

Human T-cell leukemia virus type 1 (HTLV-1) infects 15 million to 20 million people worldwide. Approximately 90% of infected individuals are asymptomatic and may remain undiagnosed, increasing the risk that they will unknowingly transmit the virus. About 5% of the HTLV-1-positive population develop adult T-cell leukemia (ATL), a fatal disease that is not highly responsive to treatment. Although ATL development remains poorly understood, two viral proteins, Tax and HBZ, have been implicated in driving disease progression by manipulating host cell signaling and transcriptional pathways. Unlike Tax, HBZ expression is consistently observed in all infected individuals, making it important to elucidate the specific role of HBZ in disease progression. Here, we present evidence that HBZ could promote the accumulation of double-stranded DNA breaks (DSBs) through the attenuation of the nonhomologous end joining (NHEJ) repair pathway. This effect may lead to genome instability, ultimately contributing to the development of ATL.
KEYWORDS: HBZ, human T cell leukemia virus, leukemia, NHEJ, Tax, viral oncogene
ABSTRACT
Adult T-cell leukemia (ATL) is a fatal malignancy of CD4+ T cells infected with human T-cell leukemia virus type 1 (HTLV-1). ATL cells often exhibit random gross chromosomal rearrangements that are associated with the induction and improper repair of double-stranded DNA breaks (DSBs). The viral oncoprotein Tax has been reported to impair DSB repair but has not been shown to be consistently expressed throughout all phases of infection. The viral oncoprotein HTLV-1 basic leucine zipper (bZIP) factor (HBZ) is consistently expressed prior to and throughout disease progression, but it is unclear whether it also influences DSB repair. We report that HBZ attenuates DSB repair by nonhomologous end joining (NHEJ), in a manner dependent upon the bZIP domain. HBZ was found to interact with two vital members of the NHEJ core machinery, Ku70 and Ku80, and to be recruited to DSBs in a bZIP-dependent manner in vitro. We observed that HBZ expression also resulted in a bZIP-dependent delay in DNA protein kinase (DNA-PK) activation following treatment with etoposide. Although Tax is reported to interact with Ku70, we did not find Tax expression to interfere with HBZ:Ku complex formation. However, as Tax was reported to saturate NHEJ, we found that this effect masked the attenuation of NHEJ by HBZ. Overall, these data suggest that DSB repair mechanisms are impaired not only by Tax but also by HBZ and show that HBZ expression may significantly contribute to the accumulation of chromosomal abnormalities during HTLV-1-mediated oncogenesis.
IMPORTANCE Human T-cell leukemia virus type 1 (HTLV-1) infects 15 million to 20 million people worldwide. Approximately 90% of infected individuals are asymptomatic and may remain undiagnosed, increasing the risk that they will unknowingly transmit the virus. About 5% of the HTLV-1-positive population develop adult T-cell leukemia (ATL), a fatal disease that is not highly responsive to treatment. Although ATL development remains poorly understood, two viral proteins, Tax and HBZ, have been implicated in driving disease progression by manipulating host cell signaling and transcriptional pathways. Unlike Tax, HBZ expression is consistently observed in all infected individuals, making it important to elucidate the specific role of HBZ in disease progression. Here, we present evidence that HBZ could promote the accumulation of double-stranded DNA breaks (DSBs) through the attenuation of the nonhomologous end joining (NHEJ) repair pathway. This effect may lead to genome instability, ultimately contributing to the development of ATL.
INTRODUCTION
Genetic instability is a prominent feature in cellular transformation and is characterized by the accumulation of abnormalities in chromosomal number and structure, which may result in changes to tumor suppressor or proto-oncogene expression (1, 2). The stability of the genome is particularly threatened by double-stranded DNA breaks (DSBs), which are acquired from both endogenous and exogenous sources (3, 4). Upon the recognition of a DSB, DNA damage response checkpoint mechanisms arrest the cell cycle, and repair is initiated. While a failure in DSB repair can lead to apoptosis, incorrect repair poses a great risk for the introduction of mutations and gross chromosomal rearrangements, which may contribute to carcinogenesis (1, 3, 5–7). Generally, two distinct repair mechanisms are used to process and seal DSBs: homologous recombination (HR) and nonhomologous end joining (NHEJ).
HR is a template-dependent repair mechanism most active during S phase, in which a damaged region is repaired using the homologous region of the intact sister chromatid as a template for DNA synthesis (8–10). Although this process may preserve the fidelity of the sequence spanning the break site, frequent repetition in the genome creates the possibility that the incorrect template sequence will be used to prime repair, which may result in genomic rearrangement (11).
NHEJ is a template-independent repair mechanism that is active during all phases of the cell cycle and consists of a classical pathway (C-NHEJ) and an alternative pathway (Alt-NHEJ) (12, 13). Alt-NHEJ is the lesser understood of the two pathways but is reported to utilize areas of microhomology to mend DNA lesions via the enzymatic activities of the MRN complex and Polϴ (13). Conversely, C-NHEJ mends DNA breaks lacking homology by either directly ligating compatible DSBs (NHEJ-C) or facilitating endonucleolytic processing prior to ligation in the case of incompatible DSBs (NHEJ-I). In each case, repair is initiated by Ku70 and Ku80, which bind and stabilize the DNA termini. The phosphatidylinositol 3-kinase-like kinase (PIKK) family member DNA protein kinase (DNA-PK), which is composed of the catalytic subunit (DNA-PKcs) and the Ku heterodimer, then bridges the break and phosphorylates a number of NHEJ repair proteins. Although DNA-PK activity is critical for repair, the significance of its kinase activity remains unclear, since the phosphorylation of NHEJ targets is not required for successful repair (14). The exception is the autophosphorylation of DNA-PK at Ser2056, which is critical for successful DSB repair (15). Ser2056 autophosphorylation induces a conformational change within the enzyme that releases it from the DNA and allows ligation enzymes, XRCC4 and ligase IV, to access and seal the break (15–19). Failure to phosphorylate Ser2056 is reported to stabilize DNA-PK on the lesion, preventing ligation and the completion of NHEJ (19). In this way, DNA-PK is hypothesized to prevent premature ligation and coordinate the later stages of NHEJ (20). In addition to serving as a critical regulator of DSB repair, DNA-PK activity is also linked to the activation of p53-dependent apoptosis in response to unrepaired DNA damage (21–27).
Defects in DSB repair pathways and cell cycle checkpoint control are clearly associated with an increased risk of developing solid tumors and hematological malignancies (3). Adult T-cell leukemia/lymphoma (ATL) is a fatal malignancy of CD4+ T cells caused by a lifelong infection with human T-cell leukemia virus type 1 (HTLV-1) (28–30). Approximately 5% of HTLV-1-positive individuals develop ATL, although the process through which this disease occurs remains poorly understood. ATL development is likely driven by a multistep oncogenic process, as evidenced by the extended length of time required for pathogenesis to occur, the limited number of infected individuals who develop the disease, and the accumulation of seemingly random genetic abnormalities in infected cells (31). Karyotyping of ATL cells isolated from patients reveals that chromosomal structural and numerical abnormalities are consistently present but that these aberrations are not necessarily conserved among patients (32).
HTLV-1 encodes two proteins that have been reported to drive oncogenesis: Tax and HTLV-1 basic leucine zipper (bZIP) factor (HBZ). Tax expression induces genetic instability by promoting multinucleation, chromosomal aneuploidy, and telomere attrition (33–37). Tax is also reported to induce DSBs through the production of reactive oxygen and nitrogen species (ROS) as a result of constitutive NF-κB activation and upregulated inducible nitric oxide synthase (iNOS) expression (38, 39). Furthermore, Tax inhibits up to 80% of HR to cause saturation of classical NHEJ and prevent the repair of newly acquired DSBs (40). Tax also promotes the dephosphorylation of the ATM kinase via the WIP1 phosphatase, which leads to a bypass of the G2/M DNA damage response checkpoint and accelerates cellular replication (41–47).
Although Tax is well established to play a significant role in creating genomic instability, and its expression is sufficient to induce leukemogenesis in Tax transgenic mouse models, it remains unclear whether oncogenesis in humans is dependent upon additional factors (48–50). Interleukin-2 (IL-2)-dependent, Tax-mediated immortalization of human primary CD4+ T cells is possible, but it is a highly inefficient and rare event (51). Furthermore, many ATL cell clones exhibit a loss of Tax expression through mutation, methylation, or deletion of the 5′ long terminal repeat (LTR), and cells that retain Tax expression exhibit only short bursts of sense proviral transactivation (52–56).
Unlike Tax, the expression of the antisense hbz gene, which is regulated by the 3′ LTR, is retained throughout infection (57–65). HBZ exhibits a variety of pro-oncogenic properties, including stimulating cellular proliferation, accelerating cell cycle progression through the upregulation of E2F-mediated transcription, and suppressing apoptosis (59, 66–69). Although little is known about whether HBZ contributes to the accumulation of chromosomal abnormalities, one study reports that ectopic HBZ expression increases the frequency of DNA breaks (70). Furthermore, HBZ-expressing cells exhibit increased sensitivity to DSBs induced by etoposide-mediated topoisomerase II inhibition and delayed onset of DNA damage-induced G2/M arrest (71, 72). These findings led us to question whether sensitivity to DSBs in HBZ-expressing cells is the result of a defect in one or more mechanisms of DSB repair.
We report that HBZ attenuates NHEJ repair of both compatible and incompatible DSBs. We observed that HBZ interacts with the NHEJ-initiating proteins Ku70 and Ku80 through its bZIP domain and that HBZ is recruited to DSB sites in vitro in a bZIP-dependent manner. Although we did not find the interaction shared between DNA-PKcs and Ku70 to be affected by HBZ, we observed that autophosphorylation at Ser2056 is delayed following treatment with etoposide, also in a bZIP-dependent manner, suggesting that HBZ interferes with the activation of DNA-PK kinase activity in response to DNA damage. Although Tax is reported to interact with Ku70, it did not interfere with the formation of the HBZ:Ku complex. Interestingly, HBZ-mediated attenuation of NHEJ was masked by the coexpression of Tax, which enhanced NHEJ activity. Together, these findings suggest that DSB repair mechanisms are impaired not only by Tax but also by HBZ and show that HBZ may contribute to the accumulation of DSBs and chromosomal abnormalities over the course of HTLV-1 infection, especially in infected cell clones that no longer express Tax consistently.
RESULTS
HBZ interacts with DNA damage repair proteins Ku70 and Ku80 via the bZIP domain.
HBZ interacts with several cellular transcription factors and coactivators, allowing this virally encoded protein to exert control over cellular gene expression. We hypothesized that these and other interactions allow HBZ to influence nuclear processes. To identify novel HBZ-binding partners, we performed a proteomic screen in which we analyzed affinity-purified HBZ protein complexes. Briefly, HBZ-containing protein complexes were immunoprecipitated from HeLa cells stably transfected with the pCMV-HBZ-FLAG expression vector. The FLAG epitope tag, located on the C-terminal end of HBZ, allowed for FLAG immunoprecipitation-mediated purification of HBZ-containing protein complexes. Eluted proteins were subsequently identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS). To account for possible nonspecific protein interactions with the anti-FLAG antibody, immunoprecipitation was repeated using protein extracts from cells containing the empty vector control (pCMV-3Tag-8). As expected, this screen produced peptide fragments unique to HBZ as well as those corresponding to several reported HBZ-interacting proteins, including p300/CBP (73–75), c-Jun (76), JunB (76), JunD (77), ATF-1 (78), ATF-7 (79), CREB-1 (78), CREM (78), C/EBPG (68), and MafG (79) (Fig. 1A). A total of 476 proteins were uniquely identified in HBZ-FLAG samples and were manually annotated based on function. Interestingly, a small fraction of these proteins had been reported to function in DNA damage repair. Among these proteins were Ku70 and Ku80, which form a dimer that binds DSBs to initiate NHEJ by recruiting the DNA-PK catalytic subunit (DNA-PKcs) to damaged sites. Given the role of NHEJ in genomic rearrangements (80), we focused on the potential interaction between HBZ and the Ku proteins.
