

Classical swine fever, is caused by classical swine fever virus (CSFV). The disease is notifiable to World Organisation for Animal Health (OIE) in most countries and causes significant financial losses to the pig industry globally. Understanding the processes of CSFV endocytosis and postinternalization will advance our knowledge of the disease and provide potential novel drug targets against CSFV. With this objective, we used systematic approaches to dissect these processes in CSFV-infected 3D4/21 cells. The data presented here demonstrate for the first time to our knowledge that CSFV is able to enter cells via caveola-mediated endocytosis that requires Rab5, Rab7 and Rab11, in addition to the previously described classical clathrin-dependent pathway that requires Rab5 and Rab7. The characterization of CSFV entry will further promote our current understanding of Pestivirus cellular entry pathways and provide novel targets for antiviral drug development.
KEYWORDS: Rab proteins, caveolae, classical swine fever virus, macrophages
ABSTRACT
The members of Flaviviridae utilize several endocytic pathways to enter a variety of host cells. Our previous work showed that classical swine fever virus (CSFV) enters porcine kidney (PK-15) cells through a clathrin-dependent pathway that requires Rab5 and Rab7. The entry mechanism for CSFV into other cell lines remains unclear, for instance, porcine alveolar macrophages (3D4/21 cells). More importantly, the trafficking of CSFV within endosomes controlled by Rab GTPases is unknown in 3D4/21 cells. In this study, entry and postinternalization of CSFV were analyzed using chemical inhibitors, RNA interference, and dominant-negative (DN) mutants. Our data demonstrated that CSFV entry into 3D4/21 cells depends on caveolae, dynamin, and cholesterol but not clathrin or macropinocytosis. The effects of DN mutants and knockdown of four Rab proteins that regulate endosomal trafficking were examined on CSFV infection, respectively. The results showed that Rab5, Rab7, and Rab11, but not Rab9, regulate CSFV endocytosis. Confocal microscopy showed that virus particles colocalize with Rab5, Rab7, or Rab11 within 30 min after virus entry and further with lysosomes, suggesting that after internalization CSFV moves to early, late, and recycling endosomes and then into lysosomes before the release of the viral genome. Our findings provide insights into the life cycle of pestiviruses in macrophages.
IMPORTANCE Classical swine fever, is caused by classical swine fever virus (CSFV). The disease is notifiable to World Organisation for Animal Health (OIE) in most countries and causes significant financial losses to the pig industry globally. Understanding the processes of CSFV endocytosis and postinternalization will advance our knowledge of the disease and provide potential novel drug targets against CSFV. With this objective, we used systematic approaches to dissect these processes in CSFV-infected 3D4/21 cells. The data presented here demonstrate for the first time to our knowledge that CSFV is able to enter cells via caveola-mediated endocytosis that requires Rab5, Rab7 and Rab11, in addition to the previously described classical clathrin-dependent pathway that requires Rab5 and Rab7. The characterization of CSFV entry will further promote our current understanding of Pestivirus cellular entry pathways and provide novel targets for antiviral drug development.
INTRODUCTION
Classical swine fever is a highly contagious and fatal disease that causes significant economic losses to the pig industry throughout the world. CSFV is a member of the genus Pestivirus within the family Flaviviridae (1, 2) and is closely related to other members of the genus, namely, bovine viral diarrhea virus 1 (BVDV-1) and BVDV-2 (3, 4), border disease virus (5, 6), an atypical pestivirus isolated from a giraffe (7), and a variety of other unclassified pestiviruses. The CSFV genome consists of a single-stranded, positive-sense RNA with a single open reading frame (ORF) encoding a polyprotein that is cleaved into 11 mature viral proteins. Of these, nucleocapsid (C) protein and the envelope glycoproteins Erns, E1, and E2 are structural proteins. E2 is the immunodominant protein in the envelope and plays an important role in virus neutralization (8, 9). E2 forms homodimers and heterodimers with glycoprotein E1. Because the formation of the heterodimer is essential for pestivirus entry into cells (10, 11), both E1 and E2 are required for virus entry via receptor-mediated endocytosis (10).
Flaviviruses utilize several endocytic pathways to enter host cells: macropinocytosis, clathrin-mediated endocytosis, caveola/cholesterol-dependent endocytosis, and clathrin- and caveola-independent endocytosis (12), although clathrin-mediated endocytosis is believed to be the major route of flavivirus entry (13). For instance, previous studies have found that Japanese encephalitis virus (JEV) enters C6/36, Vero, PK-15 cells, and neural stem cells through a clathrin-dependent pathway (14–16). Recent studies have shown that JEV infects mouse and rat neuronal cells through dynamin- and caveola-mediated endocytosis pathways (17, 18). Hepatitis C virus (HCV) entry is clathrin- and dynamin-dependent in ORL8c and HepCD81/miR122 cells, while productive entry of HCV was clathrin- and dynamin-independent in Hep3B/miR122 cells (19). Macrophages are at the frontline of defense against pathogenic microorganisms. However, little is known about the cell invasion mechanism of CSFV. Our previous work had shown that CSFV enters PK-15 cells through a clathrin-dependent pathway (20). Even though the recent work have shown that caveolin-1-mediated endocytic pathway is involved in CSFV into porcine alveolar macrophages (3D4/21 cells) (21). However, the mechanism for CSFV entry into 3D4/21 cells on the fine detail remains obscure.
The dynamics of the network of vesicles of the endocytic pathway are regulated by Rab proteins, which are small GTPases of the Ras superfamily, and their effectors (22). These proteins are involved in selection of vesicle cargos, budding, targeting, and fusion (23). Rab5 regulates the transport of newly endocytosed vesicles from the plasma membrane to early endosomes (24). Rab7, a small GTPase of the Rab family associated with both the endosome and the lysosome, was investigated extensively and well recognized to facilitate endosomal maturation, transport from the late endosome to the lysosome, and positioning of the endosome and lysosome through regulating their movement along cytoskeleton (25). Rab9 facilitates late endosome to the trans-Golgi network (26). Rab11 has previously been reported to be associated with the pericentriolar recycling compartment, post-Golgi vesicles, and the trans-Golgi network and is involved in vesicle recycling (27). Numerous reports have shown that Rab proteins are involved in the life cycle of many viruses, such as HCV (28, 29), dengue virus (30), JEV (31), and West Nile virus (32). We have previously determined that CSFV entry into PK-15 cells requires Rab5 and Rab7 (20), but which Rab protein(s) is required for CSFV entry into macrophages is largely unknown.
The aim of this study was to elucidate the mechanism of CSFV internalization into 3D4/21 cells and the roles of Rab proteins in this process. We used chemical inhibitors, RNA silencing, and the expression of dominant-negative (DN) constructs to examine which cellular molecules are involved in CSFV endocytosis in porcine alveolar macrophages. Our results indicated that CSFV entry into 3D4/21 cells is dependent on acidic endosomal pH, dynamin, cholesterol, and caveolae but not clathrin or macropinocytosis. We found that Rab5, Rab7, and Rab11 are involved in the infection process.
