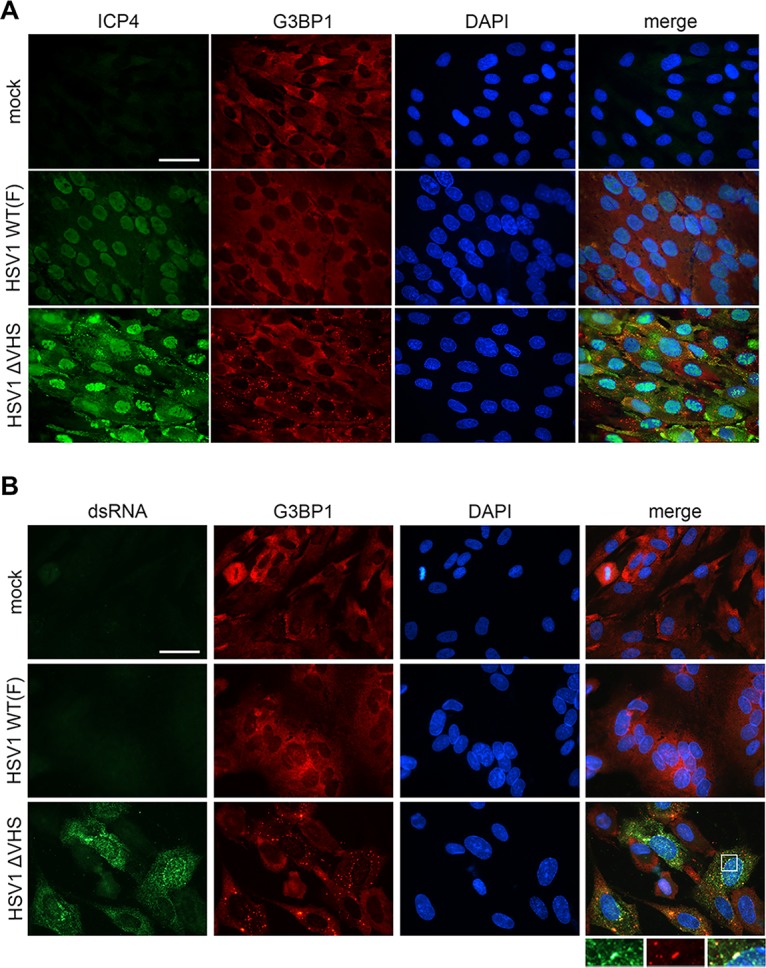
dsRNA accumulates in ΔVHS-infected cells and partially localizes to stress granules. NHDFs were mock infected or infected with WT HSV-1 (F-strain) or ΔVHS (R2621; F-strain) at an MOI of 5. At 18 hpi, the cells were fixed and analyzed by immunofluorescence for dsRNA using viral protein ICP4 (A) or the J2 monoclonal antibody (B) (green) and the SG marker G3BP1 (red). Nuclei are stained by DAPI (blue). Scale bars, 50 μm.
IFN induction precedes PKR activation and SG formation and does not require viral DNA synthesis.
To determine whether SGs could be important for PKR activation or IFN-β induction, a time course was conducted to evaluate whether each response was temporally correlated to SG assembly in ΔVHS-infected cells. Cell-free lysates for protein and RNA analysis were collected at 3-h intervals from NHDFs infected with ΔVHS. Quantitative reverse transcription-PCR (qRT-PCR) revealed that IFN-β mRNA induction peaked at 6 hpi, and the expression of two ISG mRNAs, IFIT2 and MDA5, showed similar kinetics (Fig. 2A). We next analyzed the activation of the signaling molecule TBK1, which is required for the stimulation of interferon transcription downstream of both MAVS-dependent RNA and STING-dependent DNA sensing pathways, by detecting phosphorylation at S172 by immunoblotting (50, 51). Similar to interferon mRNA levels, phospho-TBK1 was first detected at 3 hpi, with the strongest signal at 6 hpi and with decreasing intensity thereafter (Fig. 2B). In contrast, PKR activation, detected by immunoblotting for the autophosphorylated residue T446, occurred later and was strongly detected at 9 hpi, increasing to maximal activation at 12 hpi (Fig. 2B). Consistent with a dependence on PKR activity for SG formation, SGs were observed in more than 50% of cells from 9 hpi onward, increasing to a maximum of approximately 74% of cells at 12 and 15 hpi (Fig. 2C). This is consistent with the temporal pattern of PKR activation and SG assembly observed in HeLa cells infected with a ΔVHS in a KOS strain genetic background (24, 25) and the detection in human foreskin fibroblasts of SGs at 12 hpi with the same ΔVHS F-strain mutant used in our studies (23). While there are time points during infection where TBK1 activation/IFN-β mRNA are detected and SGs can be observed (6 and 9 hpi), maximal induction of TBK1 activation and IFN-β mRNA at 6 hpi precedes maximal SG detection at 12 hpi, suggesting that SGs may not play a significant role in PAMP sensing. Conversely, PKR activation correlates well with SG formation.
FIG 2.

Kinetics of induction of IFN-β and ISG mRNA, TBK1 and PKR activation, and SG assembly during ΔVHS infection. NHDFs were infected with ΔVHS at an MOI of 5 and collected at 3-h intervals. Uninfected (UI) cells were collected at 0 hpi. (A) mRNA levels of IFN-β and ISGs IFIT2 and MDA5 were analyzed by qRT-PCR from total RNA and normalized to 18S. The means ± the standard errors of the mean (SEM) of three independent experiments are plotted. (B) TBK1 (S172) and PKR (T446) phosphorylation were analyzed by immunoblotting of whole-cell lysates. (C) SG induction was quantified by counting the number of cells containing two or more G3BP1-stained SGs. More than 100 cells were counted for each time point, and the means ± the SEM of three independent experiments are plotted.