FIG 1.

HBZ interacts with Ku through its bZIP domain. (A) Table outlining previously reported HBZ-interacting partners as well as the novel HBZ-interacting proteins Ku70 and Ku80 that were recovered from HBZ-FLAG immunoprecipitations and identified by LC-MS/MS. Protein identifications were accepted at a false discovery rate of <1%, a probability of ≥95%, and a maximum of 1 missed cleavage. The numbers of peptides identified are shown. (B) HEK 293T cells were transfected with 12 μg of plasmid DNA (4 μg pEGFP-Ku70-FLAG or pEGFP-Ku80-FLAG, and 8 μg pcDNA-HBZ-Myc-His) for 48 h. Anti-FLAG antibody was used to immunoprecipitate FLAG-Ku70 and FLAG-Ku80 from 300 μg of protein extracts. Eluates (lanes 4 to 8) and 30 μg of protein input (lanes 1 to 3) were analyzed by Western blotting (WB) using the indicated antibodies. (C) Endogenous Ku70 and Ku80 were coimmunoprecipitated with HBZ from 500 μg of total protein harvested from HeLa cells stably expressing HBZ-FLAG or the empty vector. FLAG immunoprecipitation (IP) eluates (lanes 3 and 4) and 30 μg of protein inputs (lanes 1 and 2) were analyzed by Western blotting using the indicated antibodies. Immunoprecipitations with rabbit IgG were concurrently performed and are shown lanes 5 and 6. (D) MT-2 cells were electroporated with 15 μg of pCMV-HBZ-FLAG or the empty vector, and 48 h later, cells were treated for 5 h with 50 μM etoposide or DMSO. Anti-FLAG immunoprecipitations were performed with 1 mg of the cell lysate. Eluates and 2.5% protein inputs were analyzed by Western blotting using the indicated antibodies. (E) Schematic of wild-type HBZ (WT) and HBZ truncation mutants (ΔAD and ΔbZIP) used throughout this study. HBZ contains an N-terminal activation domain (AD), centrally located basic regions (BR), and a C-terminal basic leucine zipper (bZIP) domain. (F) HEK 293T cells were transfected with 12 μg of plasmid DNA (4 μg pEGFP-Ku70-FLAG and 4 μg pcDNA-HBZ-Myc-His, or 8 μg pcDNA-HBZ-ΔAD-Myc-His, or 4 μg pcDNA-HBZ-ΔbZIP-Myc-His and 4 μg empty vector as needed). Ku70-FLAG was immunoprecipitated from 300 μg of protein extracts using anti-FLAG antibody. Eluates (lanes 5 to 8) and 30 μg of protein inputs (lanes 1 to 4) were analyzed by Western blotting using the indicated antibodies. (G) HEK 293T cell lysates (800 μg) were incubated with GST alone, GST-HBZ, or GST-HBZ-bZIP. Bound proteins were eluted and analyzed by Western blotting. The membrane was first stained with Ponceau S and then incubated with the indicated antibodies.
We confirmed the interaction of HBZ with Ku70 and Ku80 using coimmunoprecipitation with lysates from HEK 293T cells transiently overexpressing HBZ-Myc-His, FLAG-Ku70, and FLAG-Ku80. Ku proteins, each of which contains an N-terminal FLAG tag, were immunoprecipitated with anti-FLAG antibody, and the eluates were analyzed by Western blotting for the presence of HBZ by probing for its C-terminal Myc tag (Fig. 1B). Additional immunoprecipitations were performed to assess the interaction between HBZ and endogenous Ku70 and Ku80. In these experiments, anti-Flag antibody was used to immunoprecipitate HBZ from HBZ-FLAG-expressing HeLa cells, and Western blots were probed with Ku70 and Ku80 antibodies (Fig. 1C). As shown, both endogenous proteins were coimmunoprecipitated with HBZ. To confirm that HBZ and Ku proteins also interact in HTLV-1-infected T cells, MT-2 cells were electroporated with pCMV-HBZ-FLAG or the empty vector, and HBZ was immunoprecipitated using anti-FLAG antibody (Fig. 1D). We observed that endogenous Ku70 and Ku80 were coimmunoprecipitated with HBZ (Fig. 1D, lane 4). To determine whether the interaction was enhanced by the induction of DNA breaks, we treated the cells with the topoisomerase II inhibitor etoposide to induce DSBs (Fig. 1D, lanes 5 to 8). We did not observe an enhancement of HBZ:Ku complex formation, suggesting that these interactions occur independently of DNA damage.
HBZ is characterized by three major regions (Fig. 1E): the N-terminal activation domain (AD) that directly interacts with cellular coactivators and histone acetyltransferases (72–75), the central basic regions (BRs) that facilitate nuclear localization (81), and the C-terminal basic leucine zipper (bZIP) motif that mediates interactions between HBZ and cellular bZIP transcription factors (68, 76–78, 82). To determine which of these regions is important for facilitating the interaction with Ku, we performed immunoprecipitations with cell lysates from HEK 293T cells that transiently overexpressed FLAG-tagged Ku70 and either wild-type HBZ (HBZ-WT), HBZ-ΔAD, or HBZ-ΔbZIP (Fig. 1F). Deletion of the HBZ AD did not prevent HBZ from interacting with Ku70 (Fig. 1F, lane 8), but deletion of the bZIP domain abolished the interaction (Fig. 1F, lane 7), indicating that the bZIP domain facilitates the HBZ:Ku interaction. To determine whether the bZIP domain of HBZ is sufficient for the interaction, we performed a glutathione S-transferase (GST) pulldown assay using GST fused to full-length HBZ (GST-HBZ) or the bZIP domain of HBZ alone (GST-bZIP). When incubated with HEK 293T cell lysates, GST-HBZ bound both Ku70 and Ku80, while GST alone did not (Fig. 1G). We observed much less Ku70/Ku80 bound to the bZIP domain alone than to full-length HBZ, which may arise from the GST portion of the protein partially obstructing the binding of the Ku proteins to the adjacent bZIP domain, as previously observed (75). It is also possible that an additional region of HBZ increases the stability of the interaction and that the bZIP domain is not sufficient for the interaction.
HBZ attenuates repair of double-stranded DNA breaks by NHEJ.
Since Ku70 and Ku80 are involved in NHEJ repair, we investigated whether HBZ impacts the repair of DSBs using previously described classical NHEJ repair reporter vectors to quantify successful repair in HBZ-expressing cells and empty vector control cells (Fig. 1A) (83, 84). Briefly, the NHEJ reporter plasmids carry one copy of the green fluorescent protein (GFP) reporter gene interrupted by a 3-kb PEM1 gene intron. Within the intron lies an adenoviral (AV) exon flanked by I-SceI restriction endonuclease sites. The NHEJ-compatible break (NHEJ-C) reporter plasmid has two I-SceI restriction sites in the direct orientation, which, upon digestion, create two compatible ends that do not require additional nucleolytic processing prior to ligation. The NHEJ-incompatible break (NHEJ-I) plasmid has two inverted I-SceI sites that generate incompatible ends, requiring nucleolytic processing prior to ligation. Excision of the AV exon during repair followed by splicing of the transcript results in GFP expression, which is quantified by flow cytometry.
To determine whether HBZ influences NHEJ of compatible DNA breaks, we transfected HEK 293T cells with the NHEJ-C reporter, the I-SceI expression vector, and either pcDNA-HBZ-Myc-His or the empty vector (Fig. 2C). Additionally, one group of cells was treated with the NHEJ inhibitor NU7441, an inhibitor of DNA-PK enzymatic activity, as a control (85). Following flow cytometry analysis of GFP-positive cells, we observed that HBZ expression significantly attenuated NHEJ-C activity, as did treatment with NU7441 (Fig. 2C). We found that attenuation was specific to the activity of the HBZ protein, since HBZ-ΔATG, which produces the HBZ transcript but not the protein, did not affect NHEJ repair. This mutant was analyzed based on previous reports indicating that the HBZ transcript is capable of deregulating transcription (59). We also confirmed that HBZ attenuates NEHJ-C in Jurkat T cells (Fig. 2D). In addition, we evaluated the efficiency of NHEJ of incompatible DNA breaks (NHEJ-I) and found that HBZ also significantly attenuates NHEJ-I activity (Fig. 2E). Finally, we assessed whether HBZ affects the HR pathway using the HR reporter pDR-GFP (Fig. 2B) (86). This reporter encodes a full-length copy and a 5′- and 3′-truncated copy of GFP separated by 3.7 kb containing a puromycin cassette. Eleven base pairs were deleted from the full-length GFP cDNA sequence and replaced with an I-SceI site and two in-frame stop codons (Sce-GFP). Once digested, the HR machinery uses the truncated internal GFP (iGFP) sequence as the template for repair, thereby replacing the missing nucleotides to allow the expression of the full-length fluorescent product. HEK 293T cells were transiently transfected with the pDR-GFP reporter plasmid and either pcDNA-HBZ-Myc-His or the empty vector. Additionally, one group of cells was treated with the HR inhibitor B02 as a control for this assay (87). We observed that HBZ failed to affect the efficiency of HR. The expression of HBZ and I-SceI was confirmed by Western blotting of the transfected cells (Fig. 2G).
FIG 2.

GFP reporter-based DSB repair vectors reveal that HBZ attenuates NHEJ-C and NHEJ-I but not HR. (A) Schematics of the reporter vectors used to measure NHEJ of compatible DSBs (NHEJ-C) and NHEJ of incompatible breaks (NHEJ-I), both of which are part of the classical NHEJ pathway. The GFP reporter gene is separated by the PEM1 gene intron containing an adenoviral (AV) exon flanked by two I-SceI restriction sites in a direct orientation (NHEJ-C) or an indirect orientation (NHEJ-I). Cleavage at these sites results in the excision of the AV exon and repair by NHEJ. The PEM1 intron is spliced out of the transcript at the splice donor (SD) site and splice acceptor (SA) site, resulting in an uninterrupted GFP-encoding transcript. CMV, cytomegalovirus. (B) Schematic of the reporter vector used to measure HR. This vector contains one complete copy of the GFP reporter gene (Sce-GFP) and a truncated copy of GFP that serves as a template for repair (iGFP), which are separated by a puromycin resistance cassette (Puro). Eleven base pairs were deleted from the Sce-GFP cDNA sequence and replaced with one I-SceI restriction site and two in-frame stop codons. This missing sequence ensures that template-dependent repair must occur to result in the production of a full-length GFP gene. (C) HEK 293T cells were transfected with a total of 6 μg of plasmid DNA (2 μg NHEJ-C reporter, 2 μg pSG-I-SceI-HA, 2 μg pcDNA-HBZ-Myc-His or pcDNA-ΔATG-Myc-His, or 2 μg empty vector) and treated with 2 μM NU7441 where indicated. Cells were collected for flow cytometric analysis of GFP expression, as outlined in Materials and Methods. Data were collected from 20,000 individual events and are shown as fold changes from the maximum repair activity (I-SceI plus reporter). HBZ data are averages of results from 10 independent experiments, ΔATG data are averages of results from three independent experiments, and NU7441 data are averages of results from 3 independent experiments. Error bars represent SEM (*, P ≤ 0.05 by Student's t test). (D) Jurkat cells were electroporated with a total of 15 μg of plasmid DNA (5 μg pcDNA-HBZ-Myc-His or empty vector and 10 μg NHEJ-C I-SceI-digested reporter) and analyzed for GFP expression. Data shown are averages of results from two independent experiments, and error bars indicate SEM (*, P ≤ 0.05 by Student's t test). (E) HEK 293T cells were transfected with a total of 6 μg of plasmid DNA (2 μg pSG-I-SceI-HA, 2 μg pcDNA-HBZ-Myc-His or empty vector, and 2 μg NHEJ-I reporter) and analyzed as outlined above. Data shown are averages of results from five independent experiments, and error bars represent SEM (*, P ≤ 0.05 by Student's t test). (F) HEK 293T cells were transfected with a total of 6 μg of plasmid DNA (2 μg pSG-I-SceI-HA, 2 μg pcDNA-HBZ-Myc-His or empty vector, and 2 μg the pDR-GFP reporter) and treated with 20 μM B02 for 48 h where indicated. Data shown are averages of results from three independent experiments, and error bars represent SEM (*, P ≤ 0.05 by Student's t test). (G) Transfected cellular protein extracts (30 to 60 μg) were analyzed by Western blotting for HBZ and/or I-SceI expression using the indicated antibodies. HA, hemagglutinin.
