

ABSTRACT
Sodium taurocholate cotransporting polypeptide (NTCP) has been identified as a hepatitis B virus (HBV) receptor, and its overexpression in HepG2 cell lines leads to efficient secretion of hepatitis B e antigen (HBeAg) following challenge with a large dose of cell culture-derived HBV (cHBV) particles. However, NTCP-reconstituted HepG2 cells are inefficiently infected by patient serum-derived HBV (sHBV) and release very little hepatitis B surface antigen (HBsAg) following cHBV infection, unlike differentiated HepaRG cells, which are naturally susceptible to both cHBV and sHBV particles. Here, we investigated whether NTCP could explain the different behaviors of the two cell types. Endogenous NTCP protein from differentiated HepaRG cells was unglycosylated despite wild-type coding sequence. HepaRG cells stably transfected with an epitope-tagged NTCP expression construct displayed higher sHBV but not cHBV susceptibility than cells transfected with the null mutant. Tagged NTCP introduced to both HepG2 and HepaRG cells was glycosylated, with N5 and N11 being sites of N-linked glycosylation. Mutating N5, N11, or both did not alter cell surface availability of NTCP or its subcellular localization, with both the singly glycosylated and nonglycosylated forms still capable of mediating cHBV infection in HepG2 cells. In conclusion, nonglycosylated NTCP is expressed by differentiated HepaRG cells and capable of mediating cHBV infection in HepG2 cells, but it cannot explain differential susceptibility of HepaRG and HepG2/NTCP cells to cHBV versus sHBV infection and different HBsAg/HBeAg ratios following cHBV infection. The responsible host factor(s) remains to be identified.
IMPORTANCE HBV can infect differentiated HepaRG cells and also HepG2 cells overexpressing NTCP, the currently accepted HBV receptor. However, HepG2/NTCP cells remain poorly susceptible to patient serum-derived HBV particles and release very little hepatitis B surface antigen following infection by cell culture-derived HBV. We found differentiated HepaRG cells expressed nonglycosylated NTCP despite a wild-type coding sequence. NTCP introduced to HepG2 cells was glycosylated at two N-linked glycosylation sites, but mutating either or both sites failed to prevent infection by cell culture-derived HBV or to confer susceptibility to serum-derived HBV. Overexpressing NTCP in HepRG cells did not increase infection by cell culture-derived HBV or distort the ratio between the two viral antigens. These findings suggest that host factors unique to HepaRG cells are required for efficient infection by serum-derived HBV, and factors other than NTCP contribute to balanced viral antigen production following infection by cell culture-derived HBV.
KEYWORDS: hepatitis B virus, hepatitis B e antigen, hepatitis B surface antigen, HepaRG, HepG2, infection, N-linked glycosylation, sodium taurocholate cotransporting polypeptide
INTRODUCTION
Although primary human hepatocytes (PHH) can be productively infected with hepatitis B virus (HBV), they suffer from limited supply and high cost, donor-to-donor variability in HBV susceptibility, and inability to perform serial passage. HepaRG is a human liver progenitor cell line. Culturing confluent HepaRG cells in medium supplemented with 2% dimethyl sulfoxide (DMSO) could trigger their differentiation into hepatocytes surrounded by biliary cells, with hepatocytes being susceptible to HBV infection (1–3). HBV expresses three coterminal envelope proteins, called large (L), middle (M), and small (S), with the pre-S1 domain in L protein interacting with the high-affinity HBV receptor. Sodium taurocholate cotransporting polypeptide (NTCP), alternatively known as solute carrier family 10 member 1 (SLC10A1), was identified as a binding partner for a myristoylated pre-S1 peptide (4). Silencing NTCP expression by short interfering RNA (siRNA) diminished HBV infectivity in PHH and HepaRG cells; conversely, NTCP overexpression in HepG2 cells, a human hepatoma cell line, conferred susceptibility to infection by cell culture-derived HBV (cHBV) particles (4). Human NTCP could also render another human hepatoma cell line (Huh7) and mice susceptible to infection with hepatitis D virus (HDV), an RNA virus employing HBV envelope proteins for virion formation and transmission (4, 5). These provocative findings established NTCP as the high-affinity HBV and HDV receptor. The NTCP-reconstituted HepG2 cell line has become a convenient cell culture model of in vitro HBV infection because, in contrast to HepaRG cells, its HBV susceptibility does not require prolonged (>2-week) induction of cellular differentiation.
Nevertheless, differentiated HepaRG cells represent a more physiological system of in vitro HBV infection, because NTCP is expressed from its normal chromosomal locus, driven by its own promoter under biological cues. In addition, differentiated HepaRG can be efficiently infected with patient serum-derived HBV (sHBV) particles, while the same sHBV isolates were much less infectious in HepG2/NTCP cells (6). Studies from others indicated that sHBV infectivity depended on high HBV DNA titer (7), separation of virions from subviral particles (8), or suspension culture (9). Moreover, infectivity was low according to hepatitis B e antigen (HBeAg) released from infected cells (7, 8). Efficient cHBV but not sHBV infection requires the addition of 4% polyethylene glycol (PEG) during virus inoculation (6, 10), a nonphysiological condition. Third, the two sensitive and convenient markers of in vitro HBV infection are HBeAg and hepatitis B surface antigen (HBsAg). The latter primarily represents S protein secreted from infected hepatocytes as subviral particles and is much more abundant than HBeAg during natural infection in humans. HepaRG, but not HepG2/NTCP, cells maintain a high HBsAg/HBeAg ratio following cHBV infection. When cultured under similar conditions (medium supplemented with 2% DMSO), HepG2/NTCP cells could release up to 100 times more HBeAg than differentiated HepaRG cells but comparable levels of HBsAg (11). Even HepG2/NTCP cells cultured in DMSO-free medium produced at least 10 times lower HBsAg/HBeAg ratios following cHBV infection than HepaRG cells cultured with 2% DMSO, and adding 1% DMSO to HepG2/NTCP cells further reduced the HBsAg/HBeAg ratio (6).
Human NTCP is a glycoprotein of 349 residues (12). It is a multitransmembrane protein localized on the basolateral side of hepatocytes, where it serves as the major importer of glycine- and taurine-conjugated bile acids (13). NTCP has two potential N-linked glycosylation sites located at its amino terminus: NAS and NFT (Fig. 1A). In the present study, we verified whether the HepaRG cell line harbors a wild-type NTCP coding sequence. We also examined the glycosylation status of endogenous NTCP protein expressed in differentiated HepaRG cells and exogenous epitope-tagged NTCP introduced to HepaRG and HepG2 cells. Finally, we investigated the impact of exogenous NTCP on cHBV infectivity in differentiated HepaRG cells and the ability of singly glycosylated and nonglycosylated NTCP mutants to mediate cHBV infection in HepG2 cells.
FIG 1.