Inhibition of viral DNA synthesis and subsequent late viral gene expression by phosphonoacetic acid (PAA) has been reported to block PKR activation and SG formation during ΔVHS (KOS) infection (24, 25), suggesting dsRNA accumulation may also be dependent on viral DNA synthesis. The early onset of IFN-β expression (Fig. 2A), however, raised the possibility that it may not require viral DNA synthesis. To more precisely delineate the relationship between SG and IFN-β mRNA accumulation, the impact of PAA upon IFN-β and ISG expression was first examined. NHDFs infected with ΔVHS were treated with PAA or vehicle control immediately after infection, and total protein lysates were collected at 12 hpi. Accumulation of two late (gamma 1) viral proteins ICP5 and glycoprotein B (gB) was sensitive to PAA, providing a positive control for its activity (Fig. 3A). The expression of two ISG-encoded proteins (MDA5 and RIG-I) was unaffected by PAA treatment, while PKR activation was inhibited (Fig. 3A). RNA was collected at 6 hpi to examine the induction of IFN-β mRNA by qRT-PCR. This was also not detectably affected by PAA (Fig. 3B). In contrast, dsRNA staining was markedly diminished by PAA, and aggregation of G3BP1 into SGs was not detected (Fig. 3C), both of which are consistent with the prevention of PKR activation by PAA treatment. Together, these data show that while dsRNA accumulation, PKR activation, and SG formation are dependent on viral DNA replication, IFN-β and ISG induction are insensitive to PAA and proceed when viral DNA synthesis is blocked. This demonstrates that IFN-β expression is separable from SG accumulation during infection and PAMP sensing and signaling are not dependent upon SG formation. Further, as dsRNA accumulates after viral DNA synthesis and IFN-β induction precedes DNA synthesis, a PAMP other than dsRNA likely triggers IFN-induced antiviral responses in ΔVHS-infected cells.
FIG 3.

IFN-β and ISG induction is not dependent on viral DNA synthesis. NHDFs were mock infected or infected with ΔVHS and treated with 300 μg/ml PAA or a vehicle control at 1 hpi. (A) PKR phosphorylation and ISG protein (MDA5, RIG-I) and late viral protein (gB, ICP5) accumulation were analyzed by immunoblotting of whole-cell lysates collected at 12 hpi. (B) IFN-β mRNA levels were analyzed by qRT-PCR from total RNA collected at 6 hpi and normalized to 18S. Means ± the SEM of three independent experiments are plotted. ns, P > 0.05 (Student t test compared to control). (C) SG induction and dsRNA accumulation were visualized at 12 hpi by immunofluorescence for G3BP1 (red) and dsRNA (green) with nuclei stained by DAPI (blue). Scale bar, 50 μm.
IFN is induced downstream of DNA sensing in ΔVHS infection.
To test the contribution of dsRNA sensing to IFN-β and ISG expression during ΔVHS infection, the sensors MDA5 or RIG-I and the adaptor signaling protein MAVS were depleted using RNA interference (RNAi) prior to infection. DNA sensors implicated in the detection of HSV-1 infection, cGAS and IFI16, and the signaling adaptor STING were also targeted (39, 41). NHDFs transfected with a nonsilencing control or gene-specific small interfering RNAs (siRNAs) were infected with ΔVHS. RNA was isolated at 6 hpi, corresponding to the peak of IFN-β mRNA expression (Fig. 2A), and analyzed by qRT-PCR, and total protein was collected at 12 hpi for analysis of induced ISG-encoded proteins by immunoblotting. Knockdown of the RNA sensors RIG-I and MDA5 had a limited impact on IFN-β mRNA levels, effecting 30 and 35% reductions, respectively, which did not reach statistical significance (Fig. 4A). Instead, the largest decrease in IFN-β mRNA accumulation was observed upon knockdown of DNA sensors cGAS (90%; P = 0.006) and IFI16 (66%; P = 0.041), suggesting that dsDNA sensing is largely responsible for signaling to induce IFN-β mRNA. Knockdown of adaptor proteins STING and MAVS resulted in a similar mean reduction in IFN-β mRNA induction (55 to 60%), but only the effect of STING depletion reached significance (P = 0.042). Accumulation of the ISG-encoded protein IFIT2 largely recapitulated the effects of each sensor/adaptor knockdown on IFN-β mRNA. While IFIT2 was induced by ΔVHS infection in control siRNA-transfected cells, it was barely detectable in cells in which cGAS was depleted and strongly reduced in cells in which IFI16 or STING was depleted (Fig. 4B). Immunoblotting for ISG-encoded proteins MDA5 and RIG-I reproduced the effects of cGAS, IFI16, and STING knockdown on IFIT2. In contrast, induction of IFIT2 protein by ΔVHS was only slightly reduced upon knockdown of MDA5 or RIG-I, with knockdown of MAVS depressing induction more obviously but less than knockdown of the DNA-sensing apparatus, consistent with IFN-β expression data. Knockdown of MDA5, RIG-I, MAVS, and STING was confirmed by immunoblotting of the same samples (Fig. 4B). Knockdown of IFI16 was confirmed in uninfected cells since it is degraded during HSV-1 infection (52) (Fig. 4C), and cGAS knockdown was confirmed by qRT-PCR due to the lack of reliable antisera (Fig. 4D). Since these data suggest that DNA sensing is driving the IFN response to ΔVHS and IFN is reported to restrict ΔVHS replication (28), we next tested whether suppression of either DNA or RNA sensing pathways by knockdown of STING or MAVS, respectively, affected ΔVHS in a multicycle growth experiment. NHDFs transfected with a nonsilencing control or gene-specific siRNAs were infected with ΔVHS at a multiplicity of infection (MOI) of 0.01. After 48 h, cultures were collected, and infectious virus particles were quantified on Vero cells. Immunoblotting verified the efficient depletion of each protein (Fig. 4E). While depletion of STING significantly increased ΔVHS titers 30-fold (P = 0.005), depletion of MAVS had no significant effect (Fig. 4F). This indicates that STING signaling is restricting ΔVHS growth and supports the importance of STING for IFN-β induction shown in Fig. 4A. Taken together with the early timing of the onset of IFN-β mRNA expression (Fig. 2) and its independence from viral DNA synthesis and late gene expression (Fig. 3), these results demonstrate that the induction of IFN-β mRNA and its restriction of ΔVHS replication largely results from DNA sensing, with RNA sensing playing a more minor role.