HBZ does not impair Ku-mediated DSB end recognition.
It was possible that HBZ inhibited NHEJ repair by binding to the Ku proteins and blocking their recruitment to DSBs. In vitro DNA-binding assays have been extensively used to characterize interactions between NHEJ core machinery and DNA termini (88–93). We previously used immobilized-DNA-binding assays to assess protein-DNA interactions (94). We tested whether HBZ influences the recruitment of Ku70 or Ku80 to DSBs using immobilized-DNA-binding assays in which a biotinylated double-stranded, blunt-end DNA fragment (35 bp) was immobilized on streptavidin-coupled beads and incubated with nuclear extracts from HEK 293T cells transiently overexpressing HBZ-WT, HBZ-ΔAD, or HBZ-ΔbZIP. Proteins captured by the immobilized DNA were eluted and analyzed by Western blotting to determine if HBZ influences Ku recruitment and, if so, which domain of HBZ is responsible for this activity (Fig. 3A). The results indicate that HBZ neither promoted nor impaired Ku recruitment to DNA termini. Interestingly, we noted that while HBZ-WT and HBZ-ΔAD were recruited to the DNA, HBZ-ΔbZIP was not, indicating that the bZIP domain of HBZ is important for the recruitment of HBZ to DSBs as well as for facilitating its interaction with Ku. These data suggest that HBZ is recruited to DNA lesions in a Ku-dependent manner.
FIG 3.

HBZ does not impair Ku-mediated DSB end recognition. (A) HEK 293T cells were transfected with the indicated plasmids, and nuclear proteins were harvested as described in Materials and Methods. Nuclear protein (10 μg) was incubated with an immobilized, blunt-ended, double-stranded DNA fragment (ITA:DSB) or with unbound resin (ITA:beads). Eluates and 5 μg of protein inputs (50% input) were analyzed by Western blotting using the indicated antibodies. ITA, immobilized template assay. (B) HeLa cells stably expressing HBZ or the empty vector were cotransfected with the NHEJ-C reporter and the I-SceI expression vector. ChIP assays were performed using Ku70 antibody. Primers located in proximity to the I-SceI cleavage site as well as an upstream, noncleaved site within the GFP gene (∼1,800 bp upstream) were used to amplify precipitated DNA and are indicated by facing arrows on the schematic. Data shown are averages of results from four independent experiments and represent fold changes in Ku70 enrichment in proximity of the I-SceI cut site (I-SceI) relative to Ku70 enrichment at the 5′ end of the GFP sequence (5′ GFP). Error bars represent SEM (*, P ≤ 0.05 by Student's t test). (C) HEK 293T cells were transfected with 12 μg of plasmid DNA (4 μg pEGFP-Ku70-FLAG and 8 μg pcDNA-HBZ-Myc-His or empty vector). Protein extracts were divided and either treated with 250 U of Benzonase for 45 min or left untreated. FLAG-Ku70 was immunoprecipitated from approximately 3 mg of protein using anti-FLAG antibodies. Eluates (lanes 5 to 8) and 10% of the input protein (lanes 1 to 4) were analyzed by Western blotting using the indicated antibodies.
To confirm that HBZ does not affect the recruitment of Ku proteins to DSBs, we performed chromatin immunoprecipitation (ChIP) assays to evaluate Ku70 recruitment to cleaved sites on the NHEJ-C plasmid, as previously reported (95–97). For these assays, stable HeLa cell lines expressing HBZ or containing the empty vector (pcDNA) were transiently transfected with the NHEJ-C reporter plasmid and the I-SceI expression plasmid. ChIP assays were performed 24 h later, at which time DNA repair proteins are highly enriched at I-SceI cleavage sites (96, 97). Quantitative PCR of ChIP samples was used to analyze Ku70 binding in proximity to the I-SceI cleavage site as well as an upstream, noncleaved site within the GFP gene (Fig. 3B). As expected, we observed significantly higher Ku70 enrichment at the I-SceI cleavage site than at the noncleaved site in both the cell line containing the empty vector and the HBZ-expressing cell line. Consistent with the biotinylation assays, HBZ did not reduce or alter Ku70 binding to the I-SceI cleavage site.
These results, and the fact that etoposide did not increase binding between HBZ and Ku proteins (Fig. 1D), suggest that the stability of this protein complex is not dependent upon DNA contact. To evaluate the importance of DNA in the formation of the HBZ:Ku complex, we immunoprecipitated overexpressed FLAG-tagged Ku70 from HEK 293T cell lysates treated with the endonuclease Benzonase (Fig. 3C). The results show that the interaction between Ku70 and HBZ is maintained in endonuclease-treated cell lysates, indicating that the stability of this protein complex is not DNA dependent and that the HBZ:Ku complex may be recruited to DNA lesions in a Ku-dependent manner.
The bZIP domain of HBZ is important for attenuation of NHEJ.
We next addressed whether the interaction between HBZ and Ku contributes to the HBZ-mediated attenuation of NHEJ. Because our findings support that HBZ interacts with Ku via its bZIP domain, we hypothesized that HBZ mutants lacking this region would be unable to attenuate NHEJ-C. HEK 293T cells were transiently transfected with the NHEJ-C reporter plasmid, the I-SceI restriction enzyme expression plasmid, and either HBZ-WT, HBZ-ΔAD, or HBZ-ΔbZIP and analyzed at 48 h posttransfection for successful NHEJ-mediated repair (Fig. 4). We observed that HBZ-ΔAD reduced repair similarly to HBZ-WT, suggesting that the activation domain is not required for the attenuation of NHEJ. HBZ-ΔbZIP was unable to attenuate repair, suggesting that the bZIP domain suppresses the activation of the NHEJ-C pathway. Because the level of expression of HBZ-ΔAD is consistently lower than the level of expression of full-length HBZ or HBZ-ΔbZIP in these types of experiments, we increased the amount of the HBZ-ΔAD expression vector that was transfected. We observed that the resulting increase in HBZ-ΔAD expression led to an even greater attenuation of NHEJ-C repair (Fig. 4, lane 5).
FIG 4.
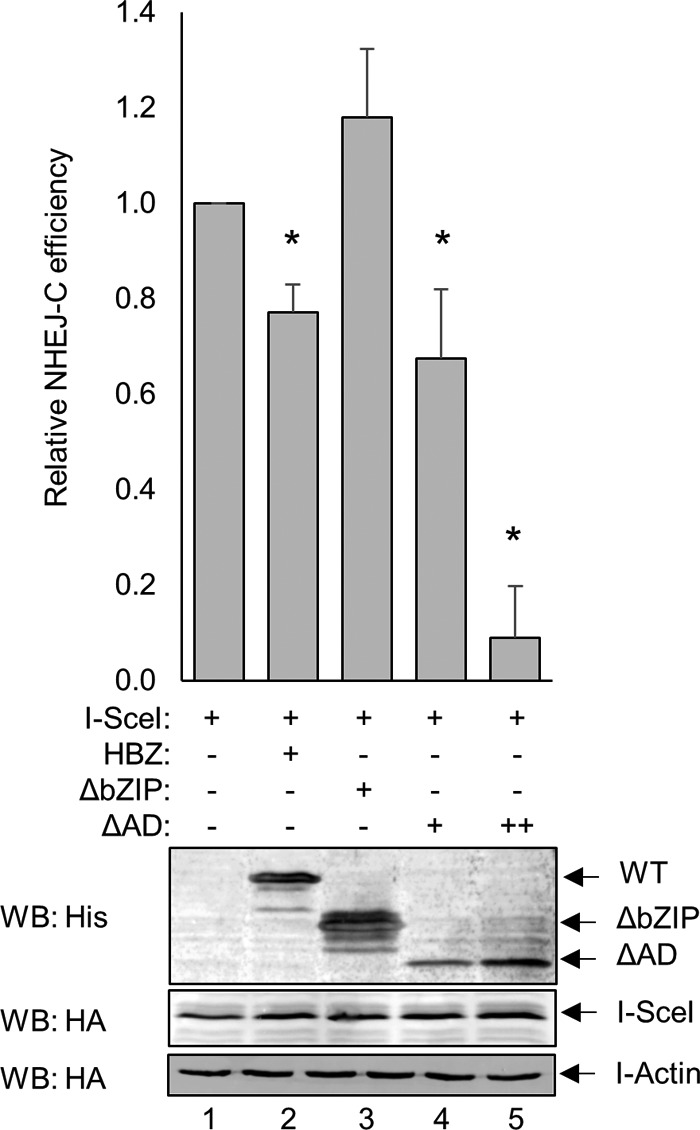
The bZIP domain of HBZ is important for the attenuation of NHEJ. HEK 293T cells were transfected with a total of 6 to 8 μg of plasmid DNA (2 μg pSG-I-SceI-HA, 2 μg pcDNA-HBZ-Myc-His or empty vector, or 2 μg pcDNA-HBZ-ΔbZIP-Myc-His or 2 to 4 μg pcDNA-HBZ-ΔAD-Myc-His, and 2 μg NHEJ-C reporter). GFP expression was detected by flow cytometry, and the fold change in NHEJ-C activity was calculated from 20,000 events. Data are averages of results from three independent experiments, and error bars represent SEM (*, P ≤ 0.05 by Student's t test). Transfected cellular protein extracts (50 μg) were analyzed by Western blotting for HBZ and I-SceI expression using the indicated antibodies.
HBZ impairs etoposide-induced DNA-PK autophosphorylation.
Because we did not find HBZ to inhibit the recruitment of Ku70 or Ku80 to DNA termini in vitro, or to reduce the level of chromatin-bound Ku70 in vivo, it is unlikely that HBZ attenuates NHEJ at the DSB recognition step. Therefore, we hypothesized that HBZ modulates Ku-dependent processes that occur after the recognition of DNA termini. DNA-PK holoenzyme recruitment and activation are believed to be important for facilitating synaptic-end bridging and preventing premature DNA processing (98). To determine if DNA-PK activities are impacted by HBZ, we first tested whether HBZ impedes the interaction between Ku70 and DNA-PKcs by immunoprecipitating Ku70 from HEK 293T cell lysates transiently overexpressing FLAG-Ku70 and HBZ-Myc-His. Prior to immunoprecipitation, cells were treated with etoposide to stimulate DNA-PK complex formation (Fig. 5A). The data show that DNA-PKcs is coimmunoprecipitated with Ku70 in cells transfected with the empty vector and in cells expressing HBZ, indicating that HBZ does not impair the formation of the DNA-PK holoenzyme. We observed that the basal interaction between Ku70 and DNA-PKcs is high in the absence of etoposide, which may be due to the overabundance of transfected Ku70.