NTCP mutants used for the present study and NTCP cDNA sequence from differentiated HepaRG cells. (A) Shown on top is the 1.6-kb NTCP mRNA according to GenBank accession number NM_003049.3, with positions 135 to 1184 (thickened portion) encoding the 349-aa NTCP protein. In the schematic representation of the 349-aa NTCP protein below, asterisks indicate the two N-linked glycosylation sites (N5 and N11). They were mutated to alanine (A) or glutamine (Q), alone or in combination, by nucleotide changes at the cDNA level as indicated. A null mutant was created by converting codon 50 from AAG to TAG. ORF, open reading frame; nt, nucleotide(s); aa, amino acid(s). (B) The NTCP mRNA produced in HepaRG cells has wild-type sequence. Shown are positions 20 to 1575 of the human NTCP cDNA sequence, with the coding region in boldface. The underlined sequences at the beginning and end represent the S5 and AS5 primers to amplify a 1.5-kb cDNA fragment from differentiated HepaRG cells. The internal underlined sequences represent the S1 and AS1 primers to amplify a shorter (1.3-kb) cDNA fragment from both HepaRG and HepG2 cells. DNA sequences of both the 1.5-kb and 1.3-kb PCR products are identical to that of NM_003049.3.
RESULTS
Rhodopsin epitope-tagged NTCP expressed in HepG2 and HepaRG cells showed different mobility due to differences in N-linked glycosylation.
The human NTCP protein contains two potential N-linked glycosylation sites: N5 (NAS) and N11 (NFT) (Fig. 1A). According to a previous report, the NTCP protein from human liver samples migrated as a broad band of 56 kDa, which was reduced to 39 kDa following N-glycanase treatment (14). In another study, a V5-tagged NTCP expression construct generated a glycosylated protein of 50 kDa in HeLa cells, which was reduced to 37 kDa after deglycosylation (15). We previously stably transfected HepG2 cells with an expression construct for full-length (349 amino acids [aa]) NTCP protein fused at the carboxyl terminus with a 9-aa epitope derived from bovine rhodopsin (6). The strong cytomegalovirus (CMV) promoter ensured NTCP overexpression, while the rhodopsin epitope enabled detection of the recombinant protein by 1D4, a murine monoclonal antibody. A broad NTCP band of 50 to 65 kDa could be detected from HepG2/NTCP cells by 1D4 as well as by a rabbit polyclonal anti-SLC10A1 antibody (Fig. 2A, lane 3, lower and upper). Treating cell lysate with PNGase F prior to Western blotting sharpened the NTCP band and shifted its position to about 32 kDa (Fig. 2A, lane 4). Culturing HepG2/NTCP cells in medium supplemented with tunicamycin, a glycosylation inhibitor, also generated the 32-kDa band (Fig. 2A, lane 5). Thus, epitope-tagged NTCP introduced to HepG2 cells was indeed modified by N-linked glycosylation. We also stably transfected the same NTCP expression construct into HepaRG cells. Western blot analysis revealed a smear with faster mobility than that in HepG2 cells (about 45 to 55 kDa) (Fig. 2B, compare lanes 2 and 3). PNGase F treatment shifted NTCP protein from both HepaRG and HepG2 cells to 32 kDa (Fig. 2B, lanes 6 and 7), suggesting that the size difference was attributable to modification by N-linked glycosylation.
FIG 2.

N-linked glycosylation of exogenous NTCP introduced to HepG2 and HepaRG cells but not endogenous NTCP expressed in HepaRG cells. Cells with or without stable transfection with epitope-tagged NTCP or the null mutant were subjected to Western blot analysis using the polyclonal anti-SLC10A1 antibody (A and B, top, and C and D) or the monoclonal 1D4 anti-tag antibody (A and B, lower). β-Actin served as a loading control. To check for N-linked glycosylation, cells were cultured in medium supplemented with tunicamycin. Alternatively, cell lysate was treated with PNGase F. (A) Similar effect of PNGase F and tunicamycin treatment in shifting NTCP mobility from HepG2/NTCP cells. (B) Faster mobility of exogenous NTCP introduced to HepaRG cells than HepG2 cells. HepaRG cells with or without stable transfection with the full-length NTCP construct or its null mutant were differentiated for 4 weeks prior to Western blotting. (C) Endogenous NTCP expressed in proliferating HepaRG cells (lane 1) versus differentiated HepaRG cells (lane 2). (D) Endogenous NTCP protein levels in differentiated HepaRG cells following lentivirus-mediated delivery of control shRNA or NTCP shRNA. Molecular sizes (in kDa) are shown to the left of each panel. Ctrl, control; RG, differentiated HepaRG cells; G2, HepG2 cells; gNTCP, glycosylated NTCP; NTCP, the nonglycosylated form; aggregate, insoluble NTCP following PNGase F treatment.
Endogenous NTCP protein produced in differentiated HepaRG cells is not modified by N-linked glycosylation.
Consistent with much reduced expression of endogenous NTCP by differentiated HepaRG cells, it was either undetectable by the rabbit polyclonal anti-SLC10A1 antibody (Fig. 2A, upper, lane 1) or faintly detected compared to the exogenous NTCP (Fig. 2B, upper, compare lanes 1 and 2). The endogenous NTCP banded at a similar position as deglycosylated epitope-tagged NTCP (Fig. 2B, compare lane 1 with lanes 6 and 7). Since the 9-aa tag increases protein size by a mere 1 kDa, this result suggests that endogenous NTCP expressed by differentiated HepaRG cells was nonglycosylated. Consistent with the reliance of NTCP expression on hepatocyte differentiation (11, 16, 17), this 32-kDa species was much more abundant in differentiated HepaRG cells than in the growing HepaRG cells (Fig. 2C). Importantly, infecting differentiated HepaRG cells with lentivirus encoding an NTCP short hairpin RNA (shRNA) greatly reduced the intensity of this protein species compared to that of transduction with control shRNA (Fig. 2D).
Sequencing NTCP cDNA from differentiated HepaRG cells revealed no mutation to abolish N-linked glycosylation.
To test whether the HepaRG cell line harbors mutations in the NTCP gene, especially the coding sequence for the sites of N-linked glycosylation, total RNA was extracted from differentiated HepaRG cells and reverse transcribed to cDNA using an oligo(dT) primer. Parallel experiment was performed on HepG2 cells, which does not express NTCP protein. Using the S1-AS1 primer pair, a 1.3-kb NTCP cDNA fragment could be amplified by PCR from differentiated HepaRG cells. A weaker product was also generated from HepG2 cells. Direct sequencing of the 1.3-kb PCR product, which covers the entire coding sequence (Fig. 1A), did not reveal any point mutation for NTCP cDNA derived from either cell type compared to the human NTCP mRNA deposited in GenBank (accession number NM_003049.3) (Fig. 1B). By modifying PCR conditions and using outside primers (S5 and AS5) (Fig. 1A), we succeeded in amplifying a 1.5-kb cDNA fragment from HepaRG cells. Sequencing of three PCR clones again revealed no difference from that of the human NTCP mRNA (Fig. 1B). Therefore, differentiated HepaRG cells express wild-type NTCP protein. We subsequently learned that human NTCP coding sequence from HepaRG cells had been deposited in GenBank as early as 2012 (4). That sequence (JQ814895) harbors two adjacent amino acid substitutions in NTCP protein, K177E and P181S, although no mutation was found elsewhere, including the two N-linked glycosylation sites.
