

Microglia contribute to many aspects of health and disease. When activated, microglia can be beneficial by repairing damage in the central nervous system (CNS) or they can turn harmful by becoming neurotoxic. In prion and prionlike diseases, the involvement of microglia in disease is unclear. Previous studies suggest that microglia can either speed up or slow down disease. In this study, we infected mice with prions and depleted microglia from the brains of mice using PLX5622, an effective CSF-1R tyrosine kinase inhibitor. Microglia were markedly reduced in brains, and prion disease was accelerated, so that mice needed to be euthanized 20 to 33 days earlier than infected control mice due to advanced clinical disease. Similar results occurred when mice were treated with PLX5622 at 80 days after infection, which was just prior to the start of clinical signs. Thus, microglia are important for removing prions, and the disease is faster when microglia are depleted.
KEYWORDS: microglia, neuroinflammation, PLX5622, CSF-1R, scrapie, prion, PrPres, PrPSc
ABSTRACT
Microglial cells in the central nervous system play important roles in neurodevelopment and resistance to infection, yet microglia can become neurotoxic under some conditions. An early event during prion infection is the activation of microglia and astrocytes in the brain prior to damage or death of neurons. Previous prion disease studies using two different strategies to manipulate signaling through the microglial receptor CSF-1R reported contrary effects on survival from prion disease. However, in these studies, reductions of microglial numbers and function were variable, thus confounding interpretation of the results. In the present work, we used oral treatment with a potent inhibitor of CSF-1R, PLX5622, to eliminate 78 to 90% of microglia from cortex early during the course of prion infection. Oral drug treatment early after infection with the RML scrapie strain significantly accelerated vacuolation, astrogliosis, and deposition of disease-associated prion protein. Furthermore, drug-treated mice had advanced clinical disease requiring euthanasia 31 days earlier than untreated control mice. Similarly, PLX5622 treatment during the preclinical phase at 80 days postinfection with RML scrapie also accelerated disease and resulted in euthanasia of mice 33 days earlier than infected controls. PLX5622 also accelerated clinical disease after infection with scrapie strains ME7 and 22L. Thus, microglia are critical in host defense during prion disease. The early accumulation of PrPSc in the absence of microglia suggested that microglia may function by clearing PrPSc, resulting in longer survival.
IMPORTANCE Microglia contribute to many aspects of health and disease. When activated, microglia can be beneficial by repairing damage in the central nervous system (CNS) or they can turn harmful by becoming neurotoxic. In prion and prionlike diseases, the involvement of microglia in disease is unclear. Previous studies suggest that microglia can either speed up or slow down disease. In this study, we infected mice with prions and depleted microglia from the brains of mice using PLX5622, an effective CSF-1R tyrosine kinase inhibitor. Microglia were markedly reduced in brains, and prion disease was accelerated, so that mice needed to be euthanized 20 to 33 days earlier than infected control mice due to advanced clinical disease. Similar results occurred when mice were treated with PLX5622 at 80 days after infection, which was just prior to the start of clinical signs. Thus, microglia are important for removing prions, and the disease is faster when microglia are depleted.
INTRODUCTION
Prion diseases are fatal transmissible diseases caused by a proteinaceous infectious agent that can infect the lymphatic and nervous systems (1). Prion diseases include Creutzfeldt-Jakob disease in humans, scrapie in sheep, chronic wasting disease in cervids, and bovine spongiform encephalopathy in cattle (2). The clinical features can vary, but common hallmarks in the central nervous system (CNS) are deposition of abnormally folded protease-resistant prion protein (PrPres or PrPSc), astrogliosis, microgliosis, neuroinflammation, and neurodegeneration (3–7). In experimental models of prion infection, activation of astroglia and microglia in the brain occurs before detectable neurodegeneration (3, 8). Thus, gliosis may be involved in the early response to the infectious agent or initial neuronal damage.
Microglia are important first responders to foreign agents and damaged cells in the CNS, but also have many essential functions in the healthy CNS, from neurodevelopment to maintaining homeostasis through surveillance and phagocytosis within the brain parenchyma (9–11). Though microglia are important in defense and maintenance of the CNS, there is evidence that their activation can lead to a dysfunctional microglial phenotype that can contribute to or exacerbate many neurological diseases, including Alzheimer's disease (12, 13), multiple sclerosis (14), Parkinson's disease (15), and HIV-associated dementia (16).
Microglia are derived early during embryogenesis from erythro-myeloid progenitors in the yolk sac that migrate to colonize the CNS rudiment (17, 18). Once in the CNS, these cells become self-renewing (19), but they are reliant for survival on continual signaling through colony-stimulating factor 1 receptor (CSF-1R), a tyrosine kinase receptor (20–22). CSF-1R has two known ligands, CSF-1 and interleukin-34 (IL-34), which are produced and secreted predominately by astrocytes and neurons in the CNS (23). In mice deficient in IL-34, microglia were reduced by 17 to 64% of normal in 5 brain regions (24). Similarly, in mutant op/op mice, which are unable to produce CSF-1, brain microglia were reduced by 34 to 47% of normal (25). Thus, manipulation of CSF-1R function might be an effective way to alter microglial function during scrapie infection.
In prion disease, the role of microglia has been studied in vivo in two different systems altering CSF-1R signaling; however, these studies appeared to give contrary effects on prion disease tempo and survival. In the first model, using IL-34−/− mice, a 14- to 21-day decrease in survival time was noted, implying that microglia may be protective during prion disease. However, microglia were not decreased in these scrapie-infected mice as they had been in uninfected mice. Thus, the effect on survival time might not be due to microglia, but rather to another effect of IL-34 deficiency (26).
In the second model, blocking of microglial proliferation by oral treatment with a CSF-1R inhibitor, GW2580, resulted in a 26-day extension of survival time. This was associated with a 50% reduction in microglia and in alteration of expression of microglial genes possibly associated with neurotoxic functions (27). The authors suggested that GW2580 blocked not only microglial proliferation, but also production of a neurotoxic substance, which resulted in the increased survival observed. Thus, microglia may have two contradictory effects in prion disease, both revealed by experiments involving the CSF-R1 receptor.
In the present studies, we used a more potent CSF-R1 inhibitor, PLX5622 (Plexxicon, Berkeley, CA), capable of causing microglial apoptosis and long-term elimination in vivo. After oral administration, this drug can reduce mature microglia in the CNS by 90% in 7 days, resulting in nearly undetectable numbers in as little as 21 days (20, 21, 28, 29). The question posed in our study was whether elimination of microglia during scrapie infection in vivo would have a positive or negative effect on survival. In PLX5622-treated mice, a large decrease in microglia was observed, and this correlated with the early appearance of PrPSc in brain and a 31- to 33-day decrease in survival time following infection with RML scrapie. The results from experiments with three different scrapie strains were similar. Thus, microglia were indispensable during scrapie infection and were critical to host defense against the disease.
RESULTS
Long-term reduction in microglia with PLX5622.