FIG 5.

HBZ interferes with etoposide-induced DNA-PK autophosphorylation. (A) HEK 293T cells were transfected with 12 μg of plasmid DNA (6 μg pEGFP-Ku70-FLAG and 6 μg pcDNA-HBZ-Myc-His or pcDNA 3.1) for 24 h. Cells were treated with 50 μM etoposide or equivalent amounts of DMSO for 5 h before protein was extracted. FLAG-Ku70 was immunoprecipitated from 1 mg of protein using anti-FLAG antibodies. Eluates (lanes 7 to 12) and 20 μg of protein inputs (lanes 1 to 6) were analyzed by Western blotting using the indicated antibodies. (B) HeLa cells stably expressing HBZ (lanes 5 to 8) or HBZ-ΔbZIP (lanes 9 to 12) or carrying an empty vector (lanes 1 to 4) were treated with 50 μM etoposide over a 90-min time course. Protein was extracted, and 30 μg of each protein was analyzed by Western blotting using the indicated antibodies. The level of DNA-PKcs autophosphorylation at Ser2056 was quantified, and the data shown are averages of results from three independent experiments. Error bars represent SEM (*, P ≤ 0.05; ns, not significant from the pcDNA value at the same time point [as determined by Student's t test]). (C) HeLa cell lines stably expressing HBZ or the empty vector were treated with 50 μM etoposide or γ-irradiated (10 Gy) and allowed to recover for the amount of time indicated before protein extracts were prepared and analyzed for DNA-PK autophosphorylation at Ser2056 or total DNA-PKcs. Protein extracts (15 μg) were analyzed by Western blotting.
The activation of DNA-PK kinase activity is dependent upon the phosphorylation of several residues and is reported to be critical for the completion of NHEJ (19, 99–103). Although DNA-PK is phosphorylated by a number of other kinases, Ser2056 has been identified as a bona fide autophosphorylation site that is important for destabilizing DNA-PK from its position at the DNA break prior to ligation, a step critical for the completion of NHEJ (15, 19). We tested whether HBZ impairs DNA-PK Ser2056 autophosphorylation by treating HeLa cells stably transfected with pcDNA-HBZ-Myc-His, pcDNA-HBZ-ΔbZIP-Myc-His, or the empty vector with etoposide over a 90-min time course (Fig. 5B). Cells were processed at 30-min intervals, and lysates were treated with phosphatase inhibitors to prevent protein dephosphorylation. From Western blot analyses, we observed both a delay and a significant reduction in DNA-PK autophosphorylation in HBZ-expressing cells relative to cells carrying the empty vector (Fig. 5B). In cells expressing the HBZ-ΔbZIP mutant, we did not observe a significant delay in DNA-PK autophosphorylation. Overall, our findings indicate that HBZ, primarily through the bZIP domain, delays and reduces DNA-PK autophosphorylation following the induction of DSBs, which may repress the maximal activation of DNA-PK.
Additionally, we tested whether HBZ influences levels of the chromatin-bound endonucleases and ligation machinery, which are also important for the completion of NHEJ. HeLa cell lines stably transfected with pcDNA-HBZ-Myc-His or the empty vector were treated with etoposide or equivalent volumes of dimethyl sulfoxide (DMSO) prior to the biochemical separation of chromatin-bound proteins. We were unable to observe noticeable changes in the levels of these downstream NHEJ enzymes within the chromatin fraction (data not shown); however, it is unknown whether they retain complete functionality in HBZ-expressing cells.
To determine whether HBZ inhibition of DNA-PK autophosphorylation applies to other mechanisms of DSB induction, we evaluated the effect of gamma irradiation on DNA-PK autophosphorylation, which also activates the phosphorylation of Ser2056 (15). HeLa cell lines expressing HBZ or carrying the empty vector were either γ-irradiated or treated with etoposide. As previously reported (15), gamma irradiation (10 Gy) caused a lower DNA-PK autophosphorylation signal than etoposide treatment (Fig. 5C). In contrast to etoposide treatment, gamma irradiation did not produce a difference in autophosphorylation between HBZ-expressing and empty-vector-expressing cells. This result suggests that HBZ inhibits DNA-PK autophosphorylation in response to DSBs with homogeneous structures (produced by etoposide) but not those with heterogeneous structures (produced by gamma irradiation) (104).
Ku simultaneously forms complexes with HBZ and Tax to differentially modulate NHEJ activity.
The HTLV-1-encoded protein Tax also dysregulates NHEJ through a diverse set of mechanisms that may impact the effect of HBZ on this repair pathway (40, 42). We addressed this possibility by first evaluating Ku70 and Ku80 protein levels in a panel of HTLV-1-infected and uninfected T-cell lines, as Tax was previously reported to reduce the expression of these proteins (Fig. 6A) (105). Western blot analysis of cell lysates shows that the virally infected cells retain the expression of the Ku proteins (Fig. 6A, lanes 8 to 14), suggesting that the effect of HBZ on NHEJ remains relevant in the presence of Tax.
FIG 6.

Ku simultaneously forms complexes with HBZ and Tax to differentially modulate NHEJ activity. (A) Protein extracts (15 μg) from activated primary CD4+ T cells (anti-CD3 and anti-CD28) as well as uninfected and HTLV-1-infected T-cell lines were analyzed by Western blotting using the indicated antibodies. (B) HEK 293T cells were transfected with 12 μg of plasmid DNA (3 μg pEGFP-Ku70-FLAG, 6 μg pcDNA-HBZ-Myc-His, 3 μg pSG-Tax-His, and empty vector as needed). FLAG-Ku70 was immunoprecipitated from 2 mg of protein using an anti-FLAG antibody. Eluates (lanes 5 to 8) and 50 μg of protein inputs (lanes 1 to 4) were analyzed by Western blotting using the indicated antibodies. (C) HEK 293T cells were transfected with 7 μg of plasmid DNA (2 μg pSG-I-SceI-HA, 2 μg pSG-HBZ-Myc, 1 μg pSG-Tax-His, 2 μg NHEJ-C reporter, and empty vector as needed). Where indicated, cells were treated with 2 μM NU7441 for 48 h. GFP expression was detected by flow cytometry, and the change in NHEJ-C activity was calculated from 20,000 events. Data for NU7441 and HBZ plus Tax plus NU7441 are averages of results from three independent experiments, and the remaining data are averages of results from six independent experiments. Error bars represent SEM (*, P ≤ 0.05 by Student's t test). Transfected cellular protein extracts (60 μg) were analyzed by Western blotting for HBZ, Tax, and I-SceI expression using the indicated antibodies.
Tax was also reported to interact with Ku70 (42), leading us to question whether Tax impedes the interaction between HBZ and Ku70 in cells coexpressing Tax and HBZ. To address this possibility, Ku70 was immunoprecipitated from HEK 293T cell lysates transiently overexpressing FLAG-Ku70 or HBZ-Myc-His and Tax tagged with a C-terminal 6×His epitope tag (Fig. 6B). We observed that both Tax and HBZ coimmunoprecipitated with Ku70 when expressed alone (Fig. 6B, lanes 6 and 7) or in combination (Fig. 6B, lane 8). Given that the Ku proteins are abundantly expressed, these data support that that neither protein influences the interaction of the other with Ku70.
Finally, Tax was reported to repress the HR repair pathway, leading to the saturation of NHEJ to compensate for the lost repair mechanism (40, 42). To determine how the coexpression of Tax and HBZ affects NHEJ repair, we evaluated the efficiency of NHEJ using the flow cytometric NHEJ reporter assay in HEK 293T cells transiently expressing the NHEJ-C reporter, the I-SceI restriction enzyme, and a combination of HBZ and Tax (Fig. 6C). Similar to the results shown in Fig. 2C, we found that HBZ attenuates NHEJ-C when expressed alone. In contrast to this effect of HBZ, we found that Tax alone enhances repair through the NHEJ-C pathway. When HBZ was coexpressed with Tax, we observed a small but significant reduction in NHEJ-C, but not below the level of repair in the absence of both viral proteins. As a control for this assay, we used the DNA-PK inhibitor NU7441 and found that it reduced NHEJ-C below the baseline level regardless of the presence or absence of the viral proteins. These data suggest that while both Tax and HBZ are able to associate with the DNA-PK holoenzyme complex, they appear to have distinct effects on the NHEJ pathway.
DISCUSSION
The accumulation of genetic abnormalities is a hallmark of ATL cells and arises, in part, from the inadequate repair of DSBs. In this study, we found that HBZ inhibits DSB repair through the NHEJ pathway. First, we discovered novel interactions between HBZ and the NHEJ-initiating proteins Ku70 and Ku80, which recognize and stabilize DNA lesions as dimers. These interactions were identified through a proteomic screen and subsequently verified in coimmunoprecipitation assays. DSBs with homogenous (compatible ends) and heterogeneous (incompatible ends) structures can be repaired through the NHEJ pathway. Interestingly, we observed that HBZ significantly attenuated NHEJ-C (compatible ends) and, to a lesser extent, NHEJ-I (incompatible ends). Further characterization revealed that the bZIP domain of HBZ, specifically, is required for successful interactions between HBZ and both Ku70 and Ku80. Moreover, the HBZ bZIP domain was also found to be required for the suppression of NHEJ-C. We observed that the HBZ:Ku complex can form independently of DNA contact and that HBZ does not affect Ku70 recruitment to DSBs but instead accompanies Ku to DNA termini in vitro. These data suggest that HBZ accesses DSBs through its association with the Ku proteins.
Ku70 and Ku80 form the DNA-binding component of the DNA-PK holoenzyme. Once bound to a DNA break, Ku proteins recruit the DNA-PK catalytic subunit, which possesses kinase activity, to further stabilize the break and coordinate repair. We observed that although HBZ forms a complex with Ku, it does not appear to disrupt the interaction between Ku70 and DNA-PKcs. However, HBZ decreased and delayed the autophosphorylation of DNA-PK at Ser2056 in response to DNA damage induced by etoposide. Phosphorylation at this residue promotes the destabilization of DNA-PK from the lesion to allow repair to be completed (19) and is therefore considered a marker of DNA-PK destabilization and the completion of NHEJ. Consequently, by reducing and delaying Ser2056 autophosphorylation in response to etoposide, HBZ may stabilize DNA-PK at DSBs to negatively impact later steps of NHEJ.
Intriguingly, HBZ did not affect Ser2056 DNA-PK autophosphorylation following gamma irradiation. This difference may reflect the fact that etoposide produces DSBs with homogeneous structures, while gamma irradiation produces DSBs with heterogeneous structures (104). This difference in DSB structure correlates with the disparity of HBZ in inhibiting repair through NHEJ-C and NHEJ-I, as NHEJ-C represents homogenous structures, while NHEJ-I represents heterogeneous structures. Differences in enzymatic processing requirements between the repair of compatible and incompatible DSBs have been reported. Therefore, in addition to inhibiting DNA-PK autophosphorylation, HBZ may target another enzymatic event that is more prominent in NHEJ-I repair.
