

Abstract
Background
X-linked ichthyosis (XLI) is a recessive keratinization condition caused by deficient activity of steroid-sulfatase due to mutations in steroid sulfatase (STS) gene located on the X chromosome. In contrast, ichthyosis vulgaris (IV) is caused by filaggrin deficiency due to semi-dominant loss-of-function mutations of filaggrin (FLG) gene. Filaggrin defects could synergize with XLI to exacerbate its phenotype.
Case presentation
We report a Chinese family with patients presenting diverse phenotype of Keratosis pilaris. A next-generation sequencing panel interrogating 25 ichthyosis related genes with sequencing coverage of the coding regions and splice site junctions, was applied to screen genetic mutations. A gross deletion encompassing the STS gene ranging from exon 1–10 and the FLG c.3321delA mutation were identified in a 31-year old male proband, one of his sister, and his mother, and all the three patients showed obvious symptom. The deletion of STS gene was confirmed by real-time quantitative PCR. The proband’s another sister and his two nephews carried only FLG c.3321delA mutation. Patients carried both mutations presented more severe symptom, while those only carried FLG c.3321delA mutation showed slight or normal phenotype.
Conclusions
In conclusion, we found that the IV phenotype was exacerbated by co-inheritance of STS and FLG mutations in a Chinese family with ichthyosis. Other genomic regions no included in the study might be also involved in phenotypic modifications.
Keywords: STS, FLG, Ichthyosis, Mutation, Co-inheritance
Background
Ichthyosis vulgaris (IV, OMIM #146700) is the most frequent genetic disorder of ichthyosis caused by filaggrin deficiency due to semi-dominant loss-of-function mutations of filaggrin (FLG) gene, which affects around 1 in 250 people [1]. IV is characterized by palmar and plantar hyperlinearity, keratosis pilaris, hyperkeratosis, xerosis, and excess scaling. The phenotype is most pronounced in winter or dry climates [2]. The onset of IV could occur at an early age, and become apparent between 3 months and 5 years of age in patients with positive family history [3]. IV is inherited as a semi-dominant manner with variable penetrance [4].
X-linked ichthyosis (XLI, OMIM#308100) is one of the second most prevalent type of ichthyosis caused by steroid sulfatase (STS) deficiency due to mutation of STS gene located on the X chromosome, affecting approximately 1:2000 to 1:6000 males worldwide. Female carriers with few exceptions do not manifest XLI [5]. XLI is clinically characterized by general scaling of the skin in which the scalp, ears, neck, trunk, and limbs are affected. It consists of mild-to-moderate polygonal, dark brown, adherent and regular scales, which is prominent on the lateral aspects of the trunk and the lower limbs. Extracutaneous signs such as corneal opacities, cryptorchidism, attention deficit hyperactivity, male baldness pattern are frequent, especially corneal opacities and cryptorchidism [6].
Clinically it may be difficult to distinguish XLI from IV [4]. Moreover, as a modifying factors, FLG mutations could exacerbate XLI phenotype, and increased prevalence of filaggrin deficiency has been observed in XLI patients [7–9]. Recently, next-generation sequencing (NGS) has been widely applied as a rapid genetic diagnosis to identify novel mutations in patients with ichthyosis [10, 11].
In the current study, we described a male proband affected XLI and IV, and female IV patients carrying heterozygous STS mutation simultaneously. Moreover, both male and female patients suffered IV were found in this family. The male proband presented the most severe ichthyosis phenotype. Female IV patients carrying heterozygous STS mutation showed more severe phenotype than IV patients without STS mutation. This study confirmed the synergic effect between XLI and IV, and female XLI carriers could also exacerbate their IV phenotype.
Case presentation
Patients
The 31-year old male proband presented with symmetrical scaling when he was young, which was more prominent on the extensor surfaces of the limbs, along with dark brown, tightly adherent polygonal scales (Fig. 1). The soles and palms were unaffected. The proband is the fourth child, and his mother had a similar but less severe phenotype. His father was unaffected. Two of the elder sister had similar phenotype with their mother, and one of them had a 4-year old boy without phenotype. Another elder sister presented slight scaling, whose 12-year old boy presented slight phenotype. In the extended family, 4 affected females had a slight phenotype. The family tree was drawn (Fig. 2).
Fig. 1.