In C57BL/10 mice, scrapie is a slow disease requiring 130 to 180 days to reach the clinical stage. Therefore, to determine whether PLX5622 therapy would be able to deplete microglia for this duration, we treated mice inoculated with normal brain homogenate (NBH) with PLX5622 for up to 181 days. Treated and age-matched untreated mice were compared to determine the drug-induced reduction in mature Iba1-positive microglia in the brain. At various times, similar regions of cortex and thalamus were sectioned, stained for Iba1, and observed by light microscopy (Fig. 1). Treatment with PLX5622 was extremely effective at reducing mature microglia in both regions of the brain at all times tested from 80 to 181 days, with losses ranging from 80% to 99% in the cortex (Fig. 1A) and 79% to 91% in the thalamus (Fig. 1D). Treatment with PLX5622 for up to 181 days did not affect neuronal densities in the hippocampus, cortex, or cerebellum when assessed by hematoxylin and eosin (H&E) staining (not shown). Furthermore, the numbers of glial fibrillary acidic protein (GFAP)-positive astrocytes were unaltered with long-term PLX5622 treatment when regions such as the cortex, thalamus, hypothalamus, or olfactory bulb were assessed by GFAP staining (not shown). Uninfected PLX5622-treated mice showed no signs of distress or weight loss and were not adversely affected by these long treatments. Our results were like those seen previously, where mice treated for up to 84 days with PLX5622 had reduced microglia with no observable astrogliosis (28). These initial studies indicated treatment with PLX5622 should allow adequate microglia ablation during the extended incubation times seen in scrapie prion infections.
FIG 1.

Long-term reduction of microglia with PLX5622 treatment. Mice injected with NBH were continually fed control chow (untreated, PLX5622 [−]) or chow supplemented with PLX5622 (+) for up to 181 days. Similar paraffin-embedded sections of cortex and thalamus were fixed and stained with antibody to Iba1. Iba1-positive cells were enumerated at 80, 98, 133, 161, and 181 days posttreatment and reported as the number of positive cells per square millimeter (A and D). The white columns represent the mean number of Iba1-positive cells in mice fed control chow (PLX5622 −), and the gray columns represent the mean numbers of Iba1-positive cells in mice fed PLX5622 chow (PLX5622 +). Each dot represents an individual mouse. Error bars represent 1 standard deviation. The percent reduction with treatment for each time point relative to control is given. Untreated and PLX5622-treated groups at each time were compared by unpaired t test: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. For a visual reference, representative images depicting approximately one-quarter of the counting area of the cerebral cortex (B and C) and thalamus (E and F) at 80 days are shown at the scale indicated in panel B.
Reduction of microglia 14 days after prion infection augments disease.
Using different experimental approaches during prion infection, microglia have been proposed to be either neuroprotective (26) or neurotoxic (27) in the brain, two diametrically opposed concepts. To better understand the influence that microglia have on prion pathogenesis, we infected mice intracranially (i.c.) with scrapie and allowed the mice to convalesce for 14 days. After this recovery period, mice were either untreated or treated with PLX5622 to eliminate microglia for the duration of the study. Three scrapie strains (RML, ME7, or 22L) were tested in independent experiments. A similar cohort of treated and untreated mice were injected i.c. with NBH as a control, which did not develop disease. The time to reach advanced clinical disease requiring euthanasia was significantly altered (P < 0.0001) in prion-infected mouse cohorts treated with PLX5622, and drug-treated mice with reduced microglia required euthanasia 20 to 31 days earlier than untreated infected mice (Fig. 2 and Table 1). Moreover, all PLX5622-treated mice regardless of scrapie strain displayed accelerated signs of scrapie infection and poor nesting behavior relative to scrapie-infected untreated mice (Fig. 3), indicating a more rapid advancement in disease with reduction of microglia in the brain.
FIG 2.
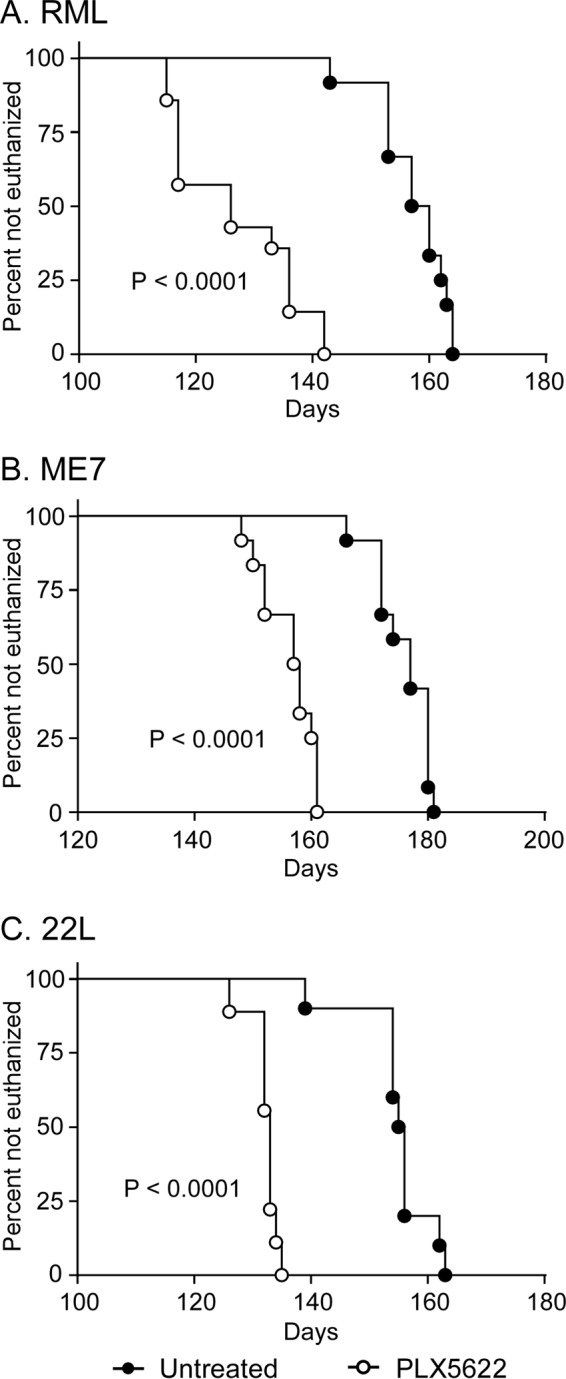
Survival of PLX5622-treated and untreated mice infected with scrapie strain RML, ME7, or 22L. Survival curves showing mice fed control chow (untreated, filled circles) or chow supplemented with PLX5622 (open circles). Mice were infected with scrapie strains RML (A), ME7 (B), or 22L (C) and monitored by observers blinded to the treatments for clinical signs of scrapie requiring euthanasia. Data are percentages of animals not euthanized versus days postinfection (days). The day each individual mouse was euthanized, the mean day of euthanasia for each mouse cohort, and the number of animals in each group are given in Table 1. Statistical analysis was performed using Mantel-Cox log rank analysis comparing PLX5622-treated to untreated animals.
TABLE 1.