Once activated, DNA-PK is demonstrated to phosphorylate a number of targets involved in NHEJ, including Artemis (106), AKT (107), H2AX (23), Ku70 and Ku80 (108), XRCC4 (109), XLF (110), and ligase IV (111); however, at this stage, the biological importance of these modifications remains largely unclear. Although the exact function of DNA-PK in NHEJ is not yet well defined, DNA-PK activity is essential for NHEJ, as the inhibition of kinase activity sensitizes cells to DNA damage-induced cell death (100, 112–116). Although we did not observe changes in levels of chromatin-bound NHEJ enzymes in HBZ-expressing cells after etoposide treatment, it is unclear whether they are present at the break site or distributed elsewhere on the chromatin. Further studies will be required to determine how NHEJ endonuclease and ligase activities are impacted by HBZ expression and the subsequent reduction in DNA-PK autophosphorylation.
Given that the viral protein Tax is known to have substantial effects on DSB repair (117, 118), it was important to understand how HBZ and Tax, together, affect the repair process. Interestingly, Tax has been shown to saturate the NHEJ repair pathway through two distinct mechanisms. The first mechanism involves Tax interactions with both DNA-PK and Ku70 and an increase in DNA-PK autophosphorylation (42). The second mechanism involves the inhibition of HR repair by Tax, which diverts DSB repair to the NHEJ pathway (40). Saturation of the NHEJ pathway has been shown to result in an inability of cells to repair subsequently acquired DSBs (119). Consistent with those previous data, we observed that NHEJ-C was enhanced in Tax-expressing cells. This effect was slightly diminished upon the coexpression of HBZ; however, in the presence of Tax, HBZ did not decrease NHEJ-C to or below the baseline level. These results suggest that the Tax-mediated enhancement of DNA-PK autophosphorylation might be stronger than the HBZ-mediated inhibition of autophosphorylation. It is important to note that the NHEJ assay used in this study provided a snapshot of DSB repair, and while Tax initially enhanced this pathway, over a more extensive time frame, Tax may ultimately impair NHEJ repair by enforcing the constitutive phosphorylation of DNA-PK (42).
The combined effects of HBZ and Tax on the NHEJ pathway are expected to be relevant to events occurring in primary HTLV-1-infected clones. A generally accepted model has been that Tax is expressed at high levels upon initial infection and later downregulated or silenced to allow the infected cell to evade a Tax-specific cytotoxic-T-cell response. However, Tax has also been reported to be highly expressed in transient, infrequent transcriptional bursts in primary HTLV-1-infected T-cell clones and in a minor subset of ATL cells (54, 55). In contrast, HBZ transcripts have been shown to be continuously present in HTLV-1-infected T cells (57–65). Therefore, given that both viral proteins have been reported to have relatively long half-lives (120–122), it is likely that HBZ and Tax are sometimes simultaneously present in primary HTLV-1-infected cells, during which time they could affect the repair of DSBs and additional processes involving DNA-PK.
In addition to modulating NHEJ repair, the activation of DNA-PK in response to DNA damage has been shown to stimulate apoptosis. Previous reports show that DNA-PK phosphorylates p53, which can activate the p53-dependent apoptosis cascade (21, 24–26). Additionally, DNA-PK was reported to phosphorylate the histone methyltransferase EZH2, leading to a reduction in its activity, which promotes apoptosis in T cells (27). Finally, DNA-PK is robustly activated in apoptotic cells and phosphorylates H2AX during apoptotic DNA fragmentation (23). It is possible that HBZ impairs DNA-PK activation to serve as a prosurvival strategy for HTLV-1-infected cells. Indeed, we previously reported that HBZ confers resistance to etoposide-induced apoptosis (72, 75). An overall reduction of DNA-PK activity in HTLV-1-infected cells may contribute to a reduction in NHEJ efficiency and lead to the accumulation of mutations while simultaneously suppressing apoptosis. These combined effects may be important for cellular transformation.
MATERIALS AND METHODS
Cell lines, cell culture, and treatments.
HEK 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin. Clonal HeLa-pCMV-3Tag8 and HeLa-pCMV-HBZ-FLAG (72) cells were cultured in spinner-flask suspensions in minimum essential medium (MEM) supplemented with 5% newborn calf serum, 3% fetal bovine serum, 2 mM l-glutamine, 1× MEM nonessential amino acids solution, 1 mM sodium pyruvate, 100 U/ml penicillin, 50 μg/ml streptomycin, and 0.2 mg/ml hygromycin B (Invitrogen). Clonal HeLa-pcDNA, HeLa-pcDNA-HBZ-Myc-His, and HeLa-pcDNA-HBZΔbZIP-Myc-His cell lines were maintained in supplemented DMEM under selection with 0.5 mg/ml Geneticin (ThermoFisher). T-cell lines were maintained in either supplemented Iscove's modified Dulbecco's medium (IMDM) or supplemented Roswell Park Memorial Institute (RPMI) medium. To inhibit NHEJ and HR repair activities, cells were treated for 48 h with 2 μM NU7441 (a DNA-PK inhibitor; Selleckchem) in DMSO or 20 μM B02 (a RAD51 inhibitor; Selleckchem) in DMSO. To induce the accumulation of DSBs, cells were treated with 50 μM etoposide (Sigma-Aldrich) in DMSO or were γ-irradiated (10 Gy) and cultured for the times indicated.
Plasmids and antibodies.
The following HBZ mammalian expression plasmids were previously described: pcDNA-HBZ Sp1-Myc-His (amino acids [aa] 1 to 206) (64), pcDNA-HBZ-ΔAD-Myc-His (aa 77 to 206) (82), pcDNA-HBZ-ΔbZIP-Myc-His (aa 1 to 130) (78), pcDNA-HBZ-ΔATG-Myc-His (123), pCMV-HBZ-FLAG (72), pSG-HBZ-Myc (123), pSG-Tax-His (65), and pCMV-HBZ-FLAG (72). Plasmids pEGFP-C1-FLAG-Ku70 and pEGFP-C1-FLAG-Ku80 were gifts from Steve Jackson (Addgene plasmids 46957 and 46958) (124). NHEJ-C and NHEJ-I reporter plasmids were provided by Vera Gorbunova (83). pDR-GFP was a gift from Maria Jasin (Addgene plasmid 26475) (86). I-SceI with a hemagglutinin (HA) tag was amplified from pCBASceI, also a gift from Maria Jasin (Addgene plasmid 26477) (125), using a 5′ BamHI primer and a 3′ BglII primer (sequences are available upon request) and subcloned into the pSG5 vector (Agilent Technologies) via BamHI and BglII digestion to construct pSG-I-SceI-HA. Empty vector plasmid pCMV-3Tag-8 was purchased from Agilent Technologies, and pcDNA3.1(+)/Myc-His A was purchased from Invitrogen.
The following antibodies were used: anti-FLAG M2 (catalog number F3165; Sigma-Aldrich), anti-Myc clone 4A6 (catalog number 05-724; EMD Millipore), anti-His H-15 (catalog number sc-803; Santa Cruz Biotechnology), anti-HA clone HA-7 (catalog number H3663; Sigma Aldrich), anti-actin clone C4 (catalog number MAB1501; EMD Millipore), anti-DNA-PKcs (catalog number 4602; Cell Signaling), anti-DNA-PKcs phospho-S2056 (catalog number ab18192; Abcam), anti-Ku70 D10A7 (catalog number 4588; Cell Signaling), anti-Ku70 (catalog number sc-5309; Santa Cruz Biotechnology), and anti-Ku80 C48E7 (catalog number 2180; Cell Signaling). Tax monoclonal antibody (hybridoma 168B17-46-92) was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program.
Identification of HBZ-binding proteins by liquid chromatography-tandem mass spectrometry.
HeLa-S3 cells expressing HBZ-FLAG or carrying the empty vector were cultured in spinner-flask suspension cultures under continual hygromycin selection. A total of 8 × 106 cells were pelleted at 350 × g for 3 min at 4°C, washed in 1× phosphate-buffered saline (PBS), and resuspended in 1 ml radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 8.0], 1% Triton X-100, 100 mM NaCl, 1 mM MgCl2, 400 nM trichostatin A, 2 μg/ml leupeptin, 5 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1 mM benzamidine). Cells were vortexed briefly and incubated on ice for 20 min. Lysates were centrifuged at 16,000 × g for 15 min at 4°C, and the soluble protein supernatant was isolated and quantified by a Bradford protein assay (Bio-Rad). Equalized cell lysates were precleared, and HBZ-FLAG protein complexes were immunoprecipitated by using anti-FLAG M2 magnetic resin (catalog number M8826; Sigma-Aldrich): 40 μl resin slurry was washed twice in 1× TBS+ (Tris-buffered saline plus 2 μg/ml leupeptin, 5 μg/ml aprotinin, 1 mM PMSF, and 1 mM benzamidine) and incubated with equalized protein extracts for approximately 16 h. Resin was washed four times in 1× TBS+, and protein complexes were eluted by FLAG peptide competition using 0.5 μg/μl FLAG peptide (catalog number F3290; Sigma-Aldrich) in 100 μl 1× TBS+ for 30 min. Eluates were run partially through an SDS-PAGE gel, and total protein bands were excised for in-gel digestion. Gel pieces were washed first in 50 mM ammonium bicarbonate (AMBIC) and then in acetonitrile. Samples were reduced with 10 mM dithiothreitol (DTT) in 50 mM AMBIC for 30 min at 56°C, washed in acetonitrile, alkylated with 55 mM iodoacetamide in 50 mM AMBIC for 20 min, washed in 50 mM AMBIC, and then washed in acetonitrile. Gel pieces were dried by speed vacuum centrifugation. Protein was digested in-gel using trypsin (Promega) in a 0.01% ProteaseMax–50 mM AMBIC solution at a 1:30 enzyme/protein ratio. Digestion was halted using formic acid, and peptides were extracted by covering the gel pieces with 60% acetonitrile–0.1% trifluoroacetic acid, followed by sonication. Peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Q-Exactive hybrid quadrupole-Orbitrap mass spectrometer; UC Davis Proteomics Core Facility). Tandem mass spectra were matched with peptide sequences by X! Tandem (Global Proteome Machine Organization) assuming the digestion enzyme trypsin. X! Tandem was searched with a fragment ion mass tolerance of 20 ppm and a parent ion tolerance of 20 ppm. Fixed modifications included cysteine carbamidomethylation. Variable modifications included Glu→pyro-Glu of the N terminus, ammonia loss of the N terminus, Gln→pyro-Glu of the N terminus, deamidated asparagine and glutamine, oxidation of methionine and tryptophan, dioxidation of methionine and tryptophan, and acetylation of the N terminus. Scaffold version Scaffold_4.7.3 (Proteome Software, Inc.) was used to validate and accept MS/MS-based protein identifications. Protein identifications were accepted at a false discovery rate of less than 1%, a probability of ≥95%, and a maximum of 1 missed cleavage. To exclude the possibility of nonspecific interactions of cellular proteins with anti-FLAG antibody, proteins that were identified in empty-vector-containing samples were excluded from the analysis, leaving proteins that were identified only in the HBZ-FLAG samples. Proteins that were uniquely identified in HBZ-FLAG samples were manually annotated based on function. Data are from a single experiment and are available upon request.
Coimmunoprecipitation and Western blotting.