FIG 4.

IFN-β and ISG induction is dependent on DNA sensing. NHDFs were transfected with siRNAs targeting dsRNA- and DNA-sensing pathway components or a nonsilencing control siRNA. At 3 days posttransfection, the cells were infected with ΔVHS at an MOI of 5. (A) Total RNA was isolated at 6 hpi and subject to qRT-PCR for IFN-β and normalized to 18S. The means ± the SEM of three independent experiments is shown. *, P < 0.05; **, P < 0.01 (Student t test compared to control). ns, nonsignificance. (B) ISG protein accumulation (IFIT2, MDA5, and RIG-I) and siRNA-mediated knockdown efficiency were analyzed by immunoblotting whole-cell lysates collected at 12 hpi. (C) Knockdown of IFI16 was verified by immunoblotting of uninfected cells 3 days after siRNA transfection. (D) Knockdown of cGAS was verified by qRT-PCR in uninfected cells 3 days after siRNA transfection. Means ± the SEM of three independent experiments are plotted. (E and F) NHDFs were transfected with siRNAs targeting STING, MAVS, or a nonsilencing control siRNA. At 3 days posttransfection, the cells were harvested and the siRNA-mediated knockdown efficiency was analyzed by immunoblotting whole-cell lysates (E) or cells infected with ΔVHS at an MOI of 0.001 (F). At 48 hpi, cultures were collected, and infectious virus was quantified by titering on Vero cells. Means ± the SEM of three independent experiments are plotted. **, P < 0.01 (Student t test compared to control).
Stress granules potentiate PKR activation.
We next sought to determine whether formation of SGs regulates PKR activation during ΔVHS infection. PKR-mediated phosphorylation of eIF2α results in translational inhibition, and thus SG formation, by increasing the affinity of eIF2α for its guanine exchange factor eIF2B, thereby sequestering the limited cellular pool of eIF2B (53). A pharmacological inhibitor of the integrated stress response, ISRIB, that acts to prevent inhibition of eIF2B and maintain translation initiation despite eIF2α phosphorylation was used to inhibit SG assembly without directly disrupting upstream signaling (54, 55). To verify ISRIB effectively suppressed SG formation in NHDFs, cells were treated with sodium arsenite in the presence or absence of 200 nM ISRIB for 30 min (Fig. 5A). Large G3BP1-stained SGs were detected by immunofluorescence in the absence of ISRIB. In contrast, G3BP1 remained diffusely cytoplasmic when cells were treated with both sodium arsenite and ISRIB, indicating the drug effectively blocks SG formation at this concentration in NHDFs. To test whether ISRIB is able to prevent ΔVHS-induced SGs, infected NHDFs were treated with ISRIB immediately following withdrawal of the infection inoculum. At 15 hpi, the cells were fixed, and SG formation was examined by staining for G3BP1 (Fig. 5B). ISRIB treatment effectively inhibited SG formation; G3BP1 remained diffusely distributed in the cytoplasm following ISRIB treatment, with no cells detected containing the multiple G3BP1-stained puncta observed in ΔVHS-infected, untreated cells. Next, we analyzed the activity of PKR in cells infected with WT or ΔVHS HSV-1 with or without ISRIB. Immunoblotting revealed that PKR autophosphorylation in ΔVHS-infected cells was much reduced by ISRIB (Fig. 5C), supporting the hypothesis that SG formation promotes PKR activation during infection. Phosphorylated eIF2α levels were also reduced by ISRIB treatment (Fig. 5C). This presumably reflects impaired PKR activation rather than a direct effect of ISRIB since eIF2α phosphorylation induced in response to ER stress by PERK is unaffected by ISRIB treatment (55). Given the significant reduction in activated PKR in response to ISRIB treatment, we verified the drug did not affect viral protein accumulation. The levels of ICP0 were slightly elevated in ΔVHS compared to WT HSV-1-infected cells, consistent with the reported extended persistence of early viral mRNAs (28), but unaffected by ISRIB in either case (Fig. 5C), indicating that ISRIB does not prevent viral entry or immediate early (IE) gene expression. The levels of the ISG-encoded protein MDA5 remained relatively unchanged by ISRIB treatment in cells infected either with WT or ΔVHS, in further support of our conclusion that SGs are dispensable for IFN signaling in HSV-1 infection.
FIG 5.