RESULTS
CSFV entry is pH dependent.
To assess whether CSFV entry is pH dependent, we first studied the effects of three lysosomotropic agents—chloroquine, NH4Cl, and bafilomycin A1 (BafA1)–on CSFV infection in 3D4/21 cells. Cells were pretreated for 1 h with the drugs (Fig. 1), followed by inoculation with CSFV for 1 h at 4°C, and then shifted to 37°C for 0 h (binding) or 1 h to (entry). The amount of virus bound or internalized was determined by quantifying viral RNA. CSFV bound to cells equally well in the presence of all three inhibitors at all concentrations tested, but entry was reduced by all three inhibitors in a dose-dependent manner (Fig. 2A to C). Viral RNA copies were reduced by 59.2% at 500 μM chloroquine, 77.9% at 200 μM NH4Cl, and 73.6% at 10 μM BafA1 compared to nontreated cells. We further examined the requirement for low pH in CSFV entry by silencing the expression of vacuolar ATPase (V-ATPase), a proton pump key to establish the low pH of endosomal compartments. Western blotting showed the efficiency of small interfering RNA (siRNA)-mediated knockdown of V-ATPase in 3D4/21 cells. Treatment with siV-ATPase significantly reduced viral RNA copies, E2 protein level, and virus titers (Fig. 2D). These data demonstrate that CSFV enters 3D4/21 cells in a pH-dependent manner.
FIG 1.

Cell viabilities upon all the drugs and siRNA duplexes were assessed by using the CellTiter 96 AQueous One Solution cell proliferation assay.
FIG 2.
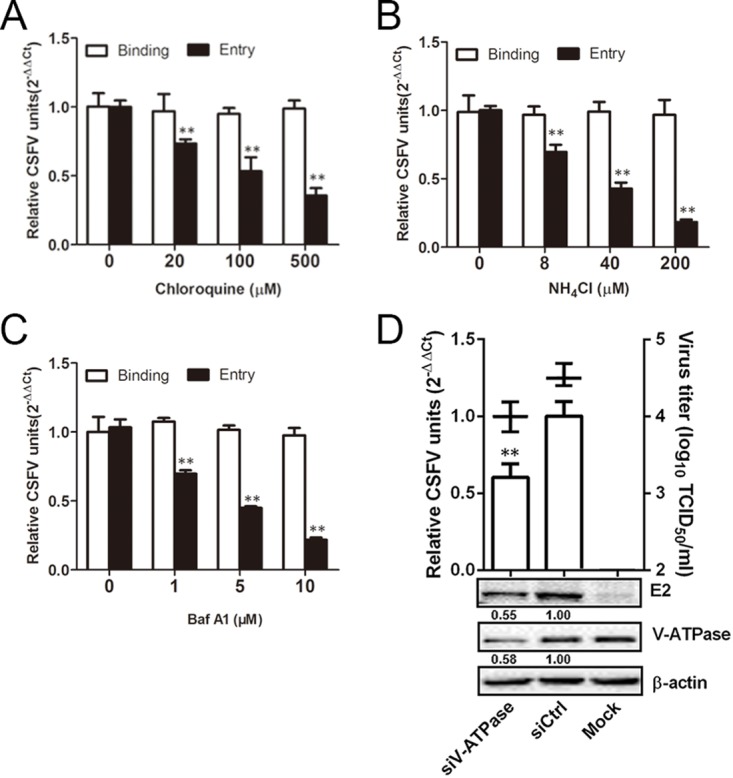
CSFV entry requires acidic endosomal pH. (A to C) Chloroquine, NH4Cl, and BafA1 inhibited CSFV entry. Cells were pretreated with subtoxic doses at 37°C for 1 h and inoculated with CSFV (MOI = 1) at 4°C for 1 h and then shifted to 37°C. At 0 (binding) or 1 (entry) hpi, infected cells were lysed to determine the viral RNA copy number by RT-qPCR. (D) V-ATPase knockdown inhibited CSFV infection. siV-ATPase or siCtrl-transfected cells were infected with CSFV (MOI = 0.1). At 24 hpi, the viral RNA levels, virus titers, and viral E2 protein levels were determined by RT-qPCR, virus titration, and Western blotting, respectively. The relative protein amount was calculated using ImageJ 7.0 software as shown in the figure. The results are presented as means ± the SD of data from three independent experiments (**, P < 0.01).
Cholesterol is required for CSFV infection.
Our previous studies have shown that cholesterol-rich membrane rafts have been shown to mediate CSFV entry in PK-15 cells (20) or JEV entry in BHK-21 cells (31). Here, we carried out a series of experiments to determine the role of cholesterol in the process of CSFV infection of 3D4/21 cells. As determined by indirect immunofluorescence, the removal of cell membrane cholesterol prior to infection, significantly inhibited CSFV replication. Red fluorescent signals were less and less, along with increasing concentrations of methyl-β-cyclodextrin (MβCD) (Fig. 3A). Quantitative reverse transcription-PCR (RT-qPCR) and virus titration both showed that in cells treated with MβCD prior to infection, viral RNA copies and viral titers were reduced in a dose-dependent manner (Fig. 3B). When viral envelope cholesterol was depleted, the decreases in viral RNA copies and virus titers were both significantly greater than in cholesterol-depleted cells at up to 5 mg/ml MβCD (Fig. 3C), indicating that viral envelope cholesterol depletion markedly affected CSFV infectivity. Cell membrane cholesterol has also been reported to be involved in other stages of the virus life cycle, such as egress (33). To examine whether CSFV egress from 3D4/21 cells is cholesterol dependent, cells were infected with CSFV for 12 h and then maintained in the presence of MβCD-treated cells for 12 to 24 h. The results showed that viral RNA copies in MβCD-treated cells decreased significantly in a dose-dependent manner (Fig. 3D). Virus titration further confirmed that the titers of progeny virus in MβCD-treated cells culture and cell supernatant decreased, along with increasing concentrations of MβCD (Fig. 3D and E), indicating that cholesterol depletion had an impact on virus egress. These data indicate that CSFV infection is dependent on cellular membrane cholesterol.
FIG 3.

Effects of cholesterol depletion on CSFV infection. (A) Cells were treated with MβCD with different concentrations for 1 h and infected with CSFV (MOI = 0.5). At 24 hpi, the cells were fixed and stained with WH303. Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). Scale bar, 50 μm. (B) Cells were pretreated with the indicated concentrations of MβCD for 1 h at 37°C and infected with CSFV (MOI = 0.1) for 24 h. (C) MβCD was mixed with CSFV (MOI = 0.1), followed by incubation at 37°C for 1 h. 3D4/21 cells were infected with the treated CSFV for 24 h. (D) Cells were infected with CSFV (MOI = 0.1) for 12 h and then treated with the indicated concentrations of MβCD for 12 h or 24 h at 37°C. Viral RNA levels and virus titers were determined by RT-qPCR and virus titration, respectively. (E) Cells were treated as for panel D. The TCID50 of the supernatant was determined. The results are presented as means ± the SD of data from three independent experiments (*, P < 0.05; **, P < 0.01).