Cells (2 × 106) were transfected using TurboFect (ThermoFisher) according to the manufacturer's instructions. Where applicable, etoposide treatment (50 μM) (Sigma-Aldrich) was carried out 4 to 5 h prior to harvesting protein extracts. Protein extracts were prepared at 24 to 48 h posttransfection. For nuclease-treated samples, 250 U Benzonase (Sigma-Aldrich) was added to cell lysates before immunoprecipitation. For coimmunoprecipitations performed with anti-FLAG M2 magnetic resin (catalog number M8826; Sigma-Aldrich), 15 μl of resin slurry was washed twice in RIPA buffer and incubated with the indicated amount of the protein extract for approximately 16 h at 4°C. For immunoprecipitations performed with other antibodies, 5 μg of the indicated antibody was preincubated with 15 μl protein G resin (Sigma-Aldrich) for 1 h and then washed in RIPA buffer and incubated with the indicated amount of the cell lysate for approximately 16 h at 4°C. For all immunoprecipitations, resin was washed four times in RIPA buffer, and bound proteins were eluted by SDS dye and resolved by SDS-PAGE and Western blotting. Immunoprecipitations using MT-2 cells were performed as follows: 3 × 106 MT-2 cells were electroporated at 200 V, 975 μF, and infinite resistance in 300 μl serum-free RPMI containing 0.1 mM DTT and 10 mM dextrose with 15 μg of pCMV-HBZ-FLAG or the empty vector in 0.4-cm cuvettes. Forty-eight hours later, where indicated, cells were treated for 5 h with 50 μM etoposide or DMSO. All blots were developed using Pierce ECL Plus (ThermoFisher) and scanned with a Typhoon 9410 imager (GE Healthcare). Quantification of DNA-PK phosphorylation was performed using Image Quant TL 1D software (version 8.1; GE Healthcare). DNA-PK phosphorylation bands were normalized against a constant, nonspecific band in the same lane on the same probed membrane.
NHEJ repair assays and flow cytometry.
HEK 293T cells (6 × 105) were transiently transfected (TurboFect) with 6 to 7 μg of DNA consisting of a mixture of a DNA repair reporter vector (NHEJ-C, NHEJ-I, or DR-GFP), pSG-I-SceI-HA, as well as the Tax and HBZ expression vectors described in the figure legends. Where indicated, cells were treated with 2 μM NU7441, 20 μM B02 (Selleckchem), or DMSO for 48 h. At 48 h posttransfection, cells were pelleted, washed twice in PBS, and resuspended in ice-cold PBS for flow cytometric analysis. Live cell populations were analyzed for GFP expression using an LSRII flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (FlowJo, LLC). Transfection groups consisted of a reporter vector alone, a reporter vector plus the I-SceI expression plasmid, and/or a reporter vector plus the I-SceI expression plasmid and expression vectors for the protein of interest (e.g., Tax and/or HBZ). For drug treatments, all samples contained the same amount of DMSO. For each experiment, background GFP levels were measured from reporter-only live cells and were subtracted from GFP levels in live cells from the other transfection groups. Maximum GFP expression (reporter plus I-SceI) was set to 1, and data are displayed as fold changes relative to GFP expression for the reporter–I-SceI group. For experiments performed with Jurkat cells, 3 × 106 cells were electroporated with the indicated expression vectors and cultured for 48 h.
In vitro GST pulldown assays.
Bacterial expression vectors for GST (pGEX-2T) and the GST-HBZ (pGEX-HBZ) and GST-bZIP (pGEX-HBZ-bZIP) fusion proteins were described previously (73, 78). pGEX plasmids were transformed in Escherichia coli BL21 codon plus(DE3) (Stratagene) and purified by glutathione-agarose affinity chromatography. GST pulldown assays were performed as previously reported, with minor modifications (78). Briefly, 20 μl of glutathione-agarose beads was equilibrated in RIPA buffer (50 mM Tris-HCl [pH 8.0], 1% Triton X-100, 100 mM NaCl, 1 mM MgCl2, 2 μg/ml leupeptin, 5 μg/ml aprotinin, 1 mM PMSF, 1 mM DTT). Purified GST fusion proteins (50 pmol) were incubated with equilibrated glutathione-agarose for 1 h at 4°C in binding buffer, followed by two washes in RIPA buffer to remove unbound protein. Cell lysates (800 μg) from HEK 293T cells were incubated with the beads for 16 h at 4°C. Beads were washed four times in RIPA buffer, and bound proteins were eluted using SDS loading dye. Proteins were analyzed by SDS-PAGE and Western blotting, using the indicated antibodies.
Nuclear protein extraction and in vitro immobilized-DNA-binding assays.
Nuclear protein extracts were prepared as follows: a total of 8 × 106 HEK 293T cells were transfected by calcium phosphate transfection with 50 μg to 100 μg of plasmid DNA, as indicated in the figure legends (50 μg pcDNA3.1, 50 μg pcDNA-HBZ-Myc-His, 100 μg pcDNA-HBZ-ΔAD-Myc-His, 50 μg pcDNA-HBZ-ΔbZIP-Myc-His, or 50 μg pcDNA-HBZ-ΔZIP), and nuclear protein was harvested at 48 h posttransfection using a previously described method (126, 127). Supernatants were collected and dialyzed for approximately 16 h against 0.1 M HM (50 mM HEPES [pH 7.9], 100 mM KCl, 20% glycerol, 12.5 mM MgCl2, 1 mM EDTA, 0.025% Tween, 1 mM DTT). The nuclear protein concentration was quantified by a Bradford protein assay (Bio-Rad).
Variations of the immobilized DNA template have been used to analyze the binding of NHEJ proteins to DNA termini (88–93). We performed our immobilized-DNA-binding assays as previously described, with only minor modifications (94). Per reaction, 2 pmol of biotinylated double-stranded DNA with a length of 35 bp was bound to 15 μl M-280 streptavidin-coupled Dynabeads (Invitrogen) according to the manufacturer's instructions. DNA-bound resin was blocked for 1 h in ITB (20 mM HEPES [pH 7.9], 0.2 mM EDTA, 100 mM KCl, 6.25 mM MgCl2, 10 mM ZnSO4, 20% glycerol, 0.01% Triton X-100, 0.2 mM PMSF, 1 mM benzamidine, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM DTT) with 5% bovine serum albumin (BSA). The resin was cleared, loaded with 10 μg of nuclear protein extracts in a total volume of 500 μl of ITB–5% BSA, and rocked for 2 h at 4°C. Resin was washed four times in ITB without BSA, and protein was eluted with SDS dye and resolved by SDS-PAGE and Western blotting.
Chromatin immunoprecipitation assays.
Cells (1 × 107) containing pcDNA 3.1 or expressing HBZ-Myc-His were transiently transfected with 10 μg of the NHEJ reporter vector and 10 μg of the I-SceI expression plasmid. At 24 h posttransfection, cells were treated with 1% formaldehyde for 10 min at 37°C and then quenched with 0.125 M glycine. Cells were harvested, and ChIP assays were performed using the Zymo Spin ChIP kit (Zymo Research) according to the manufacturer's instructions. For each ChIP reaction, 20 μg of cross-linked chromatin was diluted to a final volume of 1 ml in ChIP chromatin dilution buffer and incubated at 4°C overnight with 5 μg of anti-Ku70 A-9 antibody (catalog number sc-5309; Santa Cruz Biotechnology) or preimmune rabbit serum (IgG). Purified ChIP DNA was analyzed by real-time PCR as described previously (78). PCR primers amplified either a region of the reporter plasmid upstream and adjacent to the I-SceI cleavage site or a region at the 5′ end of the GFP gene (∼1,800 bp upstream). Primers are as follows: NHEJ I-SceI-F (5′-AACATAGCCCTGGAAAGAGAAG-3′), NHEJ I-SceI-R (5′-CTTGGAAACACCCATGTTGAAATATC-3′), NHEJ GFP-F (5′-CTGGACGGCGACGTAAAC-3′), and NHEJ GFP-R (5′-CGGTGGTGCAGATGAACTT-3′). Standard curves were generated for primer sets using 10-fold serial dilutions of each input DNA from the ChIP procedure and were included on each experimental plate. PCR efficiencies ranged from 98 to 118%, with correlation coefficients of >0.99. Quantitation was performed by comparing threshold cycle values for coimmunoprecipitated DNA to the threshold cycle value for the input DNA in each ChIP (128). Ku70 enrichment at each region of the reporter was normalized to the value for IgG. Values corresponding to the I-SceI-adjacent region were standardized to those corresponding to the GFP gene region, which were set to 1. Significance was calculated using the Student t test, and error bars represent standard errors of the means (SEM).
ACKNOWLEDGMENTS
We acknowledge Torsten Wurm for his preliminary work on this project, and we thank Stephanie Nguyen, Chyna Johnson, Kayla DeOca, and Alicia Kwon for their assistance. Additionally, we thank Vera Gorbunova for providing the NHEJ repair reporter plasmids used in this work.