Survival times of PLX5622-treated and untreated scrapie-infected C57BL/10 mice
| Treatment/strain (no. of mice in study group) | Survival times (dpi)a | Mean dpi ± SD |
|---|---|---|
| PLX5622/RML (14) | 115, 115, 117, 117, 117, 117, 126, 126, 133, 136, 136, 136, 142, 142 | 126.8 ± 10.5 |
| Untreated/RML (12) | 143, 153, 153, 153, 157, 157, 160, 160, 162, 163, 164, 164 | 157.4 ± 6.2 |
| PLX5622/ME7 (12) | 148, 150, 152, 152, 157, 157, 158, 158, 160, 161, 161, 161 | 156.3 ± 4.6 |
| Untreated/ME7 (12) | 166, 172, 172, 172, 174, 177, 177, 180, 180, 180, 180, 181 | 175.9 ± 4.7 |
| PLX5622/22L (9) | 126, 132, 132, 132, 133, 133, 133, 134, 135 | 132.2 ± 2.5 |
| Untreated/22L (10) | 139, 154, 154, 154, 155, 156, 156, 156, 162, 163 | 154.9 ± 13.0 |
Number of days postinfection when individual mice were euthanized due to the presence of advanced clinical signs.
FIG 3.

Nesting and scrapie severity scores of mice either treated or untreated with PLX5622 and infected with scrapie strain RML, ME7, or 22L. Mice infected with scrapie strain RML, ME7, or 22L were either treated with PLX5622 (open circles) or untreated (closed circles) and scored by observers blinded to the treatments for changes in nesting behavior and severity of clinical presentation (scrapie severity score) at various days postinfection (DPI). Nesting behavior was scored on a scale of 1 to 5, where 5 was a near-perfect nest and 1 was no nest. Severity of clinical presentation was scored from 0 to 3, where 0 represented no signs and 3 indicated signs requiring euthanasia (see Materials and Methods for details). All scores are representative of the average score of the study group at each day postinfection. To emphasize the differences, scrapie severity scoring is reported for each scrapie strain until the PLX5622-treatment group averaged a score of 3. The number of mice per cohort is in Table 1. Error bars represent the standard errors of the means. Multiple t test was performed comparing PLX5622-treated and untreated groups of mice at specific time points using GraphPad software. Statistical significance was determined using the Holm-Sidak method to correct for multiple comparisons, with alpha of 0.05. **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Histological examination of brain tissue from PLX5622-treated and untreated mice infected with RML scrapie indicated that mice with reduced numbers of microglia had greater PrPSc deposition and spongiform change at early time points than infected controls (Fig. 4). This was seen in some treated mice at 80 days postinfection (dpi) in the RML-infected cohort of mice, but the differences between the infected treated and untreated mice were consistent starting at 100 dpi (Fig. 4) and beyond.
FIG 4.

Representative neuropathology and immunohistochemical assessment of gliosis and PrP deposition in cerebral cortex brain sections from control and RML-infected mice either treated or untreated with PLX5622. Mice were inoculated with either normal brain homogenate (NBH) (A to D) or scrapie strain RML (E to P) and treated with PLX5622 (A to H) or untreated (I to P). Sections of cerebral cortex were probed with antibodies that recognize PrP (A, E, I, and M), GFAP (B, F, J, and N), and Iba1 (C, G, K, and O) or stained by H&E (D, H, L, and P). Representative images of the cerebral cortex are shown for all at the scale indicated in panel A. White arrowheads in panels E, I, and M indicate patches of PrPSc deposition. Black arrows in panels H and P indicate vacuoles present in the neuropil.
PLX5622-treated and untreated RML scrapie-infected mice were also compared for patterns of PrPSc deposition, aggregate morphology, and regional distribution, and no differences were observed in the brains when comparable stages of clinical disease were assessed with immunohistochemistry (Fig. 5). Similar results were seen when 22L- or ME7-infected mice either treated or untreated were compared histologically (not shown).
FIG 5.

PrPSc distribution in brains from mice with terminal RML scrapie in mice treated or untreated with PLX5622. PrP staining was performed using anti-PrP monoclonal antibody D13. (A and B) Representative images of brain from a scrapie-infected mouse treated with PLX5622 (A) and from a scrapie-infected untreated mouse euthanized at the same stage of clinical scrapie (B). The two mice have similar patterns of PrPSc distribution and aggregate morphology in the brain, demonstrated in the cerebral cortex (black arrows), hippocampus (H, upper dashed region), and thalamus (T, lower dashed region). Bar, 1 mm (this scale applies to both panels).
Additional assessment of disease-associated PrP accumulation was performed by immunoblotting. Densitometry of immunoblots of brain homogenates from RML-infected mice indicated PrPres accumulated to significantly higher levels in PLX5622-treated than in untreated mice at 80 dpi, 100 dpi, and the experimental endpoint (Fig. 6). These results suggested that reducing microglia produced faster onset of neuropathology and increased PrPres accumulation, which correlated with more-rapid clinical disease as documented above.
FIG 6.

Western blot and densitometry of PrPres in the brains of PLX5622-treated and untreated mice infected with scrapie. Mice infected with scrapie strain RML were treated with PLX5622 or untreated and euthanized at 80 dpi (A), at 100 dpi (B), or at clinical endpoint (terminal) (C). Brain homogenates were digested with proteinase K, and proteins were separated by SDS-PAGE and transferred to PVDF membranes. Blocked membranes were probed with anti-PrP (D13), and bands were visualized by chemiluminescence. (A and B) Approximately 5-s exposures to film using SuperSignal West Femto substrate; (C) 10-min exposure to film using ECL Western blotting substrate. The days postinfection of euthanasia of each mouse is given below the immunoblots in panel C. Densitometry on each lane was performed to assess differences between the groups, and plots of the adjusted volumes are shown to the right of the immunoblots. Statistical analysis was performed using a two-tailed t test comparing PLX5622-treated to untreated animals. P values are indicated.
Enumeration of Iba1-positive microglia in cortex and/or thalamus indicated PLX5622 treatment caused a decrease in microglia throughout disease (Fig. 7). However, microglial numbers gradually increased in treated infected mice as the disease progressed (Fig. 7 and 8). This was especially evident around areas of prominent vacuolation in infected PLX5622-treated mice, where microglia were seen to cluster (Fig. 8F).
FIG 7.

Iba1-positive cells in cortex and thalamus during prion disease with PLX5622 treatment beginning at 14 dpi. Mice infected with scrapie strain RML, ME7, or 22L were fed control chow (−, white columns) or chow supplemented with PLX5622 (+, gray columns). Mice were euthanized at 80 dpi, at 100 dpi, and at clinical endpoint (∼157 dpi for control or ∼127 dpi for PLX5622) for RML-infected mice; at 98 dpi, at 133 dpi, and at clinical endpoint (∼176 dpi for control or ∼156 dpi for PLX5622) for ME7-infected mice; and at 83 dpi and at clinical endpoint (∼155 dpi for control or ∼132 dpi for PLX5622) for 22L-infected mice. See Table 1 for all clinical endpoint ranges. Similar paraffin-embedded sections of cortex (A, C, and E) and thalamus (B, D, and F) were fixed and stained with antibody to Iba1, and positive cells were enumerated and reported as the number of positive cells per square millimeter. The columns are the means, and each dot in the groups represents an individual mouse. The percent reduction with treatment for each time point relative to control is given. Error bars represent 1 standard deviation. Control and PLX5622-treated groups at each time were compared by unpaired t test: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
FIG 8.