CSFV entry depends on dynamin.
Previous studies have shown that dynamin is involved in the endocytosis of flaviviruses (17) and pestiviruses (20). To determine whether dynamin is involved in CSFV infection of 3D4/21 cells, we pretreated cells with increasing concentrations of dynasore and infected them with CSFV. Viral RNA copies were determined at 0 (Binding) and 1 hpi (Entry). Dynasore had no effect on virus binding to cells but significantly reduced entry into cells in a dose-dependent manner (Fig. 4A). At 50 μM dynasore, entry was reduced by about 47%. Figure 4B illustrates, by fluorescence microscopy, the significant reduction in transferrin (Tfn) uptake and CSFV infection in cells treated with 50 μM dynasore compared to dimethyl sulfoxide (DMSO)-treated or mock-treated cells. These data demonstrate that dynasore effectively inhibits CSFV endocytosis into 3D4/21 cells. Confocal microscopy was used to further investigate the role of dynamin in CSFV endocytosis. 3D4/21 cells were transfected with constructs of wild-type (WT) and DN (K44A) dynamin II (Fig. 4C). Tfn uptake was used as a positive control. Overexpression of DN dynamin II resulted in approximately 82% inhibition of Tfn uptake compared to cells overexpressing the WT construct (Fig. 4D). CSFV infection was also reduced in DN dynamin II-transfected cells (Fig. 4C). The overexpression of DN dynamin II resulted in approximately 86% inhibition of CSFV infection (Fig. 4E). Western blotting illustrates the efficiency of siRNA-mediated knockdown of dynamin in mock- and CSFV-infected cells, as determined by viral E2 protein levels (Fig. 4F). The results from RT-PCR and viral titration indicated significant inhibition of CSFV infection in dynamin knockdown cells (Fig. 4F). These results demonstrate that CSFV entry requires dynamin.
FIG 4.

CSFV entry depends on dynamin. (A) Cells were pretreated with increasing concentrations of dynasore at 37°C for 1 h and inoculated with CSFV (MOI = 1) at 4°C for 1 h and then shifted to 37°C. At 0 or 1 hpi, the cells were lysed, and the number of viral RNA copies was determined by RT-qPCR. (B) Dynasore inhibits Tfn uptake and CSFV entry. Cells were treated with 50 μM dynasore or DMSO for 1 h at 37°C, followed by incubation with 20 μg/ml Alexa Fluor 568-labeled Tfn for 1 h at 4°C, and then shifted to 37°C for 30 min, or they were inoculated with CSFV (MOI = 0.5). The cells were fixed and stained with DAPI. (C) The effect of the dynamin DN construct on Tfn uptake was determined by confocal microscopy. Cells transfected with the GFP-tagged WT or DN dynamin were incubated with 20 μg/ml Alexa Fluor 568-labeled Tfn for 1 h at 4°C and then shifted to 37°C for 30 min. The cells were fixed and stained with DAPI. Transfected cells were infected with CSFV (MOI = 0.5); at 24 hpi, the cells were fixed with 4% PFA, reacted with WH303, and visualized by confocal microscopy. Scale bar, 50 μm. (D) A significant reduction in the signal intensity of fluorescently labeled Tfn was observed in cells transfected with the GFP-tagged dynamin DN. (E) At least 300 transfected cells were screened for CSFV infection. Values are expressed as a percentage of infected cells observed in the WT dynamin sample. (F) Dynamin knockdown inhibited CSFV infection. siDyn- or siCtrl-transfected cells were infected with CSFV (MOI = 0.1). At 24 hpi, viral RNA levels, virus titers, and viral E2 protein levels were determined by RT-qPCR, virus titration, and Western blotting, respectively. The relative protein amount was calculated using ImageJ 7.0 software as shown in the figure. All the results are presented as means ± the SD of data from three independent experiments (*, P < 0.05; **, P < 0.01).
CSFV entry is clathrin independent.
Previous work has shown that CSFV enters PK-15 cells by clathrin-mediated endocytosis (20). Here, a series of experiments were performed to examine CSFV entry into 3D4/21 cells. First, cells were treated with increasing concentrations of chlorpromazine (CPZ) for 1 h, followed by inoculation with CSFV at an MOI of 1 for 1 h at 4°C and then 37°C at 0 (binding) or 1 (entry) h postinfection (hpi). We found that pretreatment of 3D4/21 cells with CPZ had no effect on CSFV entry and binding (Fig. 5A). Immunofluorescence also demonstrated that CSFV infection (red fluorescence signals) was not inhibited at 25 μM CPZ, although Tfn uptake was significantly reduce (Fig. 5B). Next, EPS15 WT (DIIIΔ2) or DN (EΔ95/295) construct was transfected into 3D4/21 cells. We analyzed the overexpression effects on the internalization of Tfn and CSFV using confocal microscopy. EPS15 DN expression inhibited Tfn uptake (Fig. 5C) by approximately 77% compared to cells expressing the WT construct (Fig. 5D). However, the EPS15 DN protein did not inhibit CSFV infection as observed by fluorescence microscopy (Fig. 5C and E). The role of clathrin during CSFV entry was further assessed by knockdown of clathrin heavy chain (CHC). We found no reduction in viral RNA copies, E2 protein levels, or viral titers in the siCHC-transfected cells compared to that in siCtrl-transfected cells (Fig. 5F). Overall, these data demonstrate that CSFV entry into 3D4/21 cells does not depend on clathrin.
FIG 5.

Effects of EPS15Δ95/295 overexpression and chlorpromazine on CSFV infection. (A) Chlorpromazine (CPZ) did not inhibit CSFV infection. Cells were pretreated with increasing concentrations of CPZ for 1 h at 37°C, inoculated with CSFV (MOI = 1) at 4°C for 1 h, and then shifted to 37°C. At 0 or 1 hpi, the cells were lysed, and the viral RNA copy number was determined by RT-qPCR. (B) Fluorescence micrographs show that CPZ blocked transferrin uptake but not CSFV. Cells were treated with CPZ or DMSO for 1 h at 37°C, incubated with 20 μg/ml Alexa Fluor 568-labeled transferrin for 1 h at 4°C, and then shifted to 37°C for 30 min. Afterward, the cells were fixed and stained with DAPI. Alternatively, treated cells were infected with CSFV (MOI = 0.5) at 24 hpi, fixed with 4% PFA, reacted with WH303, and visualized by confocal microscopy. Scale bar, 50 μm. (C) Micrograph of cells transfected with WT or DN EPS15, incubated with 20 μg/ml Alexa Fluor 568-labeled transferrin for 1 h at 4°C, and then shifted to 37°C for 30 min. Transfected cells were infected with CSFV (MOI = 0.5). At 24 hpi, the cells were fixed with 4% PFA, reacted with WH303, and visualized by confocal microscopy. Scale bar, 50 μm. (D) Quantification of Tfn uptake shows an ∼75% reduction in signal intensity of fluorescently labeled Tfn in cells transfected with GFP-tagged DN EPS15. (E) At least 300 transfected cells were counted and scored as “+” or “−” for CSFV infection. Values are expressed as a percentage of the infected cells observed in the control sample. (F) Clathrin knockdown did not inhibit CSFV infection. siCHC- or siCtrl-transfected cells were infected with CSFV (MOI = 0.1). At 24 hpi, the viral RNA levels, virus titers, and viral E2 protein levels were determined by RT-qPCR, virus titration, and Western blotting, respectively. The relative protein amount was calculated using ImageJ 7.0 software as shown in the figure. All results are presented as means ± the SD of data from three independent experiments (**, P < 0.01).