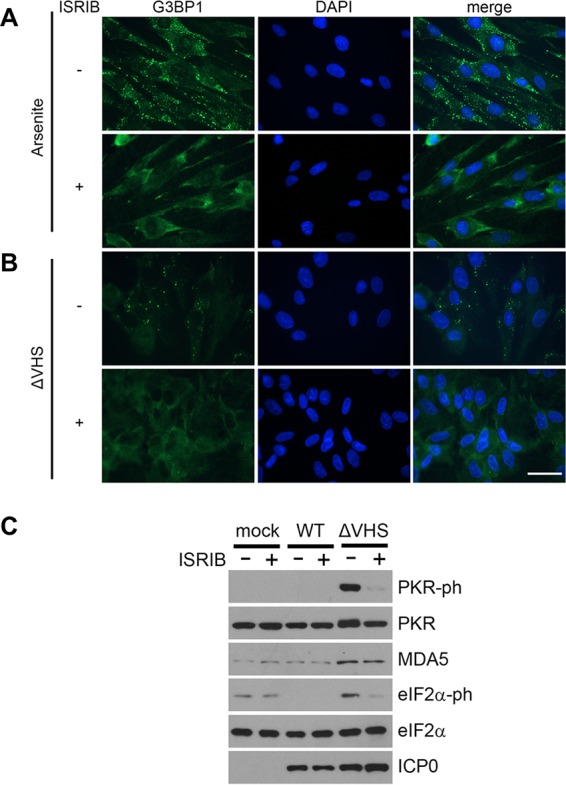
SG inhibitor ISRIB prevents PKR activation in ΔVHS-infected cells. (A) NHDFs were treated with 0.5 mM sodium arsenite and 200 nM ISRIB (+) or vehicle control (−) for 30 min before immunofluorescence staining for the SG marker G3BP1 (green). (B and C) NHDFs were infected with WT HSV-1 or ΔVHS at an MOI of 5, and infection was allowed to proceed in the presence of 200 nM ISRIB (+) or vehicle control (−) for 15 h. (B) SG induction was examined in ΔVHS cells by immunofluorescence staining for G3BP1 (green) with nuclei stained by DAPI (blue). Scale bar, 50 μm. (C) PKR phosphorylation, ISG accumulation (MDA5), viral protein accumulation (ICP0), and phosphorylated and total eIF2α were analyzed by immunoblotting whole-cell lysates.
To verify whether the inhibition of PKR activation in response to ISRIB treatment reflected SG disruption rather than a nonspecific effect of the drug on PKR, we sought to disrupt SG formation by an additional method. SG component proteins G3BP1 and TIA1 are found in ΔVHS-induced SGs (Fig. 1) (23) and are essential for SG assembly under a variety of stress conditions (56, 57). To test whether depletion of these SG scaffold proteins would affect PKR activation, we first examined whether they were required for the SG formation during ΔVHS infection. NHDFs transfected with a nonsilencing control or gene-specific siRNAs and infected with ΔVHS were fixed at 18 hpi and stained for an alternative SG marker, HuR. Quantification of cells containing HuR-stained SGs showed that 62% of control siRNA-transfected cells contained SGs (Fig. 6A) in keeping with previous data derived from G3BP1 staining (Fig. 2C). Knockdown of either G3BP1 or TIA1 significantly reduced the number of SG-containing cells, to 21 and 25%, respectively, indicating that targeting these proteins effectively interferes with SG assembly in ΔVHS-infected cells. To test whether PKR is affected by knockdown of G3BP1 or TIA1, NHDFs were transfected with two independent siRNAs targeting each gene or a control siRNA and subsequently infected with WT or ΔVHS HSV-1. Analysis of total protein collected at 18 hpi confirmed each siRNA effectively depleted its target (Fig. 6B and C). Remarkably, a substantial reduction in phosphorylated PKR was evident in ΔVHS-infected cells in which either G3BP1 (Fig. 6B) or TIA-1 (Fig. 6C) was depleted by either gene-specific siRNA. The effect of G3BP1 depletion appeared to follow knockdown efficiency since siRNA#2 elicited a greater reduction in both G3BP1 protein levels and PKR phosphorylation (Fig. 6B), indicating the effect is specific. This demonstrates that full activation of PKR is dependent upon SG core components and correlates with SG assembly. Together with the effect of ISRIB (Fig. 5), these experiments strongly argue that SG formation is important for the activation of PKR in ΔVHS infection.
FIG 6.
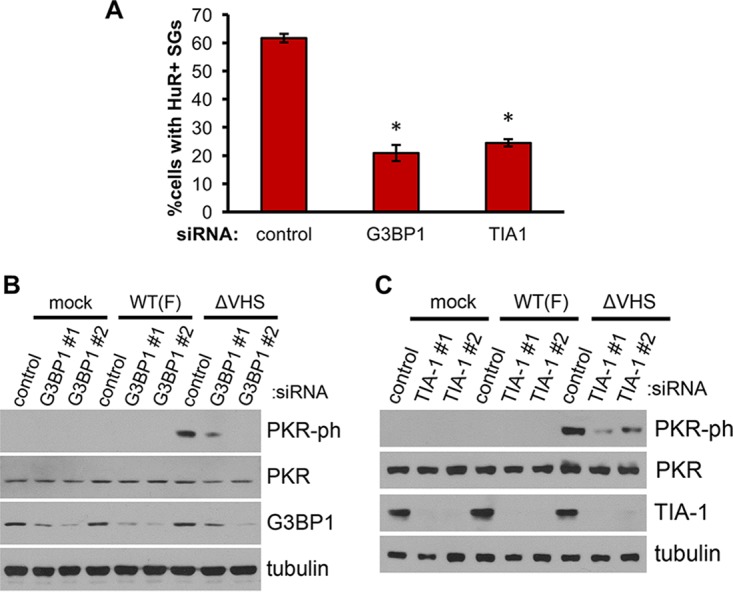
SG assembly is required for full PKR activation in ΔVHS-infected cells. NHDFs were transfected with siRNAs targeting SG scaffold proteins G3BP1 or TIA1 or a nonsilencing control siRNA. At 3 days posttransfection, the cells were infected with WT (F-strain) HSV-1 or ΔVHS at an MOI of 5, and infection was allowed to proceed for 18 h. (A) SG induction was quantified in cells transfected with control and G3BP1 (#1) and TIA1 (#1) targeting siRNAs by counting the number of cells containing two or more HuR-stained SGs. More than 100 cells were counted for each condition, and the means ± the SEM of three independent experiments are shown. *, P < 0.05 (Student t test compared to control). (B and C) PKR phosphorylation and knockdown efficiency were analyzed by immunoblotting of whole-cell lysates.