Representative neuropathology and immunohistochemical assessment of Iba1-positive cells in thalamus from control and RML-infected mice either treated or untreated with PLX5622. Mice were inoculated with either normal brain homogenate (NBH) (A to C) or scrapie strain RML (D to I) and treated with PLX5622 after 14 dpi (A to F) or untreated (G to I). Sections of thalamus were probed with antibodies that recognize Iba1. Representative images of thalamus are shown for all at the scale indicated in panel A.
Ablation of microglia by PLX5622 treatment alters proinflammatory gene expression.
Much of the inflammatory response associated with prion disease is proposed to be of microglial origin (30). Therefore, reduction in microglia using PLX5622 would be expected to reduce the expression of several proinflammatory genes during scrapie infection. We assessed the expression of several genes in the thalamus and whole brain that were previously identified as increased in the brain during scrapie at 80 dpi (5, 31). Our preliminary results from thalamus of scrapie-infected mice indicated that expression levels of several genes predominately expressed by microglia were reduced in PLX5622-treated mice relative to untreated mice (Table 2, bolded genes). A more robust analysis of transcripts confirmed many of these changes in brain, but for most genes, the difference in fold change was not as large as in thalamus. In addition, there were three genes tested (Cxcl5, Cxcl1, and Tnfsf11) that were increased in thalamus and whole brain of PLX5622-treated relative to untreated mice. Furthermore, there were several proinflammatory genes that are increased during scrapie infection at 80 dpi whose expression was unaffected by PLX5622 treatment, including Cxcl10, IL1b, and Ccl2 (Table 3, bolded genes), suggesting that their expression during prion disease was less dependent on the presence of microglia.
TABLE 2.
Effect of PLX5622 treatment on expression of proinflammatory genes at 80 days post-scrapie infectiona
| Gene | Fold change in gene expression relative to untreated RML-infected mice |
|
|---|---|---|
| Thalamus PLX5622 RML (n = 2) | Brain PLX5622 RML (n = 4) | |
| Ccr1 | −20.7 | −3.1* |
| Ccl8 | −12.7 | −28.8*** |
| Ccr5 | −11.4 | −3.3** |
| Ccr3 | −10.6 | −4.7** |
| Il1a | −8.0 | −3.8* |
| Ccl6 | −7.8 | −2.8** |
| Tnf | −7.1 | 1.1 |
| Ccl11 | −4.2 | −1.2 |
| Il1rn | −4.1 | 1.2 |
| Ccl4 | −3.8 | −1.3 |
| Ccl12 | −3.5 | −2.1 |
| Il10ra | −3.5 | −2.7** |
| Ccl9 | −2.6 | −4.8** |
| Cxcl9 | −2.6 | −1.2 |
| Il6ra | −2.6 | −2.2** |
| Ccl22 | −2.2 | −1.9 |
| Ccl3 | −2.0 | −1.5 |
| Cxcl5 | 2.3 | 3.0* |
| Cxcl1 | 2.6 | 4.8* |
| Tnfsf11 | 3.8 | 1.9 |
Bolded genes are predominately expressed by microglia in healthy brain. n, number of individual mice examined. P values were calculated using the Student t test and are indicated by asterisks: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. P values for PLX5622-treated thalamus could not be calculated because of an n of 2.
TABLE 3.
List of proinflammatory genes that were unchanged in the thalamus of mice at the 80-dpi time point when RNA from PLX5622-treated mice was compared to RNA from untreated mice
| Gene namesa |
|---|
| Aimp1, Bmp2, Ccl1, Ccl17, Ccl19, Ccl2, Ccl20, Ccl24, Ccl5, Ccl7, Ccr10, Ccr2, Ccr4, Ccr6, Ccr8, Cd40lg, Csf1, Csf2, Csf3, Cx3cl1, Cxcl10, Cxcl11, Cxcl12, Cxcl13, Cxcl15, Cxcr2, Cxcr3, Cxcr5, Fasl, Ifng, Il10rb, Il11, Il15, Il16, Il17a, Il17b, Il17f, Il1b, Il1r1, Il21, Il2rb, Il2rg, Il3, Il33, Il4, Il5, Il5ra, Il6st, Il7, Lta, Ltb, Mif, Nampt, Tnfsf10, Tnfsf13, Tnfsf13b, Tnfsf4, Vegfa |
Bolded genes were previously shown to be increased in the thalamus of RML-infected mice at 80 dpi (31). These genes in bold were similarly increased in RML-infected untreated and PLX5622-treated mice; therefore, their expression is unaffected by the reduction in microglia.
PLX5622 treatment to reduce microglia at preclinical onset also accelerates disease.
The preceding results indicated that microglia were likely important in controlling prion infection and that early elimination of microglia leads to accelerated disease. Although microglia might be beneficial at the early stages of prion disease, they might become more neurotoxic and destructive with prolonged activation during neuroinflammation. Therefore, we hypothesized that PLX5622 treatment to chemically ablate microglia just prior to clinical onset (80 dpi) might increase survival. To test this, mice were inoculated with scrapie strain RML and fed control chow for 80 dpi and then switched to PLX5622-supplemented chow until the experimental endpoint. Surprisingly, mice treated starting at 80 dpi had a more rapid disease progression and had to be euthanized due to advanced disease approximately 33 days earlier than the untreated control group (Fig. 9A and B; Table 4). Significant differences were also seen when nesting scores and scrapie severity scores were compared (Fig. 9B and C). This result was comparable to those of preceding experiments where PLX5622 therapy was begun at 14 dpi (RML panels in Fig. 2A and 3).
FIG 9.