CSFV entry is caveola dependent.
Caveolin is essential for the formation and stability of caveolae. We expressed a previously characterized caveolin DN construct (20) in 3D4/21 cells to investigate the effect of caveolin. As expected, expression of the DN protein, but not the WT protein, inhibited cholera toxin (CTB) uptake (Fig. 6A) by approximately 84% (Fig. 6B). CSFV infection in cells transfected with the DN construct was reduced approximately 80% compared to CSFV-infected cells transfected with the WT construct (Fig. 6A and C). To further investigate the role of caveolae in CSFV entry into 3D4/21 cells, we utilized siCav to knock down cellular endogenous caveolin-1. Western blotting showed that caveolin-1 in 3D4/21 cells was efficiently knocked down by siCav, as well as a significant reduction in viral E2 protein (Fig. 6D). RT-PCR and virus titration both showed that viral RNA copies and viral titers in the siCav-transfected cells was significantly reduced (Fig. 6D). Finally, cells were inoculated with purified CSFV virions (multiplicity of infection [MOI] = 10) at 4°C for 1 h to allow for attachment and shifted to 37°C for 30 min before fixation. The localization of virions with caveolin-1 and clathrin was analyzed by confocal microscopy. Virions and caveolin-1 showed significant colocalization (white arrowheads), while none was observed with virions and clathrin (Fig. 6E). The colocalization coefficients were expressed as the Pearson's correlation coefficient, measured for individual cells. As shown in Fig. 6F, the Pearson's correlation coefficient of caveolin-1 is more significant than that of clathrin during CSFV endocytosis. We concluded that CSFV entry into 3D4/21 cells is dependent on caveolae.
FIG 6.

Caveolae are required for CSFV entry. (A) The effect of DN caveolin-1 on CTB uptake and viral infection was determined by confocal microscopy. Cells transfected with constructs expressing GFP-tagged WT and DN caveolin-1 were incubated with 20 μg/ml Alexa Fluor 568-labeled CTB for 1 h at 4°C and then shifted to 37°C for 30 min. The cells were fixed and stained with DAPI. Alternatively, transfected cells were infected with CSFV (MOI = 0.5). At 24 hpi, the cells were fixed with 4% PFA, reacted with WH303, and visualized by confocal microscopy. Scale bar, 50 μm. (B) CTB uptake assay showed a significant reduction in the signal intensity of fluorescently labeled CTB was observed in GFP-tagged caveolin-1 DN cells. (C) At least 300 transfected cells were scored as “+” or “−” for CSFV infection. Values are expressed as a percentage of the infected cells observed in the control sample. (D) Caveolin-1 knockdown inhibited CSFV infection. siCaveolin-1- or siCtrl-transfected cells were infected with CSFV (MOI = 0.1). At 24 hpi, the viral RNA levels, virus titers, and viral E2 protein levels were determined by RT-qPCR, virus titration, and Western blotting, respectively. The relative protein amount was calculated using ImageJ 7.0 software as shown in the figure. (E) 3D4/21 cells grown on glass coverslips in 6-well plates were infected with CSFV (MOI = 10) at 4°C for 1 h and shifted to 37°C. After 30 min, the monolayers were fixed with 4% PFA, stained with WH303 and rabbit anti-caveolin-1, or rabbit anti-clathrin, and examined by confocal microscopy. Scale bar, 20 μm. The white arrow shows the colocalization of CSFV and caveolin-1. (F) The colocalization coefficient was expressed as a Pearson's correlation coefficient. The results are presented as means ± the SD of data from three independent experiments (**, P < 0.01).
CSFV entry is macropinocytosis independent.
Macropinocytosis also relies on phosphatidylinositol 3-kinase activation (34). Therefore, we used wortmannin, an inhibitor of this kinase (17). As shown in Fig. 7A, wortmannin had no effect on CSFV binding or entry. To address the role of macropinocytosis in CSFV entry, we examined the effect of 5-(N-ethyl-N-isopropyl) amiloride (EIPA) on CSFV binding and internalization. Pretreatment of cells with EIPA did not result in a decrease in CSFV binding or infection (Fig. 7C). In addition, as expected, there was no significant difference in viral RNA levels between cells treated with either wortmannin or EIPA and mock-treated cells (Fig. 7B and D). The viability of cells was unaffected by wortmannin or EIPA at the concentrations used in these experiments (data not shown). These findings indicate that CSFV is not internalized via macropinocytosis.
FIG 7.

Role of macropinocytosis on CSFV binding, entry, and infection. Cells were pretreated with the indicated concentrations of wortmannin or EIPA for 1 h at 37°C. Drugs were introduced into the medium during the adsorption period. (A and C) Cells were infected with CSFV at an MOI of 1 in the presence of drug at 4°C for 1 h and then shifted to 37°C for 0 (binding) or 1 (entry) h. The infected cells were then lysed, and viral RNA copies were determined by RT-qPCR. (B and D) Cells were infected with CSFV (MOI = 0.1) at 37°C. At 24 hpi, viral RNA copies were determined by RT-qPCR. The results are presented as means ± the SD of data from three independent experiments.
Role of endosomes in CSFV infection.