DISCUSSION
SGs are dynamic foci whose assembly in response to infection is subverted by many viruses. Though multiple antiviral effectors are recruited to these cytoplasmic foci, their functional role is contentious. Do viral functions that suppress SG formation represent the dismantling of important innate immune sensing and effector hubs? Or is the prevention of SG assembly merely a consequence of virally orchestrated maintenance of mRNA translation? Here, we report results that support a role for SGs in the activation of the eIF2α kinase PKR. Our data suggest that the ability of the HSV-1 endoribonuclease VHS to disrupt SG formation supports its inhibition of PKR. We fail, however, to find evidence that the formation of SGs supports IFN induction during HSV-1 infection and conclude that the suppression of IFN-β by VHS is separable from its ability to inhibit PKR activation and SG formation. Our results reveal that dsRNA, which is detected late in infection in the absence of VHS and appears to be responsible for PKR activation, is not required for IFN-β induction. Thus, the ability of VHS to control IFN is unrelated to its suppression of this dsRNA moiety.
Initial findings correlating SG formation and PKR activation were largely confined to artificial induction of SGs by G3BP1 overexpression. We directly establish that impairing SG formation in the context of a DNA virus infection can limit PKR activation showing that SGs are important modulators of an antiviral effector. Since SGs formed in ΔVHS HSV-1 (and many other) infections are PKR dependent (25), the formation of granules and further activation of PKR potentially serves as a feed forward mechanism to amplify protein synthesis inhibition. Consistent with this possibility, we found that a proportion of dsRNA in infected cells localized to SGs. Increasing the local concentration of dsRNA at SGs could therefore explain how they facilitate further activation of PKR. While not all SGs we observed colocalized with dsRNA, PKR activation was substantially impaired by inhibiting SG formation either via RNAi-mediated knockdown of a SG scaffolding protein or through using ISRIB. Activated, phosphorylated PKR is also reportedly absent from SGs induced by ectopic G3BP1 expression, but the capacity of G3BP1 to activate PKR requires PKR recruitment to granules (14, 58). Thus, it has been proposed that inactive PKR is recruited to granules, whereupon it is activated and subsequently released (14, 58). This could explain how the accumulation of dsRNA at only a subset of granules could affect a majority of PKR molecules in the cell.
The extent to which SGs broadly potentiate PKR activation in virus-infected cells remains to be investigated, though it is likely to depend on the localization and nature of the PKR ligand present. In contrast to our results, PKR activation following infection with Newcastle disease virus (NDV) is not detectably affected by disrupting SG formation via G3BP1 and G3BP2 depletion (11). However, though dsRNA was detected by immunofluorescence in NDV-infected cells, it was not localized to SGs (11). When SGs are assembled in uninfected cells treated with eIF4A chemical inhibitors, localization of PKR at SGs and eIF2α phosphorylation were not detected, and PKR activation was not reported (13, 59). Thus, mere aggregation of mRNPs to granules is insufficient to activate PKR. Whether cellular RNA is also involved in the activation of PKR when G3BP1 is overexpressed is unclear, though possible since transcriptional changes occur in G3BP1-overexpressing cells (14) and activation of PKR by self-RNAs has been described (60–62).
Surprisingly, while the PKR PAMP-responsive antiviral response pathway was regulated by SG assembly, IFN-β induction by the ΔVHS HSV-1 virus was unaffected by blocking SG formation using an inhibitor of viral DNA synthesis, PAA. In fact, accumulation of the PAMP dsRNA was blocked by PAA, suggesting that dsRNA formed in ΔVHS infection contains one or more true late viral mRNA components. This is consistent with a previous report detecting elevated levels of viral transcripts in ΔVHS compared to WT infections (28) and suggests that elevated levels of early transcripts are insufficient for formation of detectable dsRNA. It seems unlikely that late mRNAs alone are sufficient for dsRNA formation since late mRNAs were only increased to levels ∼2-fold higher ΔVHS over wild-type infections (28). More likely is a scenario where the combination of late gene mRNAs with partially complementary persistent early viral mRNAs and/or induced host mRNAs creates dsRNA; isolation and profiling of dsRNA would resolve this question.
Why precisely the dsRNA which accumulates during ΔVHS infection activates PKR but does not significantly contribute to IFN-β mRNA induction is unclear. MDA5 and RIG-I are not degraded during infection; in fact, as ISGs they are upregulated, and yet it is DNA sensing pathways that are largely responsible for signaling to induce IFN-β. Since dsRNA formation is dependent on late gene expression, potentially HSV-1 gene products able to inhibit MDA5/RIG-I signaling have accumulated to high enough levels by this point in infection to be effective and/or they are more effective than those that are reported to counter DNA-sensing components. Accumulation of the late-expressed, dsRNA-binding protein US11 is reduced in the ΔVHS mutant (24); however, even at this lower level it may provide some inhibition of RIG-I and MDA5 (63). The tegument protein UL37 deamidates RIG-I interfering with its RNA-sensing ability (64), and another tegument protein UL36 is able to deubiquitinate TRAF3, preventing MAVS-mediated activation of TBK1 and subsequent activation of IRF3 and IFN-β production (65). Together, these viral proteins likely form a significant impediment to dsRNA/MAVS-based IFN induction.