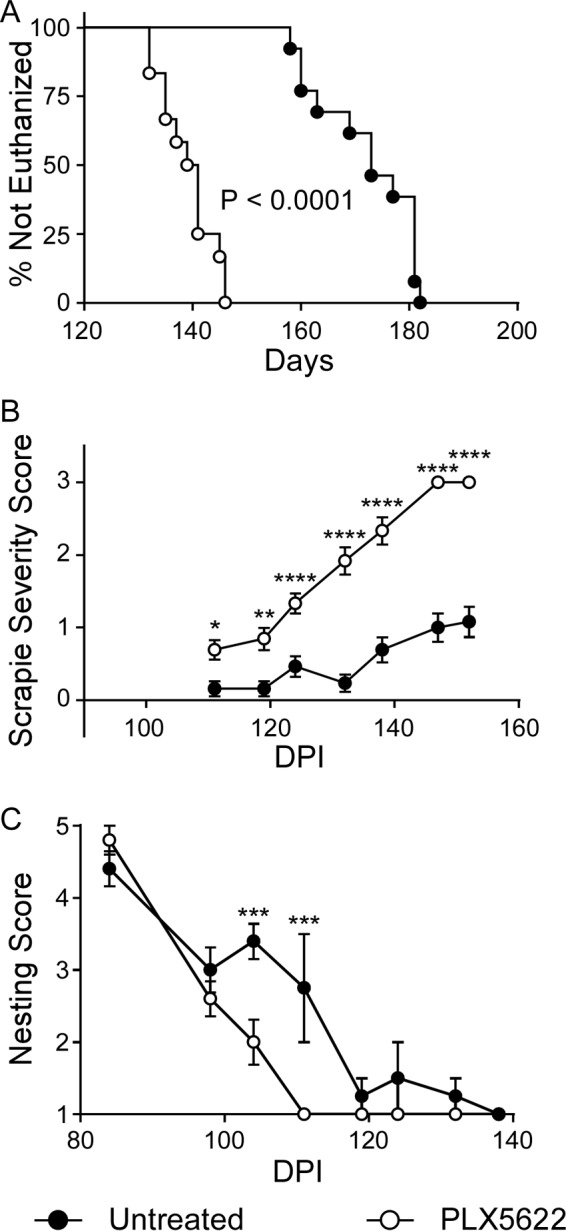
Survival, nesting scores, and scrapie severity scores of mice infected with scrapie strain RML and either treated with PLX5622 beginning at 80 dpi or left untreated. Mice were infected with scrapie strain RML and fed control chow (untreated, filled circles) or switched to chow supplemented with PLX5622 (open circles) after 80 dpi. Infected mice were monitored by observers blinded to the treatments for clinical signs of scrapie requiring euthanasia (A). Data are percentages of animals not euthanized versus days postinfection (Days). The day each individual mouse was euthanized, the median day of euthanasia for each mouse cohort, and the number of animals in each group are given in Table 4. Statistical analysis was performed using Mantel-Cox log rank analysis comparing PLX5622-treated to untreated animals. In addition, RML-infected mice were scored by observers blinded to the treatments for changes in severity of clinical presentation (scrapie severity score) (B) and nesting behavior (C) at various days postinfection (DPI). Severity of clinical presentation was scored from 0 to 3, where 0 represented no signs and 3 indicated signs requiring euthanasia, and nesting behavior was scored on a scale of 1 to 5, where 5 was a near-perfect nest and 1 was no nest (see Materials and Methods for details). All scores are representative of the average score of the study group at each day postinfection. Scrapie severity scoring stopped once the all-PLX5622-treatment group averaged a score of 3. Error bars represent the standard errors of the means. Multiple t test was performed comparing scrapie severity (B) and nesting scores (C) between PLX5622-treated and untreated groups of mice at specific time points using GraphPad software. Statistical significance was determined using the Holm-Sidak method to correct for multiple comparisons, with alpha of 0.05. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
TABLE 4.
Survival times of delayed PLX5622-treated and untreated RML-infected C57BL/10 mice
| Treatment (no. of mice in the study group) | Survival times (dpi)a | Mean dpi ± SD |
|---|---|---|
| Delayed PLX5622 (12) | 132, 132, 135, 135, 137, 139, 141, 141, 141, 145, 146, 146 | 139.2 ± 5.0 |
| Untreated (13) | 158, 160, 160, 173, 173, 181, 181, 181, 181, 177, 163, 169, 182 | 172.2 ± 9.2 |
Number of days postinfection when individual mice were euthanized due to the presence of advanced clinical signs.
We also assessed the reduction in the microglial population after delaying PLX5622 treatment. Iba1-positive cells were reduced at all time points in the cortex and the thalamus of late-treated RML-infected mice relative to untreated RML-infected mice at 104 dpi (24 days posttreatment) and at the experimental endpoint. (Fig. 10). The level of microglial reduction at 104 dpi was comparable to that of our previous experiments at a similar time point (100 dpi; Fig. 7A and B), indicating that PLX5622 treatment beginning at 80 dpi was as effective as treatment starting at 14 dpi to reduce activated microglia during prion disease. These findings suggested that microglial depletion even at the later stages of preclinical prion disease also accelerated prion pathogenesis. Therefore, microglia are likely beneficial throughout prion disease.
FIG 10.

Iba1-positive cells in cortex and thalamus during prion disease with PLX5622 treatment beginning at 80 dpi (late treatment). Mice infected with scrapie strain RML were fed control chow (−, white columns) for the duration or switched to chow supplemented with PLX5622 after 80 dpi (+, gray columns). Mice were euthanized at 104 dpi and at clinical endpoint (∼172 dpi for control or ∼139 dpi for PLX5622). See Table 4 for all clinical endpoint ranges. Similar paraffin-embedded sections of cortex (A) and thalamus (B) were fixed and stained with antibody to Iba1. Iba1-positive cells were enumerated and reported as the number of positive cells per square millimeter. The columns are the means, and each dot in the groups represents an individual mouse. The percent reduction with treatment for each time point relative to control is given. Error bars represent 1 standard deviation. Control and PLX5622-treated groups at each time were compared by unpaired t test: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
DISCUSSION
In the current work, we utilized PLX5622, an effective CSF-1R inhibitor, to eliminate microglia in scrapie-infected mice in vivo to study the role of microglia in scrapie pathogenesis. In uninfected mice, treatment with PLX5622 reduced microglia by 79 to 99% in thalamus and cerebral cortex during treatment up to 181 days in duration. Therefore, PLX5622 appeared to have excellent potential for use in long-term studies involving prion infection. In scrapie-infected mice, depletion of microglia by oral treatment with PLX5622 dramatically reduced survival (Fig. 2). The largest decrease in survival time (31 to 33 days) was seen in mice infected with the scrapie strain RML, but similar changes were also seen in mice infected with scrapie strains ME7 and 22L. In the presence of PLX5622 treatment, hallmarks of typical prion disease, including gliosis, PrPSc deposition, and vacuolation, appeared 20 to 30 days earlier than usual. The early appearance of PrPSc deposition in mice treated with PLX5622 was consistent with the hypothesis that when microglia are present, they catabolically degrade PrPSc, resulting in a more prolonged disease course.
Evidence for destruction of PrPSc by microglia was shown previously in scrapie-infected tga20 cerebellar organotypic slice cultures, where removal of microglia expressing thymidine kinase by treatment with ganciclovir resulted in higher levels of PrPSc (26, 32). However, a similar effect has been more difficult to demonstrate in vivo. Reduction of microglia in vivo during scrapie infection was attempted by preventing stimulation of CSF-1R required for microglial growth by using IL-34−/− mice. In these scrapie-infected mice, a significant decrease in survival of 14 to 21 days was observed; however, the mice did not show evidence for depletion of microglia (26). Analysis of microglia at presymptomatic and clinical time points showed no difference between wild-type and IL-34−/− mice, which was contrary to reports by others using uninfected mice (24). The authors concluded that IL-34 was not required for microglial activation in the presence of prion infection. However, this explanation leaves open the question of what caused the decrease in scrapie survival in the IL-34−/− mice that had normal levels of activated microglia. One possibility is that in the absence of IL-34, phagocytosis or catabolism of PrPSc by microglia might be impaired.
In contrast to the above-described results, the correlation between elimination of microglia and decreased survival from scrapie was very strong in our present studies using PLX5622. Throughout most of the incubation period, the microglial numbers were quite low, but near the clinical endpoint microglial numbers were higher, with values around 50% of those in untreated control mice (Fig. 7 and 10). Increased numbers of microglia at the end of the disease suggested that advanced prion infection was indeed a powerful stimulus for targeted expansion from microglial precursor cells. Nevertheless, these late-appearing activated microglia did not appear to be sufficient to halt the deteriorating clinical course in these mice.