Rab proteins orchestrate membrane traffic in cells and localize to specific subcellular compartments (35). Our previous work showed that Rab5 and Rab7 were involved in CSFV entry into PK-15 cells; the role of Rabs in CSFV entry into 3D4/21 cells is not known. In this study, three independent experiments were performed to investigate the endocytic trail, followed by CSFV. First, we examined the roles of the small GTPases, Rab5, Rab7, Rab9, and Rab11, which are key regulators in vesicular trafficking to early, late, and recycling endosomes. Rab9 mediates late endosome to trans-Golgi network trafficking. The functionality of WT and DN Rab constructs in 3D4/21 cells was tested using labeled transferrin as control. Overexpression of all DN Rabs reduced the accumulation of transferrin compared to cells expressing WT Rabs. We observed only minor fluorescence intensity of transferrin in the cytoplasm and no superposition of these proteins (data not shown). Next, the effect of the blockade in Rab-mediated transport on CSFV productive infection was examined. Overexpression of Rab5 S34N, Rab7 T22N, or Rab11 S25N reduced infection by 75, 50, and 63%, respectively, whereas the expression of Rab9 Q66L exerted no inhibitory effect on CSFV infection (Fig. 8A and B). These results demonstrated the necessity of Rab5, Rab7, and Rab11 in CSFV entry. To identify the endosomal compartment traversed by endocytosed CSFV, we used Rab5, Rab7, Rab9, Rab11, or Lamp siRNAs. The silencing efficiency of siRab5, siRab7 siRab9, siRab11, and siLamp-1 was analyzed by Western blotting. The results showed that these proteins were knocked down efficiently compared to siCtrl-transfected cells and that the reduced levels of Rab5, Rab7, Rab11, and Lamp-1 resulted in decreased CSFV (E2) protein (Fig. 8C). RT-qPCR and virus titration both showed that depletion of Rab5, Rab7, Rab11, and Lamp-1 resulted in reductions in viral RNA copies by 69, 29, 68, and 58% and in virus titers by 9.8-, 3.1-, 9.1-, and 3-fold, respectively. Western blotting, RT-qPCR, and virus titration all showed that Rab9 had no effect on CSFV infection (Fig. 8D).
FIG 8.

Effects of Rabs and Lamp-1 on CSFV infection. (A) The effect of DN Rabs on CSFV infection was determined by confocal microscopy. Cells transfected with plasmids expressing GFP-tagged WT or DN Rab5, Rab7, Rab9, or Rab11 were infected with CSFV (MOI = 0.5). At 24 hpi, the cells were fixed with 4% PFA, reacted with WH303, and visualized by confocal microscopy. Scale bar, 50 μm. (B) At least 300 transfected cells were scored as “+” or “−” for CSFV infection. Values are expressed as a percentage of the infected cells observed in the control sample. The results are presented as means ± the SD of data from three independent experiments. (C) Western blotting was used to determine the knockdown efficiency of the siRabs and siLamp-1 and their effects on the CSFV E2 protein level. Cells were transfected with either siCtrl, siRab5, siRab7, siRab9, siRab11, or siLamp-1 for 48 h and infected with CSFV (MOI = 0.1) for 24 h. The relative protein amount was calculated using ImageJ 7.0 software as shown in the figure. (D) Depletion of Rab5, Rab7, Rab11, and Lamp-1 reduced CSFV propagation. Cells transfected with the indicated siRNA were infected with CSFV (MOI = 0.1) for 24 h. Viral RNA levels and virus titers were determined by RT-qPCR and virus titration, respectively. The results are presented as means ± the SD of three independent experiments (**, P < 0.01).
CSFV and Rab colocalization during virus entry was examined by confocal microscopy. As shown in Fig. 9A, virions were seen in endosomes that were fused with Rab5, Rab7, Rab11, and Lamp-1, but not Rab9, after 30 min of internalization. The colocalization coefficients were expressed as the Pearson's correlation coefficient, measured for individual cells. The Pearson's correlation coefficient of Rab5, Rab7, Rab11, and Lamp-1 is more significant than that of Rab9 (Fig. 9B). Our results demonstrate that Rab5, Rab7, Rab11, and Lamp-1 are required for CSFV endocytosis and subsequent productive infection and indicate that CSFV fusion occurs from early endosomes to late and recycling endosomes and further into lysosomes before releasing viral RNA.
FIG 9.

Colocalization of Rabs or Lamp-1 with CSFV during the early stage of infection. (A) 3D4/21 cells grown on dishes were infected with CSFV (MOI = 10) at 4°C for 1 h and then shifted to 37°C. At 30 min, monolayers were fixed with 4% PFA, stained with WH303, rabbit anti-Rabs, or anti-Lamp-1, and then examined by confocal microscopy. Scale bar, 20 μm. The white arrowheads show the colocation of CSFV and Rab proteins. (B) The colocalization coefficients were expressed as a Pearson's correlation coefficient, measured for individual cells. The results are presented as means ± the SD of data from three independent experiments (**, P < 0.01).
DISCUSSION
The early steps of the virus life cycle, binding and entry into the host cell, are crucial determinants of infection and are potential targets for the development of antiviral therapies. A previous study showed that JEV, a Flavivirus, infects multiple cell types and utilizes multiple endocytic pathways for entry into host cells (13). Therefore, we hypothesized that CSFV also used multiple endocytic pathways, allowing it to infect different cell types. Although CSFV infects 3D4/21 cells, the mechanism of infection is unknown. All known members of Flaviviridae depend on cellular membrane cholesterol for entry (36, 37). Previous studies have shown that in cells treated with MβCD prior to virus infection, viral production was significantly decreased, whereas in cells treated after virus infection, virus production remained at control levels (31, 38). Our results showed that CSFV production was limited when cells were treated with MβCD prior to infection and almost abolished when cholesterol was depleted from the viral envelope. These data suggest that lipid rafts, cholesterol-enriched lipid-ordered membrane domains, play an important role in CSFV entry into 3D4/21 cells.
Rab GTPases control membrane identity, vesicle budding, uncoating, motility, and fusion through the recruitment of effector proteins, such as sorting adaptors, tethering factors, kinases, phosphatases, and motors, and serve as organizers of almost all membrane trafficking processes in eukaryotic cells. A large group of viruses enter cells through various endocytic pathways, and some viruses hijack Rab-dependent pathways to induce virus internalization. Of these Rabs, Rab5 and Rab7 GTPases are the key regulators of transport to early and late endosomes (20), and ablation of Rab5 and Rab7 has been used to study viral entry in dengue virus and West Nile virus. A previous study from our lab has shown that CSFV requires Rab5 and Rab7 after endocytosis in PK-15 cells (20). Here, we explored the postinternalization of CSFV in 3D4/21 cells. By overexpressing DN Rab proteins and using RNAi technology, the intracellular trafficking of CSFV particles following endocytosis was monitored. The data showed that Rab5, Rab7, and Rab11 are important for proper CSFV trafficking, while Rab9 is not. Colocalization of virions with Rab5, Rab7, or Rab11 support the conclusion that these Rabs are involved in CSFV endocytosis. Although Lamp-1 is typically associated with lysosomes (39), it has also been reported to be associated with late endosomes and prelysosomes (40). Previous studies have shown that the endolysosomal pathway is composed of at least three distinct populations of vesicles: Rab7-, Lamp-1-, and Rab7/Lamp-1-vesicles (41). The majority of endolysosomal vesicles are positive for both Rab7 and Lamp-1. Our previous work identified that Rab7 is involved in CSFV infection, but the importance of Lamp-1 had not been addressed. In this study, knockdown of Lamp-1 caused a reduction in CSFV infection, suggesting the endolysosomal pathway is involved in CSFV endocytosis. We hypothesized that CSFV particles are first transported to early endosomes in a Rab5-dependent manner, that a Rab7-dependent pathway guides CSFV to late endosomes, and that a Rab11-dependent pathway then guides CSFV to recycling endosomes. Finally, CSFV is transferred into lysosomes (Lamp-1), where viral RNA is released.