Our finding that the cGAS/IFI16/STING axis regulates IFN-β production during ΔVHS infection is consistent with a recent report showing partial rescue of ΔVHS replication with cGAS depletion (38). How IFN-β induction downstream of DNA sensing is enhanced in the absence of VHS, however, is unexplained. cGAS is reportedly activated in HSV-1 infection by mitochondrial DNA (mtDNA) following mitochondrial disruption by UL12.5 (66). VHS might regulate expression or activity of UL12.5, affecting the degree of mtDNA release and hence cGAS activation. VHS may also be required for normal expression of viral proteins that counter STING-mediated induction of IFN-β. Expression of ICP0, however, which has been linked to HSV-1 genome detection (52, 67) and reportedly limits NF-κB activation and IFN-β induction through additional mechanisms (68, 69), is not reduced in cells infected with ΔVHS (Fig. 5C). One candidate viral protein whose insufficiency might increase IFN-β induction downstream of DNA sensing is VP11/12 (UL46), a late-expressed tegument protein that reportedly binds STING and TBK1, inhibiting their ability to process cGAS-dependent signaling (46). An alternate, simpler explanation is that DNA is sensed and that IFN-β transcription activated equivalently in WT and ΔVHS infection, but in WT infection IFN-β mRNA is rapidly degraded by VHS.
Type I IFN induction has been shown to benefit from SG formation during infection with only a limited selection of viruses (7, 11, 12). However, in each study where this was reported SG disruption was achieved by RNAi-mediated knockdown of G3BP1 or PKR, proteins that may have independent roles in IFN signaling. G3BP1 overexpression was reported to activate the NF-κB pathway, which is required for IFN-β induction (14), and PKR was shown to influence MAVS signaling (70) and has also been linked to NF-κB signaling (71). Since pharmacological manipulation of SG induction has been mooted as a potential antiviral therapy (3, 72), it will be important to know how widely across viral taxa SGs regulate signaling to type I IFN and indeed whether this activity holds up to more stringent testing, using ISRIB, cells expressing nonphosphorylatable eIF2α, or targeting essential SG components not linked to interferon activation.
MATERIALS AND METHODS
Antibodies and chemicals.
Antibodies were obtained commercially as follows: α tubulin (6074; Sigma), G3BP1 (13057-2; Proteintech), TIA-1 (sc-1751; SantaCruz), Hur (sc-5261; Santa Cruz), PKR (18244-1; Proteintech), phospho-PKR T446 (32036; Abcam), TBK1 (3504; Cell Signaling), TBK1 (5483; Cell Signaling), GAPDH (2118; Cell Signaling), MDA5 (21775-1-AP; Proteintech), RIG-I (20566; Proteintech), MAVS (14341-1-AP; Proteintech), STING (19851-1-AP; Proteintech), IFIT2 (12604-1-AP; Proteintech), dsRNA (J2; SciCons, Hungary), phospho-eIF2α S51 (3398; Cell Signaling), eIF2α (5324; Cell Signaling), ICP0 (ab6513; Abcam), ICP5 (HA-018-A1242062; Eastcoast Bio), gB (HA056-100; Virusys), abd ICP4 (ab6514; Abcam). PAA and ISRIB were obtained from Sigma.
Cells and viruses.
Primary normal human dermal fibroblasts (NHDFs; Clonetics) were propagated in Dulbecco modified Eagle medium (DMEM) supplemented with 5% fetal bovine serum (FBS) plus 1% (vol/vol) penicillin-streptomycin, routinely subcultured at 1:3, and maintained until passage 24. Viruses were propagated and titered in Vero cells (American Type Culture Collection), which were grown in DMEM supplemented with 5% calf serum. WT and ΔVHS (R2621) F-strain HSV-1 viruses were described previously (73) and kindly supplied by B. Roizman (University of Chicago, Chicago, IL).
siRNA transfections and infections.
One day prior to siRNA transfection NHDF cells were seeded in a 12-well dish at 8 × 104 cells per well. The cells were transfected with siRNAs (see below) at a final concentration of 20 nM each using Lipofectamine RNAiMax (Life Technologies) at 1.25 μl/ml according to the manufacturer's instructions. G3BP1 siRNA-1 was custom generated by GeneLink as a duplex with the sequence 5′-CCAAGAUGAGGUCUUUGGUGGGUUU-3′ (74). All other siRNAs were commercially sourced as follows: AllStars negative control (SI03650318; Qiagen), cGAS (SASI_Hs01_00197467; Sigma), IFI16 (SASI_Hs01_00133540; Sigma), STING/TMEM173 (SASI_Hs02_00371843; Sigma), MDA5 (SASI_Hs01_00171929; Sigma), RIG-I (SASI_Hs01_00047980; Sigma), MAVS (SASI_Hs01_00128708; Sigma), G3BP1 siRNA −2 (SASI_Hs01_00045799; Sigma), TIA1 siRNA −1 (SASI_Hs01_00070183; Sigma), and TIA1 siRNA −2 (SASI_Hs03_00070184; Sigma). Cells were infected 3 days posttransfection after a medium change. Mock infections were performed with the same medium used to dilute the virus stocks. After multicycle growth experiments, NHDF cultures were frozen at −80°C, and after three freeze-thaw cycles infectious virus titers were quantified by plaque assay in Vero cells.