Funding for this study was provided by the National Institutes of Health National Cancer Institute (NIH/NCI grant CA128800) to I.L. The funding agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Shen Z. 2011. Genomic instability and cancer: an introduction. J Mol Cell Biol 3:1–3. doi: 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 2.Helleday T, Eshtad S, Nik-Zainal S. 2014. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khanna KK, Jackson SP. 2001. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 4.De Bont R, van Larebeke N. 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 5.Costanzo V, Chaudhuri J, Fung JC, Moran JV. 2009. Dealing with dangerous accidents: DNA double-strand breaks take centre stage. Symposium on genome instability and DNA repair. EMBO Rep 10:837–842. doi: 10.1038/embor.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoeijmakers JHJ. 2001. Genome maintenance mechanisms for preventing cancer. Nature 411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 7.Sieber OM, Heinimann K, Tomlinson IPM. 2003. Genomic instability—the engine of tumorigenesis? Nat Rev Cancer 3:701–708. doi: 10.1038/nrc1170. [DOI] [PubMed] [Google Scholar]
- 8.Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Heyer W-D. 2008. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branzei D, Foiani M. 2008. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 11.Bishop AJ, Schiestl RH. 2000. Homologous recombination as a mechanism for genome rearrangements: environmental and genetic effects. Hum Mol Genet 9:2427–2334. doi: 10.1093/hmg/9.16.2427. [DOI] [PubMed] [Google Scholar]
- 12.Mao Z, Jiang Y, Liu X, Seluanov A, Gorbunova V. 2009. DNA repair by homologous recombination, but not by nonhomologous end joining, is elevated in breast cancer cells. Neoplasia 11:683–691. doi: 10.1593/neo.09312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang HHY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neal JA, Meek K. 2011. Choosing the right path: does DNA-PK help make the decision? Mutat Res 711:73–86. doi: 10.1016/j.mrfmmm.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen BPC, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. 2005. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem 280:14709–14715. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- 16.Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiruvella KK, Liang Z, Wilson TE. 2013. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol 5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis AJ, Chen DJ. 2013. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2:130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari P-O, van Gent DC, Chen BPC, Chen DJ. 2007. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol 177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weterings E, Chen DJ. 2007. DNA-dependent protein kinase in nonhomologous end joining: a lock with multiple keys? J Cell Biol 179:183–186. doi: 10.1083/jcb.200705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S, Guo M, Ouyang H, Li X, Cordon-Cardo C, Kurimasa A, Chen DJ, Fuks Z, Ling CC, Li GC. 2000. The catalytic subunit of DNA-dependent protein kinase selectively regulates p53-dependent apoptosis but not cell-cycle arrest. Proc Natl Acad Sci U S A 97:1584–1588. doi: 10.1073/pnas.97.4.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo RA, Jack MT, Xu Y, Burma S, Chen DJ, Lee PWK. 2002. DNA damage-induced apoptosis requires the DNA-dependent protein kinase, and is mediated by the latent population of p53. EMBO J 21:3000–3008. doi: 10.1093/emboj/cdf307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjee B, Kessinger C, Kobayashi J, Chen BPC, Chen DJ, Chatterjee A, Burma S. 2006. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair (Amst) 5:575–590. doi: 10.1016/j.dnarep.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Achanta G, Pelicano H, Feng L, Plunkett W, Huang P. 2001. Interaction of p53 and DNA-PK in response to nucleoside analogues: potential role as a sensor complex for DNA damage. Cancer Res 61:8723–8729. [PubMed] [Google Scholar]
- 25.Burma S, Chen DJ. 2004. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 3:909–918. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 26.Dhanalakshmi S, Agarwal C, Singh RP, Agarwal R. 2005. Silibinin up-regulates DNA-protein kinase-dependent p53 activation to enhance UVB-induced apoptosis in mouse epithelial JB6 cells. J Biol Chem 280:20375–20383. doi: 10.1074/jbc.M414640200. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Sun H, Wang J, Wang H, Meng L, Xu C, Jin M, Wang B, Zhang Y, Zhang Y, Zhu T. 2016. DNA-PK-mediated phosphorylation of EZH2 regulates the DNA damage-induced apoptosis to maintain T-cell genomic integrity. Cell Death Dis 7:e2316. doi: 10.1038/cddis.2016.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. 1977. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood 50:481–492. [PubMed] [Google Scholar]
- 29.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particle from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 77:7415–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takatsuki K. 2005. Discovery of adult T-cell leukemia. Retrovirology 2:16. doi: 10.1186/1742-4690-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okamoto T, Ohno Y, Tsugane S, Watanabe S, Shimoyama M, Tajima K, Miwa M, Shimotohno K, Okarmoto T, Ohno Y, Tsugane S, Watanabe S, Shimoyama M, Tajima K, Miwa M, Shimotohno K. 1989. Multi-step carcinogenesis model for adult T-cell leukemia. Jpn J Cancer Res 80:191–195. doi: 10.1111/j.1349-7006.1989.tb02289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoyama T, Chaganti RSK, Yamada Y, Tsukasaki K, Atogami S, Nakamura H, Tomonaga M, Ohshima K, Kikuchi M, Sadamori N. 2001. Cytogenetic analysis and clinical significance in adult T-cell leukemia/lymphoma: a study of 50 cases from the human T-cell leukemia virus type-1 endemic area, Nagasaki. Blood 97:3612–3620. doi: 10.1182/blood.V97.11.3612. [DOI] [PubMed] [Google Scholar]
- 33.Liang M-H, Geisbert T, Yao Y, Hinrichs SH, Giam C-Z. 2002. Human T-lymphotropic virus type 1 oncoprotein Tax promotes S-phase entry but blocks mitosis. J Virol 76:4022–4033. doi: 10.1128/JVI.76.8.4022-4033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de la Fuente C, Gupta MV, Klase Z, Strouss K, Cahan P, McCaffery T, Galante A, Soteropoulos P, Pumfery A, Fujii M, Kashanchi F. 2006. Involvement of HTLV-I Tax and CREB in aneuploidy: a bioinformatics approach. Retrovirology 3:43. doi: 10.1186/1742-4690-3-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellon M, Datta A, Brown M, Pouliquen J-F, Couppie P, Kazanji M, Nicot C. 2006. Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int J Cancer 119:2090–2097. doi: 10.1002/ijc.22026. [DOI] [PubMed] [Google Scholar]
- 36.Semmes OJ, Barret JF, Dang CV, Jeang KT. 1996. Human T-cell leukemia virus type I Tax masks c-Myc function through a cAMP-dependent pathway. J Biol Chem 271:9730–9738. doi: 10.1074/jbc.271.16.9730. [DOI] [PubMed] [Google Scholar]
- 37.Majone F, Semmes OJ, Jeang K-T. 1993. Induction of micronuclei by HTLV-I Tax: a cellular assay for function. Virology 193:456–459. doi: 10.1006/viro.1993.1145. [DOI] [PubMed] [Google Scholar]
- 38.Baydoun HH, Cherian MA, Green P, Ratner L. 2015. Inducible nitric oxide synthase mediates DNA double strand breaks in human T-cell leukemia virus type 1-induced leukemia/lymphoma. Retrovirology 12:71. doi: 10.1186/s12977-015-0196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinjo T, Ham-Terhune J, Peloponese J-M Jr, Jeang K-T. 2010. Induction of reactive oxygen species by human T-cell leukemia virus type 1 Tax correlates with DNA damage and expression of cellular senescence marker. J Virol 84:5431–5437. doi: 10.1128/JVI.02460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baydoun HH, Bai XT, Shelton S, Nicot C. 2012. HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS One 7:e42226. doi: 10.1371/journal.pone.0042226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dayaram T, Lemoine FJ, Donehower LA, Marriott SJ. 2013. Activation of WIP1 phosphatase by HTLV-1 Tax mitigates the cellular response to DNA damage. PLoS One 8:e55989. doi: 10.1371/journal.pone.0055989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durkin SS, Guo X, Fryrear KA, Mihaylova VT, Gupta SK, Belgnaoui SM, Haoudi A, Kupfer GM, Semmes OJ. 2008. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J Biol Chem 283:36311–36320. doi: 10.1074/jbc.M804931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandhasin C, Ducu RI, Berkovich E, Kastan MB, Marriott SJ. 2008. Human T-cell leukemia virus type 1 tax attenuates the ATM-mediated cellular DNA damage response. J Virol 82:6952–6961. doi: 10.1128/JVI.02331-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haoudi A, Semmes OJ. 2003. The HTLV-1 Tax oncoprotein attenuates DNA damage induced G1 arrest and enhances apoptosis in p53 null cells. Virology 305:229–239. doi: 10.1006/viro.2002.1642. [DOI] [PubMed] [Google Scholar]
- 45.Kao SY, Lemoine FJ, Marriott SJ. 2000. Suppression of DNA repair by human T cell leukemia virus type 1 Tax is rescued by a functional p53 signaling pathway. J Biol Chem 275:35926–35931. doi: 10.1074/jbc.M004397200. [DOI] [PubMed] [Google Scholar]
- 46.Lemoine FJ, Marriott SJ. 2001. Accelerated G(1) phase progression induced by the human T cell leukemia virus type I (HTLV-I) Tax oncoprotein. J Biol Chem 276:31851–31857. doi: 10.1074/jbc.M105195200. [DOI] [PubMed] [Google Scholar]
- 47.Marriott SJ, Semmes OJ. 2005. Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 24:5986–5995. doi: 10.1038/sj.onc.1208976. [DOI] [PubMed] [Google Scholar]
- 48.Gao L, Deng H, Zhao H, Hirbe A, Harding J, Ratner L, Weilbaecher K. 2005. HTLV-1 Tax transgenic mice develop spontaneous osteolytic bone metastases prevented by osteoclast inhibition. Blood 106:4294–4302. doi: 10.1182/blood-2005-04-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasegawa H, Sawa H, Lewis MJ, Orba Y, Sheehy N, Yamamoto Y, Ichinohe T, Tsunetsugu-Yokota Y, Katano H, Takahashi H, Matsuda J, Sata T, Kurata T, Nagashima K, Hall WW. 2006. Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med 12:466–472. doi: 10.1038/nm1389. [DOI] [PubMed] [Google Scholar]
- 50.Ohsugi T. 2013. A transgenic mouse model of human T cell leukemia virus type 1-associated diseases. Front Microbiol 4:49. doi: 10.3389/fmicb.2013.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bellon M, Baydoun HH, Yao Y, Nicot C. 2010. HTLV-I Tax-dependent and -independent events associated with immortalization of human primary T lymphocytes. Blood 115:2441–2448. doi: 10.1182/blood-2009-08-241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga J, Nosaka K, Tanaka Y, Matsuoka M. 2004. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer 109:559–567. doi: 10.1002/ijc.20007. [DOI] [PubMed] [Google Scholar]
- 53.Azran I, Schavinsky-Khrapunsky Y, Aboud M. 2004. Role of Tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology 1:20. doi: 10.1186/1742-4690-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Billman MR, Rueda D, Bangham CRM. 2017. Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res 2:87. doi: 10.12688/wellcomeopenres.12469.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahgoub M, Yasunaga J-I, Iwami S, Nakaoka S, Koizumi Y, Shimura K, Matsuoka M. 2018. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci U S A 115:E1269–E1278. doi: 10.1073/pnas.1715724115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rauch DA, Ratner L. 2011. Targeting HTLV-1 activation of NFκB in mouse models and ATLL patients. Viruses 3:886–900. doi: 10.3390/v3060886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. 2002. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76:12813–12822. doi: 10.1128/JVI.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL. 2006. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood 107:3976–3982. doi: 10.1182/blood-2005-11-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Satou Y, Yasunaga J, Yoshida M, Matsuoka M. 2006. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A 103:720–725. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyazaki M, Yasunaga J, Taniguchi Y, Tamiya S, Nakahata T, Matsuoka M. 2007. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J Virol 81:5714–5723. doi: 10.1128/JVI.02511-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan J, Ma G, Nosaka K, Tanabe J, Satou Y, Koito A, Wain-Hobson S, Vartanian J-P, Matsuoka M. 2010. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J Virol 84:7278–7287. doi: 10.1128/JVI.02239-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barbeau B, Mesnard JM. 2011. Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs). Viruses 3:456–468. doi: 10.3390/v3050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, Ishii R, Muto S, Kotani S, Watatani Y, Takeda J, Sanada M, Tanaka H, Suzuki H, Sato Y, Shiozawa Y, Yoshizato T, Yoshida K, Makishima H, Iwanaga M, Ma G, Nosaka K, Hishizawa M, Itonaga H, Imaizumi Y, Munakata W, Ogasawara H, Sato T, Sasai K, Muramoto K, Penova M, Kawaguchi T, Nakamura H, Hama N, Shide K, Kubuki Y, Hidaka T, Kameda T, Nakamaki T, Ishiyama K, Miyawaki S, Yoon S-S, Tobinai K, Miyazaki Y, Takaori-Kondo A, Matsuda F, et al. . 2015. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 47:1304–1315. doi: 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- 64.Cavanagh M-H, Landry S, Audet B, Arpin-André C, Hivin P, Paré M-E, Thête J, Wattel E, Marriott SJ, Mesnard J-M, Barbeau B. 2006. HTLV-I antisense transcripts initiating in the 3′LTR are alternatively spliced and polyadenylated. Retrovirology 3:15. doi: 10.1186/1742-4690-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laverdure S, Polakowski N, Hoang K, Lemasson I. 2016. Permissive sense and antisense transcription from the 5′ and 3′ long terminal repeats of human T-cell leukemia virus type 1. J Virol 90:3600–3610. doi: 10.1128/JVI.02634-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanaka-Nakanishi A, Yasunaga J, Takai K, Matsuoka M. 2014. HTLV-1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res 74:188–200. doi: 10.1158/0008-5472.CAN-13-0436. [DOI] [PubMed] [Google Scholar]