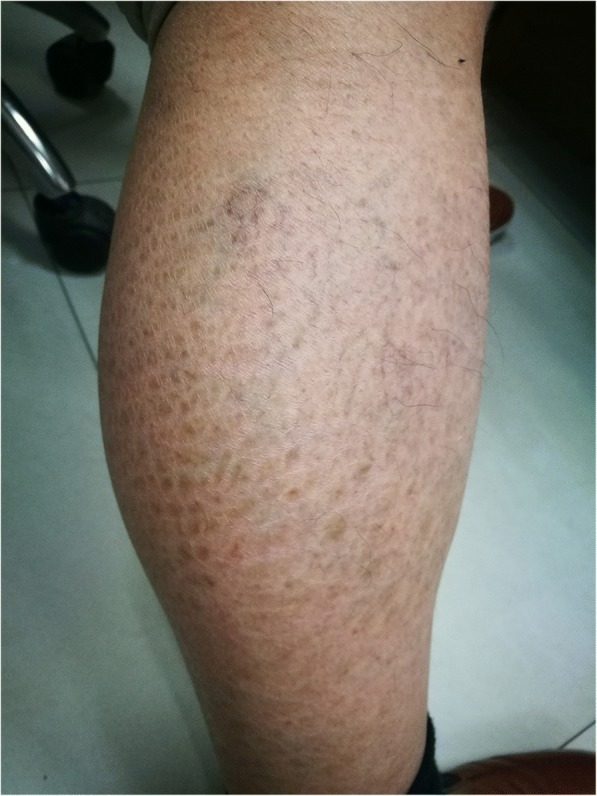
Clinical appearance of the proband co-inherited with XLI and IV. The proband had obvious, widespread and hyperpigmented ichthyosis in extensor surfaces of lower limbs even after treatment
Fig. 2.

The Chinese XLI/IV coexisting pedigree. Squares represent men, circles represent women, and arrowheads indicate the proband. Symbols in dark represent patients. I, II, III, IV represent four generations
Mutation analysis
To investigate the genetic defects for patients with ichthyosis, a panel of 25 genes (ABCA12, ALOX12B, ALOXE3, CLDN1, COL17A1, COL7A1, CYP4F22, FLG, ITGA6, ITGB4, KRT14, KRT5, LAMA3, LAMB3, LAMC2, MBTPS2, NIPAL4, PLEC, PNPLA1, SLC27A4, SNAP29, SPINK5, ST14, STS, and TGM1) underlying the most common genetic defects for ichthyosis was detected by NGS (BGI-Wuhan). Briefly, all exons with the adjacent 10 bp introns of the 25 genes covering 100,596 bp in length listed above were captured by using a microarray chip, and were further sequenced with Illumina HiSeq2000. Base calling was performed with the Illumina Pipeline. Mutations were identified using the BWA (Burrows Wheeler Aligner) software package against the hg19 human genome reference. The average sequencing depth for target region was 272.2 -fold, and the average coverage was 98.84%. 97.02% of the target region was sequenced for more than 30-fold. Mutation identified by NGS was validated by Sanger sequencing. The detection of exonic deletions using target capture and deep sequencing data was performed using the script for the detection of exonic deletions as previously described [12]. Deletion of STS gene was further validated by real-time quantitative PCR of genomic DNA isolated from peripheral blood.
STS deletion of exon 1–10 and FLG c.3321delA mutation
A total of 153 mutations were identified by NGS in the proband. After data processing and filtering referred to inherited model, minor allele frequency (MAF) in 1000G, ExAC, and gnomAD databases, splice effect, computer prediction and so on, hemizygous STS deletion of exon 1–10 (NM_000351) and heterozygous FLG NM_002016: c.3321delA (p.Ser1107SerfsTer15) frameshift mutation were identified as pathogenic mutations in the proband. The deletion of STS gene was confirmed by real-time quantitative PCR using a healthy female and a male subject as control. FLG c.3321delA mutation was confirmed by Sanger sequencing.
Several family members were included. STS deletion of exon 1–10 and FLG c.3321delA mutation were validated in these included family members (Figs 3 and 4).
Fig. 3.