Surprisingly, initiation of PLX5622 treatment at a preclinical time point, 80 dpi, caused an acceleration of clinical disease similar to that seen when treatment was initiated at 14 dpi (Fig. 9). Thus, the role of microglia in combating disease progression appeared to be more prominent in the late preclinical phase rather than in the early phase of infection between 14 and 80 dpi. The reason for this difference is not clear, but it might be related to the exponential increase in PrPSc over time after infection (33). Perhaps at early time points, when PrPSc quantities are very low, the removal of additional PrPSc has a nominal effect, whereas when levels are intermediate (i.e., during the late preclinical phase), microglia might have a more important impact on PrPSc expansion. Nevertheless, when PrPSc amounts reach a higher level nearer to the clinical endpoint, microglia are apparently no longer able to effectively control further PrPSc accumulation and the disease proceeds to a terminal state.
Other groups have previously been able to alter microglial numbers and function during prion disease in vivo, but these experiments resulted in quite different conclusions from ours. Gomez-Nicola et al. used a different inhibitor of CSF-1R, GW2580, to block microglial proliferation during prion disease (27). In these experiments, drug-treated scrapie-infected mice had a 26.5-day increase in survival time compared to untreated mice. In addition, there was a delay in various behavioral clinical signs of disease and in neurodegenerative pathology. These beneficial effects associated with blocking of microglial proliferation correlated with a 50% reduction in microglia in both hippocampus and thalamus, a decrease in expression of genes associated with the M1 phenotype, and an increase in genes associated with the M2 phenotype. These authors suggested that this switch in microglial gene expression profile might increase survival time by reducing the prion-induced neurotoxic effect of microglia.
The above-described findings are contrary to those reported in our present experiments using PLX5622 to reduce microglia. However, these two sets of experimental results might be reconciled if one considers that microglia might have multiple influences on prion disease. Based on the available evidence, we postulate two major effects of microgliosis during prion disease: (i) phagocytosis of PrPSc and (ii) production of a neurotoxic component. PLX5622 affects microglial survival, leading to microglial death through apoptosis (20, 28). Therefore, treatment with PLX5622 might prevent efficient PrPSc clearance by microglia and result in an increase in disease tempo (Tables 1 and 4; Fig. 2 and 9). Alternatively, treatment with GW2580 effectively blocks microglial proliferation but does not eliminate the microglial population. Furthermore, treatment with GW2580 also shifts the gene expression profile. This situation may result in continued destruction of PrPSc by microglia but may also reduce the neurotoxic effects of microglia due to the altered gene expression. These events might explain the increased survival time with GW2580 treatment reported by Gomez-Nicola et al. Therefore, microglia appear to demonstrate both neuroprotective and/or neurotoxic properties during prion disease, and these differing properties might be observed to various degrees depending on which CSF-1R inhibitor is used.
Microgliosis occurs prior to neuronal loss and spongiform change in the brain during prion disease (3, 8), and there is a close association of increased microgliosis in regions with greater spongiosis and astrogliosis (34). These facts suggest that microglia may contribute to prion-induced neurodegeneration. However, in our experiments microglia were not required for early spongiform change, since spongiosis was observed in the thalamus as early as 80 dpi in PLX5622-treated scrapie-infected mice that lacked normal numbers of functional microglia. In addition, astrogliosis, another hallmark of prion disease, was also present when microglia were absent or reduced. Thus, astrocytes appeared to activate directly in response to PrPSc and/or to damaged or dying neurons and did not require additional signaling from activated microglia.
Microglia exist as multiple subpopulations in the CNS that result from regional influences (35–37). With the multitude of proposed activation states (38–40), it is possible that these subpopulations of microglia may demonstrate neuroprotective characteristics, neurotoxic properties, or both at different times during prion and prionlike neurodegenerative diseases. Instead of therapies to reduce microgliosis during neurodegenerative diseases, strategies to increase specific microglial responses advantageous to the host should be considered, along with identifying and reducing microglial processes that are harmful.
MATERIALS AND METHODS
Ethics statement.
All mice were housed at the Rocky Mountain Laboratories (RML) in an AAALAC-accredited facility in compliance with guidelines provided by the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research Council). Experimentation followed RML Animal Care and Use Committee-approved protocol 2016-043-E.
Normal brain and scrapie inoculations/infections.
C57BL/10 (C57) mice were originally obtained from Jackson Laboratories and have been inbred at RML for several years. Six- to 8-week-old male and female mice were inoculated intracranially (i.c.) with 30 μl of either 1.0% NBH or 1.0% scrapie brain stock strains 22L, RML, and ME7. The titers of these scrapie stocks had been determined previously in C57 mice and contained the following 50% infective doses (ID50) in each 30-μl volume: 22L, 6.0 × 105; RML, 2.4 × 104; ME7, 5.0 × 104. At selected time points postinoculation, mice were euthanized by isoflurane anesthesia overdose followed by cervical dislocation. Brains were removed and divided into portions for histochemical, RNA, and protein analysis. Brain portions for histochemical analysis were immersed in 10% neutral buffered formalin (3.7% formaldehyde) for histology.
For determination of the scrapie severity score (41), mice were scored weekly for clinical signs beginning at 85 dpi. Observers were blinded to the PLX5622 treatment group. A single score was assigned to each individual mouse based on the extent and level of clinical signs observed. Scorers paid particular attention to the severity of somnolence, kyphosis, gait abnormalities, and decrease in body condition. Scores were as follows: 0, normal; 1, subtle clinical signs; 2, moderate clinical signs; and 3, advanced clinical signs. Mice were euthanized when they reached a score of 3.
Nesting was scored as described previously (42). Briefly, a cotton square (nestlet) was added to each cage. After a 24-hour period, observers that were blinded to the PLX5622 treatment group assessed nest building. Scoring was as follows: 1, untouched (90% intact) nestlet; 2, partially (50 to 90% intact) torn nestlet; 3, mostly (>90%) torn nestlet, but no defined nest site; 4, identifiable nest without walls; 5, nest resembles a crater with walls. Nesting is a communal activity in mice; however, in some situations where disease progression differs among mice within a box, one or more healthy mice in the box could bias the data. Even though nesting can be an early predictor of disease in mice, we feel the scrapie severity score is a more accurate indicator because mice are assessed individually.
Multiple t tests comparing the nesting and scrapie severity scores at each time point between the PLX5622-treated and untreated groups were performed using GraphPad Prism version 7.03. For a more powered analysis, the assumption was that all samples are from populations with the same scatter. The statistical significance was determined using the Holm-Sidar method for multiple comparison correction, with an alpha of 0.05.
PLX5622 treatment.
Mice were fed purified rodent diet AIN-76A (D10001; Research Diets, Inc.) with or without supplementation with compound PLX5622 (1,200 mg/kg chow; kindly provided by Plexxikon Inc., Berkeley, CA). This concentration of PLX5622 in rodent chow has been shown to reduce microglia in the brain by approximately 80% within 7 days and approximately 90% within 21 days of administration (28). In most experiments, 6- to 8-week-old male and female mice were fed control chow for 14 days after inoculation with prions or NBH to allow mice to convalesce. Then, treated mice were switched to PLX5622-supplemented chow and maintained on this diet until the experimental endpoint. In experiments where treatment with PLX5622 was delayed, 6- to 8-week-old male and female mice were inoculated with prions, fed control chow for 80 days, and then switched to chow supplemented with PLX5622 just prior to clinical signs. Mice were then maintained on supplemented chow until the experimental endpoint.