Immunofluorescence.
Cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized 0.5% Triton X-100 in PBS for 10 min. They were blocked in 4% FBS in PBS for 1 h before primary incubation in blocking solution for 2 h with the antibodies listed below and secondary detection by anti-mouse Alexa 555 (A21424; Life Technologies), anti-mouse Alexa 488 (A11029; Life Technologies), and anti-rabbit Alexa 555 (A21429; Life Technologies). Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). Cells were visualized using a Leica DM5000 microscope with a 63× objective using Leica Imaging LAS V4.3 software. J2 anti-dsRNA was used at 1:200, anti-HuR was used at 1:50, anti-ICP4 was used at 1:100, and anti-G3BP1 was used at 1:200.
qRT-PCR.
RNA was isolated using TRIzol. cDNA was synthesized using qScript (Quanta) and 500 ng of total RNA. qPCRs were conducted using Bio-Rad SsoAdvanced SYBR green supermix and a Bio-Rad CFX96 real-time system. For each biological replicate, technical duplicates were conducted. mRNA levels relative to 18S were calculated using the ΔΔCT method, and statistical analyses were performed using Microsoft Excel. The primers used were as follows: 18S (forward, AGGAATTGACGGAAGGGCACCA; reverse, TTATCGGAATTAACCAGACAAATCG), cGAS (forward, ATCTGTGGATATAACCCTGG; reverse, TCTTGGAAACCATTTCCTTC), IFN-β (forward, GAAAGAAGATTTCACCAGGG; reverse, CCTTCAGGTAATGCAGAATC), IFIT2 (forward, ACCATGAGTGAGAACAATAAG; reverse, TTAGATAGGCCAGTAGGTTG), and MDA5 (forward, GATTAAGTGGTGATACCCAAC; reverse, GTCTGACAATTGAACACCAG).
ACKNOWLEDGMENTS
We thank members of the Mohr lab and Angus Wilson for helpful discussions.
This study was supported by grants from the National Institutes of Health (NIH). H.M.B. was supported by NIH grant R21AI126102; I.M. was supported by NIH grants GM056927, AI073898, and R21AI126102. The funder had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem Sci 33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Buchan JR. 2014. mRNP granules. Assembly, function, and connections with disease. RNA Biol 11:1019–1030. doi: 10.4161/15476286.2014.972208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCormick C, Khaperskyy DA. 2017. Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol 17:647–660. doi: 10.1038/nri.2017.63. [DOI] [PubMed] [Google Scholar]
- 4.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 6.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 7.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol 87:6314–6325. doi: 10.1128/JVI.03213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixit E, Kagan JC. 2013. Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol 117:99–125. doi: 10.1016/B978-0-12-410524-9.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh SW, Onomoto K, Wakimoto M, Onoguchi K, Ishidate F, Fujiwara T, Yoneyama M, Kato H, Fujita T. 2016. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PLoS Pathog 12:e1005444. doi: 10.1371/journal.ppat.1005444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang P, Li Y, Xia J, He J, Pu J, Xie J, Wu S, Feng L, Huang X, Zhang P. 2014. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J Cell Sci 127:2471–2482. doi: 10.1242/jcs.139626. [DOI] [PubMed] [Google Scholar]
- 14.Reineke LC, Lloyd RE. 2015. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reineke LC, Dougherty JD, Pierre P, Lloyd RE. 2012. Large G3BP-induced granules trigger eIF2α phosphorylation. Mol Biol Cell 23:3499–3510. doi: 10.1091/mbc.e12-05-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li XL, Ezelle HJ, Hsi TY, Hassel BA. 2011. A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip Rev RNA 2:58–78. doi: 10.1002/wrna.32. [DOI] [PubMed] [Google Scholar]
- 17.Poblete-Duran N, Prades-Perez Y, Vera-Otarola J, Soto-Rifo R, Valiente-Echeverria F. 2016. Who regulates whom? An overview of RNA granules and viral infections. Viruses 8:E180. doi: 10.3390/v8070180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khaperskyy DA, Hatchette TF, McCormick C. 2012. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J 26:1629–1639. doi: 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- 19.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 86:10873–10879. doi: 10.1128/JVI.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2, and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus noncoding RNA. PLoS Pathog 10:e1004242. doi: 10.1371/journal.ppat.1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katsafanas GC, Moss B. 2004. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem 279:52210–52217. doi: 10.1074/jbc.M411033200. [DOI] [PubMed] [Google Scholar]
- 23.Esclatine A, Taddeo B, Roizman B. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J Virol 78:8582–8592. doi: 10.1128/JVI.78.16.8582-8592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 Vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J Virol 85:5363–5373. doi: 10.1128/JVI.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dauber B, Poon D, Dos Santos T, Duguay BA, Mehta N, Saffran HA, Smiley JR. 2016. The herpes simplex virus virion host shutoff protein enhances translation of viral true late mRNAs independently of suppressing protein kinase R and stress granule formation. J Virol 90:6049–6057. doi: 10.1128/JVI.03180-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. 2013. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J Virol 87:3271–3276. doi: 10.1128/JVI.03049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finnen RL, Hay TJ, Dauber B, Smiley JR, Banfield BW. 2014. The herpes simplex virus 2 virion-associated ribonuclease VHS interferes with stress granule formation. J Virol 88:12727–12739. doi: 10.1128/JVI.01554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasieka TJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA. 2008. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82:5527–5535. doi: 10.1128/JVI.02047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strelow LI, Leib DA. 1995. Role of the virion host shutoff (VHS) of herpes simplex virus type 1 in latency and pathogenesis. J Virol 69:6779–6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80:9341–9345. doi: 10.1128/JVI.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiflett LA, Read GS. 2013. mRNA decay during herpes simplex virus (HSV) infections: mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J Virol 87:94–109. doi: 10.1128/JVI.01557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng P, Everly DN Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J Virol 75:10272–10280. doi: 10.1128/JVI.75.21.10272-10280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng P, Everly DN Jr, Read GS. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J Virol 79:9651–9664. doi: 10.1128/JVI.79.15.9651-9664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taddeo B, Zhang W, Roizman B. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc Natl Acad Sci U S A 103:2827–2832. doi: 10.1073/pnas.0510712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulvey M, Poppers J, Sternberg D, Mohr I. 2003. Regulation of eIF2α phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J Virol 77:10917–10928. doi: 10.1128/JVI.77.20.10917-10928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dauber B, Saffran HA, Smiley JR. 2014. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J Virol 88:9624–9632. doi: 10.1128/JVI.01350-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su C, Zheng C. 2017. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J Virol 91:e02414-. doi: 10.1128/JVI.02414-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diner BA, Lum KK, Toettcher JE, Cristea IM. 2016. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio 7:e01553-. doi: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, Mogensen TH, Meyer M, Vaegter C, Nyengaard JR, Fitzgerald KA, Paludan SR. 2016. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun 7:13348. doi: 10.1038/ncomms13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA. 2014. Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289:23568–23581. doi: 10.1074/jbc.M114.554147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Almine JF, O'Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, Unterholzner L. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su C, Zhan G, Zheng C. 2016. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J 13:38. doi: 10.1186/s12985-016-0495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deschamps T, Kalamvoki M. 2017. Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J Virol 91:e00535-. doi: 10.1128/JVI.00535-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess HM, Mohr I. 2015. Cellular 5′-3′ mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe 17:332–344. doi: 10.1016/j.chom.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, Nellen W. 2000. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 6:563–570. doi: 10.1017/S1355838200992318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manche L, Green SR, Schmedt C, Mathews MB. 1992. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol 12:5238–5248. doi: 10.1128/MCB.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, Lange G, Caroll J, Reitz B, Donnelly A, Boddupalli H, Combs RG, Kretzmer K, Tripp CS. 2002. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem 277:13840–13847. doi: 10.1074/jbc.M110474200. [DOI] [PubMed] [Google Scholar]
- 51.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 52.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pavitt GD. 2005. eIF2B, a mediator of general and gene-specific translational control. Biochem Soc Trans 33:1487–1492. doi: 10.1042/BST20051487. [DOI] [PubMed] [Google Scholar]
- 54.Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, Ron D. 2015. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348:1027–1030. doi: 10.1126/science.aaa6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sidrauski C, McGeachy AM, Ingolia NT, Walter P. 2015. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015:4. doi: 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol 160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 15:5383–5398. doi: 10.1091/mbc.e04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reineke LC, Kedersha N, Langereis MA, van Kuppeveld FJ, Lloyd RE. 2015. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 6:e02486. doi: 10.1128/mBio.02486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dang Y, Kedersha N, Low WK, Romo D, Gorospe M, Kaufman R, Anderson P, Liu JO. 2006. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J Biol Chem 281:32870–32878. doi: 10.1074/jbc.M606149200. [DOI] [PubMed] [Google Scholar]
- 60.Youssef OA, Safran SA, Nakamura T, Nix DA, Hotamisligil GS, Bass BL. 2015. Potential role for snoRNAs in PKR activation during metabolic stress. Proc Natl Acad Sci U S A 112:5023–5028. doi: 10.1073/pnas.1424044112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. 2002. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell 108:221–232. doi: 10.1016/S0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 62.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, Rice CM. 2018. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell doi: 10.1016/j.cell.2017.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao J, Zeng Y, Xu S, Chen J, Shen G, Yu C, Knipe D, Yuan W, Peng J, Xu W, Zhang C, Xia Z, Feng P. 2016. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 20:770–784. doi: 10.1016/j.chom.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S, Wang K, Li J, Zheng C. 2013. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J Virol 87:11851–11860. doi: 10.1128/JVI.01211-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. 2015. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson KE, Chikoti L, Chandran B. 2013. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87:5005–5018. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taylor KE, Chew MV, Ashkar AA, Mossman KL. 2014. Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING finger are required to block interferon-stimulated gene production but not to promote viral replication. J Virol 88:8091–8101. doi: 10.1128/JVI.00944-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang J, Wang K, Wang S, Zheng C. 2013. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. J Virol 87:12935–12948. doi: 10.1128/JVI.01952-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marie IJ, Levy DE. 2016. PKR transduces MDA5-dependent signals for type I IFN induction. PLoS Pathog 12:e1005489. doi: 10.1371/journal.ppat.1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. 2000. NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB-inducing kinase and IκB kinase. Mol Cell Biol 20:1278–1290. doi: 10.1128/MCB.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rozelle DK, Filone CM, Kedersha N, Connor JH. 2014. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol Cell Biol 34:2003–2016. doi: 10.1128/MCB.01630-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poon AP, Roizman B. 1997. Differentiation of the shutoff of protein synthesis by virion host shutoff and mutant gamma(1)34.5 genes of herpes simplex virus 1. Virology 229:98–105. doi: 10.1006/viro.1996.8425. [DOI] [PubMed] [Google Scholar]
- 74.Winslow S, Leandersson K, Larsson C. 2013. Regulation of PMP22 mRNA by G3BP1 affects cell proliferation in breast cancer cells. Mol Cancer 12:156. doi: 10.1186/1476-4598-12-156. [DOI] [PMC free article] [PubMed] [Google Scholar]