Our results demonstrate that CSFV enters 3D4/21 cells through caveola-dependent endocytosis and, after endocytosis, CSFV is transported from early to late and recycling endosomes but not to the trans-Golgi network before an acidic pH-dependent step, presumably resulting in the release of CSFV nucleocapsid into the cytoplasm, leading to successful infection.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
Porcine alveolar macrophages (3D4/21 cells) were maintained at 37°C in 5% CO2 in Dulbecco modified eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco/Invitrogen), 0.2% NaHCO3, 100 μg/ml streptomycin, and 100 IU/ml penicillin (Gibco). The highly virulent CSFV Shimen strain (GenBank accession number AF092448) used in this study was described previously (20). Total RNA was extracted using TRIzol reagent (Invitrogen, USA), and CSFV viral RNA was measured using RT-qPCR as described previously (42). Data are presented as 2−ΔΔCT values from quadruplicate samples (43). Virus titers were analyzed based on limited dilution by determination of the 50% tissue culture infective doses (TCID50) as described previously (42).
EPS15 is an adaptor for clathrin-mediated endocytosis. Plasmids expressing green fluorescent protein (GFP)-tagged WT EPS15 (DIIIΔ2) and dominant-negative (DN) EPS15 (EΔ95/295) were kindly provided by Sandrine Belouzard (Institut Pasteur de Lille, Lille, France) (44). The plasmids expressing GFP-tagged WT and DN (K44A) dynamin II and the plasmid constructs expressing GFP-tagged WT and DN caveolin-1 were provided by Gary R. Whittaker (Cornell University, USA). The plasmid constructs for WT and DN Rab5 (S34N), WT and DN Rab7 (T22N), and WT and DN Rab11 (S25N) were obtained from Collin R. Parrish (Cornell University, USA). The plasmid constructs for WT and DN Rab9 (Q66L) were from Kui Yang (Louisiana State University, USA).
Cell infection and drug treatments.
The drugs used in this study included chloroquine (Sigma-Aldrich, no. PHR1258), an endosomal acidification inhibitor; NH4Cl (Sigma-Aldrich, no. A9434); bafilomycin A1 (BafA1; Cayman, no. 11038), a potent inhibitor of V-ATPase and a specific inhibitor of acidification of endosomal vesicles (44); chlorpromazine (CPZ; Sigma-Aldrich, no. C0982), an inhibitor of clathrin lattice polymerization; dynasore (Abcam, no. ab120192), a noncompetitive dynamin GTPase inhibitor; methyl-β-cyclodextrin (MβCD; Sigma-Aldrich, no. C4767), which disrupts lipid rafts by depleting the cholesterol component; wortmannin (Cayman, no. 10010591), an inhibitor of phosphatidylinositol 3-kinase activation; and EIPA (Santa Cruz Biotechnology, sc-202458), an inhibitor of macropinocytosis that blocks Na/H exchange.
To test the effects of these drugs on CSFV infection, 3D4/21 cells were seeded in 12-well plates. After reaching 90% confluence, the cells were treated with the indicated concentrations of drug for 1 h at 37°C. For binding and entry experiments, cells pretreated with chloroquine, NH4Cl, or BafA1 were inoculated at an MOI of 1 in the presence of the drug at 4°C for 1 h and then shifted to 37°C. At 0 (binding) or 1 (entry) hpi, the cells were lysed by three freeze-thaw cycles, total RNA was extracted using TRIzol reagent, and viral RNA was measured using RT-qPCR as described previously (42). Data are presented as 2−ΔΔCT from quadruplicate samples. For the replication experiments, cells pretreated with the indicated drugs, were infected at an MOI of 0.1 in the presence of the drug at 4°C for 1 h. Cells were washed with phosphate-buffered saline (PBS) and incubated in maintenance media without the drug for 24 h at 37°C. The cells were lysed, the total RNA was extracted, and the viral RNA was measured as described above.
Binding and entry assay.
Cells were pretreated with subtoxic doses (Fig. 1) at 37°C for 1 h and inoculated with CSFV (MOI = 1) at 4°C for 1 h to allow virus attachment without internalization, and then the cells were washed with ice-cold PBS three times so that unbound viruses were removed. The culture medium was replaced with fresh serum-free DMEM, and the cells were subsequently shifted to 37°C with 5% CO2 to allow virus internalization. After 1 h, the cells were washed with citrate buffer solution (pH 3) to remove the noninternalized visions on the surface of cells, and then the cells were washed three times with ice-cold PBS.
Cell viability assay.
The cytotoxic effect of the drugs on 3D4/21 cells was evaluated using a CellTiter 96 AQueous One Solution cell proliferation assay kit (Promega) in accordance with the manufacturer's instructions. No cytotoxicity was observed in cells treated with the indicated concentrations of the drugs or siRNA duplexes (Fig. 1).
Cholesterol depletion.
We used three approaches to identify the role of cholesterol in CSFV infection. (i) The cell membrane was depleted of cholesterol with MβCD for 1 h prior to CSFV infection. (ii) The cell membrane was depleted of cholesterol with MβCD for 1 h postinfection. In both cases, the cells were washed twice with medium after the drug treatment. (iii) The viral membrane was depleted of cholesterol with MβCD. CSFV was treated with MβCD for 1 h prior to infection. The treated virus was centrifuged through a 20% sucrose cushion at 70,000 × g for 2 h to remove the MβCD, resuspended in medium containing 10% FBS, and filtered before being used to infect cells. At 24 hpi, infected cells were incubated with MβCD for 12 h and either lysed and analyzed by RT-qPCR or fixed for confocal fluorescence microscopy. Cells for microscopy were incubated with an anti-CSFV E2 monoclonal antibody (WH303) for 1 h at room temperature, washed three times with PBS, and incubated with Alexa Fluor 488-conjugated anti-mouse antibody for 30 min at room temperature.
Plasmids and siRNA transfections.
To measure the replication of CSFV in cells transfected with DN mutants, cells were grown to 70% confluence on coverslip dishes and transfected with 2.5 μg of the indicated plasmid using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. For RNA knockdown, 3D4/21 cells were seeded in 6-well plates at 2.5 × 105 cells/well before transfection with 100 nM siRNA using Lipofectamine 3000 according to the manufacturer's instructions. The siRNA duplexes used in study are as follows: clathrin heavy chain (siCHC, sc-35067), caveolin-1 (siCav, sc-29241), V-ATPase B1 (siV-ATPase, sc-36787), Dynamin (siDyn, sc-43736), Lamp-1 (siLamp-1, sc-29389), siRab5 (sc-36344), siRab7 (sc-29460), siRab9 (sc-44065), siRab11 (Invitrogen, no. 317644A10), and the negative control (siCtrl, sc-37007; Santa Cruz Biotechnology). At 48 h posttransfection, cells were infected with CSFV. At 24 hpi, CSFV replication was measured by RT-qPCR (MOI = 0.1) or confocal fluorescence microscopy (MOI = 0.5).