Immunoblot detection of PrPSc.
Dissected brain tissue was homogenized in phosphate-buffered saline (PBS) to create a 20%, wt/vol, brain homogenate (BH). Homogenization was done using a mini-bead beater homogenization system for 45 s on the homogenate setting. For proteinase K (PK) treatment, samples were incubated with detergents and PK as follows: 20 μl of a 20% tissue homogenate was adjusted to 100 mM Tris HCl (pH 8.3), 1% Triton X-100, 1% sodium deoxycholate, and 50 μg/ml of proteinase K (Roche Diagnostics) in a total volume of 31 μl. Samples were digested for 45 min at 37°C. The reaction was stopped by adding 2 μl of 100 mM Pefabloc (Roche Diagnostics) and placed on ice for 5 min. An equal volume of 2× Laemmli sample buffer (Bio-Rad, Hercules, CA) was added, and then tubes were boiled 5 min. Samples were run immediately or frozen at −20°C until electrophoresed on a 16% Tris-glycine SDS-PAGE gel (Life Technologies, CA), and gels were transferred to polyvinylidene difluoride (PVDF) membranes using the 7-min transfer program 3 of the iBlot transfer system (Life Technologies). The PLX5622-treated and untreated mouse samples at 80 dpi, at 100 dpi, or at the terminal endpoint were transferred to the same PVDF membrane and blocked overnight in 2.5% dry milk in Tris-buffered saline. Membranes were probed with a 1:100 dilution of monoclonal human anti-PrP antibody D13 derived from cell culture supernatants produced in our laboratory from CHO cells expressing the D13 antibody construct (43) that were kindly provided by R. Anthony Williamson. Monoclonal antibody D13 recognizes residues 94 to 105 in PrP (44) derived from mouse, hamster, and squirrel monkey and has been extensively used for detection of PrP in immunoblots and immunohistochemistry. The secondary antibody was peroxidase-conjugated anti-human IgG, used at 1:5,000 (Sigma), and immunoreactive bands were visualized using the SuperSignal West Femto substrate detection system (Thermo Scientific) for the 80- and 100-dpi sample immunoblots or the ECL Western blotting substrate detection system (Thermo Scientific) for the terminal sample immunoblots. Densitometry on unsaturated immunoreactive bands was performed on exposed film using the Bio-Rad ChemiDoc MP system. Adjusted volumes for immunoreactive bands were calculated by taking the total band volume and subtracting the global background using Image Lab software version 5.0 (Bio-Rad). Statistical analysis of densitometry by two-tailed Student t test was performed using GraphPad Prism.
Immunohistochemical detection of PrPSc, microglia, and astroglia.
Portions of brain were removed and placed in 10% neutral buffered formalin for 3 to 5 days. Tissues were then processed and embedded in paraffin. Sections were cut using a standard Leica microtome, placed on positively charged glass slides, and air dried overnight at room temperature. The following day, slides were heated in an oven at 60°C for 30 min.
Deparaffinization, antigen retrieval, and staining were performed using the Ventana automated Discovery XT stainer. Because of the intense aggregation of PrPSc, immunostaining PrPSc requires stringent antigen retrieval using high temperatures. In the present experiments, PrPSc antigens were exposed by incubation in CC1 buffer (Ventana) containing Tris-borate-EDTA, pH 8.0, for 100 min at 95°C. Staining for PrP was done using human anti-PrP monoclonal antibody D13, described above. For immunohistochemistry, D13 culture fluid was used at a dilution of 1:100 for 2 h at 37°C. The secondary antibody was biotinylated goat anti-human IgG at a 1:250 dilution (Jackson ImmunoResearch, West Grove, PA), and streptavidin-biotin peroxidase was used with 3,3′-diaminobenzidine (DAB) as chromogen (DAB Map kit; Ventana Medical Systems, Tucson, AZ). Hematoxylin was used as a counterstain for all slides.
To stain microglia, rabbit anti-Iba1 was used at a 1:2,000 dilution and was a gift from John Portis, RML, Hamilton, MT. To stain astrocytes, rabbit anti-glial fibrillary acidic protein (GFAP) (Dako number Z0334) was used at a dilution of 1:3,500. Primary antibodies were diluted in PBS containing stabilizing protein and 0.1% Proclin 300 (Ventana antibody dilution buffer). Diluent without antibody was used as a negative control. The Ventana streptavidin-biotin alkaline phosphatase system was used to detect Iba1 and GFAP (45) with the exceptions that the secondary antibody was goat anti-rabbit Ig (HK336-9R; Biogenex) and Fast Red chromogen was used. Slides were examined, and photomicrographs were taken and observed using an Olympus BX51 microscope and Microsuite FIVE software. Comparisons of the numbers of Iba1-positive microglia between PLX5622-treated and untreated cohorts were made by unpaired Student's t test using GraphPad Prism.
RNA isolation.
Total RNA was isolated from the left hemisphere of mouse brain by dissociation of tissue in 2 ml TRI-reagent (Sigma) following the manufacturer's protocol. Isolated RNA was rinsed in 2 ml 75% ethanol, centrifuged for 10 min at 13,000 × g, and air dried. Total RNA was suspended in 100 μl of DNase reaction buffer (Ambion) and digested with 6 units of DNase I (Ambion) for 30 min at room temperature. RNA was reisolated and cleaned using the RNA Clean & Concentrator-25 column kit (Zymo Research), eluted with 150 μl nuclease-free water with 1× RNase inhibitor (SUPERase-In; Ambion), and stored at −80°C until use.
qRT-PCR analysis.
For quantitative analysis of changes in transcription using real-time quantitative reverse transcription-PCR (qRT-PCR) arrays, 400 ng of high-quality RNA from each sample was reverse transcribed to synthesize cDNA using the RT2 First Strand kit per the manufacturer's instruction (Qiagen). Each cDNA reaction mixture was mixed with 2× RT2 SYBR green Mastermix purchased from Qiagen with RNase-free water to a final volume of 1.3 ml. Ten microliters of the mixture was then added to each well of a 384-well format plate of the mouse inflammatory cytokine & receptors Super-array PAMM-011ZE (Qiagen). The analysis was carried out on an Applied Biosystems ViiA 7 real-time PCR system with a 384-well block using the following conditions: 1 cycle for 10 min at 95°C, 40 cycles for 15 s at 95°C, and then 1 min at 60°C, with fluorescence data collection. Melting curves were generated at the end of the completed run to determine the quality of the reaction products. Raw threshold cycle (CT) data were collected with a CT of 35 as the cutoff. CT data were analyzed using the web-based RT2 Profiler PCR Array Data Analysis (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). All CT values were normalized to the average of the CT values for the housekeeping genes Actb, Gapdh, and Hsp90ab1. Changes in transcription were calculated by the software using the ΔΔCT-based method (46). Statistical analysis was performed using unpaired Student's t test to compare the replicate ΔCT values for each gene in the control group versus infected groups. A mean of ≥2.0-fold change and P values of ≤0.05 were considered significant. For qRT-PCR data, no effort was made to adjust P values for multiple comparisons, since we were interested in controlling only for the individual error rate, where an adjustment for multiple tests is deemed unnecessary.