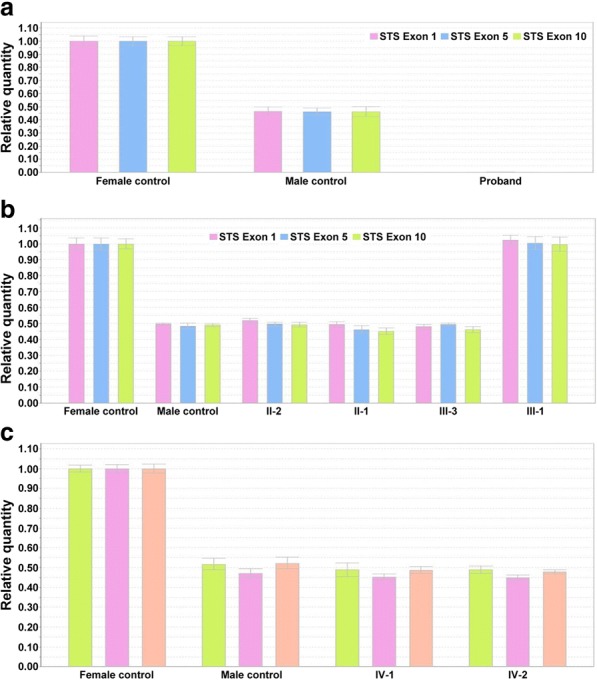
Expression of STS gene in a Chinese family with ichthyosis. The expression of STS was detected by real-time quantitative PCR. Primers were designed to amplify exon 1, 5, and 10 of STS gene. a Proband, b Proband’s parents and sisters, c Proband’s nephew
Fig. 4.
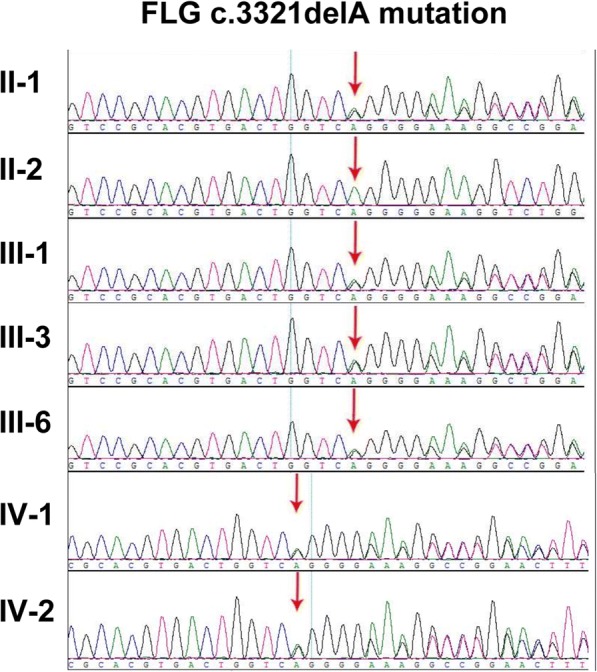
FLG c.3321delA mutation in a Chinese family with ichthyosis. Sanger sequencing was performed for this family members
Genotype and phenotype correlation
In this family, the male proband presented the most severe scaling (Table 1). He carried hemizygous STS deletion of exon 1–10 and heterozygous FLG c.3321delA mutation. His mother and one elder sister showed obvious but less sever symptom than the proband harbored heterozygous STS deletion of exon 1–10 and heterozygous FLG c.3321delA mutation. Another elder sister and 12-year old nephew showed slight phenotype carried only heterozygous FLG c.3321delA mutation. Another 4-year old nephew carried only heterozygous FLG c.3321delA mutation had no clinical symptom yet. The proband’s father was unaffected and neither mutation was detected.
Table 1.
STS and FLG mutation in a Chinese family with ichthyosis
| No. | Relation | Gender | Age | Symptom | FLG c.3321delA | STS Exon 1–10 Del |
|---|---|---|---|---|---|---|
| II-1 | Mother | Female | 70 | Obvious | Heterozygous | Heterozygous |
| II-2 | Father | Male | 69 | Normal | Wild type | Wild type |
| III-1 | First sister | Female | 40 | Slight | Heterozygous | Wild type |
| III-3 | Second sister | Female | 37 | Obvious | Heterozygous | Heterozygous |
| III-5 | Third sister | Female | 33 | Obvious | Not available | Not available |
| III-6 | Proband | Male | 31 | Obvious | Heterozygous | Hemizygous |
| IV-1 | Son of III-1 | Male | 12 | Slight | Heterozygous | Wild type |
| IV-4 | Son of III-3 | Male | 4 | Normal | Heterozygous | Wild type |
Discussion and conclusions
STS deficiency results in accumulation of cholesterol sulfate in the outer layers of the skin, inducing intercellular cohesion and scaling in XLI [13]. Up to 90% of the XLI patients presented complete deletion of the entire STS gene, and deletions could even extend to neighboring genes sometimes, leading to continuous gene syndromes [6, 14]. Less frequent point mutations have been reported as well [15, 16]. In this study, STS deletion of exon 1–10 has been identified by NGS, and further confirmed by real-time PCR via amplifying exon 1, 5, 10 in genomic DNA, which were widely used to detect STS deletion of exon 1–10 [6, 17]. The proband was hemizygote, one of his elder sister and their mother were heterozygotes. They all showed entire deletion of the STS gene, consistent with previous studies.