Human NTCP protein is glycosylated at N5 and N11.
To verify whether N-linked glycosylation of NTCP affects its subcellular localization and ability to mediate cHBV infection, we introduced into the epitope-tagged NTCP construct the 5A, 5Q, 11A, and 11Q single mutations as well as the 5A/11A and 5Q/11Q double mutations (Fig. 1A). The mutant constructs and a null mutant capable of translating just the first 49 residues of NTCP protein were stably transfected to HepG2 cells. Western blot analysis revealed faster migration of the 5A, 5Q, 11A, and 11Q single mutants (Fig. 3B, lanes 3 to 6) than the wild-type NTCP construct (lane 2) and even faster (about 32 kDa) migration of the 5A/11A and 5Q/11Q double mutants (lanes 7 and 8), consistent with a nonglycosylated form. These findings confirm N5 and N11 as the only glycosylation sites in human NTCP, as reported by others (18, 19). Some NTCP mutants retaining a single N-linked glycosylation site, such as N11A and N11Q, produced small amounts of the nonglycosylated form as well (Fig. 3B, upper, lanes 5 and 6).
FIG 3.

Stable expression of NTCP glycosylation mutants in HepG2 cells and cHBV infectivity in the presence or absence of 2% DMSO. Transfected cells seeded in 96-well plates were cultured in medium with or without 2% DMSO. Duplicate wells were infected with cHBV at 1,400 genome equivalent/cell, while the other two wells were uninfected. Cells and culture supernatant were harvested 17 days later. (A) HBeAg and HBsAg were measured from culture supernatant of both infected wells. (C) Cell lysate was pooled from infected cells cultured in DMSO-free and DMSO-containing medium for Southern blot analysis of HBV DNA. Cloned HBV DNA cleaved with restriction enzymes (200 pg) served as size markers. (B) Cell lysate from the two noninfected wells was combined for Western blot analysis of NTCP (using 1D4 antibody) followed by β-actin. Double, single, and non indicate doubly and singly glycosylated as well as nonglycosylated forms of NTCP protein. WT, wild type.
Lack of N-linked glycosylation does not alter NTCP subcellular localization.
Immunofluorescent (IF) staining revealed that, similar to the wild-type NTCP protein, the single and double glycosylation mutants all were available on the cell surface (Fig. 4A). Cell fractionation experiments revealed that the majority of the wild-type NTCP protein was associated with plasma, mitochondrial, and/or endoplasmic reticulum (ER)/Golgi membranes (Fig. 4B, lane 3). It was also found in soluble nuclear extract and cytoskeleton but poorly detectable in cytosol and absent from chromatin-bound nuclear extract. The presence of NTCP in the cytoskeletal fraction is consistent with previous reports that NTCP traffics between an intracellular vesicular pool and the cell surface through microfilament- and microtubule-based cytoskeletons (20, 21). A very similar distribution pattern was observed for 5A/11A, a nonglycosylated NTCP mutant (Fig. 4B, compare lanes 7 to 12 with 1 to 6). Thus, lack of N-linked glycosylation does not prevent NTCP from cell surface targeting or subcellular trafficking.
FIG 4.
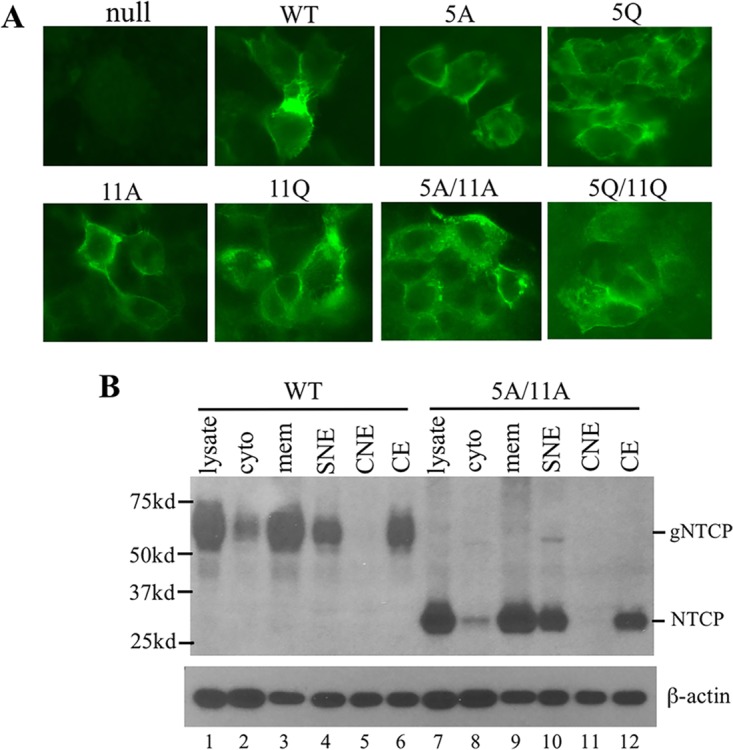
Subcellular localization of the NTCP glycosylation mutants. (A) IF staining. HepG2 cells grown on coverslips were transfected with wild-type (WT) NTCP or various glycosylation mutants and fixed 2 days later for staining with the 1D4 antibody. Images were taken with focus on cell surface. Cells transfected with the null mutant served as a negative control. (B) Subcellular fractionation. HepG2 cells stably transfected with WT NTCP or the 5A/11A nonglycosylated mutant were subject to subcellular fractionation using a commercial kit. NTCP in the unfractionated total lysate, the cytoplasmic (cyto) and membrane (mem) fractions, as well as soluble nuclear extract (SNE), chromatin-bound nuclear extract (CNE), and cytoskeletal extract (CE) were detected in Western blotting by the 1D4 antibody. The same blot was reprobed with β-actin antibody followed stripping. gNTCP, doubly glycosylated form of NTCP. NTCP, the nonglycosylated form.
Nonglycosylated NTCP mutants could mediate cHBV infection in HepG2 cells.