ACKNOWLEDGMENTS
Compound PLX5622 was kindly provided by Plexxikon Inc. We thank Jeffrey Severson for assistance with animal husbandry, Nancy Kurtz and Lori Lubke for assistance with immunohistochemical procedures, and Sue Priola, Allison Kraus, and Clayton Winkler for critical readings of the manuscript.
This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Prusiner SB. 1991. Molecular biology of prion diseases. Science 252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 2.Aguzzi A, Sigurdson C, Heikenwaelder M. 2008. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- 3.Williams A, Lucassen PJ, Ritchie D, Bruce M. 1997. PrP deposition, microglial activation, and neuronal apoptosis in murine scrapie. Exp Neurol 144:433–438. doi: 10.1006/exnr.1997.6424. [DOI] [PubMed] [Google Scholar]
- 4.Caughey B, Baron GS, Chesebro B, Jeffrey M. 2009. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll JA, Striebel JF, Race B, Phillips K, Chesebro B. 2015. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J Virol 89:2388–2404. doi: 10.1128/JVI.02952-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booth S, Bowman C, Baumgartner R, Sorensen G, Robertson C, Coulthart M, Phillipson C, Somorjai RL. 2004. Identification of central nervous system genes involved in the host response to the scrapie agent during preclinical and clinical infection. J Gen Virol 85:3459–3471. doi: 10.1099/vir.0.80110-0. [DOI] [PubMed] [Google Scholar]
- 7.Aguzzi A, Zhu C. 2017. Microglia in prion diseases. J Clin Invest 127:3230–3239. doi: 10.1172/JCI90605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Betmouni S, Perry VH, Gordon JL. 1996. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience 74:1–5. doi: 10.1016/0306-4522(96)00212-6. [DOI] [PubMed] [Google Scholar]
- 9.Hong S, Stevens B. 2016. Microglia: phagocytosing to clear, sculpt, and eliminate. Dev Cell 38:126–128. doi: 10.1016/j.devcel.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Hong S, Dissing-Olesen L, Stevens B. 2016. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36:128–134. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bilimoria PM, Stevens B. 2015. Microglia function during brain development: new insights from animal models. Brain Res 1617:7–17. doi: 10.1016/j.brainres.2014.11.032. [DOI] [PubMed] [Google Scholar]
- 12.Salter MW, Stevens B. 2017. Microglia emerge as central players in brain disease. Nat Med 23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- 13.Mandrekar-Colucci S, Landreth GE. 2010. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets 9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muzio L, Martino G, Furlan R. 2007. Multifaceted aspects of inflammation in multiple sclerosis: the role of microglia. J Neuroimmunol 191:39–44. doi: 10.1016/j.jneuroim.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A. 2007. Neuroinflammation in Alzheimer's disease and Parkinson's disease: are microglia pathogenic in either disorder? Int Rev Neurobiol 82:235–246. doi: 10.1016/S0074-7742(07)82012-5. [DOI] [PubMed] [Google Scholar]
- 16.Garden GA. 2002. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 40:240–251. doi: 10.1002/glia.10155. [DOI] [PubMed] [Google Scholar]
- 17.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. 2015. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. 2010. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. 2007. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 20.Elmore MR, Lee RJ, West BL, Green KN. 2015. Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology, and neuroinflammation. PLoS One 10:e0122912. doi: 10.1371/journal.pone.0122912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. 2014. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. 2011. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6:e26317. doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER. 2016. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci 39:378–393. doi: 10.1016/j.tins.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. 2012. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wegiel J, Wisniewski HM, Dziewiatkowski J, Tarnawski M, Kozielski R, Trenkner E, Wiktor-Jedrzejczak W. 1998. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res 804:135–139. doi: 10.1016/S0006-8993(98)00618-0. [DOI] [PubMed] [Google Scholar]
- 26.Zhu C, Herrmann US, Falsig J, Abakumova I, Nuvolone M, Schwarz P, Frauenknecht K, Rushing EJ, Aguzzi A. 2016. A neuroprotective role for microglia in prion diseases. J Exp Med 213:1047–1059. doi: 10.1084/jem.20151000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH. 2013. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33:2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL, Green KN. 2015. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation 12:139–152. doi: 10.1186/s12974-015-0366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. 2014. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep 9:2124–2138. doi: 10.1016/j.celrep.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vincenti JE, Murphy L, Grabert K, McColl BW, Cancellotti E, Freeman TC, Manson JC. 2015. Defining the microglia response during the time course of chronic neurodegeneration. J Virol 90:3003–3017. doi: 10.1128/JVI.02613-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll JA, Striebel JF, Rangel A, Woods T, Phillips K, Peterson KE, Race B, Chesebro B. 2016. Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: comparison of three prion strains. PLoS Pathog 12:e1005551. doi: 10.1371/journal.ppat.1005551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A. 2008. A versatile prion replication assay in organotypic brain slices. Nat Neurosci 11:109–117. doi: 10.1038/nn2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tribouillard-Tanvier D, Race B, Striebel JF, Carroll JA, Phillips K, Chesebro B. 2012. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: no effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J Virol 86:10377–10383. doi: 10.1128/JVI.01340-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruce ME, McBride PA, Farquhar CF. 1989. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett 102:1–6. doi: 10.1016/0304-3940(89)90298-X. [DOI] [PubMed] [Google Scholar]
- 35.Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J, Degos V, Jacotot E, Hagberg H, Savman K, Mallard C, Gressens P, Fleiss B. 2013. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun 32:70–85. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. 2014. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ransohoff RM, Perry VH. 2009. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 38.Streit WJ, Walter SA, Pennell NA. 1999. Reactive microgliosis. Prog Neurobiol 57:563–581. doi: 10.1016/S0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 39.Nelson PT, Soma LA, Lavi E. 2002. Microglia in diseases of the central nervous system. Ann Med 34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- 40.Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai LH. 2017. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep 21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carroll JA, Race B, Phillips K, Striebel JF, Chesebro B. 2017. Statins are ineffective at reducing neuroinflammation or prolonging survival in scrapie-infected mice. J Gen Virol 98:2190–2199. doi: 10.1099/jgv.0.000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP, Scott H, Rawlins JN, Perry VH. 2003. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci 17:2147–2155. doi: 10.1046/j.1460-9568.2003.02662.x. [DOI] [PubMed] [Google Scholar]
- 43.Rangel A, Race B, Phillips K, Striebel J, Kurtz N, Chesebro B. 2014. Distinct patterns of spread of prion infection in brains of mice expressing anchorless or anchored forms of prion protein. Acta Neuropathol Commun 2:8. doi: 10.1186/2051-5960-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsunaga Y, Peretz D, Williamson A, Burton D, Mehlhorn I, Groth D, Cohen FE, Prusiner SB, Baldwin MA. 2001. Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: dependence of conformation on pH. Proteins 44:110–118. doi: 10.1002/prot.1077. [DOI] [PubMed] [Google Scholar]
- 45.Kercher L, Favara C, Striebel JF, LaCasse R, Chesebro B. 2007. Prion protein expression differences in microglia and astroglia influence scrapie-induced neurodegeneration in the retina and brain of transgenic mice. J Virol 81:10340–10351. doi: 10.1128/JVI.00865-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]