Genetic linkage analyses on IV patients mapped the FLG gene to the epidermal differentiation complex on chromosome 1q21 [18]. Loss-of-function mutations of the FLG gene have been identified to underlie IV, which is inherited in a semi-dominant model with 83–96% penetrance where heterozygotes have mild sub-clinical phenotype compared with homozygotes who with more prominent ichthyosis [1]. FLG mutations tend to be population specific. S2554X and 3321delA mutations were prevalent mutations in Asians, including Japanese and Chinese IV patients [19]. In this study, heterozygous FLG 3321delA mutation was observed in several patients. Some patients only carried heterozygous FLG 3321delA mutation, and some patients harbored both FLG 3321delA mutation and STS deletion.
Accumulating evidence indicates that FLG mutations may act as modifying factors of STS mutation, which could exacerbate XLI phenotype. Süßmuth K et al. found that the prevalence of FLG mutations was significantly increased in XLI patients compared to a population-based control cohort [8]. Liao H et al. suggested that different pathways disrupting epidermal differentiation may increase phenotypic severity [20]. Zhang Q et al. reported that filaggrin defects may synergize with deficiency of STS to exacerbate the XLI phenotype [9]. Ramesh R et al. also demonstrated the modifying function of FLG null alleles on XLI [7].
In this family, the male patients carried both heterozygous FLG 3321delA mutation and hemizygous STS deletion was the most severe affected, indicating the synergic effect of FLG and STS mutation.
Moreover, among the female patients, those carried two mutations were more severe affected than those harboring only heterozygous FLG 3321delA mutation, although previous studies reported that female carriers of STS mutation were seldom affected. The second elder sister who was 37-year old and carried both mutations presented more sever scaling than the first elder sister who was 40-year old and only harbored FLG mutation. These results suggest that female XLI carriers could also exacerbate their IV phenotype, and the effect of age was excluded.
In conclusion, we found that the IV phenotype was exacerbated by co-inheritance of STS and FLG mutations in a Chinese family with ichthyosis. Other genomic regions no included in the study might be also involved in phenotypic modifications.
Acknowledgements
We thank Ms. Li Wang (BGI-Wuhan) for the help in the analysis of sequencing results.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81500925).
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- 1000G
1000 Genomes Project
- BWA
Burrows Wheeler Aligner
- ExAC
The Exome Aggregation Consortium
- FLG
filaggrin
- gnomAD
The Genome Aggregation Database
- IV
Ichthyosis vulgaris
- MAF
Minor allele frequency
- NGS
Next-generation sequencing
- STS
Steroid sulfatase
- XLI
X-linked ichthyosis
Authors’ contributions
XW and LT conceived the experiments. NS and YL collected clinical information. YZ provided patient samples. XW wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved guidelines by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. Written informed consent were obtained from all family members.
Consent for publication
Written informed consent for publication of medical data and genetic data were obtained from all family members.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Xiong Wang and Lu Tan contributed equally to this work.
References
- 1.Thyssen JP, Godoy-Gijon E, Elias PM. Ichthyosis vulgaris: the filaggrin mutation disease. Br J Dermatol. 2013;168(6):1155–1166. doi: 10.1111/bjd.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeichi T, Akiyama M. Inherited ichthyosis: non-syndromic forms. J Dermatol. 2016;43(3):242–251. doi: 10.1111/1346-8138.13243. [DOI] [PubMed] [Google Scholar]
- 3.Okulicz JF, Schwartz RA. Hereditary and acquired ichthyosis vulgaris. Int J Dermatol. 2003;42(2):95–98. doi: 10.1046/j.1365-4362.2003.01308.x. [DOI] [PubMed] [Google Scholar]
- 4.Smith FJ, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, Liao H, Evans AT, Goudie DR, Lewis-Jones S, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–342. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
- 5.Wu B, Paller AS: Ichthyosis, X-linked. In: StatPearls. Treasure Island (FL); 2017.