HepG2 cells stably transfected with each NTCP construct were seeded in 8 replicate wells of 96-well plates, with 4 wells cultured in regular medium and the other 4 wells in medium supplemented with 2% DMSO. Two wells from each group were infected with cHBV, and all 8 wells were harvested 17 days later. Both HBeAg and HBsAg were measured from culture supernatant at day 9 (data not shown) and day 17 postinfection (Fig. 3A), with similar results. Based on Western blot analysis of uninfected cells cultured under identical conditions, the 5A mutant produced less NTCP protein than the wild-type construct, especially in the presence of 2% DMSO (Fig. 3B, lane 3). This correlated with reduced HBeAg and HBsAg titers following cHBV infection (Fig. 3A). In medium supplemented with 2% DMSO, the nonglycosylated mutants (5A/11A and 5Q/11Q) also produced somewhat less NTCP protein than the wild-type construct (Fig. 3B, lower, lanes 7 and 8), which may explain the lower HBeAg secretion from cells transfected with the 5A/11A mutant (Fig. 3A, lower). Consistent with our previous report (6), adding 2% DMSO to culture medium markedly reduced HBsAg but not HBeAg secretion (Fig. 3A, compare upper and lower). Importantly, replicative HBV DNA could be detected from lysate of cells expressing any of the NTCP glycosylation mutants, including 5A/11A and 5Q/11Q (Fig. 3C; here, cells cultured in DMSO-free and DMSO-containing medium were combined). Therefore, NTCP mutants with single or no N-linked glycosylation site continued to support cHBV infection in HepG2 cells, with the cHBV infectivity correlated primarily with NTCP protein level rather than the glycosylation status. We also attempted to infect such HepG2 cells with three sHBV isolates, which could efficiently infect differentiated HepaRG cells (6). Viral/subviral particles concentrated by ultracentrifugation through sucrose cushion were inoculated to HepG2 cells in the absence of 4% PEG. HBeAg measurement suggested no productive infection in cells expressing wild-type NTCP or any of the glycosylation mutants (data not shown).
Stable transfection with either full-length NTCP expression construct or a null mutant diminished HBV infectivity in HepaRG cells.
Considering the much lower NTCP expression level in differentiated HepaRG cells than HepG2/NTCP cells, we wondered whether introducing exogenous NTCP into HepaRG cells could augment HBV infectivity. Toward this end, subconfluent HepaRG cells were transfected with a rhodopsin epitope-tagged NTCP expression construct, and stable transfectants were selected by blasticidin as a population. A null mutant capable of expressing just the N-terminal 49 residues was similarly transfected to serve as a negative control. Differentiated HepaRG/NTCP cells overproduced glycosylated epitope-tagged NTCP compared to the level of production of the parental HepaRG cells or HepaRG/null cells (Fig. 2B). Similar levels of HBeAg and HBsAg were released by HepaRG/NTCP cells and HepaRG/null cells following challenge with cHBV in the presence of 4% PEG (Fig. 5A). However, parallel infection experiments revealed much higher levels of both HBeAg and HBsAg from parental HepaRG cells (Fig. 5B). In this regard, we found that HepaRG cells stably transfected with both full-length NTCP and its null mutant displayed slower growth than parental cells. Furthermore, following differentiation clustered hepatocytes are flattened and enlarged, with less distinct island-like morphology relative to biliary cells (data not shown). Compared with parental HepaRG cells, stably transfected HepaRG cells were poorly infectible with sHBV in the absence of PEG. Still, higher HBeAg and HBsAg titers were produced from HepaRG/NTCP cells than HepaRG/null cells (Fig. 5C).
FIG 5.

Impact of stable transfection with the NTCP expression construct or its null mutant on cHBV and sHBV infectivity in differentiated HepaRG cells. Proliferating HepaRG cells were transfected with epitope-tagged NTCP construct or its null mutant. Stable transfectants were selected by blasticidin as a population. After differentiation, cells were infected with cHBV in the presence of 4% PEG (A and B) or sHBV in the absence of PEG (C). HBeAg and HBsAg were measured from the indicated volume of culture supernatant at various time points postinfection. Panels A and B represent two separate infection experiments, with cells seeded under similar conditions and differentiated for the same duration prior to infection. NTCP protein levels in differentiated HepaRG cells with or without stable transfection with NTCP expression construct or the null mutant are shown in Fig. 2B (lanes 1, 2, and 4). O.D.450, optical density at 450 nm.
DISCUSSION
Differentiated HepaRG cells, and especially HepG2 cells, stably transfected with NTCP cDNA have become the most popular in vitro systems to conduct HBV infection experiments. The two systems have their respective advantages and drawbacks. HepaRG cells require prolonged (>2-week) culture in medium supplemented with DMSO to induce hepatocyte differentiation, with batch-to-batch variability in HBV susceptibility and higher susceptibility often correlated with prolonged induction period. In HepG2/NTCP but not HepaRG cells, the role of NTCP in mediating HBV infectivity is self-evident, because the parental HepG2 cell line is clearly resistant to HBV infection. Certainly, knockdown experiments implicated endogenous NTCP as a mediator of HBV infection in differentiated HepaRG cells (4, 11). The drawbacks of HepG2/NTCP cells include NTCP overexpression driven by a foreign promoter (which precludes transcriptional regulation postinfection), poor susceptibility to sHBV infection, and a distorted HBsAg/HBeAg ratio following cHBV infection (6, 11). In the present study, we investigated whether differences in NTCP protein sequence, expression level, or glycosylation status account for some of the contrasting features between differentiated HepaRG cells and NTCP-reconstituted HepG2 cells.
Human NTCP is anchored on cell surfaces through 9 transmembrane segments. Of the five potential N-linked glycosylation sites located at positions 5, 11, 103, 117, and 336, only N5 and N11 are accessible for N-linked glycosylation. Indeed, rat NTCP is glycosylated at N5 and N11 only (12), and another study suggested N5 and N11 as the glycosylation sites for human NTCP (19). NTCP from human liver samples had a reported molecular weight of 56 kDa, which was reduced to 39 kDa following deglycosylation (14). We found that in HepG2 cells, the epitope-tagged NTCP construct generated a broad band of 50 to 65 kDa, which was converted to a 32-kDa species by either PNGase F treatment of cell lysate or tunicamycin treatment of living cells (Fig. 2A). A similar effect of PNGase treatment on accelerating mobility of NTCP introduced to HepG2 cells has been described by others (9, 18), confirming efficient glycosylation of NTCP protein by HepG2 cells. Compared with the wild-type NTCP construct, our single glycosylation mutants (5A, 5Q, 11A, and 11Q) all showed reduced protein size, while the double mutants (5A/11A and 5Q/11Q) expressed an even smaller NTCP protein of 32 kDa (Fig. 3B). These findings confirm that in HepG2 cells, wild-type human NTCP is glycosylated at both N5 and N11 but no additional positions.