- 67.Ma G, Yasunaga J, Matsuoka M. 2016. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 13:16. doi: 10.1186/s12977-016-0249-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao T, Coutts A, Xu L, Yu J, Ohshima K, Matsuoka M. 2013. HTLV-1 bZIP factor supports proliferation of adult T cell leukemia cells through suppression of C/EBPα signaling. Retrovirology 10:159. doi: 10.1186/1742-4690-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kawatsuki A, Yasunaga J-I, Mitobe Y, Green PL, Matsuoka M. 2016. HTLV-1 bZIP factor protein targets the Rb/E2F-1 pathway to promote proliferation and apoptosis of primary CD4(+) T cells. Oncogene 35:4509–4517. doi: 10.1038/onc.2015.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vernin C, Thenoz M, Pinatel C, Gessain A, Gout O, Delfau-Larue M-H, Nazaret N, Legras-Lachuer C, Wattel E, Mortreux F. 2014. HTLV-1 bZIP factor HBZ promotes cell proliferation and genetic instability by activating OncomiRs. Cancer Res 74:6082–6093. doi: 10.1158/0008-5472.CAN-13-3564. [DOI] [PubMed] [Google Scholar]
- 71.Wurm T, Lemasson I. 2011. Potential interference of HTLV-1 HBZ protein with the DNA damage response pathway. Retrovirology 8:A203. doi: 10.1186/1742-4690-8-S1-A203. [DOI] [Google Scholar]
- 72.Wright DG, Marchal C, Hoang K, Ankney JA, Nguyen ST, Rushing AW, Polakowski N, Miotto B, Lemasson I. 2016. Human T-cell leukemia virus type-1-encoded protein HBZ represses p53 function by inhibiting the acetyltransferase activity of p300/CBP and HBO1. Oncotarget 7:1687–1706. doi: 10.18632/oncotarget.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Clerc I, Polakowski N, Andre-Arpin C, Cook P, Barbeau B, Mesnard JM, Lemasson I. 2008. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem 283:23903–23913. doi: 10.1074/jbc.M803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cook PR, Polakowski N, Lemasson I. 2012. HTLV-1 HBZ protein deregulates interactions between cellular factors and the KIX domain of p300/CBP. J Mol Biol 409:384–398. doi: 10.1016/j.jmb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wurm T, Wright DG, Polakowski N, Mesnard J-M, Lemasson I. 2012. The HTLV-1-encoded protein HBZ directly inhibits the acetyl transferase activity of p300/CBP. Nucleic Acids Res 40:5910–5925. doi: 10.1093/nar/gks244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Basbous J, Arpin C, Gaudray G, Piechaczyk M, Devaux C, Mesnard JM. 2003. The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J Biol Chem 278:43620–46327. doi: 10.1074/jbc.M307275200. [DOI] [PubMed] [Google Scholar]
- 77.Kuhlmann A-SS, Villaudy J, Gazzolo L, Castellazzi M, Mesnard J-M, Duc Dodon M. 2007. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 4:92. doi: 10.1186/1742-4690-4-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanagh M-H, Thébault S, Barbeau B, Nyborg JK, Mesnard J-M. 2007. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol 81:1543–1553. doi: 10.1128/JVI.00480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reinke AW, Grigoryan G, Keating AE. 2010. Identification of bZIP interaction partners of viral proteins HBZ, MEQ, BZLF1, and K-bZIP using coiled-coil arrays. Biochemistry 49:1985–1997. doi: 10.1021/bi902065k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bunting SF, Nussenzweig A. 2013. End-joining, translocations and cancer. Nat Rev Cancer 13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hivin P, Frederic M, Arpin-Andre C, Basbous J, Gay B, Thebault S, Mesnard JM. 2005. Nuclear localization of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J Cell Sci 118:1355–1362. doi: 10.1242/jcs.01727. [DOI] [PubMed] [Google Scholar]
- 82.Thebault S, Basbous J, Hivin P, Devaux C, Mesnard JM. 2004. HBZ interacts with JunD and stimulates its transcriptional activity. FEBS Lett 562:165–170. doi: 10.1016/S0014-5793(04)00225-X. [DOI] [PubMed] [Google Scholar]
- 83.Mao Z, Bozzella M, Seluanov A, Gorbunova V. 2008. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seluanov A, Mao Z, Gorbunova V. 2010. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp 2010:e2002. doi: 10.3791/2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC. 2004. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett 14:6083–6087. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 86.Pierce AJ, Johnson RD, Thompson LH, Jasin M. 1999. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. 2011. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 6:628–635. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Labhart P. 1999. Ku-dependent nonhomologous DNA end joining in Xenopus egg extracts. Mol Cell Biol 19:2585–2593. doi: 10.1128/MCB.19.4.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Di Virgilio M, Gautier J. 2005. Repair of double-strand breaks by nonhomologous end joining in the absence of Mre11. J Cell Biol 171:765–771. doi: 10.1083/jcb.200506029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Härer C, Marchetti C, Morrice N, Jeggo PA, Lees-Miller SP. 2006. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J 25:3880–3889. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA. 2010. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem 285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee K-J, Saha J, Sun J, Fattah KR, Wang S-C, Jakob B, Chi L, Wang S-Y, Taucher-Scholz G, Davis AJ, Chen DJ. 2016. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res 44:1732–1745. doi: 10.1093/nar/gkv1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Polakowski N, Han H, Lemasson I. 2011. Direct inhibition of RNAse T2 expression by the HTLV-1 viral protein Tax. Viruses 3:1485–1500. doi: 10.3390/v3081485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. 2011. SIRT6 promotes DNA repair under stress by activating PARP1. Science 332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T. 2011. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 30:2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- 97.Tuzon CT, Spektor T, Kong X, Congdon LM, Wu S, Schotta G, Yokomori K, Rice JC. 2014. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep 8:430–438. doi: 10.1016/j.celrep.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Davis AJ, Chen BPC, Chen DJ. 2014. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 17:21–29. doi: 10.1016/j.dnarep.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chan DW, Chen BPC, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, Chen DJ. 2002. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 16:2333–2338. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang W, Crowe JL, Lan L, Zha S, Liu X, Nakajima S, Wang Y, Li C, Lee BJ, Dubois RL, Liu C, Yu X. 2015. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol Cell 58:172–185. doi: 10.1016/j.molcel.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cui X, Yu Y, Gupta S, Cho Y-M, Lees-Miller SP, Meek K. 2005. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol 25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ding Q, Reddy YVR, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K. 2003. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol 23:5836–5848. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dobbs TA, Tainer JA, Lees-Miller SP. 2010. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst) 9:1307–1314. doi: 10.1016/j.dnarep.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Álvarez-Quilón A, Serrano-Benítez A, Lieberman JA, Quintero C, Sánchez-Gutiérrez D, Escudero LM, Cortés-Ledesma F. 2014. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat Commun 5:3347. doi: 10.1038/ncomms4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ducu RI, Dayaram T, Marriott SJ. 2011. The HTLV-1 Tax oncoprotein represses Ku80 gene expression. Virology 416:1–8. doi: 10.1016/j.virol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ma Y, Pannicke U, Lu H, Niewolik D, Schwarz K, Lieber MR. 2005. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human artemis. J Biol Chem 280:33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- 107.Stronach EA, Chen M, Maginn EN, Agarwal R, Mills GB, Wasan H, Gabra H. 2011. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 13:1069–1080. doi: 10.1593/neo.111032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan DW, Ye R, Veillette CJ, Lees-Miller SP. 1999. DNA-dependent protein kinase phosphorylation sites in Ku 70/80 heterodimer. Biochemistry 38:1819–1828. doi: 10.1021/bi982584b. [DOI] [PubMed] [Google Scholar]
- 109.Lee K-J, Jovanovic M, Udayakumar D, Bladen CL, Dynan WS. 2004. Identification of DNA-PKcs phosphorylation sites in XRCC4 and effects of mutations at these sites on DNA end joining in a cell-free system. DNA Repair (Amst) 3:267–276. doi: 10.1016/j.dnarep.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 110.Yu Y, Mahaney BL, Yano KI, Ye R, Fang S, Douglas P, Chen DJ, Lees-Miller SP. 2008. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 7:1680–1692. doi: 10.1016/j.dnarep.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Y-G, Nnakwe C, Lane WS, Modesti M, Frank KM. 2004. Phosphorylation and regulation of DNA ligase IV stability by DNA-dependent protein kinase. J Biol Chem 279:37282–37290. doi: 10.1074/jbc.M401217200. [DOI] [PubMed] [Google Scholar]
- 112.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GCM, Curtin NJ. 2006. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res 66:5354–5362. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 113.Gao Y, Chaudhuri J, Zhu C, Davidson L, Weaver DT, Alt FW. 1998. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity 9:367–376. doi: 10.1016/S1074-7613(00)80619-6. [DOI] [PubMed] [Google Scholar]
- 114.Hosoi Y, Miyachi H, Matsumoto Y, Ikehata H, Komura J, Ishii K, Zhao H-J, Yoshida M, Takai Y, Yamada S, Suzuki N, Ono T. 1998. A phosphatidylinositol 3-kinase inhibitor wortmannin induces radioresistant DNA synthesis and sensitizes cells to bleomycin and ionizing radiation. Int J Cancer 78:642–647. doi:. [DOI] [PubMed] [Google Scholar]
- 115.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. 1999. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol 19:3877–3884. doi: 10.1128/MCB.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Neal JA, Sugiman-Marangos S, VanderVere-Carozza P, Wagner M, Turchi J, Lees-Miller SP, Junop MS, Meek K. 2014. Unraveling the complexities of DNA-dependent protein kinase autophosphorylation. Mol Cell Biol 34:2162–2175. doi: 10.1128/MCB.01554-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Giam C-Z, Semmes OJ. 2016. HTLV-1 infection and adult T-cell leukemia/lymphoma—a tale of two proteins: Tax and HBZ. Viruses 8:161. doi: 10.3390/v8060161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nicot C. 2015. HTLV-I Tax-mediated inactivation of cell cycle checkpoints and DNA repair pathways contribute to cellular transformation: “a random mutagenesis model.” J Cancer Sci 2:6. doi: 10.13188/2377-9292.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wechsler T, Chen BPC, Harper R, Morotomi-Yano K, Huang BCB, Meek K, Cleaver JE, Chen DJ, Wabl M. 2004. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci U S A 101:1247–1252. doi: 10.1073/pnas.0307765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Panfil AR, Dissinger NJ, Howard CM, Murphy BM, Landes K, Fernandez SA, Green PL. 2016. Functional comparison of HBZ and the related APH-2 protein provides insight into human T-cell leukemia virus type 1 pathogenesis. J Virol 90:3760–3772. doi: 10.1128/JVI.03113-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yoshida M, Satou Y, Yasunaga J, Fujisawa J, Matsuoka M. 2008. Transcriptional control of spliced and unspliced human T-cell leukemia virus type 1 bZIP factor (HBZ) gene. J Virol 82:9359–9368. doi: 10.1128/JVI.00242-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hemelaar J, Bex F, Booth B, Cerundolo V, McMichael A, Daenke S. 2001. Human T-cell leukemia virus type 1 Tax protein binds to assembled nuclear proteasomes and enhances their proteolytic activity. J Virol 75:11106–11115. doi: 10.1128/JVI.75.22.11106-11115.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Polakowski N, Gregory H, Mesnard J-M, Lemasson I. 2010. Expression of a protein involved in bone resorption, Dkk1, is activated by HTLV-1 bZIP factor through its activation domain. Retrovirology 7:61. doi: 10.1186/1742-4690-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Britton S, Coates J, Jackson SP. 2013. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol 202:579–595. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Richardson C, Moynahan ME, Jasin M. 1998. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kingston RE, Chen CA, Rose JK, Kingston RE, Chen CA, Rose JK. 2003. Calcium phosphate transfection. Curr Protoc Mol Biol Chapter 9:Unit 9.1 doi: 10.1002/0471142727.mb0901s63. [DOI] [PubMed] [Google Scholar]
- 127.Abmayr SM, Yao T, Parmely T, Workman JL. 2006. Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr Protoc Mol Biol Chapter 12:Unit 12.1. doi: 10.1002/0471142727.mb1201s75. [DOI] [PubMed] [Google Scholar]
- 128.Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. 2001. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]