Confocal fluorescence microscopy.
3D4/21 cells grown on dishes were infected with CSFV at an MOI of 5 at 4°C for 1 h and shifted to 37°C. After incubation, the monolayers were fixed with 4% paraformaldehyde (PFA) in PBS and permeabilized with 0.1% Triton X-100. For visualization of clathrin and caveolin-1, cells were stained with a mouse anti-CSFV E2 antibody (WH303), rabbit anti-clathrin antibody (P1663), or rabbit anti-caveolin-1 antibody (D46G3). For visualization of the Rab and Lamp-1 proteins, cells were stained with WH303, rabbit anti-Rab5 (ab18211), rabbit anti-Rab7 (ab74906), rabbit anti-Rab9 (D52G8), or rabbit anti-Rab11 (ab3612). The colocalization coefficients were calculated using professional quantitative colocalization analysis software (Nikon A1; Nikon, Japan) included with a Nikon A1 confocal microscope and expressed as a Pearson's correlation coefficient (45).
Western blotting.
Cells were washed three times with PBS and lysed in cold lysis buffer (1% Triton X-100 and 1 mM phenylmethylsulfonyl fluoride in PBS) for 30 min. Lysates were clarified by centrifugation at 12,000 × g for 10 min. Proteins in the lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and then probed with the indicated antibodies. β-Actin was used as a loading control. In order to determine levels of viral E2 proteins, the corresponding protein/actin quantity is used to calculate the grayscale using ImageJ 7.0 software.
Quantification of transferrin and the B subunit of cholera toxin uptake.
Alexa 568-labeled human transferrin (Tfn) or the B subunit of CTB (Molecular Probes) was used in uptake assays as described previously (19). Briefly, 3D4/21 cells were seeded in 6-well plates and transfected with WT or DN plasmid constructs or siRNAs or treated with the indicated drugs. Cells were incubated with Tfn- or CTB-AF568 at 20 μg/ml for 1 h at 4°C and then washed and transferred to 37°C for 30 min. The cells were visualized by confocal fluorescence microscopy. Uptake is represented as the mean and the standard error of the mean of the integrated fluorescence intensity from two independent experiments.
Statistical analysis.
All data are presented as means ± the standard deviations (SD). A Student t test was used to compare the data from pairs of treated and untreated groups. Statistical significance is indicated by asterisks (*, P < 0.05; **, P < 0.01) in the figures. All statistical analyses and calculations were performed using Prism 5 (GraphPad Software, Inc., La Jolla, CA).
ACKNOWLEDGMENTS
This study was supported by the State Key Laboratory of the Veterinary Biotechnology Foundation (SKLVBF201608) and by a grant from the National Natural Science Foundation of China (31572554).
We thank Elizabeth Wills, Cornell University, for critically reading and editing the manuscript.
REFERENCES
- 1.Meyers G, Thiel HJ, Rumenapf T. 1996. Classical swine fever virus: recovery of infectious viruses from cDNA constructs and generation of recombinant cytopathogenic defective interfering particles. J Virol 70:1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paton DJ, McGoldrick A, Greiser-Wilke I, Parchariyanon S, Song JY, Liou PP, Stadejek T, Lowings JP, Bjorklund H, Belak S. 2000. Genetic typing of classical swine fever virus. Vet Microbiol 73:137–157. doi: 10.1016/S0378-1135(00)00141-3. [DOI] [PubMed] [Google Scholar]
- 3.Passler T, Riddell KP, Edmondson MA, Chamorro MF, Neill JD, Brodersen BW, Walz HL, Galik PK, Zhang Y, Walz PH. 2014. Experimental infection of pregnant goats with bovine viral diarrhea virus (BVDV) 1 or 2. Vet Res 45:38. doi: 10.1186/1297-9716-45-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuta A, Wozniakowski G, Polak MP. 2015. Cross-priming amplification for detection of bovine viral diarrhoea virus species 1 and 2. J Appl Microbiol 119:632–639. doi: 10.1111/jam.12859. [DOI] [PubMed] [Google Scholar]
- 5.Mao L, Liu X, Li W, Yang L, Zhang W, Jiang J. 2015. Characterization of one sheep border disease virus in China. Virol J 12:15. doi: 10.1186/s12985-014-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tautz N, Tews BA, Meyers G. 2015. The molecular biology of pestiviruses. Adv Virus Res 93:47–160. doi: 10.1016/bs.aivir.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Schirrmeier H, Strebelow G, Depner K, Hoffmann B, Beer M. 2004. Genetic and antigenic characterization of an atypical pestivirus isolate, a putative member of a novel pestivirus species. J Gen Virol 85:3647–3652. doi: 10.1099/vir.0.80238-0. [DOI] [PubMed] [Google Scholar]
- 8.Zhou B, Liu K, Jiang Y, Wei JC, Chen PY. 2011. Multiple linear B-cell epitopes of classical swine fever virus glycoprotein E2 expressed in Escherichia coli as multiple epitope vaccine induces a protective immune response. Virol J 8:378. doi: 10.1186/1743-422X-8-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Mao L, Zhou B, Liu X, Yang L, Zhang W, Jiang J. 2015. The swine CD81 enhances E2-based DNA vaccination against classical swine fever. Vaccine 33:3542–3548. doi: 10.1016/j.vaccine.2015.05.055. [DOI] [PubMed] [Google Scholar]
- 10.Hulst MM, Moormann RJ. 1997. Inhibition of pestivirus infection in cell culture by envelope proteins Erns and E2 of classical swine fever virus: Erns and E2 interact with different receptors. J Gen Virol 78(Pt 11):2779–2787. doi: 10.1099/0022-1317-78-11-2779. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Nie Y, Wang P, Ding M, Deng H. 2004. Characterization of classical swine fever virus entry by using pseudotyped viruses: E1 and E2 are sufficient to mediate viral entry. Virology 330:332–341. doi: 10.1016/j.virol.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 12.Yamauchi Y, Helenius A. 2013. Virus entry at a glance. J Cell Sci 126:1289–1295. doi: 10.1242/jcs.119685. [DOI] [PubMed] [Google Scholar]
- 13.Nain M, Abdin MZ, Kalia M, Vrati S. 2016. Japanese encephalitis virus invasion of cell: allies and alleys. Rev Med Virol 26:129–141. doi: 10.1002/rmv.1868. [DOI] [PubMed] [Google Scholar]
- 14.Nawa M, Takasaki T, Yamada K, Kurane I, Akatsuka T. 2003. Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J Gen Virol 84:1737–1741. doi: 10.1099/vir.0.18883-0. [DOI] [PubMed] [Google Scholar]
- 15.Yang S, He M, Liu X, Li X, Fan B, Zhao S. 2013. Japanese encephalitis virus infects porcine kidney epithelial PK15 cells via clathrin- and cholesterol-dependent endocytosis. Virol J 10:258. doi: 10.1186/1743-422X-10-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuang CK, Yang TH, Chen TH, Yang CF, Chen WJ. 2015. Heat shock cognate protein 70 isoform D is required for clathrin-dependent endocytosis of Japanese encephalitis virus in C6/36 cells. J Gen Virol 96:793–803. doi: 10.1099/jgv.0.000015. [DOI] [PubMed] [Google Scholar]