Intriguingly, introducing the identical epitope-tagged NTCP construct into the HepaRG cell line generated faster-migrating species of 45 to 55 kDa, although PNGase F treatment converted exogenous NTCP protein from both HepG2 and differentiated HepaRG cells into 32 kDa (Fig. 2B). One possible explanation is that in HepaRG cells the recombinant NTCP protein is glycosylated at just one of the two glycosylation sites. Alternatively, both N5 and N11 were glycosylated, but there was a difference in N glycan trimming and modifications between the two cell types. More surprisingly, endogenous NTCP protein expressed by differentiated HepaRG cells appeared nonglycosylated (Fig. 2B). In this regard the 32-kDa species was much more abundant in differentiated than growing HepaRG cells, supporting its identity as NTCP (Fig. 2C). Further, shRNA-mediated knockdown experiments confirmed this protein band as NTCP (Fig. 2D). Since cDNA sequencing of differentiated HepaRG cells from both Wenhui Li's group (JQ814895) and from us (Fig. 1B) revealed no mutation in the NTCP glycosylation site, the lack of N-linked glycosylation cannot be explained by NTCP itself. Enzymes involved in protein glycosylation, such as galactosyl transferase and sialyl transferase, undergo ontogenic regulation (22, 23). Rat NTCP produced in the first 4 weeks of life was underglycosylated as a 39-kDa species rather than the 50-kDa species found in adult liver, with both species converted to a 33-kDa form following treatment with N-glycanase (24). HepaRG is a human progenitor cell line (1, 25) and may display features of fetal hepatocytes in terms of protein glycosylation. Given that exogenous NTCP did get glycosylated in HepaRG cells (although to a smaller size than in HepG2 cells), the rhodopsin epitope might promote N-linked glycosylation. Alternatively, constitutive NTCP expression in growing HepaRG cells driven by the CMV promoter contributes to its sustained glycosylation even after differentiation.
Plasma membrane proteins often undergo N-linked glycosylation in the endoplasmic reticulum and are further glycosylated in the Golgi apparatus before trafficking to the cell surface. When human NTCP was expressed in HeLa cells, two protein bands of approximately 50 kDa and 40 kDa were detectable in total cell lysate, but only the 50-kDa species was found on the cell surface (15). In another study employing the 293 (human embryonic kidney) cell line, the FLAG-tagged NTCP expression construct generated both the 50-kDa smear and a 37-kDa core glycosylated form, but an R252H mutant produced exclusively the 37-kDa species (26). The wild-type NTCP rather than the R252H mutant was available on the cell surface and could take up conjugated bile salts. Likewise, eliminating three or all four glycosylation sites in the rat bile salt export pump diminished protein half-life and reduced bile acid transport activity (27). We introduced both N-to-A and N-to-Q mutations into the N5 and/or N11 sites so as to separate the effect of specific amino acid changes from altered glycosylation status. All six NTCP mutants, including the nonglycosylated mutants, were available on the cell surface (Fig. 4A). Cell fractionation experiments revealed that both the wild-type NTCP and the nonglycosylated N5A/N11A mutant were associated with the membrane fraction, cytoskeleton, and soluble nuclear extract rather than the cytosolic fraction or chromatin (Fig. 4B). Consequently, all six NTCP mutants could mediate cHBV infection when stably transfected to HepG2 cells (Fig. 3A and C). Besides data shown in Fig. 3, we have conducted several additional infection experiments. The N11A single mutant tended to display higher protein levels than wild-type NTCP and, consequently, higher HBeAg and HBsAg titers following cHBV infection. The double mutants, especially 5Q/11Q, often showed low levels of NTCP protein expression. However, if corrected for the NTCP protein level, the nonglycosylated NTCP mutants (5A/11A and 5Q/11Q) were at least as efficient as the wild-type protein in mediating cHBV infection.
Our finding is inconsistent with a very recent report by Appelman and colleagues (18). These authors introduced 5Q, 11Q, and 5Q/11Q mutations into an NTCP expression construct with a hemagglutinin (HA) tag placed at the amino terminus and used a lentiviral vector to deliver the NTCP mutants into HepG2 cells. Based on HBeAg measurement and PCR quantification of covalently closed circular DNA (cccDNA), the single mutants conferred more or less wild-type levels of cHBV infectivity in HepG2 cells. However, HepG2 cells transduced with the 5Q/11Q double mutant failed to support cHBV infection and were severely impaired in bile acid transport. In Western blotting, both single mutants showed higher NTCP protein levels than the wild-type construct, whereas the double mutant had extremely low protein levels as a consequence of accelerated degradation (18). We suspect that failure of the nonglycosylated N5Q/N11Q double mutant to mediate HBV infection was primarily a consequence of its extremely low protein level, which could be affected by the vector used to drive NTCP expression, the cellular background (cell density and differentiation), which could also alter NTCP protein level (6). Alternatively, juxtaposition of the 9-aa HA tag with the 5Q/11Q double mutation might affect NTCP function or stability. Our positive results with both 5A/11A and 5Q/11Q mutants indicate that nonglycosylated NTCP retains its function as an HBV receptor so long as a decent protein level is maintained. This is consistent with the cHBV susceptibility of differentiated HepaRG cells, which expresses a nonglycosylated form of wild-type NTCP protein.
One may wonder whether the lack of NTCP glycosylation is responsible for the much higher sHBV susceptibility of differentiated HepaRG cells and if NTCP overexpression in HepG2/NTCP cells accounts for their higher cHBV susceptibility. Unfortunately, HepG2 cells stably transfected with wild-type NTCP or its glycosylation mutants failed to be infected by any of the three sHBV isolates that we tested so far. Screening of additional high-titer sHBV isolates might overcome this hurdle (7). The overexpression approach in HepaRG cells turned out to be more complicated, because stable transfection with the epitope-tagged NTCP rather diminished both cHBV and sHBV infectivity (Fig. 5B and data not shown). One possibility is that stable plasmid DNA transfection and/or drug selection interferes with cellular differentiation. Indeed, we found retarded growth of HepaRG cells transfected with either NTCP or its null mutant and altered cell morphology following their differentiation. Thus, comparison should be made between HepaRG/NTCP cells and HepaRG/null cells. In this regard NTCP overexpression moderately increased sHBV but not cHBV infectivity (Fig. 5B and C), raising the possibility that glycosylated NTCP with a rhodopsin epitope could mediate sHBV infection.
Besides their different cHBV and sHBV susceptibilities, HepG2/NTCP cells differ from HepaRG cells in producing up to 100-fold-reduced HBsAg/HBeAg ratios following cHBV infection (6, 11). Such a difference can be reproduced following HBV genome delivery by adeno-associated virus (AAV) or transfection and is attributable to genotype D inoculum, NTCP overexpression, and DMSO added to cell medium. Indeed, a genotype D genome delivered by AAV produced a much lower HBsAg/HBeAg ratio in HepG2/NTCP cells than a genotype C genome but a higher HBsAg/HBeAg ratio than the genotype C genome in differentiated HepaRG cells (6). Genotype D isolates have a weaker SPII promoter than genotype A isolates, leading to reduced S protein expression and HBsAg secretion in both HepG2 and Huh7 cells following transient transfection with dimeric HBV DNA constructs (28). NTCP may affect HBsAg/HBeAg ratios at two levels. First, cHBV-infected HepG2/NTCP cells overproduce the 3.5-kb RNAs responsible for genome replication and HBeAg expression relative to the 2.4-kb and 2.1-kb RNAs for envelope protein expression (6, 11, 29). Second, overexpressed NTCP could inhibit HBsAg secretion, leading to an increased intracellular HBsAg/extracellular HBsAg ratio (6). In the present study, we found that HepG2 cells stably transfected with nonglycosylated NTCP mutants did not produce a higher HBsAg/HBeAg ratio than cells expressing wild-type NTCP following cHBV infection (Fig. 3A). Overexpressing rhodopsin epitope-tagged NTCP in HepaRG cells did not markedly reduce the HBsAg/HBeAg ratio, whether compared to parental HepaRG cells or to HepaRG null cells (Fig. 5A and B).