- 6.Canueto J, Ciria S, Hernandez-Martin A, Unamuno P, Gonzalez-Sarmiento R. Analysis of the STS gene in 40 patients with recessive X-linked ichthyosis: a high frequency of partial deletions in a Spanish population. J Eur Acad Dermatol Venereol. 2010;24(10):1226–1229. doi: 10.1111/j.1468-3083.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 7.Ramesh R, Chen H, Kukula A, Wakeling EL, Rustin MH, McLean WH. Exacerbation of X-linked ichthyosis phenotype in a female by inheritance of filaggrin and steroid sulfatase mutations. J Dermatol Sci. 2011;64(3):159–162. doi: 10.1016/j.jdermsci.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Sussmuth K, Gruber R, Rodriguez E, Traupe H, Amler S, Sanchez-Guijo A, Valentin F, Tarinski T, Straub N, Metze D, et al. Increased prevalence of Filaggrin deficiency in 51 patients with recessive X-linked ichthyosis presenting for dermatological examination. J Investigative Dermatol. 2018;138(3):709–11. [DOI] [PubMed]
- 9.Zhang Q, Si N, Liu Y, Zhang D, Wang R, Zhang Y, Wang S, Liu X, Deng X, Ma Y, et al. Steroid sulfatase and filaggrin mutations in a boy with severe ichthyosis, elevated serum IgE level and moyamoya syndrome. Gene. 2017;628:103–108. doi: 10.1016/j.gene.2017.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Kong X, Wei X, Sun Y, Yin D, Zhang Q, Du L, Man J, Mao L, Li H, et al. Targeted next-generation sequencing identifies nine novel filaggrin gene variants in Chinese Han patients with ichthyosis vulgaris. Br J Dermatol. 2017;177(5):e202–e203. [DOI] [PubMed]
- 11.Youssefian L, Vahidnezhad H, Saeidian AH, Sotoudeh S, Mahmoudi H, Daneshpazhooh M, Aghazadeh N, Adams R, Ghanadan A, Zeinali S, et al. Autosomal recessive congenital ichthyosis: CERS3 mutations identified by a next generation sequencing panel targeting ichthyosis genes. Eur J Human Genet. 2017;25(11):1282–1285. doi: 10.1038/ejhg.2017.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, Chen D, Wang GL, Zhang VW, Wong LJ. Improved molecular diagnosis by the detection of exonic deletions with target gene capture and deep sequencing. Genet Med. 2015;17(2):99–107. doi: 10.1038/gim.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alperin ES, Shapiro LJ. Characterization of point mutations in patients with X-linked ichthyosis. Effects on the structure and function of the steroid sulfatase protein. J Biol Chem. 1997;272(33):20756–20763. doi: 10.1074/jbc.272.33.20756. [DOI] [PubMed] [Google Scholar]
- 14.Nagai K, Shima H, Kamimura M, Kanno J, Suzuki E, Ishiguro A, Narumi S, Kure S, Fujiwara I, Fukami M. Xp22.31 microdeletion due to microhomology-mediated break-induced replication in a boy with contiguous gene deletion syndrome. Cytogenet Genome Res. 2017;151(1):1–4. doi: 10.1159/000458469. [DOI] [PubMed] [Google Scholar]
- 15.Oyama N, Matsuda M, Hamada T, Numata S, Teye K, Hashimoto T, Hasegawa M. Two novel missense mutations of STS gene underlie X-linked recessive ichthyosis: understanding of the mutational and structural spectrum. J Eur Acad Dermatol Venereol. 2016;30(9):1629–1631. doi: 10.1111/jdv.13231. [DOI] [PubMed] [Google Scholar]
- 16.del Refugio Rivera Vega M, Murillo-Vilches MR, Toral-Lopez J, Sanchez EG, Sanchez AT, Gonzalez-Huerta LM, Cuevas-Covarrubias SA. X-linked ichthyosis in a patient with a novel nonsense mutation in the STS gene. J Dermatol Sci. 2015;80(2):160–162. doi: 10.1016/j.jdermsci.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Murtaza G, Siddiq S, Khan S, Hussain S, Naeem M. Molecular study of X-linked ichthyosis: report of a novel 2-bp insertion mutation in the STS and a very rare case of homozygous female patient. J Dermatol Sci. 2014;74(2):165–167. doi: 10.1016/j.jdermsci.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Zhong W, Cui B, Zhang Y, Jiang H, Wei S, Bu L, Zhao G, Hu L, Kong X. Linkage analysis suggests a locus of ichthyosis vulgaris on 1q22. J Hum Genet. 2003;48(7):390–392. doi: 10.1007/s10038-003-0043-1. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Guo Y, Wang W, Yu X, Yao Z. Associations of FLG mutations between ichthyosis vulgaris and atopic dermatitis in Han Chinese. Allergy. 2011;66(9):1253–1254. doi: 10.1111/j.1398-9995.2011.02597.x. [DOI] [PubMed] [Google Scholar]
- 20.Liao H, Waters AJ, Goudie DR, Aitken DA, Graham G, Smith FJ, Lewis-Jones S, McLean WH. Filaggrin mutations are genetic modifying factors exacerbating X-linked ichthyosis. J invest Dermatol. 2007;127(12):2795–2798. doi: 10.1038/sj.jid.5700971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.