- 17.Zhu YZ, Xu QQ, Wu DG, Ren H, Zhao P, Lao WG, Wang Y, Tao QY, Qian XJ, Wei YH, Cao MM, Qi ZT. 2012. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J Virol 86:13407–13422. doi: 10.1128/JVI.00903-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalia M, Khasa R, Sharma M, Nain M, Vrati S. 2013. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol 87:148–162. doi: 10.1128/JVI.01399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuda M, Suzuki R, Kataoka C, Watashi K, Aizaki H, Kato N, Matsuura Y, Suzuki T, Wakita T. 2014. Alternative endocytosis pathway for productive entry of hepatitis C virus. J Gen Virol 95:2658–2667. doi: 10.1099/vir.0.068528-0. [DOI] [PubMed] [Google Scholar]
- 20.Shi BJ, Liu CC, Zhou J, Wang SQ, Gao ZC, Zhang XM, Zhou B, Chen PY. 2016. Entry of classical swine fever virus into PK-15 cells via a pH-, dynamin-, and cholesterol-dependent, clathrin-mediated endocytic pathway that requires Rab5 and Rab7. J Virol 90:9194–9208. doi: 10.1128/JVI.00688-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ning P, Gao L, Zhou Y, Hu C, Lin Z, Gong C, Guo K, Zhang X. 2016. Caveolin-1-mediated endocytic pathway is involved in classical swine fever virus Shimen infection of porcine alveolar macrophages. Vet Microbiol 195:81–86. doi: 10.1016/j.vetmic.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Jordens I, Marsman M, Kuijl C, Neefjes J. 2005. Rab proteins, connecting transport and vesicle fusion. Traffic 6:1070–1077. doi: 10.1111/j.1600-0854.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 23.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 24.Mendoza P, Diaz J, Torres VA. 2014. On the role of Rab5 in cell migration. Curr Mol Med 14:235–245. doi: 10.2174/1566524014666140128111347. [DOI] [PubMed] [Google Scholar]
- 25.Vonderheit A, Helenius A. 2005. Rab7 associates with early endosomes to mediate sorting and transport of Semliki Forest virus to late endosomes. PLoS Biol 3:e233. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murray JL, Mavrakis M, McDonald NJ, Yilla M, Sheng J, Bellini WJ, Zhao L, Le Doux JM, Shaw MW, Luo CC, Lippincott-Schwartz J, Sanchez A, Rubin DH, Hodge TW. 2005. Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J Virol 79:11742–11751. doi: 10.1128/JVI.79.18.11742-11751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilcke M, Johannes L, Galli T, Mayau V, Goud B, Salamero J. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J Cell Biol 151:1207–1220. doi: 10.1083/jcb.151.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salloum S, Wang H, Ferguson C, Parton RG, Tai AW. 2013. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog 9:e1003513. doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen TC, Hsieh CH, Sarnow P. 2015. Supporting role for GTPase Rab27a in hepatitis C virus RNA replication through a novel miR-122-mediated effect. PLoS Pathog 11:e1005116. doi: 10.1371/journal.ppat.1005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. 2008. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog 4:e1000244. doi: 10.1371/journal.ppat.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu CC, Zhang YN, Li ZY, Hou JX, Zhou J, Kan L, Zhou B, Chen PY. 2017. Rab5 and Rab11 are required for clathrin-dependent endocytosis of Japanese encephalitis virus in BHK-21 cells. J Virol 91:e01113-. doi: 10.1128/JVI.01113-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krishnan MN, Sukumaran B, Pal U, Agaisse H, Murray JL, Hodge TW, Fikrig E. 2007. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J Virol 81:4881–4885. doi: 10.1128/JVI.02210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brogden G, Adamek M, Proepsting MJ, Ulrich R, Naim HY, Steinhagen D. 2015. Cholesterol-rich lipid rafts play an important role in the Cyprinid herpesvirus 3 replication cycle. Vet Microbiol 179:204–212. doi: 10.1016/j.vetmic.2015.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan L, Zhang Y, Zhan Y, Yuan Y, Sun Y, Qiu X, Meng C, Song C, Liao Y, Ding C. 2016. Newcastle disease virus employs macropinocytosis and Rab5a-dependent intracellular trafficking to infect DF-1 cells. Oncotarget 7:86117–86133. doi: 10.18632/oncotarget.10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stenmark H. 2009. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 36.Pan J, Lai CB, Scott WR, Straus SK. 2010. Synthetic fusion peptides of tick-borne encephalitis virus as models for membrane fusion. Biochemistry 49:287–296. doi: 10.1021/bi9017895. [DOI] [PubMed] [Google Scholar]
- 37.Olmstead AD, Knecht W, Lazarov I, Dixit SB, Jean F. 2012. Human subtilase SKI-1/S1P is a master regulator of the HCV Lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog 8:e1002468. doi: 10.1371/journal.ppat.1002468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu YZ, Cao MM, Wang WB, Wang W, Ren H, Zhao P, Qi ZT. 2012. Association of heat-shock protein 70 with lipid rafts is required for Japanese encephalitis virus infection in Huh7 cells. J Gen Virol 93:61–71. doi: 10.1099/vir.0.034637-0. [DOI] [PubMed] [Google Scholar]
- 39.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. 2007. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Humphries WHt, Szymanski CJ, Payne CK. 2011. Endo-lysosomal vesicles positive for Rab7 and LAMP1 are terminal vesicles for the transport of dextran. PLoS One 6:e26626. doi: 10.1371/journal.pone.0026626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng EL, Gan BQ, Ng F, Tang BL. 2012. Rab GTPases regulating receptor trafficking at the late endosome-lysosome membranes. Cell Biochem Funct 30:515–523. doi: 10.1002/cbf.2827. [DOI] [PubMed] [Google Scholar]
- 42.He DN, Zhang XM, Liu K, Pang R, Zhao J, Zhou B, Chen PY. 2014. In vitro inhibition of the replication of classical swine fever virus by porcine Mx1 protein. Antiviral Res 104:128–135. doi: 10.1016/j.antiviral.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Lecot S, Belouzard S, Dubuisson J, Rouille Y. 2005. Bovine viral diarrhea virus entry is dependent on clathrin-mediated endocytosis. J Virol 79:10826–10829. doi: 10.1128/JVI.79.16.10826-10829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou J, Chen J, Zhang XM, Gao ZC, Liu CC, Zhang YN, Hou JX, Li ZY, Kan L, Li WL, Zhou B. 2018. Porcine Mx1 protein inhibits classical swine fever virus replication by targeting nonstructural protein NS5B. J Virol 92:e02147-17. doi: 10.1128/JVI.02147-17. [DOI] [PMC free article] [PubMed] [Google Scholar]