In conclusion, exogenous epitope-tagged NTCP is glycosylated in both HepG2 and HepaRG cells, albeit to different extents. Nonglycosylated NTCP is expressed by differentiated HepaRG cells and is capable of mediating cHBV infection in HepG2 cells. So far there is no evidence for the nonglycosylated form of NTCP to be more efficient at mediating sHBV infection or for overexpressed NTCP to reduce the HBsAg/HBeAg ratio in HepaRG cells. The different cHBV and sHBV susceptibilities of HepG2/NTCP cells versus HepaRG cells, as well as their different HBsAg/HBeAg ratios following cHBV infection, cannot be explained by the status of NTCP glycosylation, and future studies should be directed at other host factors.
MATERIALS AND METHODS
NTCP glycosylation mutants and the null mutant.
The pcDNA6-NTCP expression construct has been described (6). It produces, under the transcriptional control of the CMV promoter, the 349-aa human NTCP protein fused with a 9-aa bovine rhodopsin epitope at the carboxyl terminus. In the present study, codons 5, 11, or both were mutated from asparagine to alanine or glutamine (Fig. 1A). The 5A, 5Q, 11A, and 11Q single mutations and the 5A/11A and 5Q/11Q double mutations were introduced by overlap extension PCR using Phusion DNA polymerase (New England BioLabs) with an NheI restriction site attached to the outside sense primer and an AgeI site attached to the outside antisense primer. The final 1.1-kb PCR product was doubly digested with NheI and AgeI for replacement of the cognate DNA fragment in the pcDNA6-NTCP construct. An NTCP null mutant was generated by converting codon 50 from AAG to TAG, a stop codon.
Cell culture and transfection with NTCP expression constructs.
HepG2 cells were cultured in minimum essential medium Eagle (MEME) supplemented with 10% fetal bovine serum (FBS) and 1% nonessential amino acids. The HepG2/NTCP cell line, which has the pcDNA6-NTCP expression construct stably transfected to HepG2 cells (6), was cultured in MEME supplemented with 10% FBS and 10 μg/ml blasticidin. In the present study, the glycosylation mutants and the null mutant were also transfected to HepG2 cells, with stable transfectants (as a pool) selected by blasticidin. HepaRG cells were grown in William's E medium supplemented with hydrocortisone, insulin, and 10% FBS. To induce cellular differentiation, confluent cells were maintained in medium supplemented with 2% DMSO for a minimum of 2 weeks. Subconfluent HepaRG cells were transfected with the pcDNA6-NTCP expression construct or the null mutant, followed by blasticidin selection of stable transfectants (as a pool rather than single cell clones).
NTCP detection and verification of its glycosylation status.
Endogenous NTCP protein expressed from differentiated HepaRG cells was detected by a rabbit polyclonal anti-SLC10A1 antibody raised against the carboxyl terminus of NTCP (Sigma-Aldrich). A mouse monoclonal antibody against the 9-aa rhodopsin epitope (1D4; Santa Cruz) was employed to selectively detect exogenous NTCP. For IF staining, cells were fixed in ethanol-acetic acid (95%:5%) solution at −20°C and incubated with 1D4 antibody at a 1:100 dilution, followed by incubation with a 1:300 dilution of Alexa-488 goat anti-mouse IgG (Invitrogen). For Western blot analysis, cells were lysed in a buffer containing 10 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40. An aliquot of lysate containing 20 μg of proteins was separated in SDS-10% PAGE. After transfer, the blot was incubated at 4°C overnight with anti-SLC10A1 or 1D4 antibody at 1:2,000 dilution, followed by wash with phosphate-buffered saline with Tween 20 (PBST). The blot was further incubated at room temperature for 1 h with goat anti-rabbit antibody (1:40,000 dilution) conjugated with horseradish peroxidase (HRP) or HRP-conjugated rabbit anti-mouse antibody (1:40,000 dilution). After further wash, signals were revealed by enhanced chemiluminescence (ECL). For a loading control, the blot was stripped, followed by reprobing with mouse anti-β actin antibody (1:3,000 dilution; Sigma) and rabbit anti-mouse secondary antibody. To remove N-linked glycans prior to Western blotting, cell lysate containing 20 μg proteins was treated with 1 U of PNGase F (New England BioLabs) at 37°C overnight. Alternatively, cells were cultured in medium supplemented with 1 μg/ml tunicamycin (Sigma-Aldrich) overnight prior to harvesting.
Silencing NTCP expression in differentiated HepaRG cells.
Lentiviruses encoding four 29mer shRNAs against human NTCP (TL309420V) or scrambled control shRNA were purchased from OriGene. Differentiated HepaRG cells seeded in 96-well plates were infected overnight with lentivirus at a multiplicity of infection of 100 in the presence of 8 μg/ml Polybrene. Medium was changed the next day, and cells were harvested 4 days postinfection. NTCP protein was detected by Western blotting from cell lysate using anti-SLC10A1 antibody, followed by detection of β-actin from the same blot. Of the four shRNA constructs provided, construct D (TGCTCTCTTCTGCCTCAATGGACGGTGCA) could efficiently reduce NTCP expression, thus being used in the present study.
Subcellular fractionation.
A subcellular fractionation kit for cultured cells (Thermo Scientific) was used to determine the subcellular distribution of wild-type NTCP and its glycosylation mutants stably transfected to HepG2 cells. The exact protocol was followed (with protease inhibitors added) to sequentially obtain the cytoplasmic extract, membrane fraction (plasma, mitochondria, and ER/Golgi membranes but not nuclear membrane), nuclear extract, chromatin-bound nuclear proteins, and finally cytoskeletal proteins. NTCP in these various fractions was detected by Western blotting using the 1D4 antibody.
Sequencing of NTCP cDNA from HepaRG and HepG2 cells.
HepG2 and differentiated HepaRG cells were lysed in TRIzol solution. The lysate was extracted with chloroform and RNA was precipitated with isopropanol and dissolved in RNase-free water supplemented with RNasin (1 U/μl). cDNA was synthesized at 42°C for 1 h by avian myeloblastosis virus (AMV) reverse transcriptase using oligo(dT) primer. A nearly 1.3-kb NTCP cDNA was amplified by 35 cycles of PCR using primers S1 (5′-GAACTGCACAAGAAACGGAGTCAGC-3′) and AS1 (5′-GTAGACACCCTGTCTGTGTTCCC-3′) and Q5 DNA polymerase (New England BioLabs). The PCR conditions were initial denaturation at 98°C for 30 s, 35 cycles of 98°C for 10 s, 69°C for 20 s, and 72°C for 40 s, and a final extension at 72°C for 10 min. The PCR product was sequenced directly using internal primers S3 (5′-GGCCATCTTGGTCTGTGGCTG-3′) and AS3 (5′-GCACCGTCCATTGAGGCAGAAG-3′). Alternatively, a larger (1.5-kb) NTCP cDNA fragment was amplified from HepaRG cells using primer pair S5 (5′-gtcaagcTTGTCCAGAAACTCTCTGTCCTGAG-3′; HindIII site is underlined; lowercase letters are non-NTCP sequence added) and AS5 (5′-gtcgaattcTGGGTCACAAAACTTGGAATGGG-3′; EcoRI site is underlined). The Q5 enzyme was added after heating the remaining PCR contents at 80°C for 2 min. The PCR conditions were initial denaturation at 98°C for 3 min, followed by 35 cycles of 98°C for 15 s, 69°C for 20 s, and 72°C for 1 min. This was followed by a final extension at 72°C for 10 min. The PCR product was double digested with EcoRI and HindIII for cloning to pUC18 vector, and three PCR clones were sequenced.
cHBV and sHBV infection of differentiated HepaRG cells.
The single cHBV isolate used for infection experiments originated from a HepDE19 cell line stably transfected with an HBV genome of genotype D (30), with viral and subviral particles concentrated from culture supernatant by PEG precipitation. All sHBV isolates came from precollected HBeAg- and HBsAg-positive serum samples from Shanghai, China (31), with viral/subviral particles concentrated by ultracentrifugation through a 10 to 20% sucrose cushion (6). Parental HepaRG cells and those stably transfected with NTCP or its null mutant were seeded in 96-well plates, and differentiation was achieved by further culturing confluent cells in medium supplemented with 2% DMSO for >2 months. Cells were incubated overnight with cHBV (at about 1,400 genome equivalents/cell) or sHBV (at about 500 genome equivalents/cell) in FBS-free medium in the presence (cHBV) or absence (sHBV) of 4% PEG 8000. The inoculum was removed followed by extensive wash, and HBeAg and HBsAg released to culture supernatant were measured by enzyme-linked immunosorbent assay (ELISA) using commercial kits (KHB, Shanghai, China).
sHBV and cHBV infection of HepG2 cells.
HepG2 cells stably transfected with wild-type NTCP or the glycosylation mutants were seeded in 96-well plates and were incubated overnight with sHBV in FBS-free medium in the absence of PEG. Only sHBV isolates proven to be infectious in differentiated HepaRG cells were used, and HBV infectivity was monitored by ELISA detection of HBeAg and HBsAg. For cHBV infection experiments shown in Fig. 3, four wells of cells in 96-well plates were cultured in regular medium, while the other four wells were maintained in medium supplemented with 2% DMSO. Two days later two out of the four wells were incubated overnight with cHBV at 1,400 genome equivalents/cell in FBS-free medium supplemented with 4% PEG. Cells were washed and further incubated in medium with or without 2% DMSO. HBeAg and HBsAg released to culture supernatant were measured by ELISA. Both wells of infected cells were harvested by trypsinization at day 17 postinfection and combined for Southern blot analysis of replicative HBV DNA, while the two uninfected wells were lysed at the same time point by lysis buffer (10 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM EDTA, and 1% NP-40). They were pooled for Western blot analysis of NTCP expression.
For Southern blot analysis, lysate from cells cultured in regular medium was pooled with that of cells cultured in 2% DMSO medium. The combined lysate (60 μl) was incubated at 37°C for 2 h with 0.5 mg/ml of proteinase K in a total volume of 400 μl, with the remaining volume filled up with proteinase K buffer (25 mM Tris, pH 7.5, 10 mM EDTA, 100 mM NaCl, 0.5% SDS). After phenol extraction, DNA was precipitated by ethanol and resuspended in Tris-EDTA buffer. DNA was separated in 1.5% agarose gel, with cloned HBV DNA digested with different restriction enzymes serving as size markers. After transfer to nylon membrane, the blot was hybridized overnight with 32P-labeled HBV DNA probe of genotype D, washed at 65°C with 2×SSC (1× SSC is is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% SDS, and exposed to X-ray films (6, 32, 33).
ACKNOWLEDGMENTS
We are grateful to Christian Trepo for the HepaRG cell line, to Camille Sureau for instructions on culturing of HepaRG cells, to Wenhui Li for the human NTCP cDNA, and to Haitao Guo for the HepDE19 cell line.
This work was supported by NIH grants R21AI107618, R01AI116639, and R21AI113394.
REFERENCES
- 1.Cerec V, Glaise D, Garnier D, Morosan S, Turlin B, Drenou B, Gripon P, Kremsdorf D, Guguen-Guillouzo C, Corlu A. 2007. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 45:957–967. doi: 10.1002/hep.21536. [DOI] [PubMed] [Google Scholar]
- 2.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A 99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parent R, Marion MJ, Furio L, Trepo C, Petit MA. 2004. Origin and characterization of a human bipotent liver progenitor cell line. Gastroenterology 126:1147–1156. doi: 10.1053/j.gastro.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He W, Ren B, Mao F, Jing Z, Li Y, Liu Y, Peng B, Yan H, Qi Y, Sun Y, Guo JT, Sui J, Wang F, Li W. 2015. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog 11:e1004840. doi: 10.1371/journal.ppat.1004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Zong L, Sureau C, Barker L, Wands JR, Tong S. 2016. Unusual features of sodium taurocholate cotransporting polypeptide as a hepatitis B virus receptor. J Virol 90:8302–8313. doi: 10.1128/JVI.01153-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choijilsuren G, Jhou RS, Chou SF, Chang CJ, Yang HI, Chen YY, Chuang WL, Yu ML, Shih C. 2017. Heparin at physiological concentration can enhance PEG-free in vitro infection with human hepatitis B virus. Sci Rep 7:14461. doi: 10.1038/s41598-017-14573-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rydell GE, Prakash K, Norder H, Lindh M. 2017. Hepatitis B surface antigen on subviral particles reduces the neutralizing effect of anti-HBs antibodies on hepatitis B viral particles in vitro. Virology 509:67–70. doi: 10.1016/j.virol.2017.05.017. [DOI] [PubMed] [Google Scholar]
- 9.Okuyama-Dobashi K, Kasai H, Tanaka T, Yamashita A, Yasumoto J, Chen W, Okamoto T, Maekawa S, Watashi K, Wakita T, Ryo A, Suzuki T, Matsuura Y, Enomoto N, Moriishi K. 2015. Hepatitis B virus efficiently infects non-adherent hepatoma cells via human sodium taurocholate cotransporting polypeptide. Sci Rep 5:17047. doi: 10.1038/srep17047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gripon P, Diot C, Guguen-Guillouzo C. 1993. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology 192:534–540. doi: 10.1006/viro.1993.1069. [DOI] [PubMed] [Google Scholar]
- 11.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 12.Hagenbuch B, Meier PJ. 1994. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Investig 93:1326–1331. doi: 10.1172/JCI117091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doring B, Lutteke T, Geyer J, Petzinger E. 2012. The SLC10 carrier family: transport functions and molecular structure. Curr Top Membr 70:105–168. doi: 10.1016/B978-0-12-394316-3.00004-1. [DOI] [PubMed] [Google Scholar]
- 14.Shneider BL, Fox VL, Schwarz KB, Watson CL, Ananthanarayanan M, Thevananther S, Christie DM, Hardikar W, Setchell KD, Mieli-Vergani G, Suchy FJ, Mowat AP. 1997. Hepatic basolateral sodium-dependent-bile acid transporter expression in two unusual cases of hypercholanemia and in extrahepatic biliary atresia. Hepatology 25:1176–1183. doi: 10.1002/hep.510250521. [DOI] [PubMed] [Google Scholar]
- 15.Ho RH, Leake BF, Roberts RL, Lee W, Kim RB. 2004. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem 279:7213–7222. doi: 10.1074/jbc.M305782200. [DOI] [PubMed] [Google Scholar]
- 16.Gerloff T, Geier A, Stieger B, Hagenbuch B, Meier PJ, Matern S, Gartung C. 1999. Differential expression of basolateral and canalicular organic anion transporters during regeneration of rat liver. Gastroenterology 117:1408–1415. doi: 10.1016/S0016-5085(99)70291-X. [DOI] [PubMed] [Google Scholar]
- 17.Liang D, Hagenbuch B, Stieger B, Meier PJ. 1993. Parallel decrease of Na(+)-taurocholate cotransport and its encoding mRNA in primary cultures of rat hepatocytes. Hepatology 18:1162–1166. [PubMed] [Google Scholar]
- 18.Appelman MD, Chakraborty A, Protzer U, McKeating JA, van de Graaf SF. 2017. N-glycosylation of the Na+-taurocholate cotransporting polypeptide (NTCP) determines its trafficking and stability and is required for hepatitis B virus infection. PLoS One 12:e0170419. doi: 10.1371/journal.pone.0170419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallen S, Mareninova O, Branden M, Sachs G. 2002. Organization of the membrane domain of the human liver sodium/bile acid cotransporter. Biochemistry 41:7253–7266. doi: 10.1021/bi012152s. [DOI] [PubMed] [Google Scholar]
- 20.Dranoff JA, McClure M, Burgstahler AD, Denson LA, Crawford AR, Crawford JM, Karpen SJ, Nathanson MH. 1999. Short-term regulation of bile acid uptake by microfilament-dependent translocation of rat ntcp to the plasma membrane. Hepatology 30:223–229. doi: 10.1002/hep.510300136. [DOI] [PubMed] [Google Scholar]
- 21.Sarkar S, Bananis E, Nath S, Anwer MS, Wolkoff AW, Murray JW. 2006. PKCzeta is required for microtubule-based motility of vesicles containing the ntcp transporter. Traffic 7:1078–1091. doi: 10.1111/j.1600-0854.2006.00447.x. [DOI] [PubMed] [Google Scholar]
- 22.Kato S, Oda-Tamai S, Akamatsu N. 1988. Postnatal changes in N-linked oligosaccharides of glycoproteins in rat liver. Biochem J 253:59–66. doi: 10.1042/bj2530059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda-Tamai S, Kato S, Akamatsu N. 1991. Postnatal changes in sialylation of glycoproteins in rat liver. Biochem J 280(Part 1):179–185. doi: 10.1042/bj2800179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardikar W, Ananthanarayanan M, Suchy FJ. 1995. Differential ontogenic regulation of basolateral and canalicular bile acid transport proteins in rat liver. J Biol Chem 270:20841–20846. [DOI] [PubMed] [Google Scholar]
- 25.Kanninen LK, Porola P, Niklander J, Malinen MM, Corlu A, Guguen-Guillouzo C, Urtti A, Yliperttula ML, Lou YR. 2016. Hepatic differentiation of human pluripotent stem cells on human liver progenitor HepaRG-derived acellular matrix. Exp Cell Res 341:207–217. doi: 10.1016/j.yexcr.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Vaz FM, Paulusma CC, Huidekoper H, de Ru M, Lim C, Koster J, Ho-Mok K, Bootsma AH, Groen AK, Schaap FG, Oude Elferink RP, Waterham HR, Wanders RJ. 2015. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology 61:260–267. doi: 10.1002/hep.27240. [DOI] [PubMed] [Google Scholar]
- 27.Mochizuki K, Kagawa T, Numari A, Harris MJ, Itoh J, Watanabe N, Mine T, Arias IM. 2007. Two N-linked glycans are required to maintain the transport activity of the bile salt export pump (ABCB11) in MDCK II cells. Am J Physiol Gastrointest Liver Physiol 292:G818–G828. doi: 10.1152/ajpgi.00415.2006. [DOI] [PubMed] [Google Scholar]
- 28.Zhang F, Tang X, Garcia T, Lok AS, Wang Y, Jia H, Qin Y, Chen C, Wen Y, Li J, Tong S. 2017. Characterization of contrasting features between hepatitis B virus genotype A and genotype D in small envelope protein expression and surface antigen secretion. Virology 503:52–61. doi: 10.1016/j.virol.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tropberger P, Mercier A, Robinson M, Zhong W, Ganem DE, Holdorf M. 2015. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc Natl Acad Sci U S A 112:E5715–E5724. doi: 10.1073/pnas.1518090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin Y, Tang X, Garcia T, Hussain M, Zhang J, Lok A, Wands J, Li J, Tong S. 2011. Hepatitis B virus genotype C isolates with wild-type core promoter sequence replicate less efficiently than genotype B isolates but possess higher virion secretion capacity. J Virol 85:10167–10177. doi: 10.1128/JVI.00819-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwei K, Tang X, Lok AS, Sureau C, Garcia T, Li J, Wands J, Tong S. 2013. Impaired virion secretion by hepatitis B virus immune escape mutants and its rescue by wild-type envelope proteins or a second-site mutation. J Virol 87:2352–2357. doi: 10.1128/JVI.02701-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zong L, Qin Y, Jia H, Ye L, Wang Y, Zhang J, Wands JR, Tong S, Li J. 2017. Differential regulation of hepatitis B virus core protein expression and genome replication by a small upstream open reading frame and naturally occurring mutations in the precore region. Virology 505:155–161. doi: 10.1016/j.virol.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]